
The Epigenetic Reader Methyl-CpG-Binding Protein 2 (MeCP2) Is an Emerging Oncogene in Cancer Biology



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Interplay between DNA Methylation and Cancer
MeCP2 and DNA Methylation
3. An Overview of the Genetics of Cancer: Tumor Suppressor Genes and Oncogenes
3.1. A Brief History
3.2. Tumor-Suppressor Genes and Oncogenes: Mechanism of Action
4. Connections between MeCP2 and Human Cancer
4.1. MeCP2 and Breast Cancer

4.2. MeCP2 and Colorectal Cancer
4.3. MeCP2 and Pancreatic Cancer
4.4. MeCP2 and Gastric Cancer

4.5. MeCP2 and Hepatocellular Carcinoma (HCC)

4.6. MeCP2 and Prostate Cancer
4.7. MeCP2 and Glioma
4.8. MeCP2 and Oral Squamous-Cell Carcinoma
4.9. MeCP2 and Renal Cell Cancer
4.10. MeCP2 and Lung Cancer
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Delcuve, G.P.; Rastegar, M.; Davie, J.R. Epigenetic control. J. Cell Physiol. 2009, 219, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Barber, B.A.; Rastegar, M. Epigenetic control of Hox genes during neurogenesis, development, and disease. Ann. Anat. 2010, 192, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Rastegar, M. Editorial (Thematic Issue: NeuroEpigenetics and Neurodevelopmental Disorders: From Molecular Mechanisms to Cell Fate Commitments of the Brain Cells and Human Disease). Curr. Top. Med. Chem. 2017, 17, 769–770. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Abotaleb, M.; Samuel, S.M.; Varghese, E.; Varghese, S.; Kubatka, P.; Liskova, A.; Büsselberg, D. Flavonoids in Cancer and Apoptosis. Cancers 2018, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- Canadian Cancer Society. Advisory in collaboration with the Canadian Cancer Society SC, Canada atPHAo. In Canadian Cancer Statistics: A 2022 Special Report on Cancer Prevalence; Canadian Cancer Society: Toronto, ON, Canada, 2022. [Google Scholar]
- Baxter, E.; Windloch, K.; Gannon, F.; Lee, J.S. Epigenetic regulation in cancer progression. Cell. Biosci. 2014, 4, 45. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef]
- Jiang, W.; Xia, T.; Liu, C.; Li, J.; Zhang, W.; Sun, C. Remodeling the Epigenetic Landscape of Cancer—Application Potential of Flavonoids in the Prevention and Treatment of Cancer. Front. Oncol. 2021, 11, 705903. [Google Scholar] [CrossRef]
- Yu, X.; Li, M.; Guo, C.; Wu, Y.; Zhao, L.; Shi, Q.; Song, J.; Song, B. Therapeutic Targeting of Cancer: Epigenetic Homeostasis. Front. Oncol. 2021, 11, 747022. [Google Scholar] [CrossRef]
- Bin Akhtar, G.; Buist, M.; Rastegar, M. MeCP2 and transcriptional control of eukaryotic gene expression. Eur. J. Cell Biol. 2022, 101, 151237. [Google Scholar] [CrossRef]
- Liyanage, V.R.B.; Jarmasz, J.S.; Murugeshan, N.; Del Bigio, M.R.; Rastegar, M.; Davie, J.R. DNA Modifications: Function and Applications in Normal and Disease States. Biology 2014, 3, 670–723. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA methylation landscape of cancer. Trends Genet. 2021, 37, 1012–1027. [Google Scholar] [CrossRef] [PubMed]
- Vuu, Y.M.; Roberts, C.T.; Rastegar, M. MeCP2 Is an Epigenetic Factor That Links DNA Methylation with Brain Metabolism. Int. J. Mol. Sci. 2023, 24, 4218. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, N.; Rabbani, S.A. DNA Methylation Readers and Cancer: Mechanistic and Therapeutic Applications. Front. Oncol. 2019, 9, 489. [Google Scholar] [CrossRef]
- McCabe, M.T.; Brandes, J.C.; Vertino, P.M. Cancer DNA methylation: Molecular mechanisms and clinical implications. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 3927–3937. [Google Scholar] [CrossRef]
- Herman, J.G.; Baylin, S.B. Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med. 2003, 349, 2042–2054. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Xu, X. DNA Methyltransferases, DNA Methylation, and Age-Associated Cognitive Function. Int. J. Mol. Sci. 2018, 19, 1315. [Google Scholar] [CrossRef]
- Hervouet, E.; Peixoto, P.; Delage-Mourroux, R.; Boyer-Guittaut, M.; Cartron, P.-F. Specific or not specific recruitment of DNMTs for DNA methylation, an epigenetic dilemma. Clin. Epigenetics 2018, 10, 17. [Google Scholar] [CrossRef]
- Nowacka-Zawisza, M.; Wiśnik, E. DNA methylation and histone modifications as epigenetic regulation in prostate cancer. Oncol. Rep. 2017, 38, 2587–2596. [Google Scholar] [CrossRef]
- Subramaniam, D.; Thombre, R.; Dhar, A.; Anant, S. DNA Methyltransferases: A Novel Target for Prevention and Therapy. Front. Oncol. 2014, 4, 80. [Google Scholar] [CrossRef]
