

Short-Term TERT Inhibition Impairs Cellular Proliferation via a Telomere Length-Independent Mechanism and Can Be Exploited as a Potential Anticancer Approach
 , ,
, ,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Compounds
2.2. Cell Cultures
2.3. Animals
2.4. Plasmids and Transfection
2.5. Reverse Transcription and Quantitative Real-Time PCR
2.6. Immunoblot and Co-Immunoprecipitation
2.7. Nuclear and Cytoplasmic Fraction
2.8. Immunofluorescence
2.9. Cell Viability and Cell Cycle Analysis
2.10. Telomere Length Measurement
2.11. Xenotransplantation of LCL and BL Cells in Zebrafish Embryos
2.12. Embryo Dissociation and Flow Cytometric Analysis
2.13. Statistical Analyses
3. Results
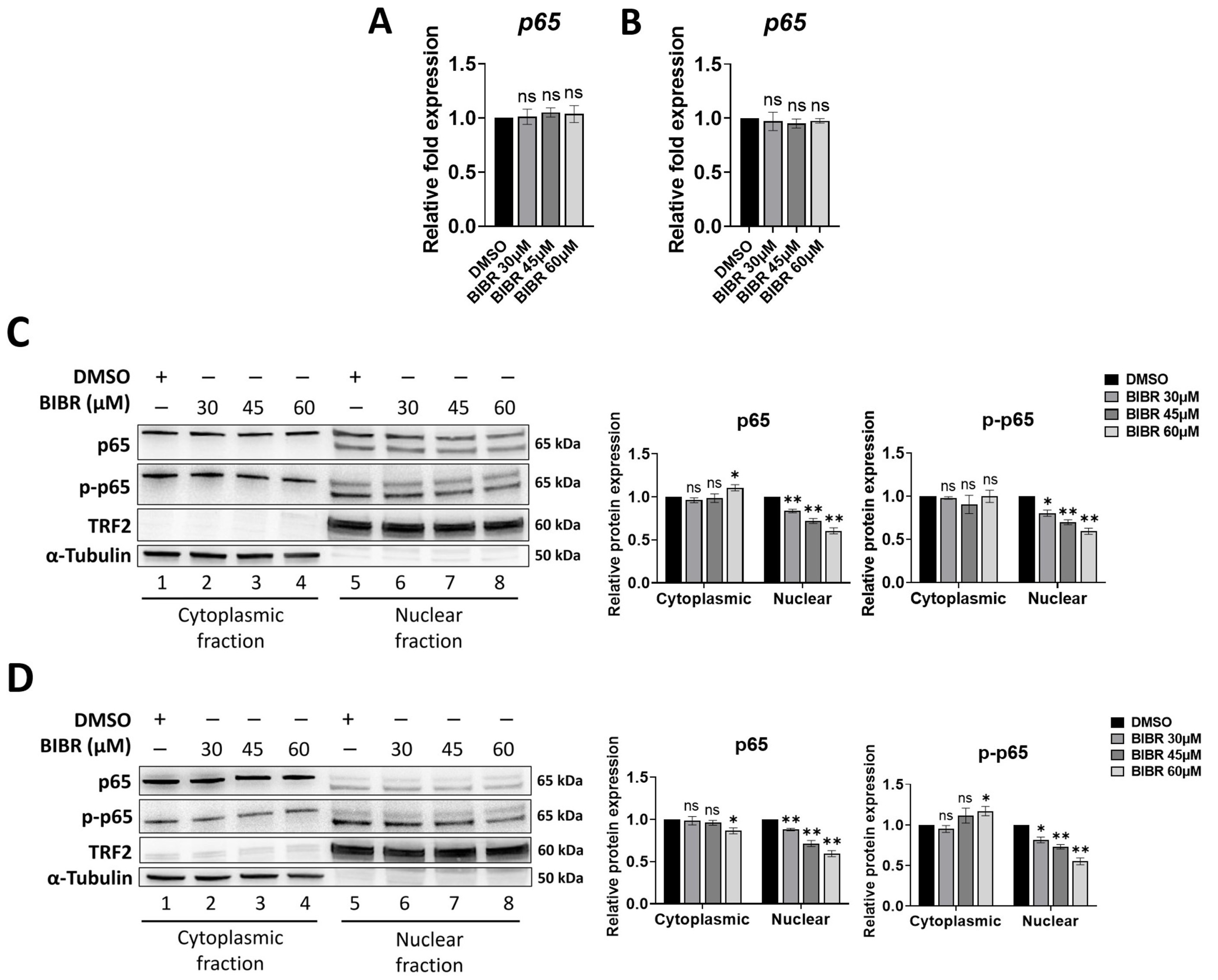
3.1. TERT Inhibition Reduced Nuclear Levels of p65
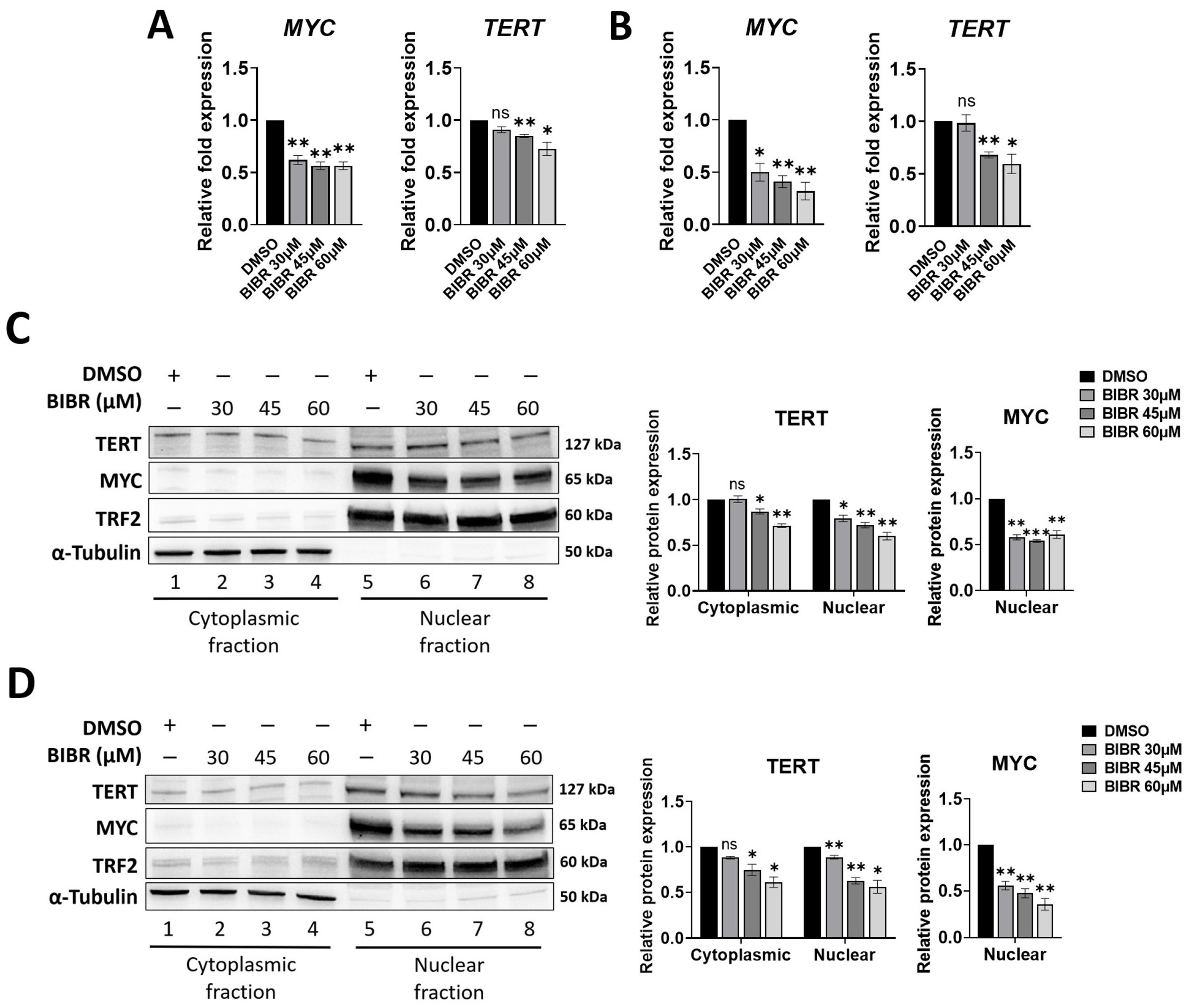
3.2. TERT Inhibition by BIBR Suppressed Transcription of a Subset of NF-κB Target Genes, including MYC
3.3. MYC Deregulation Mediated by TERT Inhibition Was Independent of WNT/β-Catenin Signalling, and TERT and MYC Did Not Interact at the Protein Level
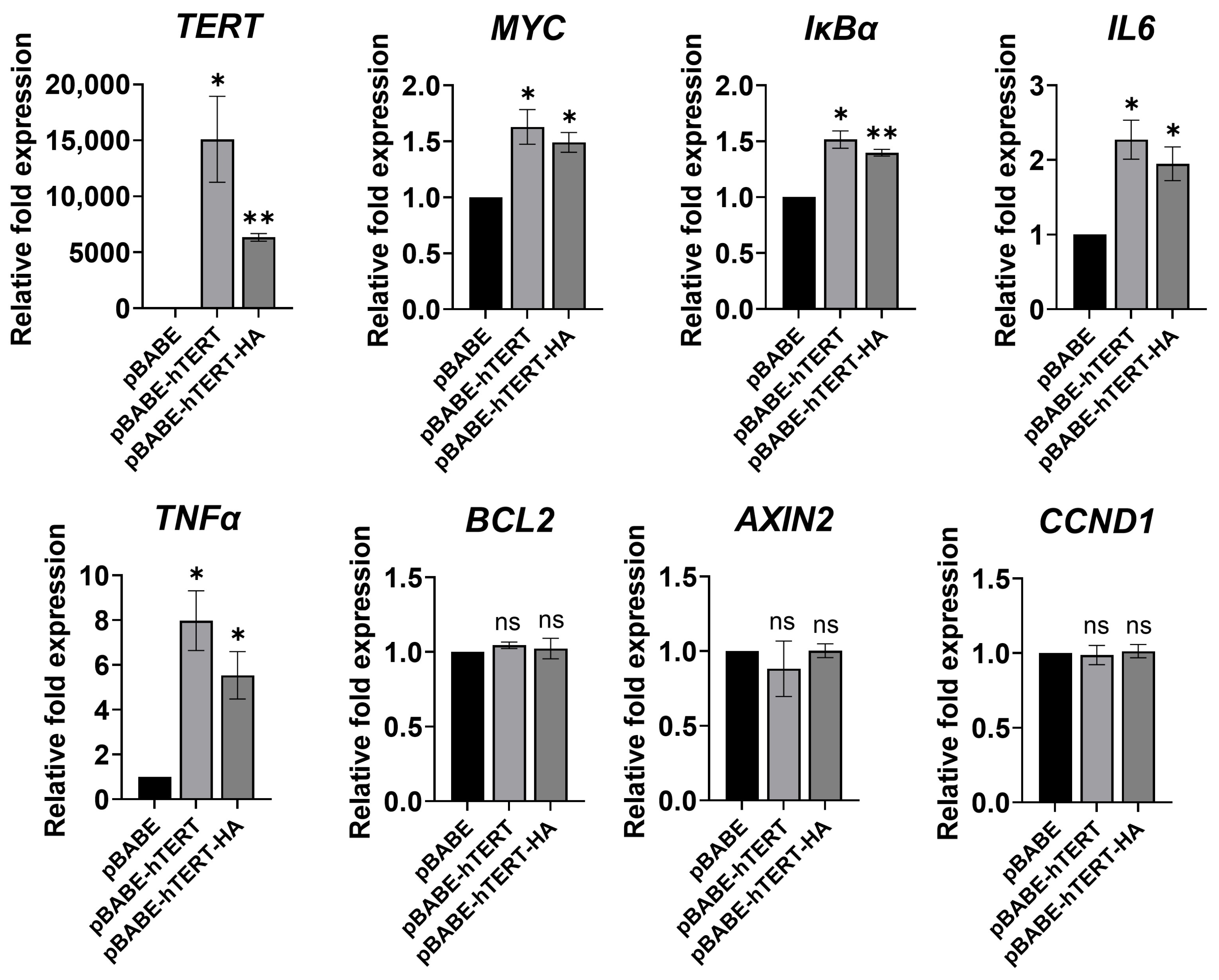
3.4. Ectopic TERT Expression Activated Transcription of NF-κB Target Genes
3.5. p65 Inhibition Recapitulated the Effects of TERT Inhibition
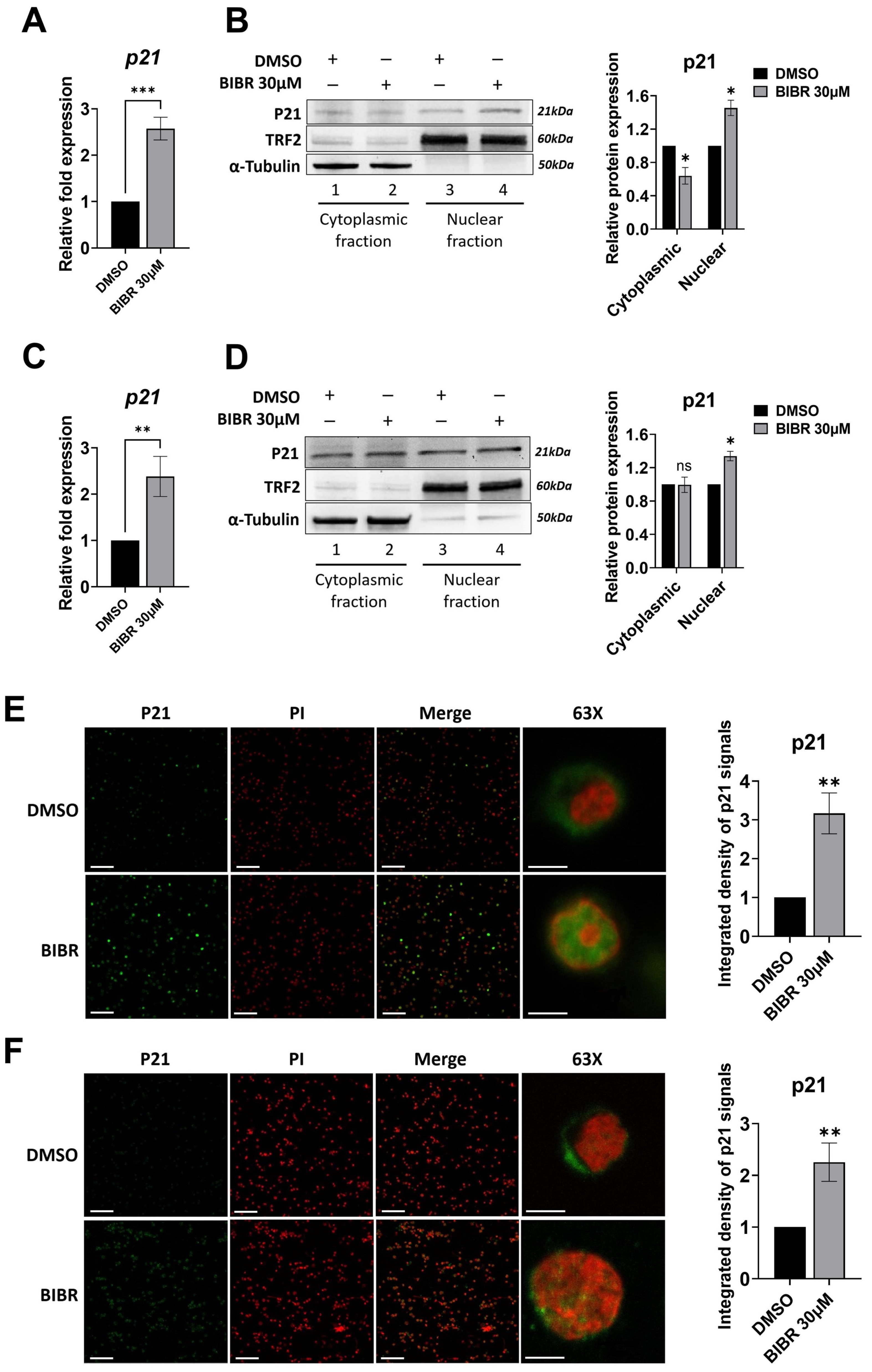
3.6. TERT Inhibition Promoted P21 Expression and Nuclear Localization
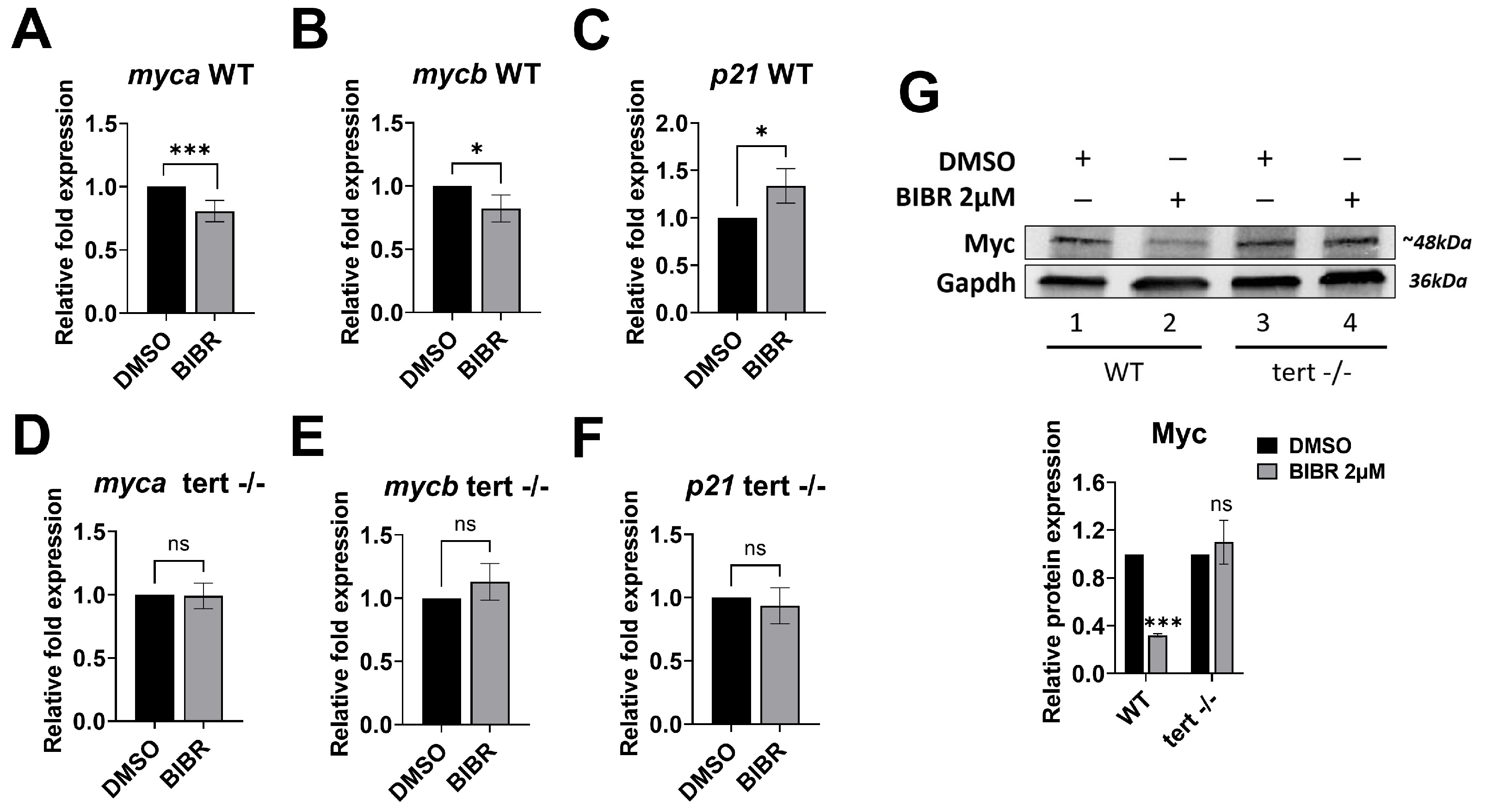
3.7. Short-Term Tert Inhibition by BIBR in Zebrafish Reduced Myc and Increased p21 Expression
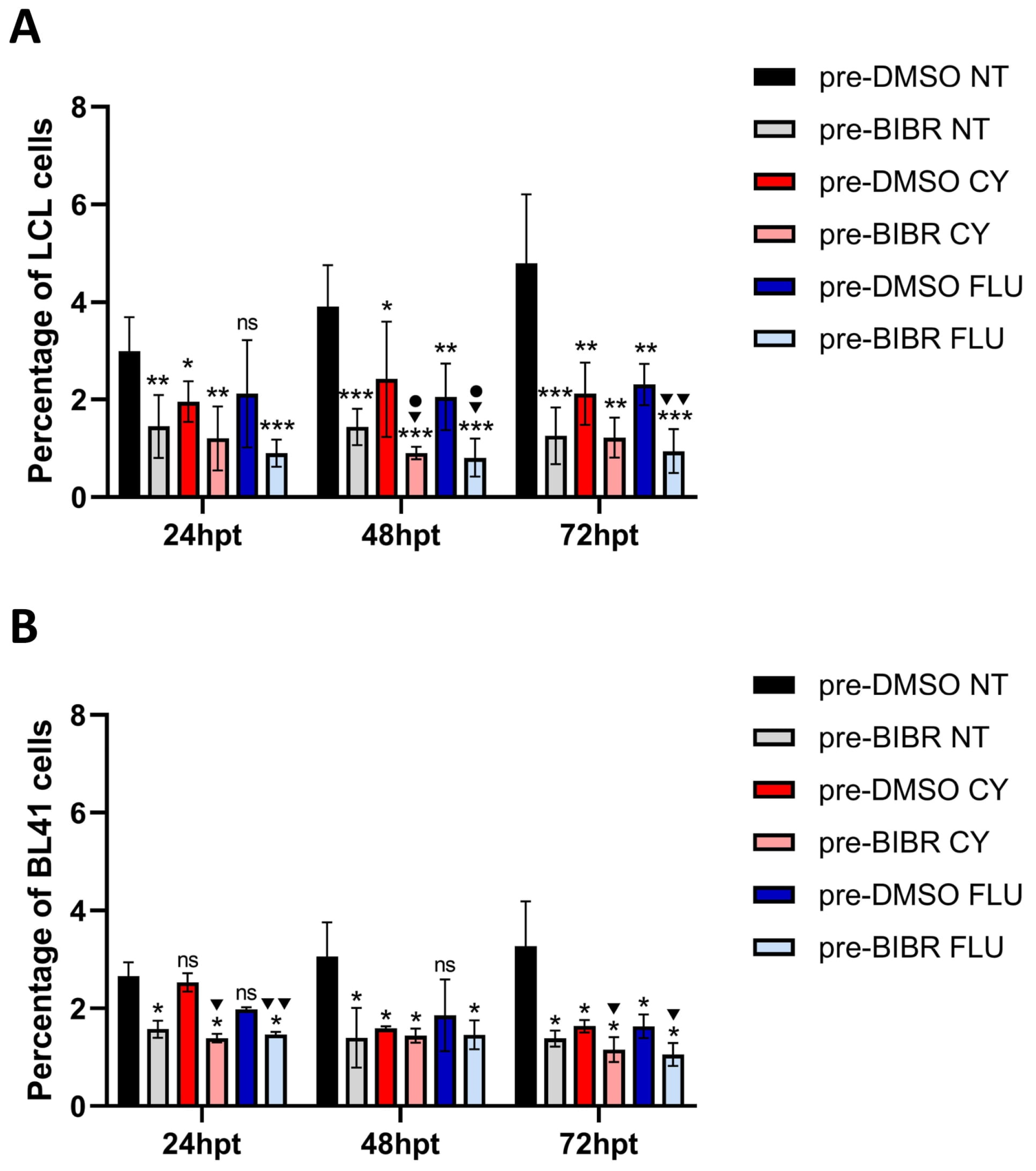
3.8. Anti-Proliferative Effects of Combined Treatment with BIBR and FLU or CY in EBV-Immortalized and Fully Transformed B Cells Xenografted in Zebrafish
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roake, C.M.; Artandi, S.E. Regulation of human telomerase in homeostasis and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 384–397. [Google Scholar] [CrossRef]
- de Lange, T. How telomeres solve the end-protection problem. Science 2009, 326, 948–952. [Google Scholar] [CrossRef]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef]
- Nakamura, T.M.; Morin, G.B.; Chapman, K.B.; Weinrich, S.L.; Andrews, W.H.; Lingner, J.; Harley, C.B.; Cech, T.R. Telomerase catalytic subunit homologs from fission yeast and human. Science 1997, 277, 955–959. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Harley, C.B. Telomerase and cancer therapeutics. Nat. Rev. Cancer 2008, 8, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.H.; Greider, C.W.; Szostak, J.W. Telomeres and telomerase: The path from maize, Tetrahymena and yeast to human cancer and aging. Nat. Med. 2006, 12, 1133–1138. [Google Scholar] [CrossRef]
- Li, Y.; Tergaonkar, V. Noncanonical functions of telomerase: Implications in telomerase-targeted cancer therapies. Cancer Res. 2014, 74, 1639–1644. [Google Scholar] [CrossRef]
- Choi, J.; Southworth, L.K.; Sarin, K.Y.; Venteicher, A.S.; Ma, W.; Chang, W.; Cheung, P.; Jun, S.; Artandi, M.K.; Shah, N.; et al. TERT promotes epithelial proliferation through transcriptional control of a Myc- and Wnt-related developmental program. PLoS Genet. 2008, 4, e10. [Google Scholar] [CrossRef] [PubMed]
