
Kinase Inhibitors in the Treatment of Ovarian Cancer: Current State and Future Promises
 , , , and
, , , and 
Abstract
Simple Summary
Abstract
1. Background
1.1. Epithelial- and High Grade Serous Ovarian Carcinoma
1.2. Development of the Current Treatment
1.3. Challenges in Developing New Treatments
1.4. Kinase Inhibitors as Cancer Treatments in General
2. Current Progress with Small-Molecule Kinase Inhibitors as Targeted Treatment for HGSC
2.1. Many Less and Few More Promising Attempts
2.2. Targeting Receptor Tyrosine Kinases (RTKs)
2.2.1. Aiming at Upregulated ErbB Family Receptors
2.2.2. Exploiting High Angiogenic Drive
2.2.3. Exploring Oncogenic Potential of FGFR
2.2.4. Probing the Complex Network of IGF Signaling
2.3. Targeting Intracellular Signaling Cascades
2.3.1. PI3K-AKT-mTOR Arm
2.3.2. RAS-RAF-MEK-ERK (MAPK) Arm
2.3.3. Targeting Cell-Cycle Machinery
2.4. Kinase Inhibitors in Recently Concluded Clinical Trials—What Is Promising?
2.4.1. Multi-Targeted Anti-Angiogenic TKIs
2.4.2. Targeting Intracellular Pathways
3. Kinase Inhibitors in Ongoing Clinical Trials—What to Expect?
4. Promising Preclinical Studies Using Ovarian Cancer Organoids and Mouse Models—New Arising, Promising Treatments?
4.1. Patient-Derived Organoid Cultures as Indicative Model Systems for Preclinical Drug Validation
4.1.1. Kinase Inhibition Responses Differ among PDO Cultures
4.1.2. Synergistic Effect of Kinase Inhibition and SOC on PARP- or Platinum-Resistant PDOs
4.2. Lessons to Be Learned from Recent In Vivo Studies Conducted with Xenograft Models
4.2.1. Ingenious and Rational Drug Combinations with Kinase Inhibition Should Be Explored
4.2.2. Targeting Focal Adhesion Kinase (FAK) and Anaplastic Lymphoma Kinase (ALK)
4.2.3. Multiple Targeting of Cell-Cycle, Cell-Proliferation and Survival Pathways
5. Concluding Remarks and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Tomao, F.; D’Incalci, M.; Biagioli, E.; Peccatori, F.A.; Colombo, N. Restoring platinum sensitivity in recurrent ovarian cancer by extending the platinum-free interval: Myth or reality? Cancer 2017, 123, 3450–3459. [Google Scholar] [CrossRef]
- Loret, N.; Denys, H.; Tummers, P.; Berx, G. The Role of Epithelial-to-Mesenchymal Plasticity in Ovarian Cancer Progression and Therapy Resistance. Cancers 2019, 11, 838. [Google Scholar] [CrossRef] [PubMed]
- Kenny, H.A.; Nieman, K.M.; Mitra, A.K.; Lengyel, E. The first line of intra-abdominal metastatic attack: Breaching the mesothelial cell layer. Cancer Discov. 2011, 1, 100–102. [Google Scholar] [CrossRef]
- Bhatla, N.; Jones, A. The World Ovarian Cancer Coalition Atlas. 2018, 1, 1–39. Available online: https://worldovariancancercoalition.org/wp-content/uploads/2018/10/THE-WORLD-OVARIAN-CANCER-COALITION-ATLAS-2018.pdf (accessed on 3 October 2022).
- Wiltshaw, E.; Kroner, T. Phase II study of cis-dichlorodiammineplatinum(II) (NSC-119875) in advanced adenocarcinoma of the ovary. Cancer Treat. Rep. 1976, 60, 55–60. [Google Scholar]
- Piccart, M.J.; Bertelsen, K.; James, K.; Cassidy, J.; Mangioni, C.; Simonsen, E.; Stuart, G.; Kaye, S.; Vergote, I.; Blom, R.; et al. Randomized intergroup trial of cisplatin-paclitaxel versus cisplatin-cyclophosphamide in women with advanced epithelial ovarian cancer: Three-year results. J. Natl. Cancer Inst. 2000, 92, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Muggia, F.M.; Braly, P.S.; Brady, M.F.; Sutton, G.; Niemann, T.H.; Lentz, S.L.; Alvarez, R.D.; Kucera, P.R.; Small, J.M. Phase III randomized study of cisplatin versus paclitaxel versus cisplatin and paclitaxel in patients with suboptimal stage III or IV ovarian cancer: A gynecologic oncology group study. J. Clin. Oncol. 2000, 18, 106–115. [Google Scholar] [CrossRef]
- Mahmood, R.D.; Morgan, R.D.; Edmondson, R.J.; Clamp, A.R.; Jayson, G.C. First-Line Management of Advanced High-Grade Serous Ovarian Cancer. Curr. Oncol. Rep. 2020, 22, 64. [Google Scholar] [CrossRef]
- Baert, T.; Ferrero, A.; Sehouli, J.; O’Donnell, D.M.; Gonzalez-Martin, A.; Joly, F.; van der Velden, J.; Blecharz, P.; Tan, D.S.P.; Querleu, D.; et al. The systemic treatment of recurrent ovarian cancer revisited. Ann. Oncol. 2021, 32, 710–725. [Google Scholar] [CrossRef]
- Liu, J.F.; Barry, W.T.; Birrer, M.; Lee, J.M.; Buckanovich, R.J.; Fleming, G.F.; Rimel, B.; Buss, M.K.; Nattam, S.; Hurteau, J.; et al. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: A randomised phase 2 study. Lancet Oncol. 2014, 15, 1207–1214. [Google Scholar] [CrossRef]
- Alvarez Secord, A.; O’Malley, D.M.; Sood, A.K.; Westin, S.N.; Liu, J.F. Rationale for combination PARP inhibitor and antiangiogenic treatment in advanced epithelial ovarian cancer: A review. Gynecol. Oncol. 2021, 162, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, R.; Shao, W.; Sellers, W.R. Oncogene addiction: Pathways of therapeutic response, resistance, and road maps toward a cure. EMBO Rep. 2015, 16, 280–296. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.A.; Etemadmoghadam, D.; Temple, J.; Lynch, A.G.; Riad, M.; Sharma, R.; Stewart, C.; Fereday, S.; Caldas, C.; Defazio, A.; et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J. Pathol. 2010, 221, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Cao, J.; Topatana, W.; Juengpanich, S.; Li, S.; Zhang, B.; Shen, J.; Cai, L.; Cai, X.; Chen, M. Targeting mutant p53 for cancer therapy: Direct and indirect strategies. J. Hematol. Oncol. 2021, 14, 157. [Google Scholar] [CrossRef]
- Chardin, L.; Leary, A. Immunotherapy in Ovarian Cancer: Thinking Beyond PD-1/PD-L1. Front. Oncol. 2021, 11, 795547. [Google Scholar] [CrossRef]