- Bird, A.P. CpG-rich islands and the function of DNA methylation. Nature 1986, 321, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Santini, V.; Kantarjian, H.M.; Issa, J.P. Changes in DNA methylation in neoplasia: Pathophysiology and therapeutic implications. Ann. Intern. Med. 2001, 134, 573–586. [Google Scholar] [CrossRef]
- Ellis, J.; Hotta, A.; Rastegar, M. Retrovirus silencing by an epigenetic TRIM. Cell 2007, 131, 13–14. [Google Scholar] [CrossRef] [PubMed]
- Zachariah, R.M.; Rastegar, M. Linking epigenetics to human disease and Rett syndrome: The emerging novel and challenging concepts in MeCP2 research. Neural Plast. 2012, 2012, 415825. [Google Scholar] [CrossRef] [PubMed]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.; Qin, J.; Zoghbi, H.Y. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, V.R.; Zachariah, R.M.; Davie, J.R.; Rastegar, M. Ethanol deregulates Mecp2/MeCP2 in differentiating neural stem cells via interplay between 5-methylcytosine and 5-hydroxymethylcytosine at the Mecp2 regulatory elements. Exp. Neurol. 2015, 265, 102–117. [Google Scholar] [CrossRef]
- Liyanage, V.R.B.; Olson, C.O.; Zachariah, R.M.; Davie, J.R.; Rastegar, M. DNA Methylation Contributes to the Differential Expression Levels of Mecp2 in Male Mice Neurons and Astrocytes. Int. J. Mol. Sci. 2019, 20, 1845. [Google Scholar] [CrossRef]
- Olson, C.O.; Zachariah, R.M.; Ezeonwuka, C.D.; Liyanage, V.R.; Rastegar, M. Brain region-specific expression of MeCP2 isoforms correlates with DNA methylation within Mecp2 regulatory elements. PLoS ONE. 2014, 9, e90645. [Google Scholar] [CrossRef]
- Xu, W.; Liyanage, V.R.B.; MacAulay, A.; Levy, R.D.; Curtis, K.; Olson, C.O.; Zachariah, R.M.; Amiri, S.; Buist, M.; Hicks, G.G.; et al. Genome-Wide Transcriptome Landscape of Embryonic Brain-Derived Neural Stem Cells Exposed to Alcohol with Strain-Specific Cross-Examination in BL6 and CD1 Mice. Sci. Rep. 2019, 9, 206. [Google Scholar] [CrossRef]
- Liyanage, V.R.; Zachariah, R.M.; Rastegar, M. Decitabine alters the expression of Mecp2 isoforms via dynamic DNA methylation at the Mecp2 regulatory elements in neural stem cells. Mol. Autism. 2013, 4, 46. [Google Scholar] [CrossRef]
- Buist, M.; El Tobgy, N.; Shevkoplyas, D.; Genung, M.; Sher, A.A.; Pejhan, S.; Rastegar, M. Differential Sensitivity of the Protein Translation Initiation Machinery and mTOR Signaling to MECP2 Gain- and Loss-of-Function Involves MeCP2 Isoform-Specific Homeostasis in the Brain. Cells 2022, 11, 1442. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.O.; Pejhan, S.; Kroft, D.; Sheikholeslami, K.; Fuss, D.; Buist, M.; Ali Sher, A.; Del Bigio, M.R.; Sztainberg, Y.; Siu, V.M.; et al. MECP2 Mutation Interrupts Nucleolin-mTOR-P70S6K Signaling in Rett Syndrome Patients. Front. Genet. 2018, 9, 635. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, H.; Muffat, J.; Cheng, A.W.; Orlando, D.A.; Loven, J.; Kwok, S.M.; Feldman, D.A.; Bateup, H.S.; Gao, Q.; et al. Global transcriptional and translational repression in human-embryonic-stem-cell-derived Rett syndrome neurons. Cell Stem Cell 2013, 13, 446–458. [Google Scholar] [CrossRef]
- Tong, D.; Zhang, J.; Wang, X.; Li, Q.; Liu, L.Y.; Yang, J.; Guo, B.; Ni, L.; Zhao, L. MeCP2 facilitates breast cancer growth via promoting ubiquitination-mediated P53 degradation by inhibiting RPL5/RPL11 transcription. Oncogenesis 2020, 9, 56. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Pruitt, K. Functional assessment of MeCP2 in Rett syndrome and cancers of breast, colon, and prostate. Biochem. Cell Biol. 2017, 95, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Razin, A.; Shemer, R. DNA methylation in early development. Hum. Mol. Genet. 1995, 4, 1751–1755. [Google Scholar] [CrossRef]
- Olynik, B.M.; Rastegar, M. The genetic and epigenetic journey of embryonic stem cells into mature neural cells. Front. Genet. 2012, 3, 81. [Google Scholar] [CrossRef]
- Rastegar, M.; Yasui, D.H. Editorial: Epigenetic Mechanisms and Their Involvement in Rare Diseases. Front. Genet. 2021, 12, 755076. [Google Scholar] [CrossRef]
- Liyanage, V.R.; Curtis, K.; Zachariah, R.M.; Chudley, A.E.; Rastegar, M. Overview of the Genetic Basis and Epigenetic Mechanisms that Contribute to FASD Pathobiology. Curr. Top. Med. Chem. 2017, 17, 808–828. [Google Scholar] [CrossRef] [PubMed]
- Bestor, T.H.; Verdine, G.L. DNA methyltransferases. Curr. Opin. Cell Biol. 1994, 6, 380–389. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef]