- Geserick, C.; Tejera, A.; González-Suárez, E.; Klatt, P.; Blasco, M.A. Expression of mTert in primary murine cells links the growth-promoting effects of telomerase to transforming growth factor-beta signaling. Oncogene 2006, 25, 4310–4319. [Google Scholar] [CrossRef]
- Masutomi, K.; Possemato, R.; Wong, J.M.; Currier, J.L.; Tothova, Z.; Manola, J.B.; Ganesan, S.; Lansdorp, P.M.; Collins, K.; Hahn, W.C. The telomerase reverse transcriptase regulates chromatin state and DNA damage responses. Proc. Natl. Acad. Sci. USA 2005, 102, 8222–8227. [Google Scholar] [CrossRef] [PubMed]
- Celeghin, A.; Giunco, S.; Freguja, R.; Zangrossi, M.; Nalio, S.; Dolcetti, R.; De Rossi, A. Short-term inhibition of TERT induces telomere length-independent cell cycle arrest and apoptotic response in EBV-immortalized and transformed B cells. Cell Death Dis. 2016, 7, e2562. [Google Scholar] [CrossRef]
- Lee, J.; Sung, Y.H.; Cheong, C.; Choi, Y.S.; Jeon, H.K.; Sun, W.; Hahn, W.C.; Ishikawa, F.; Lee, H.W. TERT promotes cellular and organismal survival independently of telomerase activity. Oncogene 2008, 27, 3754–3760. [Google Scholar] [CrossRef]
- Del Bufalo, D.; Rizzo, A.; Trisciuoglio, D.; Cardinali, G.; Torrisi, M.R.; Zangemeister-Wittke, U.; Zupi, G.; Biroccio, A. Involvement of hTERT in apoptosis induced by interference with Bcl-2 expression and function. Cell Death Differ. 2005, 12, 1429–1438. [Google Scholar] [CrossRef]
- Bryan, C.; Rice, C.; Hoffman, H.; Harkisheimer, M.; Sweeney, M.; Skordalakes, E. Structural Basis of Telomerase Inhibition by the Highly Specific BIBR1532. Structure 2015, 23, 1934–1942. [Google Scholar] [CrossRef]
- Shirgahi Talari, F.; Bagherzadeh, K.; Golestanian, S.; Jarstfer, M.; Amanlou, M. Potent Human Telomerase Inhibitors: Molecular Dynamic Simulations, Multiple Pharmacophore-Based Virtual Screening, and Biochemical Assays. J. Chem. Inf. Model. 2015, 55, 2596–2610. [Google Scholar] [CrossRef] [PubMed]
- Damm, K.; Hemmann, U.; Garin-Chesa, P.; Hauel, N.; Kauffmann, I.; Priepke, H.; Niestroj, C.; Daiber, C.; Enenkel, B.; Guilliard, B.; et al. A highly selective telomerase inhibitor limiting human cancer cell proliferation. EMBO J. 2001, 20, 6958–6968. [Google Scholar] [CrossRef] [PubMed]
- Pascolo, E.; Wenz, C.; Lingner, J.; Hauel, N.; Priepke, H.; Kauffmann, I.; Garin-Chesa, P.; Rettig, W.J.; Damm, K.; Schnapp, A. Mechanism of human telomerase inhibition by BIBR1532, a synthetic, non-nucleosidic drug candidate. J. Biol. Chem. 2002, 277, 15566–15572. [Google Scholar] [CrossRef]
- Bashash, D.; Ghaffari, S.H.; Mirzaee, R.; Alimoghaddam, K.; Ghavamzadeh, A. Telomerase inhibition by non-nucleosidic compound BIBR1532 causes rapid cell death in pre-B acute lymphoblastic leukemia cells. Leuk. Lymphoma 2013, 54, 561–568. [Google Scholar] [CrossRef]
- Doğan, F.; Özateş, N.P.; Bağca, B.G.; Abbaszadeh, Z.; Söğütlü, F.; Gasımlı, R.; Gündüz, C.; Biray Avcı, Ç. Investigation of the effect of telomerase inhibitor BIBR1532 on breast cancer and breast cancer stem cells. J. Cell Biochem. 2019, 120, 1282–1293. [Google Scholar] [CrossRef]
- Giunco, S.; Zangrossi, M.; Dal Pozzolo, F.; Celeghin, A.; Ballin, G.M.; Petrara, M.R.; Amin, A.; Argenton, F.; Godinho Ferreira, M.; De Rossi, A. Anti-Proliferative and Pro-Apoptotic Effects of Short-Term Inhibition of Telomerase In Vivo and in Human Malignant B Cells Xenografted in Zebrafish. Cancers 2020, 12, 2052. [Google Scholar] [CrossRef]