- Palaia, I.; Tomao, F.; Sassu, C.M.; Musacchio, L.; Benedetti Panici, P. Immunotherapy For Ovarian Cancer: Recent Advances And Combination Therapeutic Approaches. OncoTargets Ther. 2020, 13, 6109–6129. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Cross, D.; Janne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Yap, T.A.; Carden, C.P.; Kaye, S.B. Beyond chemotherapy: Targeted therapies in ovarian cancer. Nat. Rev. Cancer 2009, 9, 167–181. [Google Scholar] [CrossRef]
- Kohler, M.; Janz, I.; Wintzer, H.O.; Wagner, E.; Bauknecht, T. The expression of EGF receptors, EGF-like factors and c-myc in ovarian and cervical carcinomas and their potential clinical significance. Anticancer Res. 1989, 9, 1537–1547. [Google Scholar]
- Kohler, M.; Bauknecht, T.; Grimm, M.; Birmelin, G.; Kommoss, F.; Wagner, E. Epidermal growth factor receptor and transforming growth factor alpha expression in human ovarian carcinomas. Eur. J. Cancer 1992, 28A, 1432–1437. [Google Scholar] [CrossRef]
- Bonello, M.; Sims, A.H.; Langdon, S.P. Human epidermal growth factor receptor targeted inhibitors for the treatment of ovarian cancer. Cancer Biol. Med. 2018, 15, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Q.; Liu, J. The therapeutic potential of targeting the EGFR family in epithelial ovarian cancer. Br. J. Cancer 2011, 104, 1241–1245. [Google Scholar] [CrossRef]
- Chelariu-Raicu, A.; Levenback, C.F.; Slomovitz, B.M.; Wolf, J.; Bodurka, D.C.; Kavanagh, J.J.; Morrison, C.; Gershenson, D.M.; Coleman, R.L. Phase Ib/II study of weekly topotecan and daily gefitinib in patients with platinum resistant ovarian, peritoneal, or fallopian tube cancer. Int. J. Gynecol. Cancer 2020, 30, 1768–1774. [Google Scholar] [CrossRef]
- Tuefferd, M.; Couturier, J.; Penault-Llorca, F.; Vincent-Salomon, A.; Broet, P.; Guastalla, J.P.; Allouache, D.; Combe, M.; Weber, B.; Pujade-Lauraine, E.; et al. HER2 status in ovarian carcinomas: A multicenter GINECO study of 320 patients. PLoS ONE 2007, 2, e1138. [Google Scholar] [CrossRef] [PubMed]
- Oikkonen, J.; Zhang, K.; Salminen, L.; Schulman, I.; Lavikka, K.; Andersson, N.; Ojanpera, E.; Hietanen, S.; Grenman, S.; Lehtonen, R.; et al. Prospective Longitudinal ctDNA Workflow Reveals Clinically Actionable Alterations in Ovarian Cancer. JCO Precis. Oncol. 2019, 3, 1–12. [Google Scholar] [CrossRef]
- Wilken, J.A.; Badri, T.; Cross, S.; Raji, R.; Santin, A.D.; Schwartz, P.; Branscum, A.J.; Baron, A.T.; Sakhitab, A.I.; Maihle, N.J. EGFR/HER-targeted therapeutics in ovarian cancer. Future Med. Chem. 2012, 4, 447–469. [Google Scholar] [CrossRef]
- Davies, S.; Holmes, A.; Lomo, L.; Steinkamp, M.P.; Kang, H.; Muller, C.Y.; Wilson, B.S. High incidence of ErbB3, ErbB4, and MET expression in ovarian cancer. Int. J. Gynecol. Pathol. 2014, 33, 402–410. [Google Scholar] [CrossRef]
- Saglam, O.; Xiong, Y.; Marchion, D.C.; Strosberg, C.; Wenham, R.M.; Johnson, J.J.; Saeed-Vafa, D.; Cubitt, C.; Hakam, A.; Magliocco, A.M. ERBB4 Expression in Ovarian Serous Carcinoma Resistant to Platinum-Based Therapy. Cancer Control 2017, 24, 89–95. [Google Scholar] [CrossRef][Green Version]
- Herr, D.; Sallmann, A.; Bekes, I.; Konrad, R.; Holzheu, I.; Kreienberg, R.; Wulff, C. VEGF induces ascites in ovarian cancer patients via increasing peritoneal permeability by downregulation of Claudin 5. Gynecol. Oncol. 2012, 127, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Burger, R.A.; Brady, M.F.; Bookman, M.A.; Fleming, G.F.; Monk, B.J.; Huang, H.; Mannel, R.S.; Homesley, H.D.; Fowler, J.; Greer, B.E.; et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N. Engl. J. Med. 2011, 365, 2473–2483. [Google Scholar] [CrossRef] [PubMed]
- Perren, T.J.; Swart, A.M.; Pfisterer, J.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; Kurzeder, C.; et al. A phase 3 trial of bevacizumab in ovarian cancer. N. Engl. J. Med. 2011, 365, 2484–2496. [Google Scholar] [CrossRef]
- Jin, M.; Cai, J.; Wang, X.; Zhang, T.; Zhao, Y. Successful maintenance therapy with apatinib inplatinum-resistant advanced ovarian cancer and literature review. Cancer Biol. Ther. 2018, 19, 1088–1092. [Google Scholar] [CrossRef] [PubMed]
- Orbegoso, C.; Marquina, G.; George, A.; Banerjee, S. The role of Cediranib in ovarian cancer. Expert Opin. Pharmacother. 2017, 18, 1637–1648. [Google Scholar] [CrossRef]
- Coleman, R.L.; Broaddus, R.R.; Bodurka, D.C.; Wolf, J.K.; Burke, T.W.; Kavanagh, J.J.; Levenback, C.F.; Gershenson, D.M. Phase II trial of imatinib mesylate in patients with recurrent platinum- and taxane-resistant epithelial ovarian and primary peritoneal cancers. Gynecol. Oncol. 2006, 101, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Schilder, R.J.; Sill, M.W.; Lee, R.B.; Shaw, T.J.; Senterman, M.K.; Klein-Szanto, A.J.; Miner, Z.; Vanderhyden, B.C. Phase II evaluation of imatinib mesylate in the treatment of recurrent or persistent epithelial ovarian or primary peritoneal carcinoma: A Gynecologic Oncology Group Study. J. Clin. Oncol. 2008, 26, 3418–3425. [Google Scholar] [CrossRef] [PubMed]
- Matei, D.; Emerson, R.E.; Schilder, J.; Menning, N.; Baldridge, L.A.; Johnson, C.S.; Breen, T.; McClean, J.; Stephens, D.; Whalen, C.; et al. Imatinib mesylate in combination with docetaxel for the treatment of patients with advanced, platinum-resistant ovarian cancer and primary peritoneal carcinomatosis: A Hoosier Oncology Group trial. Cancer 2008, 113, 723–732. [Google Scholar] [CrossRef]
- Posadas, E.M.; Kwitkowski, V.; Kotz, H.L.; Espina, V.; Minasian, L.; Tchabo, N.; Premkumar, A.; Hussain, M.M.; Chang, R.; Steinberg, S.M.; et al. A prospective analysis of imatinib-induced c-KIT modulation in ovarian cancer: A phase II clinical study with proteomic profiling. Cancer 2007, 110, 309–317. [Google Scholar] [CrossRef]
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR Landscape in Cancer: Analysis of 4,853 Tumors by Next-Generation Sequencing. Clin. Cancer Res. 2016, 22, 259–267. [Google Scholar] [CrossRef]