- Sakai, T.; Toguchida, J.; Ohtani, N.; Yandell, D.W.; Rapaport, J.M.; Dryja, T.P. Allele-specific hypermethylation of the retinoblastoma tumor-suppressor gene. Am. J. Hum. Genet. 1991, 48, 880–888. [Google Scholar] [PubMed]
- Wajed, S.A.; Laird, P.W.; DeMeester, T.R. DNA methylation: An alternative pathway to cancer. Ann. Surg. 2001, 234, 10–20. [Google Scholar] [CrossRef]
- Hentze, J.L.; Høgdall, C.K.; Høgdall, E.V. Methylation and ovarian cancer: Can DNA methylation be of diagnostic use? Mol. Clin. Oncol. 2019, 10, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.; Frigola, J.; Vendrell, E.; Risques, R.A.; Fraga, M.F.; Morales, C.; Moreno, V.; Esteller, M.; Capellà, G.; Ribas, M.; et al. Chromosomal instability correlates with genome-wide DNA demethylation in human primary colorectal cancers. Cancer Res. 2006, 66, 8462–9468. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.D.; Meehan, R.R.; Henzel, W.J.; Maurer-Fogy, I.; Jeppesen, P.; Klein, F.; Bird, A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 1992, 69, 905–914. [Google Scholar] [CrossRef]
- Meehan, R.R.; Lewis, J.D.; Bird, A.P. Characterization of MeCP2, a vertebrate DNA binding protein with affinity for methylated DNA. Nucleic Acids Res. 1992, 20, 5085–5092. [Google Scholar] [CrossRef]
- Della Ragione, F.; Filosa, S.; Scalabrì, F.; D’Esposito, M. MeCP2 as a genome-wide modulator: The renewal of an old story. Front. Genet. 2012, 3, 181. [Google Scholar]
- Good, K.V.; Vincent, J.B.; Ausió, J. MeCP2: The Genetic Driver of Rett Syndrome Epigenetics. Front. Genet. 2021, 12, 620859. [Google Scholar] [CrossRef]
- Bianciardi, L.; Fichera, M.; Failla, P.; Di Marco, C.; Grozeva, D.; Mencarelli, M.A.; Spiga, O.; Mari, F.; Meloni, I.; Raymond, L.; et al. MECP2 missense mutations outside the canonical MBD and TRD domains in males with intellectual disability. J. Hum. Genet. 2016, 61, 95–101. [Google Scholar] [CrossRef]
- Shevkoplyas, D.; Vuu, Y.M.; Davie, J.R.; Rastegar, M. The Chromatin Structure at the MECP2 Gene and In Silico Prediction of Potential Coding and Non-Coding MECP2 Splice Variants. Int. J. Mol. Sci. 2022, 23, 15643. [Google Scholar] [CrossRef] [PubMed]
- Kriaucionis, S.; Bird, A. The major form of MeCP2 has a novel N-terminus generated by alternative splicing. Nucleic Acids Res. 2004, 32, 1818–1823. [Google Scholar] [CrossRef] [PubMed]
- Pejhan, S.; Del Bigio, M.R.; Rastegar, M. The MeCP2E1/E2-BDNF-miR132 Homeostasis Regulatory Network Is Region-Dependent in the Human Brain and Is Impaired in Rett Syndrome Patients. Front. Cell Dev. Biol. 2020, 8, 763. [Google Scholar] [CrossRef] [PubMed]
- Buist, M.; Fuss, D.; Rastegar, M. Transcriptional Regulation of MECP2E1-E2 Isoforms and BDNF by Metformin and Simvastatin through Analyzing Nascent RNA Synthesis in a Human Brain Cell Line. Biomolecules. 2021, 11, 1253. [Google Scholar] [CrossRef]
- Sheikholeslami, K.; Ali Sher, A.; Lockman, S.; Kroft, D.; Ganjibakhsh, M.; Nejati-Koshki, K.; Shojaei, S.; Ghavami, S.; Rastegar, M. Simvastatin Induces Apoptosis in Medulloblastoma Brain Tumor Cells via Mevalonate Cascade Prenylation Substrates. Cancers 2019, 11, 994. [Google Scholar] [CrossRef]
- Dastidar, S.G.; Bardai, F.H.; Ma, C.; Price, V.; Rawat, V.; Verma, P.; Narayanan, V.; D’Mello, S.R. Isoform-specific toxicity of Mecp2 in postmitotic neurons: Suppression of neurotoxicity by FoxG1. J. Neurosci. 2012, 32, 2846–2855. [Google Scholar] [CrossRef]
- Lipsick, J. A History of Cancer Research: Tumor Suppressor Genes. Cold Spring Harb. Perspect. Biol. 2020, 12, a035907. [Google Scholar] [CrossRef]
- Osborne, C.; Wilson, P.; Tripathy, D. Oncogenes and tumor suppressor genes in breast cancer: Potential diagnostic and therapeutic applications. Oncologist 2004, 9, 361–377. [Google Scholar] [CrossRef]
- Arizmendi-Izazaga, A.; Martinez-Baltazar, R.; Liborio-Bautista, A.; Olea-Flores, M.; Ortiz-Ortiz, J.; Navarro-Tito, N. The NRSF/REST transcription factor in hallmarks of cancer: From molecular mechanisms to clinical relevance. Biochimie 2023, 206, 116–134. [Google Scholar] [CrossRef]
- Talia, K.L.; Banet, N.; Buza, N. The role of HER2 as a therapeutic biomarker in gynaecological malignancy: Potential for use beyond uterine serous carcinoma. Pathology 2023, 55, 8–18. [Google Scholar] [CrossRef]