- Akiyama, M.; Hideshima, T.; Hayashi, T.; Tai, Y.T.; Mitsiades, C.S.; Mitsiades, N.; Chauhan, D.; Richardson, P.; Munshi, N.C.; Anderson, K. Nuclear factor-kappaB p65 mediates tumor necrosis factor alpha-induced nuclear translocation of telomerase reverse transcriptase protein. Cancer Res. 2003, 63, 18–21. [Google Scholar]
- Ghosh, A.; Saginc, G.; Leow, S.C.; Khattar, E.; Shin, E.M.; Yan, T.D.; Wong, M.; Zhang, Z.; Li, G.; Sung, W.K.; et al. Telomerase directly regulates NF-κB-dependent transcription. Nat. Cell Biol. 2012, 14, 1270–1281. [Google Scholar] [CrossRef]
- Ding, D.; Xi, P.; Zhou, J.; Wang, M.; Cong, Y.S. Human telomerase reverse transcriptase regulates MMP expression independently of telomerase activity via NF-κB-dependent transcription. FASEB J. 2013, 27, 4375–4383. [Google Scholar] [CrossRef]
- Deacon, K.; Knox, A.J. PINX1 and TERT Are Required for TNF-α-Induced Airway Smooth Muscle Chemokine Gene Expression. J. Immunol. 2018, 200, 1283–1294. [Google Scholar] [CrossRef]
- Kong, W.; Lv, N.; Wysham, W.Z.; Roque, D.R.; Zhang, T.; Jiao, S.; Song, D.; Chen, J.; Bae-Jump, V.L.; Zhou, C. Knockdown of hTERT and Treatment with BIBR1532 Inhibit Cell Proliferation and Invasion in Endometrial Cancer Cells. J. Cancer 2015, 6, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Bian, C.; Zhen, C.; Liu, L.; Lin, Z.; Nisar, M.F.; Wang, M.; Bartsch, J.W.; Huang, E.; Ji, P.; et al. Telomerase reverse transcriptase mediates EMT through NF-κB signaling in tongue squamous cell carcinoma. Oncotarget 2017, 8, 85492–85503. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.J.; Zeng, S.; Xie, R.; Hu, C.J.; Wang, S.M.; Wu, Y.Y.; Xiao, Y.F.; Yang, S.M. hTERT promotes gastric intestinal metaplasia by upregulating CDX2 via NF-κB signaling pathway. Oncotarget 2017, 8, 26969–26978. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.M.; Khattar, E.; Leow, S.C.; Liu, C.Y.; Muller, J.; Ang, W.X.; Li, Y.; Franzoso, G.; Li, S.; Guccione, E.; et al. Telomerase regulates MYC-driven oncogenesis independent of its reverse transcriptase activity. J. Clin. Investig. 2015, 125, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Xie, R.; Qin, Y.; Xiao, Y.F.; Yong, X.; Zheng, L.; Dong, H.; Yang, S.M. Human telomerase reverse transcriptase (hTERT) promotes gastric cancer invasion through cooperating with c-Myc to upregulate heparanase expression. Oncotarget 2016, 7, 11364–11379. [Google Scholar] [CrossRef]
- Shkreli, M.; Sarin, K.Y.; Pech, M.F.; Papeta, N.; Chang, W.; Brockman, S.A.; Cheung, P.; Lee, E.; Kuhnert, F.; Olson, J.L.; et al. Reversible cell-cycle entry in adult kidney podocytes through regulated control of telomerase and Wnt signaling. Nat. Med. 2011, 18, 111–119. [Google Scholar] [CrossRef]
- Chen, K.; Chen, L.; Li, L.; Qu, S.; Yu, B.; Sun, Y.; Wan, F.; Chen, X.; Liang, R.; Zhu, X. A positive feedback loop between Wnt/β-catenin signaling and hTERT regulates the cancer stem cell-like traits in radioresistant nasopharyngeal carcinoma cells. J. Cell Biochem. 2020, 121, 4612–4622. [Google Scholar] [CrossRef]
- Park, J.I.; Venteicher, A.S.; Hong, J.Y.; Choi, J.; Jun, S.; Shkreli, M.; Meng, Z.; Cheung, P.; Ji, H.; McLaughlin, M.; et al. Telomerase modulates Wnt signalling by association with target gene chromatin. Nature 2009, 460, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Tergaonkar, V. NFκB drives TERT promoter reactivation in cancer. Cell Cycle 2016, 15, 156–157. [Google Scholar] [CrossRef]