- Steele, I.A.; Edmondson, R.J.; Bulmer, J.N.; Bolger, B.S.; Leung, H.Y.; Davies, B.R. Induction of FGF receptor 2-IIIb expression and response to its ligands in epithelial ovarian cancer. Oncogene 2001, 20, 5878–5887. [Google Scholar] [CrossRef]
- Byron, S.A.; Gartside, M.G.; Wellens, C.L.; Goodfellow, P.J.; Birrer, M.J.; Campbell, I.G.; Pollock, P.M. FGFR2 mutations are rare across histologic subtypes of ovarian cancer. Gynecol. Oncol. 2010, 117, 125–129. [Google Scholar] [CrossRef]
- Oosterhuis, G.J.; Vermes, I.; Lambalk, C.B.; Michgelsen, H.W.; Schoemaker, J. Insulin-like growth factor (IGF)-I and IGF binding protein-3 concentrations in fluid from human stimulated follicles. Hum. Reprod. 1998, 13, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Yee, D.; Morales, F.R.; Hamilton, T.C.; Von Hoff, D.D. Expression of insulin-like growth factor I, its binding proteins, and its receptor in ovarian cancer. Cancer Res. 1991, 51, 5107–5112. [Google Scholar]
- Resnicoff, M.; Ambrose, D.; Coppola, D.; Rubin, R. Insulin-like growth factor-1 and its receptor mediate the autocrine proliferation of human ovarian carcinoma cell lines. Lab. Investig. 1993, 69, 756–760. [Google Scholar] [PubMed]
- Pejovic, T.; Pande, N.T.; Mori, M.; Mhawech-Fauceglia, P.; Harrington, C.; Mongoue-Tchokote, S.; Dim, D.; Andrews, C.; Beck, A.; Tarumi, Y.; et al. Expression profiling of the ovarian surface kinome reveals candidate genes for early neoplastic changes. Transl. Oncol. 2009, 2, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Tap, W.D.; Demetri, G.; Barnette, P.; Desai, J.; Kavan, P.; Tozer, R.; Benedetto, P.W.; Friberg, G.; Deng, H.; McCaffery, I.; et al. Phase II study of ganitumab, a fully human anti-type-1 insulin-like growth factor receptor antibody, in patients with metastatic Ewing family tumors or desmoplastic small round cell tumors. J. Clin. Oncol. 2012, 30, 1849–1856. [Google Scholar] [CrossRef]
- Hubbard, R.D.; Wilsbacher, J.L. Advances towards the development of ATP-competitive small-molecule inhibitors of the insulin-like growth factor receptor (IGF-IR). Chem. Med. Chem. 2007, 2, 41–46. [Google Scholar] [CrossRef]
- Liefers-Visser, J.A.L.; Meijering, R.A.M.; Reyners, A.K.L.; van der Zee, A.G.J.; de Jong, S. IGF system targeted therapy: Therapeutic opportunities for ovarian cancer. Cancer Treat. Rev. 2017, 60, 90–99. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Montalto, G.; Cervello, M.; Nicoletti, F.; Fagone, P.; Malaponte, G.; Mazzarino, M.C.; et al. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget 2012, 3, 954–987. [Google Scholar] [CrossRef]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Levine, D.A.; Bogomolniy, F.; Yee, C.J.; Lash, A.; Barakat, R.R.; Borgen, P.I.; Boyd, J. Frequent mutation of the PIK3CA gene in ovarian and breast cancers. Clin. Cancer Res. 2005, 11, 2875–2878. [Google Scholar] [CrossRef]
- Campbell, I.G.; Russell, S.E.; Choong, D.Y.; Montgomery, K.G.; Ciavarella, M.L.; Hooi, C.S.; Cristiano, B.E.; Pearson, R.B.; Phillips, W.A. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res. 2004, 64, 7678–7681. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Hirsch, M.; Palescandolo, E.; Kim, E.; Liu, J.; van Hummelen, P.; MacConaill, L.; Drapkin, R.; Hahn, W.C. High throughput interrogation of somatic mutations in high grade serous cancer of the ovary. PLoS ONE 2011, 6, e24433. [Google Scholar] [CrossRef]
- Shayesteh, L.; Lu, Y.; Kuo, W.L.; Baldocchi, R.; Godfrey, T.; Collins, C.; Pinkel, D.; Powell, B.; Mills, G.B.; Gray, J.W. PIK3CA is implicated as an oncogene in ovarian cancer. Nat. Genet. 1999, 21, 99–102. [Google Scholar] [CrossRef]
- Dolly, S.O.; Wagner, A.J.; Bendell, J.C.; Kindler, H.L.; Krug, L.M.; Seiwert, T.Y.; Zauderer, M.G.; Lolkema, M.P.; Apt, D.; Yeh, R.F.; et al. Phase I Study of Apitolisib (GDC-0980), Dual Phosphatidylinositol-3-Kinase and Mammalian Target of Rapamycin Kinase Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2016, 22, 2874–2884. [Google Scholar] [CrossRef]
- Wicki, A.; Brown, N.; Xyrafas, A.; Bize, V.; Hawle, H.; Berardi, S.; Cmiljanovic, N.; Cmiljanovic, V.; Stumm, M.; Dimitrijevic, S.; et al. First-in human, phase 1, dose-escalation pharmacokinetic and pharmacodynamic study of the oral dual PI3K and mTORC1/2 inhibitor PQR309 in patients with advanced solid tumors (SAKK 67/13). Eur. J. Cancer 2018, 96, 6–16. [Google Scholar] [CrossRef]
- Mahadevan, D.; Chiorean, E.G.; Harris, W.B.; Von Hoff, D.D.; Stejskal-Barnett, A.; Qi, W.; Anthony, S.P.; Younger, A.E.; Rensvold, D.M.; Cordova, F.; et al. Phase I pharmacokinetic and pharmacodynamic study of the pan-PI3K/mTORC vascular targeted pro-drug SF1126 in patients with advanced solid tumours and B-cell malignancies. Eur. J. Cancer 2012, 48, 3319–3327. [Google Scholar] [CrossRef]
- Markman, B.; Tabernero, J.; Krop, I.; Shapiro, G.I.; Siu, L.; Chen, L.C.; Mita, M.; Melendez Cuero, M.; Stutvoet, S.; Birle, D.; et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of the oral phosphatidylinositol-3-kinase and mTOR inhibitor BGT226 in patients with advanced solid tumors. Ann. Oncol. 2012, 23, 2399–2408. [Google Scholar] [CrossRef]
- Shapiro, G.I.; Bell-McGuinn, K.M.; Molina, J.R.; Bendell, J.; Spicer, J.; Kwak, E.L.; Pandya, S.S.; Millham, R.; Borzillo, G.; Pierce, K.J.; et al. First-in-Human Study of PF-05212384 (PKI-587), a Small-Molecule, Intravenous, Dual Inhibitor of PI3K and mTOR in Patients with Advanced Cancer. Clin. Cancer Res. 2015, 21, 1888–1895. [Google Scholar] [CrossRef]
- Wu, P.K.; Becker, A.; Park, J.I. Growth Inhibitory Signaling of the Raf/MEK/ERK Pathway. Int. J. Mol. Sci. 2020, 21, 5436. [Google Scholar] [CrossRef]
- Spaans, V.M.; Trietsch, M.D.; Crobach, S.; Stelloo, E.; Kremer, D.; Osse, E.M.; Haar, N.T.; van Eijk, R.; Muller, S.; van Wezel, T.; et al. Designing a high-throughput somatic mutation profiling panel specifically for gynaecological cancers. PLoS ONE 2014, 9, e93451. [Google Scholar] [CrossRef]