- Jang, H.; Seo, A.N.; Kim, M. Clinicopathological Characteristics of Advanced Epstein-Barr Virus-associated Gastric Cancer Highlighting Aberrant p53 Expression. Anticancer. Res. 2022, 42, 4955–4962. [Google Scholar] [CrossRef] [PubMed]
- Lhermitte, B.; Blandin, A.F.; Coca, A.; Guerin, E.; Durand, A.; Entz-Werle, N. Signaling pathway deregulation and molecular alterations across pediatric medulloblastomas. Neurochirurgie 2021, 67, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Vriend, J.; Rastegar, M. Ubiquitin ligases and medulloblastoma: Genetic markers of the four consensus subgroups identified through transcriptome datasets. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165839. [Google Scholar] [CrossRef] [PubMed]
- Boveri, T. Zur Frage der Entstehung Maligner Tumoren; Gustav Fischer: Jena, Germany, 1914. [Google Scholar]
- Nowell, P.C. The minute chromosome (Phl) in chronic granulocytic leukemia. Blut 1962, 8, 65–66. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA. 1971, 68, 820–823. [Google Scholar] [CrossRef]
- Cavenee, W.K.; Dryja, T.P.; Phillips, R.A.; Benedict, W.F.; Godbout, R.; Gallie, B.L.; Murphree, A.L.; Strong, L.C.; White, R.L. Expression of recessive alleles by chromosomal mechanisms in retinoblastoma. Nature 1983, 305, 779–784. [Google Scholar] [CrossRef]
- Friend, S.H.; Bernards, R.; Rogelj, S.; Weinberg, R.A.; Rapaport, J.M.; Albert, D.M.; Dryja, T.P. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 1986, 323, 643–646. [Google Scholar] [CrossRef]
- Stehelin, D.; Varmus, H.E.; Bishop, J.M. Vogt PKDNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian, D.N.A. Nature 1976, 260, 170–173. [Google Scholar] [CrossRef]
- Reddy, E.P.; Reynolds, R.K.; Santos, E.; Barbacid, M. A point mutation is responsible for the acquisition of transforming properties by the T24 human bladder carcinoma oncogene. Nature 1982, 300, 149–152. [Google Scholar] [CrossRef]
- Bell, D.W. Our changing view of the genomic landscape of cancer. J. Pathol. 2010, 220, 231–243. [Google Scholar] [CrossRef]
- Wu, C.H.; Gordon, J.; Rastegar, M.; Ogretmen, B.; Safa, A.R. Proteinase-3, a serine protease which mediates doxorubicin-induced apoptosis in the HL-60 leukemia cell line, is downregulated in its doxorubicin-resistant variant. Oncogene 2002, 21, 5160–5174. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; Rastegar, M.; Gordon, J.; Safa, A.R. beta(2)-microglobulin induces apoptosis in HL-60 human leukemia cell line and its multidrug resistant variants overexpressing MRP1 but lacking Bax or overexpressing P-glycoprotein. Oncogene 2001, 20, 7006–7020. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.; Wu, C.H.; Rastegar, M.; Safa, A.R. Beta2-microglobulin induces caspase-dependent apoptosis in the CCRF-HSB-2 human leukemia cell line independently of the caspase-3, -8 and -9 pathways but through increased reactive oxygen species. Int. J. Cancer 2003, 103, 316–327. [Google Scholar] [CrossRef]
- Karpel, H.; Slomovitz, B.; Coleman, R.L.; Pothuri, B. Biomarker-driven therapy in endometrial cancer. Int. J. Gynecol. Cancer 2023, 33, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. Principles of tumor suppression. Cell 2004, 116, 235–246. [Google Scholar] [CrossRef]
- Loboda, A.P.; Adonin, L.S.; Zvereva, S.D.; Guschin, D.Y.; Korneenko, T.V.; Telegina, A.V.; Kondratieva, O.K.; Frolova, S.E.; Pestov, N.B.; Barlev, N.A. BRCA Mutations-The Achilles Heel of Breast, Ovarian and Other Epithelial Cancers. Int. J. Mol. Sci. 2023, 24, 4982. [Google Scholar] [CrossRef]
- Steffen, C.L.; Kaya, P.; Schaffner-Reckinger, E.; Abankwa, D. Eliminating oncogenic RAS: Back to the future at the drawing board. Biochem. Soc. Trans. 2023, 51, 447–456. [Google Scholar] [CrossRef]
- Nambiar, M.; Kari, V.; Raghavan, S.C. Chromosomal translocations in cancer. Biochim. Biophys. Acta 2008, 1786, 139–152. [Google Scholar] [CrossRef]
- Storlazzi, C.T.; Lonoce, A.; Guastadisegni, M.C.; Trombetta, D.; D’Addabbo, P.; Daniele, G.; L’Abbate, A.; Macchia, G.; Surace, C.; Kok, K.; et al. Gene amplification as double minutes or homogeneously staining regions in solid tumors: Origin and structure. Genome Res. 2010, 20, 1198–1206. [Google Scholar] [CrossRef]
- Freudenberg, J.A.; Wang, Q.; Katsumata, M.; Drebin, J.; Nagatomo, I.; Greene, M.I. The role of HER2 in early breast cancer metastasis and the origins of resistance to HER2-targeted therapies. Exp. Mol. Pathol. 2009, 87, 1–11. [Google Scholar] [CrossRef]
- Bornkamm, G.W.; Polack, A.; Eick, D.; Berger, R.; Lenoir, G.M. [Chromosome translocations and Epstein-Barr virus in Burkitt’s lymphoma]. Onkologie. 1987, 10, 196–204. [Google Scholar]