- Bu, D.X.; Johansson, M.E.; Ren, J.; Xu, D.W.; Johnson, F.B.; Edfeldt, K.; Yan, Z.Q. Nuclear factor {kappa}B-mediated transactivation of telomerase prevents intimal smooth muscle cell from replicative senescence during vascular repair. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2604–2610. [Google Scholar] [CrossRef]
- Hoffmeyer, K.; Raggioli, A.; Rudloff, S.; Anton, R.; Hierholzer, A.; Del Valle, I.; Hein, K.; Vogt, R.; Kemler, R. Wnt/β-catenin signaling regulates telomerase in stem cells and cancer cells. Science 2012, 336, 1549–1554. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Toh, L.; Lau, P.; Wang, X. Human telomerase reverse transcriptase (hTERT) is a novel target of the Wnt/β-catenin pathway in human cancer. J. Biol. Chem. 2012, 287, 32494–32511. [Google Scholar] [CrossRef] [PubMed]
- Latil, A.; Vidaud, D.; Valéri, A.; Fournier, G.; Vidaud, M.; Lidereau, R.; Cussenot, O.; Biàche, I. htert expression correlates with MYC over-expression in human prostate cancer. Int. J. Cancer 2000, 89, 172–176. [Google Scholar] [CrossRef]
- Wu, K.J.; Grandori, C.; Amacker, M.; Simon-Vermot, N.; Polack, A.; Lingner, J.; Dalla-Favera, R. Direct activation of TERT transcription by c-MYC. Nat. Genet. 1999, 21, 220–224. [Google Scholar] [CrossRef]
- Kar, A.; Saha, D.; Purohit, G.; Singh, A.; Kumar, P.; Yadav, V.K.; Kumar, P.; Thakur, R.K.; Chowdhury, S. Metastases suppressor NME2 associates with telomere ends and telomerase and reduces telomerase activity within cells. Nucleic Acids Res. 2012, 40, 2554–2565. [Google Scholar] [CrossRef]
- Faumont, N.; Durand-Panteix, S.; Schlee, M.; Grömminger, S.; Schuhmacher, M.; Hölzel, M.; Laux, G.; Mailhammer, R.; Rosenwald, A.; Staudt, L.M.; et al. c-Myc and Rel/NF-kappaB are the two master transcriptional systems activated in the latency III program of Epstein-Barr virus-immortalized B cells. J. Virol. 2009, 83, 5014–5027. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.M.; Kanda, K.; Zhang, L.; Boxer, L.M. Activation of the c-myc p1 promoter in Burkitt’s lymphoma by the hs3 immunoglobulin heavy-chain gene enhancer. Leukemia 2007, 21, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Kanda, K.; Hu, H.M.; Zhang, L.; Grandchamps, J.; Boxer, L.M. NF-kappa B activity is required for the deregulation of c-myc expression by the immunoglobulin heavy chain enhancer. J. Biol. Chem. 2000, 275, 32338–32346. [Google Scholar] [CrossRef]
- Sheng, W.Y.; Chen, Y.R.; Wang, T.C. A major role of PKC theta and NFkappaB in the regulation of hTERT in human T lymphocytes. FEBS Lett. 2006, 580, 6819–6824. [Google Scholar] [CrossRef]
- Zhao, S.; Huang, J.; Ye, J. A fresh look at zebrafish from the perspective of cancer research. J Exp Clin. Cancer Res. 2015, 34, 80. [Google Scholar] [CrossRef]
- Imamura, S.; Uchiyama, J.; Koshimizu, E.; Hanai, J.; Raftopoulou, C.; Murphey, R.D.; Bayliss, P.E.; Imai, Y.; Burns, C.E.; Masutomi, K.; et al. A non-canonical function of zebrafish telomerase reverse transcriptase is required for developmental hematopoiesis. PLoS ONE 2008, 3, e3364. [Google Scholar] [CrossRef] [PubMed]