- Janku, F.; Lee, J.J.; Tsimberidou, A.M.; Hong, D.S.; Naing, A.; Falchook, G.S.; Fu, S.; Luthra, R.; Garrido-Laguna, I.; Kurzrock, R. PIK3CA mutations frequently coexist with RAS and BRAF mutations in patients with advanced cancers. PLoS ONE 2011, 6, e22769. [Google Scholar] [CrossRef]
- Holderfield, M.; Deuker, M.M.; McCormick, F.; McMahon, M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat. Rev. Cancer 2014, 14, 455–467. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Champer, M.; Miller, D.; Kuo, D.Y. Response to trametinib in recurrent low-grade serous ovarian cancer with NRAS mutation: A case report. Gynecol. Oncol. Rep. 2019, 28, 26–28. [Google Scholar] [CrossRef]
- Farley, J.; Brady, W.E.; Vathipadiekal, V.; Lankes, H.A.; Coleman, R.; Morgan, M.A.; Mannel, R.; Yamada, S.D.; Mutch, D.; Rodgers, W.H.; et al. Selumetinib in women with recurrent low-grade serous carcinoma of the ovary or peritoneum: An open-label, single-arm, phase 2 study. Lancet Oncol. 2013, 14, 134–140. [Google Scholar] [CrossRef]
- Liu, S.; Zou, Q.; Chen, J.P.; Yao, X.; Guan, P.; Liang, W.; Deng, P.; Lai, X.; Yin, J.; Chen, J.; et al. Targeting enhancer reprogramming to mitigate MEK inhibitor resistance in preclinical models of advanced ovarian cancer. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Campbell, R.M.; Anderson, B.D.; Brooks, N.A.; Brooks, H.B.; Chan, E.M.; De Dios, A.; Gilmour, R.; Graff, J.R.; Jambrina, E.; Mader, M.; et al. Characterization of LY2228820 dimesylate, a potent and selective inhibitor of p38 MAPK with antitumor activity. Mol. Cancer Ther. 2014, 13, 364–374. [Google Scholar] [CrossRef]
- Ishitsuka, K.; Hideshima, T.; Neri, P.; Vallet, S.; Shiraishi, N.; Okawa, Y.; Shen, Z.; Raje, N.; Kiziltepe, T.; Ocio, E.M.; et al. p38 mitogen-activated protein kinase inhibitor LY2228820 enhances bortezomib-induced cytotoxicity and inhibits osteoclastogenesis in multiple myeloma; therapeutic implications. Br. J. Haematol. 2008, 141, 598–606. [Google Scholar] [CrossRef]
- Aesoy, R.; Sanchez, B.C.; Norum, J.H.; Lewensohn, R.; Viktorsson, K.; Linderholm, B. An autocrine VEGF/VEGFR2 and p38 signaling loop confers resistance to 4-hydroxytamoxifen in MCF-7 breast cancer cells. Mol. Cancer Res. 2008, 6, 1630–1638. [Google Scholar] [CrossRef]
- Patnaik, A.; Haluska, P.; Tolcher, A.W.; Erlichman, C.; Papadopoulos, K.P.; Lensing, J.L.; Beeram, M.; Molina, J.R.; Rasco, D.W.; Arcos, R.R.; et al. A First-in-Human Phase I Study of the Oral p38 MAPK Inhibitor, Ralimetinib (LY2228820 Dimesylate), in Patients with Advanced Cancer. Clin. Cancer Res. 2016, 22, 1095–1102. [Google Scholar] [CrossRef]
- Vergote, I.; Heitz, F.; Buderath, P.; Powell, M.; Sehouli, J.; Lee, C.M.; Hamilton, A.; Fiorica, J.; Moore, K.N.; Teneriello, M.; et al. A randomized, double-blind, placebo-controlled phase 1b/2 study of ralimetinib, a p38 MAPK inhibitor, plus gemcitabine and carboplatin versus gemcitabine and carboplatin for women with recurrent platinum-sensitive ovarian cancer. Gynecol. Oncol. 2020, 156, 23–31. [Google Scholar] [CrossRef]
- Lin, Z.P.; Zhu, Y.L.; Ratner, E.S. Targeting Cyclin-Dependent Kinases for Treatment of Gynecologic Cancers. Front. Oncol. 2018, 8, 303. [Google Scholar] [CrossRef]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef]
- Dall’Acqua, A.; Bartoletti, M.; Masoudi-Khoram, N.; Sorio, R.; Puglisi, F.; Belletti, B.; Baldassarre, G. Inhibition of CDK4/6 as Therapeutic Approach for Ovarian Cancer Patients: Current Evidences and Future Perspectives. Cancers 2021, 13, 3035. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef]
- Sausville, E.; Lorusso, P.; Carducci, M.; Carter, J.; Quinn, M.F.; Malburg, L.; Azad, N.; Cosgrove, D.; Knight, R.; Barker, P.; et al. Phase I dose-escalation study of AZD7762, a checkpoint kinase inhibitor, in combination with gemcitabine in US patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 539–549. [Google Scholar] [CrossRef]
- Daud, A.I.; Ashworth, M.T.; Strosberg, J.; Goldman, J.W.; Mendelson, D.; Springett, G.; Venook, A.P.; Loechner, S.; Rosen, L.S.; Shanahan, F.; et al. Phase I dose-escalation trial of checkpoint kinase 1 inhibitor MK-8776 as monotherapy and in combination with gemcitabine in patients with advanced solid tumors. J. Clin. Oncol. 2015, 33, 1060–1066. [Google Scholar] [CrossRef]
- Slipicevic, A.; Holth, A.; Hellesylt, E.; Trope, C.G.; Davidson, B.; Florenes, V.A. Wee1 is a novel independent prognostic marker of poor survival in post-chemotherapy ovarian carcinoma effusions. Gynecol. Oncol. 2014, 135, 118–124. [Google Scholar] [CrossRef]
- Lheureux, S.; Cristea, M.C.; Bruce, J.P.; Garg, S.; Cabanero, M.; Mantia-Smaldone, G.; Olawaiye, A.B.; Ellard, S.L.; Weberpals, J.I.; Wahner Hendrickson, A.E.; et al. Adavosertib plus gemcitabine for platinum-resistant or platinum-refractory recurrent ovarian cancer: A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2021, 397, 281–292. [Google Scholar] [CrossRef]
- Oza, A.M.; Estevez-Diz, M.; Grischke, E.M.; Hall, M.; Marme, F.; Provencher, D.; Uyar, D.; Weberpals, J.I.; Wenham, R.M.; Laing, N.; et al. A Biomarker-enriched, Randomized Phase II Trial of Adavosertib (AZD1775) Plus Paclitaxel and Carboplatin for Women with Platinum-sensitive TP53-mutant Ovarian Cancer. Clin. Cancer Res. 2020, 26, 4767–4776. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Wang, T.; Tang, J.; Yang, H.; Yin, R.; Zhang, J.; Zhou, Q.; Liu, Z.; Cao, L.; Li, L.; Huang, Y.; et al. Effect of Apatinib Plus Pegylated Liposomal Doxorubicin vs. Pegylated Liposomal Doxorubicin Alone on Platinum-Resistant Recurrent Ovarian Cancer: The APPROVE Randomized Clinical Trial. JAMA Oncol. 2022, 8, 1169–1176. [Google Scholar] [CrossRef]