- Howell, G.M.; Hodak, S.P.; Yip, L. RAS mutations in thyroid cancer. Oncologist 2013, 18, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Pejhan, S.; Rastegar, M. Role of DNA Methyl-CpG-Binding Protein MeCP2 in Rett Syndrome Pathobiology and Mechanism of Disease. Biomolecules 2021, 11, 75. [Google Scholar] [CrossRef]
- Rastegar, M.; Hotta, A.; Pasceri, P.; Makarem, M.; Cheung, A.Y.; Elliott, S.; Park, K.J.; Adachi, M.; Jones, F.S.; Clarke, I.D.; et al. MECP2 isoform-specific vectors with regulated expression for Rett syndrome gene therapy. PLoS ONE 2009, 4, e6810. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Ezeonwuka, C.D.; Rastegar, M. MeCP2-Related Diseases and Animal Models. Diseases 2014, 2, 45–70. [Google Scholar] [CrossRef]
- Yasui, D.H.; Gonzales, M.L.; Aflatooni, J.O.; Crary, F.K.; Hu, D.J.; Gavino, B.J.; Golub, M.S.; Vincent, J.B.; Carolyn Schanen, N.; Olson, C.O.; et al. Mice with an isoform-ablating Mecp2 exon 1 mutation recapitulate the neurologic deficits of Rett syndrome. Hum. Mol. Genet. 2014, 23, 2447–2458. [Google Scholar] [CrossRef]
- Neupane, M.; Clark, A.P.; Landini, S.; Birkbak, N.J.; Eklund, A.C.; Lim, E.; Culhane, A.C.; Barry, W.T.; Schumacher, S.E.; Beroukhim, R.; et al. MECP2 Is a Frequently Amplified Oncogene with a Novel Epigenetic Mechanism That Mimics the Role of Activated RAS in Malignancy. Cancer Discov. 2016, 6, 45–58. [Google Scholar] [CrossRef]
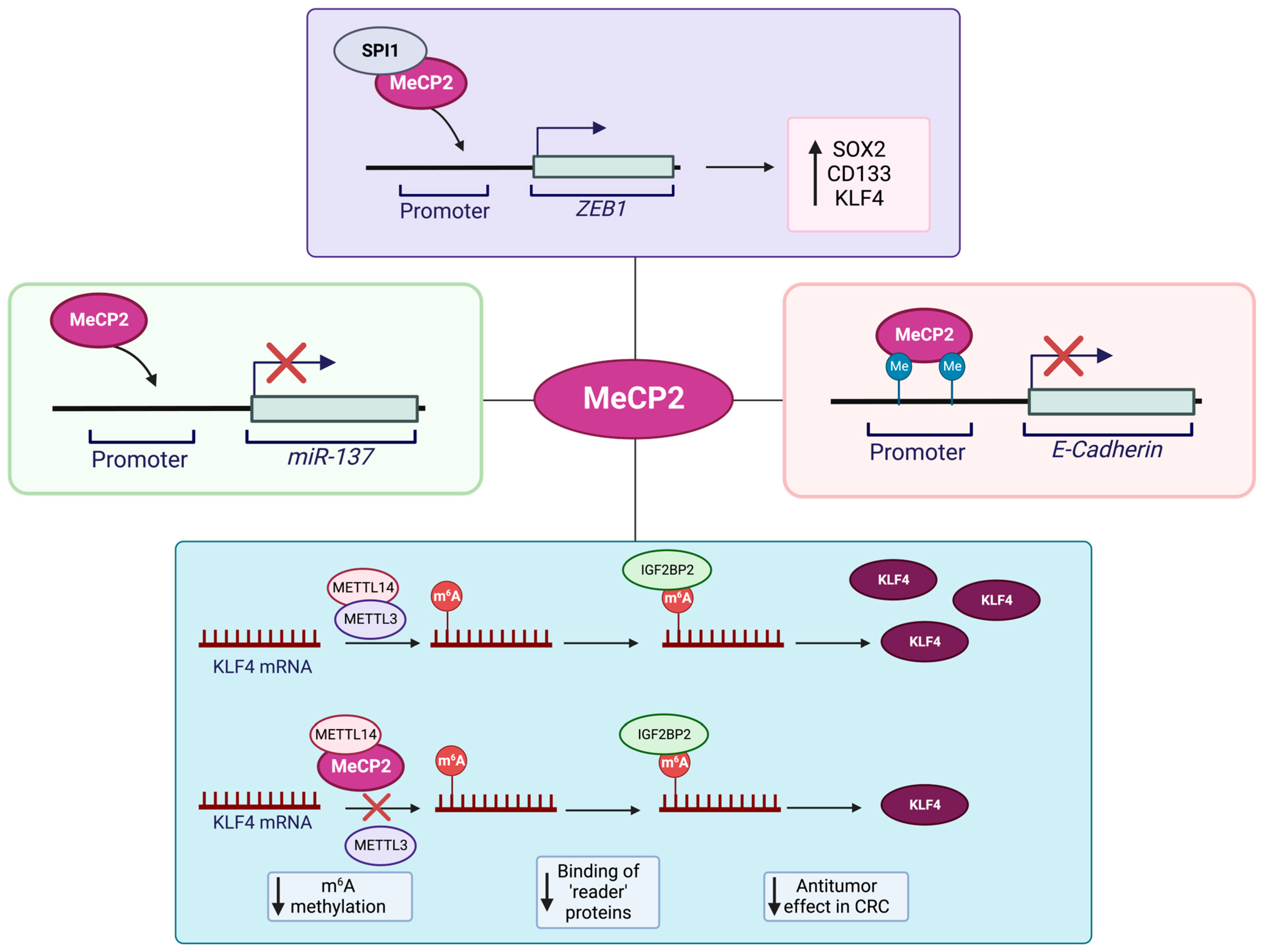
- Luo, D.; Ge, W. MeCP2 Promotes Colorectal Cancer Metastasis by Modulating ZEB1 Transcription. Cancers 2020, 12, 758. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, J.; Gao, N.; Wang, Y.; Chen, Y.; Han, J. MECP2 expression in gastric cancer and its correlation with clinical pathological parameters. Medicine 2017, 96, e7691. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, Y.; Tong, D.; Qin, Y.; Yang, J.; Xue, M.; Du, N.; Liu, L.; Guo, B.; Hou, N.; et al. MeCP2 Promotes Gastric Cancer Progression Through Regulating FOXF1/Wnt5a/β-Catenin and MYOD1/Caspase-3 Signaling Pathways. EBioMedicine 2017, 16, 87–100. [Google Scholar] [CrossRef]
- Fang, J.Y.; Cheng, Z.H.; Chen, Y.X.; Lu, R.; Yang, L.; Zhu, H.Y.; Lu, L.G. Expression of Dnmt1, demethylase, MeCP2 and methylation of tumor-related genes in human gastric cancer. World J. Gastroenterol. 2004, 10, 3394–3398. [Google Scholar] [CrossRef] [PubMed]
- Patra, S.K.; Patra, A.; Zhao, H.; Carroll, P.; Dahiya, R. Methyl-CpG-DNA binding proteins in human prostate cancer: Expression of CXXC sequence containing MBD1 and repression of MBD2 and MeCP2. Biochem. Biophys. Res. Commun. 2003, 302, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Müller, H.M.; Fiegl, H.; Goebel, G.; Hubalek, M.M.; Widschwendter, A.; Müller-Holzner, E.; Marth, C.; Widschwendter, M. MeCP2 and MBD2 expression in human neoplastic and non-neoplastic breast tissue and its association with oestrogen receptor status. Br. J. Cancer 2003, 89, 1934–1939. [Google Scholar] [CrossRef]
- Song, N.; Li, K.; Wang, Y.; Chen, Z.; Shi, L. Lentivirus-mediated knockdown of MeCP2 inhibits the growth of colorectal cancer cells in vitro. Mol. Med. Rep. 2016, 13, 860–866. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Wu, H.-J.; Chen, S.-Y.; Hou, M.-F.; Lin, C.-S.; Chu, P.-Y. Epigenetic therapy combination of UNC0638 and CI-994 suppresses breast cancer via epigenetic remodeling of BIRC5 and GADD45A. Biomed. Pharmacother. 2022, 145, 112431. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Jiang, Z.; Wang, M.; Ma, L. Current understandings and clinical translation of nanomedicines for breast cancer therapy. Adv. Drug Deliv. Rev. 2022, 180, 114034. [Google Scholar] [CrossRef]
- Castro-Piedras, I.; Vartak, D.; Sharma, M.; Pandey, S.; Casas, L.; Molehin, D.; Rasha, F.; Fokar, M.; Nichols, J.; Almodovar, S.; et al. Identification of Novel MeCP2 Cancer-Associated Target Genes and Post-Translational Modifications. Front. Oncol. 2020, 10, 576362. [Google Scholar] [CrossRef]
- Billard, L.-M.; Magdinier, F.; Lenoir, G.M.; Frappart, L.; Dante, R. MeCP2 and MBD2 expression during normal and pathological growth of the human mammary gland. Oncogene 2002, 21, 2704–2712. [Google Scholar] [CrossRef]
- Rasti, M.; Arabsolghar, R.; Khatooni, Z.; Mostafavi-Pour, Z. p53 Binds to estrogen receptor 1 promoter in human breast cancer cells. Pathol. Oncol. Res. 2012, 18, 169–175. [Google Scholar] [CrossRef]