- Henriques, C.M.; Carneiro, M.C.; Tenente, I.M.; Jacinto, A.; Ferreira, M.G. Telomerase is required for zebrafish lifespan. PLoS Genet. 2013, 9, e1003214. [Google Scholar] [CrossRef] [PubMed]
- Cassar, S.; Adatto, I.; Freeman, J.L.; Gamse, J.T.; Iturria, I.; Lawrence, C.; Muriana, A.; Peterson, R.T.; Van Cruchten, S.; Zon, L.I. Use of Zebrafish in Drug Discovery Toxicology. Chem. Res. Toxicol. 2020, 33, 95–118. [Google Scholar] [CrossRef]
- Savarese, T.; Abate, A.; Basnet, R.M.; Lorini, L.; Gurizzan, C.; Tomasoni, M.; Lombardi, D.; Tomasini, D.; Zizioli, D.; Memo, M.; et al. Cytotoxic effects of targeted agent alone or with chemotherapy in the treatment of adenoid cystic carcinoma: A preclinical study. Sci. Rep. 2022, 12, 9951. [Google Scholar] [CrossRef] [PubMed]
- Terrin, L.; Dolcetti, R.; Corradini, I.; Indraccolo, S.; Dal Col, J.; Bertorelle, R.; Bonaldi, L.; Esposito, G.; De Rossi, A. hTERT inhibits the Epstein-Barr virus lytic cycle and promotes the proliferation of primary B lymphocytes: Implications for EBV-driven lymphomagenesis. Int. J. Cancer 2007, 121, 576–587. [Google Scholar] [CrossRef]
- Giunco, S.; Dolcetti, R.; Keppel, S.; Celeghin, A.; Indraccolo, S.; Dal Col, J.; Mastorci, K.; De Rossi, A. hTERT inhibition triggers Epstein-Barr virus lytic cycle and apoptosis in immortalized and transformed B cells: A basis for new therapies. Clin. Cancer Res. 2013, 19, 2036–2047. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, C.B.; Ballard, W.W.; Kimmel, S.R.; Ullmann, B.; Schilling, T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995, 203, 253–310. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Terrin, L.; Rampazzo, E.; Pucciarelli, S.; Agostini, M.; Bertorelle, R.; Esposito, G.; DelBianco, P.; Nitti, D.; De Rossi, A. Relationship between tumor and plasma levels of hTERT mRNA in patients with colorectal cancer: Implications for monitoring of neoplastic disease. Clin. Cancer Res. 2008, 14, 7444–7451. [Google Scholar] [CrossRef] [PubMed]
- Tsang, M.; Gantchev, J.; Ghazawi, F.M.; Litvinov, I.V. Protocol for adhesion and immunostaining of lymphocytes and other non-adherent cells in culture. Biotechniques 2017, 63, 230–233. [Google Scholar] [CrossRef]
- Cawthon, R.M. Telomere length measurement by a novel monochrome multiplex quantitative PCR method. Nucleic Acids Res. 2009, 37, e21. [Google Scholar] [CrossRef]
- Boscolo-Rizzo, P.; Giunco, S.; Rampazzo, E.; Brutti, M.; Spinato, G.; Menegaldo, A.; Stellin, M.; Mantovani, M.; Bandolin, L.; Rossi, M.; et al. TERT promoter hotspot mutations and their relationship with TERT levels and telomere erosion in patients with head and neck squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2020, 146, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Rampazzo, E.; Bertorelle, R.; Serra, L.; Terrin, L.; Candiotto, C.; Pucciarelli, S.; Del Bianco, P.; Nitti, D.; De Rossi, A. Relationship between telomere shortening, genetic instability, and site of tumour origin in colorectal cancers. Br. J. Cancer 2010, 102, 1300–1305. [Google Scholar] [CrossRef]
- David, A.; Arnaud, N.; Fradet, M.; Lascaux, H.; Ouk-Martin, C.; Gachard, N.; Zimber-Strobl, U.; Feuillard, J.; Faumont, N. c-Myc dysregulation is a co-transforming event for nuclear factor-κB activated B cells. Haematologica 2017, 102, 883–894. [Google Scholar] [CrossRef]