- Miao, M.; Deng, G.; Luo, S.; Zhou, J.; Chen, L.; Yang, J.; He, J.; Li, J.; Yao, J.; Tan, S.; et al. A phase II study of apatinib in patients with recurrent epithelial ovarian cancer. Gynecol. Oncol. 2018, 148, 286–290. [Google Scholar] [CrossRef]
- Liu, J.F.; Brady, M.F.; Matulonis, U.; Miller, A.; Kohn, E.C.; Swisher, E.M.; Cella, D.; Tew, W.P.; Cloven, N.G.; Muller, C.Y.; et al. Olaparib With or Without Cediranib Versus Platinum-Based Chemotherapy in Recurrent Platinum-Sensitive Ovarian Cancer (NRG-Gy004): A Randomized, Open-Label, Phase III Trial. Clin. Oncol. 2022, 40, 2138–2147. [Google Scholar] [CrossRef]
- Colombo, N.; Tomao, F.; Benedetti Panici, P.; Nicoletto, M.O.; Tognon, G.; Bologna, A.; Lissoni, A.A.; DeCensi, A.; Lapresa, M.; Mancari, R.; et al. Randomized phase II trial of weekly paclitaxel vs. cediranib-olaparib (continuous or intermittent schedule) in platinum-resistant high-grade epithelial ovarian cancer. Gynecol. Oncol. 2022, 164, 505–513. [Google Scholar] [CrossRef]
- Lheureux, S.; Oaknin, A.; Garg, S.; Bruce, J.P.; Madariaga, A.; Dhani, N.C.; Bowering, V.; White, J.; Accardi, S.; Tan, Q.; et al. EVOLVE: A Multicenter Open-Label Single-Arm Clinical and Translational Phase II Trial of Cediranib Plus Olaparib for Ovarian Cancer after PARP Inhibition Progression. Clin. Cancer Res. 2020, 26, 4206–4215. [Google Scholar] [CrossRef]
- Zimmer, A.S.; Nichols, E.; Cimino-Mathews, A.; Peer, C.; Cao, L.; Lee, M.J.; Kohn, E.C.; Annunziata, C.M.; Lipkowitz, S.; Trepel, J.B.; et al. A phase I study of the PD-L1 inhibitor, durvalumab, in combination with a PARP inhibitor, olaparib, and a VEGFR1-3 inhibitor, cediranib, in recurrent women’s cancers with biomarker analyses. J. Immunother. Cancer 2019, 7, 197. [Google Scholar] [CrossRef]
- Ledermann, J.A.; Embleton, A.C.; Raja, F.A.; Perren, T.J.; Jayson, G.C.; Rustin, G.J.S.; Kaye, S.B.; Hirte, H.; Eisenhauer, E.A.; Vaughan, M.; et al. Cediranib in patients with relapsed platinum-sensitive ovarian cancer (ICON6): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2016, 387, 1066–1074. [Google Scholar] [CrossRef]
- Hirte, H.; Lheureux, S.; Fleming, G.F.; Sugimoto, A.; Morgan, R.; Biagi, J.; Wang, L.; McGill, S.; Ivy, S.P.; Oza, A.M. A phase 2 study of cediranib in recurrent or persistent ovarian, peritoneal or fallopian tube cancer: A trial of the Princess Margaret, Chicago and California Phase II Consortia. Gynecol. Oncol. 2015, 138, 55–61. [Google Scholar] [CrossRef]
- Liu, J.F.; Barry, W.T.; Birrer, M.; Lee, J.M.; Buckanovich, R.J.; Fleming, G.F.; Rimel, B.J.; Buss, M.K.; Nattam, S.R.; Hurteau, J.; et al. Overall survival and updated progression-free survival outcomes in a randomized phase II study of combination cediranib and olaparib versus olaparib in relapsed platinum-sensitive ovarian cancer. Ann. Oncol. 2019, 30, 551–557. [Google Scholar] [CrossRef]
- Cowan, M.; Swetzig, W.M.; Adorno-Cruz, V.; Pineda, M.J.; Neubauer, N.L.; Berry, E.; Lurain, J.R.; Shahabi, S.; Taiym, D.; Nelson, V.; et al. Efficacy and safety of tivozanib in recurrent, platinum-resistant ovarian, fallopian tube or primary peritoneal cancer, an NCCN phase II trial. Gynecol. Oncol. 2021, 163, 57–63. [Google Scholar] [CrossRef]
- Hall, M.R.; Dehbi, H.M.; Banerjee, S.; Lord, R.; Clamp, A.; Ledermann, J.A.; Nicum, S.; Lilleywhite, R.; Bowen, R.; Michael, A.; et al. A phase II randomised, placebo-controlled trial of low dose (metronomic) cyclophosphamide and nintedanib (BIBF1120) in advanced ovarian, fallopian tube or primary peritoneal cancer. Gynecol. Oncol. 2020, 159, 692–698. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Cibula, D.; Mirza, M.R.; Reuss, A.; Ricci, C.; Colombo, N.; Koch, H.; Goffin, F.; Gonzalez-Martin, A.; Ottevanger, P.B.; et al. Final results from GCIG/ENGOT/AGO-OVAR 12, a randomised placebo-controlled phase III trial of nintedanib combined with chemotherapy for newly diagnosed advanced ovarian cancer. Int. J. Cancer 2020, 146, 439–448. [Google Scholar] [CrossRef]
- du Bois, A.; Kristensen, G.; Ray-Coquard, I.; Reuss, A.; Pignata, S.; Colombo, N.; Denison, U.; Vergote, I.; Del Campo, J.M.; Ottevanger, P.; et al. Standard first-line chemotherapy with or without nintedanib for advanced ovarian cancer (AGO-OVAR 12): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2016, 17, 78–89. [Google Scholar] [CrossRef]
- Xia, L.; Peng, J.; Lou, G.; Pan, M.; Zhou, Q.; Hu, W.; Shi, H.; Wang, L.; Gao, Y.; Zhu, J.; et al. Antitumor activity and safety of camrelizumab plus famitinib in patients with platinum-resistant recurrent ovarian cancer: Results from an open-label, multicenter phase 2 basket study. J. Immunother. Cancer 2022, 10, e003831. [Google Scholar] [CrossRef]
- Vergote, I.; du Bois, A.; Floquet, A.; Rau, J.; Kim, J.W.; Del Campo, J.M.; Friedlander, M.; Pignata, S.; Fujiwara, K.; Colombo, N.; et al. Overall survival results of AGO-OVAR16: A phase 3 study of maintenance pazopanib versus placebo in women who have not progressed after first-line chemotherapy for advanced ovarian cancer. Gynecol. Oncol. 2019, 155, 186–191. [Google Scholar] [CrossRef]
- Richardson, D.L.; Sill, M.W.; Coleman, R.L.; Sood, A.K.; Pearl, M.L.; Kehoe, S.M.; Carney, M.E.; Hanjani, P.; Van Le, L.; Zhou, X.C.; et al. Paclitaxel With and Without Pazopanib for Persistent or Recurrent Ovarian Cancer: A Randomized Clinical Trial. JAMA Oncol. 2018, 4, 196–202. [Google Scholar] [CrossRef]
- Kim, J.W.; Mahner, S.; Wu, L.Y.; Shoji, T.; Kim, B.G.; Zhu, J.Q.; Takano, T.; Park, S.Y.; Kong, B.H.; Wu, Q.; et al. Pazopanib Maintenance Therapy in East Asian Women With Advanced Epithelial Ovarian Cancer: Results From AGO-OVAR16 and an East Asian Study. Int. J. Gynecol. Cancer 2018, 28, 2–10. [Google Scholar] [CrossRef]