- Sharma, D.; Blum, J.; Yang, X.; Beaulieu, N.; Macleod, A.R.; Davidson, N.E. Release of methyl CpG binding proteins and histone deacetylase 1 from the estrogen receptor α (ER) promoter upon reactivation in ER-negative human breast cancer cells. Mol. Endocrinol. 2005, 19, 1740–1751. [Google Scholar] [CrossRef]
- Jiang, W.; Liang, Y.-L.; Liu, Y.; Chen, Y.-Y.; Yang, S.-T.; Li, B.-R.; Yu, Y.-X.; Lyu, Y.; Wang, R. MeCP2 inhibits proliferation and migration of breast cancer via suppression of epithelial-mesenchymal transition. J. Cell Mol. Med. 2020, 24, 7959–7967. [Google Scholar] [CrossRef]
- Pampalakis, G.; Prosnikli, E.; Agalioti, T.; Vlahou, A.; Zoumpourlis, V.; Sotiropoulou, G. A Tumor-Protective Role for Human Kallikrein-Related Peptidase 6 in Breast Cancer Mediated by Inhibition of Epithelial-to-Mesenchymal Transition. Cancer Res. 2009, 69, 3779–3787. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Simmons, G.E.; Jr Malyarchuk, S.; Calhoun, T.N.; Pruitt, K. A novel MeCP2 acetylation site regulates interaction with ATRX and HDAC1. Genes Cancer 2015, 6, 408–421. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jin, X.; Li, Y.; Ruan, Y.; Lu, Y.; Yang, M.; Lin, D.; Song, P.; Guo, Y.; Zhao, S.; et al. DNA methylation of claudin-6 promotes breast cancer cell migration and invasion by recruiting MeCP2 and deacetylating H3Ac and H4Ac. J. Exp. Clin. Cancer Res. 2016, 35, 120. [Google Scholar] [CrossRef]
- Zhou, Q.; Guo, J.; Huang, W.; Yu, X.; Xu, C.; Long, X. Linc-ROR promotes the progression of breast cancer and decreases the sensitivity to rapamycin through miR-194-3p targeting MECP2. Mol Oncol. 2020, 14, 2231–2250. [Google Scholar] [CrossRef] [PubMed]
- Ballestar, E.; Paz, M.F.; Valle, L.; Wei, S.; Fraga, M.F.; Espada, J.; Cigudosa, J.C.; Huang, T.H.-M.; Esteller, M. Methyl-CpG binding proteins identify novel sites of epigenetic inactivation in human cancer. EMBO J. 2003, 22, 6335–6345. [Google Scholar] [CrossRef]
- Yilmaz, T.U.; Güneş, A.; Pösteki, G.; Okay, E. Rett syndrome with colon cancer presented with sigmoid volvulus: Report of a case. Int. J. Surg. Case Rep. 2014, 5, 577–579. [Google Scholar] [CrossRef]
- Wang, S.; Gan, M.; Chen, C.; Zhang, Y.; Kong, J.; Zhang, H.; Lai, M. Methyl CpG binding protein 2 promotes colorectal cancer metastasis by regulating N(6)-methyladenosine methylation through methyltransferase-like 14. Cancer Sci. 2021, 112, 3243–3254. [Google Scholar] [CrossRef]
- Darwanto, A.; Kitazawa, R.; Maeda, S.; Kitazawa, S. MeCP2 and promoter methylation cooperatively regulate E-cadherin gene expression in colorectal carcinoma. Cancer Sci. 2003, 94, 442–447. [Google Scholar] [CrossRef]
- Chen, T.; Cai, S.-L.; Li, J.; Qi, Z.-P.; Li, X.-Q.; Ye, L.-C.; Xie, X.-F.; Hou, Y.-Y.; Yao, L.-Q.; Xu, M.-D.; et al. Mecp2-mediated Epigenetic Silencing of miR-137 Contributes to Colorectal Adenoma-Carcinoma Sequence and Tumor Progression via Relieving the Suppression of c-Met. Sci. Rep. 2017, 7, 44543. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Gao, J. Expression and clinical significance of methyl-CpG-binding protein 2 in pancreas cancer and surrounding tissue. Chin. J. Pancreatol. 2021, 112–116. [Google Scholar]
- Wang, H.; Li, J.; He, J.; Liu, Y.; Feng, W.; Zhou, H.; Zhou, M.; Wei, H.; Lu, Y.; Peng, W.; et al. Methyl-CpG-binding protein 2 drives the Furin/TGF-β1/Smad axis to promote epithelial–mesenchymal transition in pancreatic cancer cells. Oncogenesis 2020, 9, 76. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Bian, S.; Li, J.; He, J.; Chen, H.; Ge, L.; Jiao, Z.; Zhang, Y.; Peng, W.; Du, F.; et al. MeCP2 suppresses LIN28A expression via binding to its methylated-CpG islands in pancreatic cancer cells. Oncotarget 2016, 7, 14476–14485. [Google Scholar] [CrossRef]
- Dandrea, M.; Donadelli, M.; Costanzo, C.; Scarpa, A.; Palmieri, M. MeCP2/H3meK9 are involved in IL-6 gene silencing in pancreatic adenocarcinoma cell lines. Nucleic Acids Res. 2009, 37, 6681–6690. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, X.; Yang, J.; Jiang, Q.; Zhang, J.; Qin, Y.; Wang, L.; Liu, L.; Ni, L.; Tong, D. MECP2 Promotes Migration and Invasion of Gastric Cancer Cells via Modulating the Notch1/c-Myc/mTOR Signaling Pathways by Suppressing FBXW7 Transcription. 2022. Available online: https://assets.researchsquare.com/files/rs-48700/v1/ca06d91a-1674-4ad5-9c79-3a5ba919317a.pdf?c=1631847938 (accessed on 9 April 2023).