- Cahir-McFarland, E.D.; Davidson, D.M.; Schauer, S.L.; Duong, J.; Kieff, E. NF-kappa B inhibition causes spontaneous apoptosis in Epstein-Barr virus-transformed lymphoblastoid cells. Proc. Natl. Acad. Sci. USA 2000, 97, 6055–6060. [Google Scholar] [CrossRef] [PubMed]
- Dirmeier, U.; Hoffmann, R.; Kilger, E.; Schultheiss, U.; Briseño, C.; Gires, O.; Kieser, A.; Eick, D.; Sugden, B.; Hammerschmidt, W. Latent membrane protein 1 of Epstein-Barr virus coordinately regulates proliferation with control of apoptosis. Oncogene 2005, 24, 1711–1717. [Google Scholar] [CrossRef] [PubMed]
- Shkoda, A.; Town, J.A.; Griese, J.; Romio, M.; Sarioglu, H.; Knöfel, T.; Giehler, F.; Kieser, A. The germinal center kinase TNIK is required for canonical NF-κB and JNK signaling in B-cells by the EBV oncoprotein LMP1 and the CD40 receptor. PLoS Biol. 2012, 10, e1001376. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, S.; Yang, T.; Zhu, F.; Zhu, J.; Huang, Y.; Wu, L.; Chen, L.; Xu, Z. CD40L-mediated inhibition of NF-kappaB in CA46 Burkitt lymphoma cells promotes apoptosis. Leuk. Lymphoma 2008, 49, 1792–1799. [Google Scholar] [CrossRef]
- Maguire, O.; O’Loughlin, K.; Minderman, H. Simultaneous assessment of NF-κB/p65 phosphorylation and nuclear localization using imaging flow cytometry. J. Immunol. Methods 2015, 423, 3–11. [Google Scholar] [CrossRef]
- Yin, L.; Hubbard, A.K.; Giardina, C. NF-kappa B regulates transcription of the mouse telomerase catalytic subunit. J. Biol. Chem. 2000, 275, 36671–36675. [Google Scholar] [CrossRef]
- Xu, J.; Chen, Y.; Huo, D.; Khramtsov, A.; Khramtsova, G.; Zhang, C.; Goss, K.H.; Olopade, O.I. β-catenin regulates c-Myc and CDKN1A expression in breast cancer cells. Mol. Carcinog. 2016, 55, 431–439. [Google Scholar] [CrossRef]
- Zhao, T.; Hu, F.; Qiao, B.; Chen, Z.; Tao, Q. Telomerase reverse transcriptase potentially promotes the progression of oral squamous cell carcinoma through induction of epithelial-mesenchymal transition. Int. J. Oncol. 2015, 46, 2205–2215. [Google Scholar] [CrossRef]
- Zhang, K.; Guo, Y.; Wang, X.; Zhao, H.; Ji, Z.; Cheng, C.; Li, L.; Fang, Y.; Xu, D.; Zhu, H.H.; et al. WNT/β-Catenin Directs Self-Renewal Symmetric Cell Division of hTERThigh Prostate Cancer Stem Cells. Cancer Res. 2017, 77, 2534–2547. [Google Scholar] [CrossRef]
- Metcalf, R.A.; Zhao, S.; Anderson, M.W.; Lu, Z.S.; Galperin, I.; Marinelli, R.J.; Cherry, A.M.; Lossos, I.S.; Natkunam, Y. Characterization of D-cyclin proteins in hematolymphoid neoplasms: Lack of specificity of cyclin-D2 and D3 expression in lymphoma subtypes. Mod. Pathol. 2010, 23, 420–433. [Google Scholar] [CrossRef]
- Vera, E.; Canela, A.; Fraga, M.F.; Esteller, M.; Blasco, M.A. Epigenetic regulation of telomeres in human cancer. Oncogene 2008, 27, 6817–6833. [Google Scholar] [CrossRef]
- Lecarpentier, Y.; Schussler, O.; Hébert, J.L.; Vallée, A. Multiple Targets of the Canonical WNT/β-Catenin Signaling in Cancers. Front. Oncol. 2019, 9, 1248. [Google Scholar] [CrossRef]
- Pate, K.T.; Stringari, C.; Sprowl-Tanio, S.; Wang, K.; TeSlaa, T.; Hoverter, N.P.; McQuade, M.M.; Garner, C.; Digman, M.A.; Teitell, M.A.; et al. Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J. 2014, 33, 1454–1473. [Google Scholar] [CrossRef]