- Lee, J.M.; Annunziata, C.M.; Hays, J.L.; Cao, L.; Choyke, P.; Yu, M.; An, D.; Turkbey, I.B.; Minasian, L.M.; Steinberg, S.M.; et al. Phase II trial of bevacizumab and sorafenib in recurrent ovarian cancer patients with or without prior-bevacizumab treatment. Gynecol. Oncol. 2020, 159, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.A.; Sill, M.W.; Makker, V.; Mutch, D.G.; Carlson, J.W.; Darus, C.J.; Mannel, R.S.; Bender, D.P.; Crane, E.K.; Aghajanian, C. A randomized phase II study of cabozantinib versus weekly paclitaxel in the treatment of persistent or recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer: An NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol. 2019, 152, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Vergote, I.B.; Smith, D.C.; Berger, R.; Kurzrock, R.; Vogelzang, N.J.; Sella, A.; Wheler, J.; Lee, Y.; Foster, P.G.; Weitzman, R.; et al. A phase 2 randomised discontinuation trial of cabozantinib in patients with ovarian carcinoma. Eur. J. Cancer 2017, 83, 229–236. [Google Scholar] [CrossRef]
- Backes, F.J.; Wei, L.; Chen, M.; Hill, K.; Dzwigalski, K.; Poi, M.; Phelps, M.; Salani, R.; Copeland, L.J.; Fowler, J.M.; et al. Phase I evaluation of lenvatinib and weekly paclitaxel in patients with recurrent endometrial, ovarian, fallopian tube, or primary peritoneal Cancer. Gynecol. Oncol. 2021, 162, 619–625. [Google Scholar] [CrossRef]
- Chan, J.K.; Brady, W.; Monk, B.J.; Brown, J.; Shahin, M.S.; Rose, P.G.; Kim, J.H.; Secord, A.A.; Walker, J.L.; Gershenson, D.M. A phase II evaluation of sunitinib in the treatment of persistent or recurrent clear cell ovarian carcinoma: An NRG Oncology/Gynecologic Oncology Group Study (GOG-254). Gynecol. Oncol. 2018, 150, 247–252. [Google Scholar] [CrossRef]
- Monk, B.J.; Grisham, R.N.; Banerjee, S.; Kalbacher, E.; Mirza, M.R.; Romero, I.; Vuylsteke, P.; Coleman, R.L.; Hilpert, F.; Oza, A.M.; et al. MILO/ENGOT-ov11: Binimetinib Versus Physician’s Choice Chemotherapy in Recurrent or Persistent Low-Grade Serous Carcinomas of the Ovary, Fallopian Tube, or Primary Peritoneum. J. Clin. Oncol. 2020, 38, 3753–3762. [Google Scholar] [CrossRef] [PubMed]
- Grisham, R.N.; Moore, K.N.; Gordon, M.S.; Harb, W.; Cody, G.; Halpenny, D.F.; Makker, V.; Aghajanian, C.A. Phase Ib Study of Binimetinib with Paclitaxel in Patients with Platinum-Resistant Ovarian Cancer: Final Results, Potential Biomarkers, and Extreme Responders. Clin. Cancer Res. 2018, 24, 5525–5533. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Tinker, A.; Clarke, B.; Ghatage, P.; Welch, S.; Weberpals, J.I.; Dhani, N.C.; Butler, M.O.; Tonkin, K.; Tan, Q.; et al. A Clinical and Molecular Phase II Trial of Oral ENMD-2076 in Ovarian Clear Cell Carcinoma (OCCC): A Study of the Princess Margaret Phase II Consortium. Clin. Cancer Res. 2018, 24, 6168–6174. [Google Scholar] [CrossRef] [PubMed]
- Westin, S.N.; Labrie, M.; Litton, J.K.; Blucher, A.; Fang, Y.; Vellano, C.P.; Marszalek, J.R.; Feng, N.; Ma, X.; Creason, A.; et al. Phase Ib Dose Expansion and Translational Analyses of Olaparib in Combination with Capivasertib in Recurrent Endometrial, Triple-Negative Breast, and Ovarian Cancer. Clin. Cancer Res. 2021, 27, 6354–6365. [Google Scholar] [CrossRef]
- Blagden, S.P.; Hamilton, A.L.; Mileshkin, L.; Wong, S.; Michael, A.; Hall, M.; Goh, J.C.; Lisyanskaya, A.S.; DeSilvio, M.; Frangou, E.; et al. Phase IB Dose Escalation and Expansion Study of AKT Inhibitor Afuresertib with Carboplatin and Paclitaxel in Recurrent Platinum-resistant Ovarian Cancer. Clin. Cancer Res. 2019, 25, 1472–1478. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Wulf, G.M.; Barry, W.T.; Birrer, M.; Westin, S.N.; Farooq, S.; Bell-McGuinn, K.M.; Obermayer, E.; Whalen, C.; Spagnoletti, T.; et al. Phase I dose escalation study of the PI3kinase pathway inhibitor BKM120 and the oral poly (ADP ribose) polymerase (PARP) inhibitor olaparib for the treatment of high-grade serous ovarian and breast cancer. Ann. Oncol. 2017, 28, 512–518. [Google Scholar] [CrossRef]
- Arend, R.C.; Davis, A.M.; Chimiczewski, P.; O’Malley, D.M.; Provencher, D.; Vergote, I.; Ghamande, S.; Birrer, M.J. EMR 20006-012: A phase II randomized double-blind placebo controlled trial comparing the combination of pimasertib (MEK inhibitor) with SAR245409 (PI3K inhibitor) to pimasertib alone in patients with previously treated unresectable borderline or low grade ovarian cancer. Gynecol. Oncol. 2020, 156, 301–307. [Google Scholar] [CrossRef]
- Farley, J.H.; Brady, W.E.; O’Malley, D.; Fujiwara, K.; Yonemori, K.; Bonebrake, A.; Secord, A.A.; Stephan, J.M.; Walker, J.L.; Nam, J.H.; et al. A phase II evaluation of temsirolimus with carboplatin and paclitaxel followed by temsirolimus consolidation in clear cell ovarian cancer: An NRG oncology trial. Gynecol. Oncol. 2022, 167, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Emons, G.; Kurzeder, C.; Schmalfeldt, B.; Neuser, P.; de Gregorio, N.; Pfisterer, J.; Park-Simon, T.W.; Mahner, S.; Schroder, W.; Luck, H.J.; et al. Temsirolimus in women with platinum-refractory/resistant ovarian cancer or advanced/recurrent endometrial carcinoma. A phase II study of the AGO-study group (AGO-GYN8). Gynecol. Oncol. 2016, 140, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.E.; Chu, T.; Elvin, J.A.; Edwards, R.P.; Zorn, K.K. Phase II study of everolimus and bevacizumab in recurrent ovarian, peritoneal, and fallopian tube cancer. Gynecol. Oncol. 2020, 156, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Tew, W.P.; Sill, M.W.; Walker, J.L.; Secord, A.A.; Bonebrake, A.J.; Schilder, J.M.; Stuckey, A.; Rice, L.; Tewari, K.S.; Aghajanian, C.A. Randomized phase II trial of bevacizumab plus everolimus versus bevacizumab alone for recurrent or persistent ovarian, fallopian tube or peritoneal carcinoma: An NRG oncology/gynecologic oncology group study. Gynecol. Oncol. 2018, 151, 257–263. [Google Scholar] [CrossRef]