- Zhang, X.Y.; Xu, Y.Y.; Chen, W.Y. MicroRNA-1324 inhibits cell proliferative ability and invasiveness by targeting MECP2 in gastric cancer. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 4766–4774. [Google Scholar]
- Zhao, L.Y.; Tong, D.D.; Xue, M.; Ma, H.L.; Liu, S.Y.; Yang, J.; Liu, Y.X.; Guo, B.; Ni, L.; Liu, L.Y.; et al. MeCP2, a target of miR-638, facilitates gastric cancer cell proliferation through activation of the MEK1/2-ERK1/2 signaling pathway by upregulating GIT1. Oncogenesis 2017, 6, e368. [Google Scholar] [CrossRef]
- Wada, R.; Akiyama, Y.; Hashimoto, Y.; Fukamachi, H.; Yuasa, Y. miR-212 is downregulated and suppresses methyl-CpG-binding protein MeCP2 in human gastric cancer. Int. J. Cancer 2010, 127, 1106–1114. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Wu, Q.; Ni, Z.; Lei, C.; Li, T.; Shi, Y. miR-19a/b and MeCP2 repress reciprocally to regulate multidrug resistance in gastric cancer cells. Int. J. Mol. Med. 2018, 42, 228–236. [Google Scholar] [CrossRef]
- Qin, Y.; Ma, X.; Guo, C.; Cai, S.; Ma, H.; Zhao, L. MeCP2 confers 5-fluorouracil resistance in gastric cancer via upregulating the NOX4/PKM2 pathway. Cancer Cell Int. 2022, 22, 86. [Google Scholar] [CrossRef]
- Tong, D.; Zhang, J.; Wang, X.; Li, Q.; Liu, L.; Lu, A.; Guo, B.; Yang, J.; Ni, L.; Qin, H.; et al. MiR-22, regulated by MeCP2, suppresses gastric cancer cell proliferation by inducing a deficiency in endogenous S-adenosylmethionine. Oncogenesis 2020, 9, 99. [Google Scholar] [CrossRef]
- Tong, D.; Zhao, L.; He, K.; Sun, H.; Cai, D.; Ni, L.; Sun, R.; Chang Se Song, T.; Huang, C. MECP2 promotes the growth of gastric cancer cells by suppressing miR-338-mediated antiproliferative effect. Oncotarget 2016, 7, 34845–34859. [Google Scholar] [CrossRef] [PubMed]
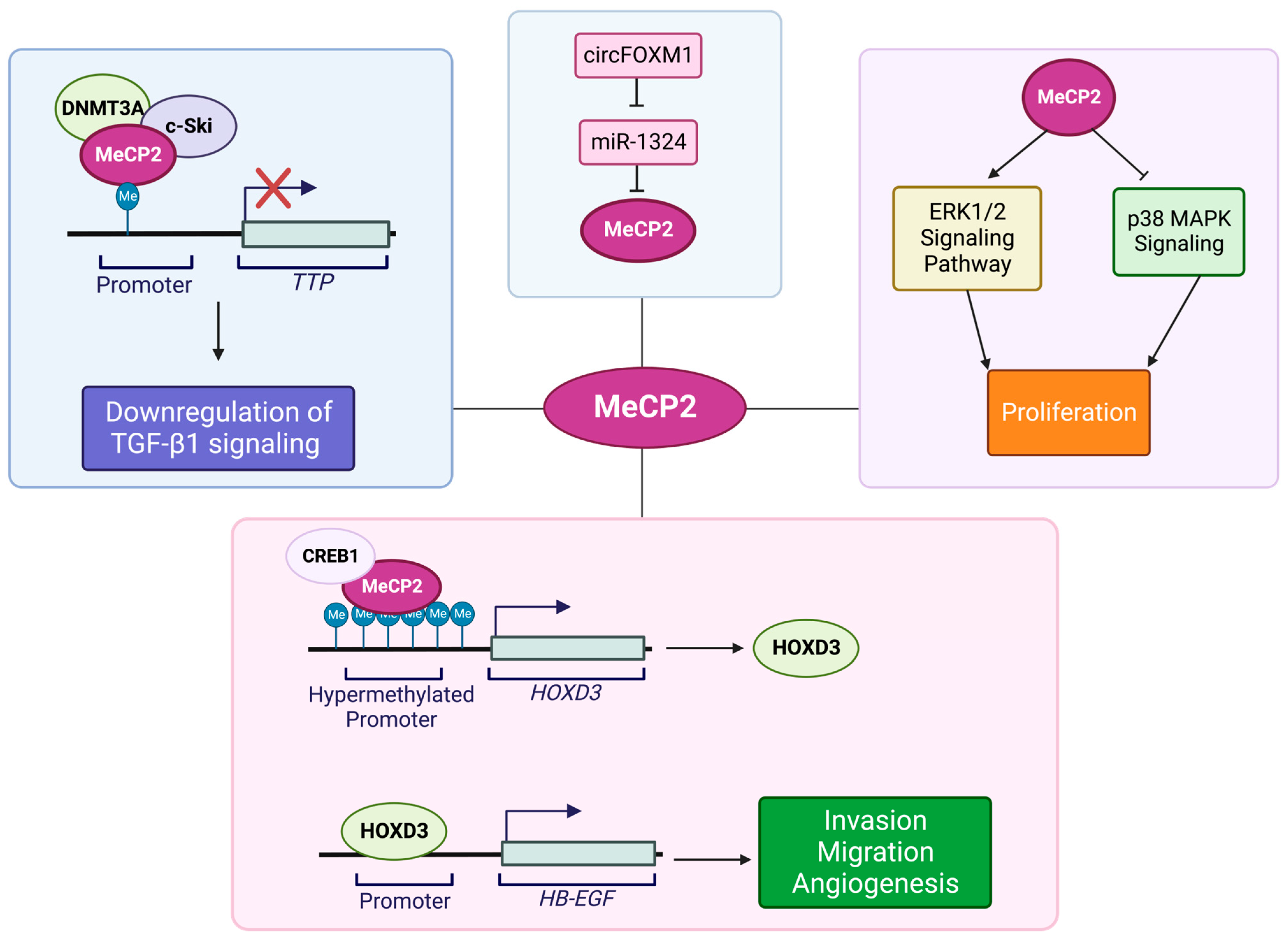
- Sohn, B.H.; Park, I.Y.; Lee, J.J.; Yang, S.J.; Jang, Y.J.; Park, K.C.; Kim, D.J.; Lee, D.C.; Sohn, H.A.; Kim, T.W.; et al. Functional switching of TGF-beta1 signaling in liver cancer via epigenetic modulation of a single CpG site in TTP promoter. Gastroenterology 2010, 138, 1898–1908. [Google Scholar] [CrossRef]
- Zhao, L.Y.; Zhang, J.; Guo, B.; Yang, J.; Han, J.; Zhao, X.G.; Wang, X.F.; Liu, L.Y.; Li, Z.F.; Song, T.S.; et al. MECP2 promotes cell proliferation by activating ERK1/2 and inhibiting p38 activity in human hepatocellular carcinoma HEPG2 cells. Cell. Mol. Biol. 2013, 59, 1876–1881. [Google Scholar]
- Wu, X.; Zhao, B.; Cheng, Y.; Yang, Y.; Huang, C.; Meng, X.; Wu, B.; Zhang, L.; Lv, X.; Li, J. Melittin induces PTCH1 expression by down-regulating MeCP2 in human hepatocellular carcinoma SMMC-7721 cells. Toxicol. Appl. Pharmacol. 2015, 288, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Gao, Y.; Tong, D.; Wang, X.; Guo, C.; Guo, B.; Yang, Y.; Zhao, L.; Zhang, J.; Yang, J.; et al. MeCP2 drives hepatocellular carcinoma progression via enforcing HOXD3 promoter methylation and expression through the HB-EGF/EGFR pathway. Mol. Oncol. 2021, 15, 3147–3163. [Google Scholar] [CrossRef]
- Weng, H.; Zeng, L.; Cao, L.; Chen, T.; Li, Y.; Xu, Y.; Peng, Y.; Ye, Y. circFOXM1 contributes to sorafenib resistance of hepatocellular carcinoma cells by regulating MECP2 via miR-1324. Mol. Ther. Nucleic Acids 2021, 23, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Bernard, D.; Gil, J.; Dumont, P.; Rizzo, S.; Monté, D.; Quatannens, B.; Hudson, D.; Visakorpi, T.; Fuks, F.; de Launoit, Y. The methyl-CpG-binding protein MECP2 is required for prostate cancer cell growth. Oncogene 2006, 25, 1358–1366. [Google Scholar] [CrossRef]