- Counter, C.M.; Hahn, W.C.; Wei, W.; Caddle, S.D.; Beijersbergen, R.L.; Lansdorp, P.M.; Sedivy, J.M.; Weinberg, R.A. Dissociation among in vitro telomerase activity, telomere maintenance, and cellular immortalization. Proc. Natl. Acad. Sci. USA 1998, 95, 14723–14728. [Google Scholar] [CrossRef] [PubMed]
- Ouellette, M.M.; Aisner, D.L.; Savre-Train, I.; Wright, W.E.; Shay, J.W. Telomerase activity does not always imply telomere maintenance. Biochem. Biophys. Res. Commun. 1999, 254, 795–803. [Google Scholar] [CrossRef]
- Wang, H.; Xu, L.; Zhao, J.; Wang, D.; Guo, R.; Wang, J.; Gong, W.; Liu, T.; Zhang, Y.; Dong, L. Regulatory mechanism of pyrrolidine dithiocarbamate is mediated by nuclear factor-κB and inhibits neutrophil accumulation in ARDS mice. Exp. Ther. Med. 2014, 8, 614–622. [Google Scholar] [CrossRef]
- Miao, C.; Lv, Y.; Zhang, W.; Chai, X.; Feng, L.; Fang, Y.; Liu, X.; Zhang, X. Pyrrolidine Dithiocarbamate (PDTC) Attenuates Cancer Cachexia by Affecting Muscle Atrophy and Fat Lipolysis. Front. Pharmacol. 2017, 8, 915. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Zhou, X.; Ji, T.; Chen, G. NF-κB p65-dependent transcriptional regulation of histone deacetylase 2 contributes to the chronic constriction injury-induced neuropathic pain via the microRNA-183/TXNIP/NLRP3 axis. J. Neuroinflammation 2020, 17, 225. [Google Scholar] [CrossRef] [PubMed]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Fan, J.; Wray, J.; Meng, X.; Shen, Z. BCCIP is required for the nuclear localization of the p21 protein. Cell Cycle 2009, 8, 3019–3024. [Google Scholar] [CrossRef]
- Meijer, A.H.; van der Sar, A.M.; Cunha, C.; Lamers, G.E.; Laplante, M.A.; Kikuta, H.; Bitter, W.; Becker, T.S.; Spaink, H.P. Identification and real-time imaging of a myc-expressing neutrophil population involved in inflammation and mycobacterial granuloma formation in zebrafish. Dev. Comp. Immunol. 2008, 32, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, M.; Nandakumar, J.; Sullivan, K.D.; Espinosa, J.M.; Cech, T.R. Inhibition of telomerase recruitment and cancer cell death. J. Biol. Chem. 2013, 288, 33171–33180. [Google Scholar] [CrossRef]
- Ward, R.J.; Autexier, C. Pharmacological telomerase inhibition can sensitize drug-resistant and drug-sensitive cells to chemotherapeutic treatment. Mol. Pharmacol. 2005, 68, 779–786. [Google Scholar] [CrossRef]
- Wu, X.Q.; Yang, Y.; Li, W.X.; Cheng, Y.H.; Li, X.F.; Huang, C.; Meng, X.M.; Wu, B.M.; Liu, X.H.; Zhang, L.; et al. Telomerase reverse transcriptase acts in a feedback loop with NF-κB pathway to regulate macrophage polarization in alcoholic liver disease. Sci. Rep. 2016, 6, 18685. [Google Scholar] [CrossRef] [PubMed]
- Rafat, A.; Dizaji Asl, K.; Mazloumi, Z.; Movassaghpour, A.A.; Talebi, M.; Shanehbandi, D.; Farahzadi, R.; Nejati, B.; Nozad Charoudeh, H. Telomerase inhibition on acute myeloid leukemia stem cell induced apoptosis with both intrinsic and extrinsic pathways. Life Sci. 2022, 295, 120402. [Google Scholar] [CrossRef] [PubMed]
- Giridharan, S.; Srinivasan, M. Mechanisms of NF-κB p65 and strategies for therapeutic manipulation. J. Inflamm. Res. 2018, 11, 407–419. [Google Scholar] [CrossRef]
- Thornburg, N.J.; Kulwichit, W.; Edwards, R.H.; Shair, K.H.; Bendt, K.M.; Raab-Traub, N. LMP1 signaling and activation of NF-kappaB in LMP1 transgenic mice. Oncogene 2006, 25, 288–297. [Google Scholar] [CrossRef]