- Shah, P.D.; Wethington, S.L.; Pagan, C.; Latif, N.; Tanyi, J.; Martin, L.P.; Morgan, M.; Burger, R.A.; Haggerty, A.; Zarrin, H.; et al. Combination ATR and PARP Inhibitor (CAPRI): A phase 2 study of ceralasertib plus olaparib in patients with recurrent, platinum-resistant epithelial ovarian cancer. Gynecol. Oncol. 2021, 163, 246–253. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Cheng, S.-C.; Wahner Hendrickson, A.E.; Penson, R.T.; Schumer, S.T.; Doyle, L.A.; Lee, E.K.; Kohn, E.C.; Duska, L.R.; Crispens, M.A.; et al. Berzosertib plus gemcitabine versus gemcitabine alone in platinum-resistant high-grade serous ovarian cancer: A multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 957–968. [Google Scholar] [CrossRef]
- Do, K.T.; Kochupurakkal, B.; Kelland, S.; de Jonge, A.; Hedglin, J.; Powers, A.; Quinn, N.; Gannon, C.; Vuong, L.; Parmar, K.; et al. Phase 1 Combination Study of the CHK1 Inhibitor Prexasertib and the PARP Inhibitor Olaparib in High-grade Serous Ovarian Cancer and Other Solid Tumors. Clin. Cancer Res. 2021, 27, 4710–4716. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Selle, F.; Weber, B.; Ray-Coquard, I.L.; Vergote, I.; Sufliarsky, J.; Del Campo, J.M.; Lortholary, A.; Lesoin, A.; Follana, P.; et al. Volasertib Versus Chemotherapy in Platinum-Resistant or -Refractory Ovarian Cancer: A Randomized Phase II Groupe des Investigateurs Nationaux pour l’Etude des Cancers de l’Ovaire Study. J. Clin. Oncol. 2016, 34, 706–713. [Google Scholar] [CrossRef]
- Bowles, D.W.; Ma, W.W.; Senzer, N.; Brahmer, J.R.; Adjei, A.A.; Davies, M.; Lazar, A.J.; Vo, A.; Peterson, S.; Walker, L.; et al. A multicenter phase 1 study of PX-866 in combination with docetaxel in patients with advanced solid tumours. Br. J. Cancer 2013, 109, 1085–1092. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Barry, W.T.; Birrer, M.; Westin, S.N.; Cadoo, K.A.; Shapiro, G.I.; Mayer, E.L.; O’Cearbhaill, R.E.; Coleman, R.L.; Kochupurakkal, B.; et al. Olaparib and alpha-specific PI3K inhibitor alpelisib for patients with epithelial ovarian cancer: A dose-escalation and dose-expansion phase 1b trial. Lancet Oncol. 2019, 20, 570–580. [Google Scholar] [CrossRef]
- Behbakht, K.; Sill, M.W.; Darcy, K.M.; Rubin, S.C.; Mannel, R.S.; Waggoner, S.; Schilder, R.J.; Cai, K.Q.; Godwin, A.K.; Alpaugh, R.K. Phase II trial of the mTOR inhibitor, temsirolimus and evaluation of circulating tumor cells and tumor biomarkers in persistent and recurrent epithelial ovarian and primary peritoneal malignancies: A Gynecologic Oncology Group study. Gynecol. Oncol. 2011, 123, 19–26. [Google Scholar] [CrossRef]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernandez-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef]
- Nanki, Y.; Chiyoda, T.; Hirasawa, A.; Ookubo, A.; Itoh, M.; Ueno, M.; Akahane, T.; Kameyama, K.; Yamagami, W.; Kataoka, F.; et al. Patient-derived ovarian cancer organoids capture the genomic profiles of primary tumours applicable for drug sensitivity and resistance testing. Sci. Rep. 2020, 10, 12581. [Google Scholar] [CrossRef]
- Kaipio, K.; Chen, P.; Roering, P.; Huhtinen, K.; Mikkonen, P.; Ostling, P.; Lehtinen, L.; Mansuri, N.; Korpela, T.; Potdar, S.; et al. ALDH1A1-related stemness in high-grade serous ovarian cancer is a negative prognostic indicator but potentially targetable by EGFR/mTOR-PI3K/aurora kinase inhibitors. J. Pathol. 2020, 250, 159–169. [Google Scholar] [CrossRef]
- Phan, N.; Hong, J.J.; Tofig, B.; Mapua, M.; Elashoff, D.; Moatamed, N.A.; Huang, J.; Memarzadeh, S.; Damoiseaux, R.; Soragni, A. A simple high-throughput approach identifies actionable drug sensitivities in patient-derived tumor organoids. Commun. Biol. 2019, 2, 78. [Google Scholar] [CrossRef]
- de Witte, C.J.; Espejo Valle-Inclan, J.; Hami, N.; Lohmussaar, K.; Kopper, O.; Vreuls, C.P.H.; Jonges, G.N.; van Diest, P.; Nguyen, L.; Clevers, H.; et al. Patient-Derived Ovarian Cancer Organoids Mimic Clinical Response and Exhibit Heterogeneous Inter- and Intrapatient Drug Responses. Cell. Rep. 2020, 31, 107762. [Google Scholar] [CrossRef]
- Chen, H.; Gotimer, K.; De Souza, C.; Tepper, C.G.; Karnezis, A.N.; Leiserowitz, G.S.; Chien, J.; Smith, L.H. Short-term organoid culture for drug sensitivity testing of high-grade serous carcinoma. Gynecol. Oncol. 2020, 157, 783–792. [Google Scholar] [CrossRef]
- Mullen, M.M.; Lomonosova, E.; Toboni, M.D.; Oplt, A.; Cybulla, E.; Blachut, B.; Zhao, P.; Noia, H.; Wilke, D.; Rankin, E.B.; et al. GAS6/AXL Inhibition Enhances Ovarian Cancer Sensitivity to Chemotherapy and PARP Inhibition through Increased DNA Damage and Enhanced Replication Stress. Mol. Cancer Res. 2022, 20, 265–279. [Google Scholar] [CrossRef]
- Parashar, D.; Geethadevi, A.; Mittal, S.; McAlarnen, L.A.; George, J.; Kadamberi, I.P.; Gupta, P.; Uyar, D.S.; Hopp, E.E.; Drendel, H.; et al. Patient-Derived Ovarian Cancer Spheroids Rely on PI3K-AKT Signaling Addiction for Cancer Stemness and Chemoresistance. Cancers 2022, 14, 958. [Google Scholar] [CrossRef]
- Roering, P.; Siddiqui, A.; Heuser, V.D.; Potdar, S.; Mikkonen, P.; Oikkonen, J.; Li, Y.; Pikkusaari, S.; Wennerberg, K.; Hynninen, J.; et al. Effects of Wee1 inhibitor adavosertib on patient-derived high-grade serous ovarian cancer cells are multiple and independent of homologous recombination status. Front. Oncol. 2022, 12, 954430. [Google Scholar] [CrossRef]
- Hill, S.J.; Decker, B.; Roberts, E.A.; Horowitz, N.S.; Muto, M.G.; Worley, M.J., Jr.; Feltmate, C.M.; Nucci, M.R.; Swisher, E.M.; Nguyen, H.; et al. Prediction of DNA Repair Inhibitor Response in Short-Term Patient-Derived Ovarian Cancer Organoids. Cancer Discov. 2018, 8, 1404–1421. [Google Scholar] [CrossRef]