- Lee, J.; Lee, M.S.; Jeoung, D.I.; Kim, Y.M.; Lee, H. Promoter CpG-Site Methylation of the KAI1 Metastasis Suppressor Gene Contributes to Its Epigenetic Repression in Prostate Cancer. Prostate 2017, 77, 350–360. [Google Scholar] [CrossRef]
- Pulukuri, S.M.; Patibandla, S.; Patel, J.; Estes, N.; Rao, J.S. Epigenetic inactivation of the tissue inhibitor of metalloproteinase-2 (TIMP-2) gene in human prostate tumors. Oncogene 2007, 26, 5229–5237. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, K.; Gopisetty, G.; Gordian, E.; Navarro, L.; Hader, C.; Reis, I.M.; Schulz, W.A.; Singal, R. Methylation-Mediated Repression of GADD45α in Prostate Cancer and Its Role as a Potential Therapeutic Target. Cancer Res. 2009, 69, 1527–1535. [Google Scholar] [CrossRef]
- Guan, Y.; Guan, X.; An, H.; Baihetiya, A.; Wang, W.; Shao, W.; Yang, H.; Wang, Y. Epigenetic silencing of miR-137 induces resistance to bicalutamide by targeting TRIM24 in prostate cancer cells. Am. J. Transl. Res. 2019, 11, 3226–3237. [Google Scholar] [PubMed]
- Sharma, K.; Singh, J.; Frost, E.E.; Pillai, P.P. MeCP2 overexpression inhibits proliferation, migration and invasion of C6 glioma by modulating ERK signaling and gene expression. Neurosci. Lett. 2018, 674, 42–48. [Google Scholar] [CrossRef]
- Bian, E.; Chen, X.; Xu, Y.; Ji, X.; Cheng, M.; Wang, H.; Fang, Z.; Zhao, B. A central role for MeCP2 in the epigenetic repression of miR-200c during epithelial-to-mesenchymal transition of glioma. J. Exp. Clin. Cancer Res. 2019, 38, 366. [Google Scholar] [CrossRef]
- Zhang, N.; Wei, Z.-L.; Yin, J.; Zhang, L.; Wang, J.; Jin, Z.-L. MiR-106a* inhibits oral squamous cell carcinoma progression by directly targeting MeCP2 and suppressing the Wnt/β-Catenin signaling pathway. Am. J. Transl. Res. 2018, 10, 3542–3554. [Google Scholar] [PubMed]
- Liu, H.; Liu, Q.-L.; Zhai, T.-S.; Lu, J.; Dong, Y.-Z.; Xu, Y.-F. Silencing miR-454 suppresses cell proliferation, migration and invasion via directly targeting MECP2 in renal cell carcinoma. Am. J. Transl. Res. 2020, 12, 4277–4289. [Google Scholar] [PubMed]
- Lin, R.K.; Hsu, H.S.; Chang, J.W.; Chen, C.Y.; Chen, J.T.; Wang, Y.C. Alteration of DNA methyltransferases contributes to 5′CpG methylation and poor prognosis in lung cancer. Lung Cancer 2007, 55, 205–213. [Google Scholar] [CrossRef]
- Han, X.; Wu, J.; Zhang, Y.; Song, J.; Shi, Z.; Chang, H. LINC00518 Promotes Cell Proliferation by Regulating the Cell Cycle of Lung Adenocarcinoma Through miR-185-3p Targeting MECP2. Front. Oncol. 2021, 11, 646559. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nejati-Koshki, K.; Roberts, C.-T.; Babaei, G.; Rastegar, M. The Epigenetic Reader Methyl-CpG-Binding Protein 2 (MeCP2) Is an Emerging Oncogene in Cancer Biology. Cancers 2023, 15, 2683. https://doi.org/10.3390/cancers15102683
Nejati-Koshki K, Roberts C-T, Babaei G, Rastegar M. The Epigenetic Reader Methyl-CpG-Binding Protein 2 (MeCP2) Is an Emerging Oncogene in Cancer Biology. Cancers. 2023; 15(10):2683. https://doi.org/10.3390/cancers15102683
Chicago/Turabian StyleNejati-Koshki, Kazem, Chris-Tiann Roberts, Ghader Babaei, and Mojgan Rastegar. 2023. "The Epigenetic Reader Methyl-CpG-Binding Protein 2 (MeCP2) Is an Emerging Oncogene in Cancer Biology" Cancers 15, no. 10: 2683. https://doi.org/10.3390/cancers15102683
APA StyleNejati-Koshki, K., Roberts, C.-T., Babaei, G., & Rastegar, M. (2023). The Epigenetic Reader Methyl-CpG-Binding Protein 2 (MeCP2) Is an Emerging Oncogene in Cancer Biology. Cancers, 15(10), 2683. https://doi.org/10.3390/cancers15102683