- Song, Y.J.; Jen, K.Y.; Soni, V.; Kieff, E.; Cahir-McFarland, E. IL-1 receptor-associated kinase 1 is critical for latent membrane protein 1-induced p65/RelA serine 536 phosphorylation and NF-kappaB activation. Proc. Natl. Acad. Sci. USA 2006, 103, 2689–2694. [Google Scholar] [CrossRef]
- Giunco, S.; Celeghin, A.; Gianesin, K.; Dolcetti, R.; Indraccolo, S.; De Rossi, A. Cross talk between EBV and telomerase: The role of TERT and NOTCH2 in the switch of latent/lytic cycle of the virus. Cell Death Dis. 2015, 6, e1774. [Google Scholar] [CrossRef]
- Gartel, A.L.; Shchors, K. Mechanisms of c-myc-mediated transcriptional repression of growth arrest genes. Exp. Cell Res. 2003, 283, 17–21. [Google Scholar] [CrossRef]
- Brenner, C.; Deplus, R.; Didelot, C.; Loriot, A.; Viré, E.; De Smet, C.; Gutierrez, A.; Danovi, D.; Bernard, D.; Boon, T.; et al. Myc represses transcription through recruitment of DNA methyltransferase corepressor. EMBO J. 2005, 24, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Koundrioukoff, S.; Carignon, S.; Técher, H.; Letessier, A.; Brison, O.; Debatisse, M. Stepwise activation of the ATR signaling pathway upon increasing replication stress impacts fragile site integrity. PLoS Genet. 2013, 9, e1003643. [Google Scholar] [CrossRef] [PubMed]
- Chiappori, A.A.; Kolevska, T.; Spigel, D.R.; Hager, S.; Rarick, M.; Gadgeel, S.; Blais, N.; Von Pawel, J.; Hart, L.; Reck, M.; et al. A randomized phase II study of the telomerase inhibitor imetelstat as maintenance therapy for advanced non-small-cell lung cancer. Ann. Oncol. 2015, 26, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.A.; Drissi, R.; Muscal, J.A.; Panditharatna, E.; Fouladi, M.; Ingle, A.M.; Ahern, C.H.; Reid, J.M.; Lin, T.; Weigel, B.J.; et al. A phase I trial of imetelstat in children with refractory or recurrent solid tumors: A Children’s Oncology Group Phase I Consortium Study (ADVL1112). Clin. Cancer Res. 2013, 19, 6578–6584. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amin, A.; Morello, M.; Petrara, M.R.; Rizzo, B.; Argenton, F.; De Rossi, A.; Giunco, S. Short-Term TERT Inhibition Impairs Cellular Proliferation via a Telomere Length-Independent Mechanism and Can Be Exploited as a Potential Anticancer Approach. Cancers 2023, 15, 2673. https://doi.org/10.3390/cancers15102673
Amin A, Morello M, Petrara MR, Rizzo B, Argenton F, De Rossi A, Giunco S. Short-Term TERT Inhibition Impairs Cellular Proliferation via a Telomere Length-Independent Mechanism and Can Be Exploited as a Potential Anticancer Approach. Cancers. 2023; 15(10):2673. https://doi.org/10.3390/cancers15102673
Chicago/Turabian StyleAmin, Aamir, Marzia Morello, Maria Raffaella Petrara, Beatrice Rizzo, Francesco Argenton, Anita De Rossi, and Silvia Giunco. 2023. "Short-Term TERT Inhibition Impairs Cellular Proliferation via a Telomere Length-Independent Mechanism and Can Be Exploited as a Potential Anticancer Approach" Cancers 15, no. 10: 2673. https://doi.org/10.3390/cancers15102673
APA StyleAmin, A., Morello, M., Petrara, M. R., Rizzo, B., Argenton, F., De Rossi, A., & Giunco, S. (2023). Short-Term TERT Inhibition Impairs Cellular Proliferation via a Telomere Length-Independent Mechanism and Can Be Exploited as a Potential Anticancer Approach. Cancers, 15(10), 2673. https://doi.org/10.3390/cancers15102673