- Maenhoudt, N.; Defraye, C.; Boretto, M.; Jan, Z.; Heremans, R.; Boeckx, B.; Hermans, F.; Arijs, I.; Cox, B.; Van Nieuwenhuysen, E.; et al. Developing Organoids from Ovarian Cancer as Experimental and Preclinical Models. Stem. Cell Rep. 2020, 14, 717–729. [Google Scholar] [CrossRef]
- Kopper, O.; de Witte, C.J.; Lohmussaar, K.; Valle-Inclan, J.E.; Hami, N.; Kester, L.; Balgobind, A.V.; Korving, J.; Proost, N.; Begthel, H.; et al. An organoid platform for ovarian cancer captures intra- and interpatient heterogeneity. Nat. Med. 2019, 25, 838–849. [Google Scholar] [CrossRef]
- Senkowski, W.; Gall-Mas, L.; Falco, M.M.; Li, Y.; Lavikka, K.; Kriegbaum, M.C.; Oikkonen, J.; Bulanova, D.; Pietras, E.J.; Voßgröne, K.; et al. A platform for efficient establishment, expansion and drug response profiling of high-grade serous ovarian cancer organoids. bioRxiv 2022. bioRxiv: 2022.2004.2021.489027. [Google Scholar] [CrossRef]
- Dong, R.; Qiang, W.; Guo, H.; Xu, X.; Kim, J.J.; Mazar, A.; Kong, B.; Wei, J.J. Histologic and molecular analysis of patient derived xenografts of high-grade serous ovarian carcinoma. J. Hematol. Oncol. 2016, 9, 92. [Google Scholar] [CrossRef]
- Ricci, F.; Bizzaro, F.; Cesca, M.; Guffanti, F.; Ganzinelli, M.; Decio, A.; Ghilardi, C.; Perego, P.; Fruscio, R.; Buda, A.; et al. Patient-derived ovarian tumor xenografts recapitulate human clinicopathology and genetic alterations. Cancer Res. 2014, 74, 6980–6990. [Google Scholar] [CrossRef]
- Gopinathan, G.; Berlato, C.; Lakhani, A.; Szabova, L.; Pegrum, C.; Pedrosa, A.R.; Laforets, F.; Maniati, E.; Balkwill, F.R. Immune Mechanisms of Resistance to Cediranib in Ovarian Cancer. Mol. Cancer. Ther. 2022, 21, 1030–1043. [Google Scholar] [CrossRef]
- Ravoori, M.K.; Singh, S.P.; Lee, J.; Bankson, J.A.; Kundra, V. In Vivo Assessment of Ovarian Tumor Response to Tyrosine Kinase Inhibitor Pazopanib by Using Hyperpolarized (13)C-Pyruvate MR Spectroscopy and (18)F-FDG PET/CT Imaging in a Mouse Model. Radiology 2017, 285, 830–838. [Google Scholar] [CrossRef]
- Tyulyandina, A.; Harrison, D.; Yin, W.; Stepanova, E.; Kochenkov, D.; Solomko, E.; Peretolchina, N.; Daeyaert, F.; Joos, J.B.; Van Aken, K.; et al. Alofanib, an allosteric FGFR2 inhibitor, has potent effects on ovarian cancer growth in preclinical studies. Investig. New Drugs 2017, 35, 127–133. [Google Scholar] [CrossRef]
- Harris, F.R.; Zhang, P.; Yang, L.; Hou, X.; Leventakos, K.; Weroha, S.J.; Vasmatzis, G.; Kovtun, I.V. Targeting HER2 in patient-derived xenograft ovarian cancer models sensitizes tumors to chemotherapy. Mol. Oncol. 2019, 13, 132–152. [Google Scholar] [CrossRef]
- Huo, X.; Zhang, W.; Zhao, G.; Chen, Z.; Dong, P.; Watari, H.; Narayanan, R.; Tillmanns, T.D.; Pfeffer, L.M.; Yue, J. FAK PROTAC Inhibits Ovarian Tumor Growth and Metastasis by Disrupting Kinase Dependent and Independent Pathways. Front. Oncol. 2022, 12, 851065. [Google Scholar] [CrossRef]
- Fang, D.D.; Tao, R.; Wang, G.; Li, Y.; Zhang, K.; Xu, C.; Zhai, G.; Wang, Q.; Wang, J.; Tang, C.; et al. Discovery of a novel ALK/ROS1/FAK inhibitor, APG-2449, in preclinical non-small cell lung cancer and ovarian cancer models. BMC Cancer 2022, 22, 752. [Google Scholar] [CrossRef]
- Kanakkanthara, A.; Hou, X.; Ekstrom, T.L.; Zanfagnin, V.; Huehls, A.M.; Kelly, R.L.; Ding, H.; Larson, M.C.; Vasmatzis, G.; Oberg, A.L.; et al. Repurposing Ceritinib Induces DNA Damage and Enhances PARP Inhibitor Responses in High-Grade Serous Ovarian Carcinoma. Cancer Res. 2022, 82, 307–319. [Google Scholar] [CrossRef]
- Xu, J.; Gao, Y.; Luan, X.; Li, K.; Wang, J.; Dai, Y.; Kang, M.; Lu, C.; Zhang, M.; Lu, C.X.; et al. An effective AKT inhibitor-PARP inhibitor combination therapy for recurrent ovarian cancer. Cancer Chemother. Pharmacol. 2022, 89, 683–695. [Google Scholar] [CrossRef]
- Wang, L.; Wang, L.; Cybula, M.; Drumond-Bock, A.L.; Moxley, K.M.; Bieniasz, M. Multi-kinase targeted therapy as a promising treatment strategy for ovarian tumors expressing sfRon receptor. Genes Cancer 2020, 11, 106–121. [Google Scholar] [CrossRef]
- Qi, G.; Ma, H.; Li, Y.; Peng, J.; Chen, J.; Kong, B. TTK inhibition increases cisplatin sensitivity in high-grade serous ovarian carcinoma through the mTOR/autophagy pathway. Cell Death Dis. 2021, 12, 1135. [Google Scholar] [CrossRef]
- Chesnokov, M.S.; Khan, I.; Park, Y.; Ezell, J.; Mehta, G.; Yousif, A.; Hong, L.J.; Buckanovich, R.J.; Takahashi, A.; Chefetz, I. The MEK1/2 Pathway as a Therapeutic Target in High-Grade Serous Ovarian Carcinoma. Cancers 2021, 13, 1369. [Google Scholar] [CrossRef]
- Parmar, K.; Kochupurakkal, B.S.; Lazaro, J.B.; Wang, Z.C.; Palakurthi, S.; Kirschmeier, P.T.; Yang, C.; Sambel, L.A.; Farkkila, A.; Reznichenko, E.; et al. The CHK1 Inhibitor Prexasertib Exhibits Monotherapy Activity in High-Grade Serous Ovarian Cancer Models and Sensitizes to PARP Inhibition. Clin. Cancer Res. 2019, 25, 6127–6140. [Google Scholar] [CrossRef]
- Au-Yeung, G.; Lang, F.; Azar, W.J.; Mitchell, C.; Jarman, K.E.; Lackovic, K.; Aziz, D.; Cullinane, C.; Pearson, R.B.; Mileshkin, L.; et al. Selective Targeting of Cyclin E1-Amplified High-Grade Serous Ovarian Cancer by Cyclin-Dependent Kinase 2 and AKT Inhibition. Clin. Cancer Res. 2017, 23, 1862–1874. [Google Scholar] [CrossRef]
- Zhou, J.; Alfraidi, A.; Zhang, S.; Santiago-O’Farrill, J.M.; Yerramreddy Reddy, V.K.; Alsaadi, A.; Ahmed, A.A.; Yang, H.; Liu, J.; Mao, W.; et al. A Novel Compound ARN-3236 Inhibits Salt-Inducible Kinase 2 and Sensitizes Ovarian Cancer Cell Lines and Xenografts to Paclitaxel. Clin. Cancer Res. 2017, 23, 1945–1954. [Google Scholar] [CrossRef]
- Bizzaro, F.; Fuso Nerini, I.; Taylor, M.A.; Anastasia, A.; Russo, M.; Damia, G.; Guffanti, F.; Guana, F.; Ostano, P.; Minoli, L.; et al. VEGF pathway inhibition potentiates PARP inhibitor efficacy in ovarian cancer independent of BRCA status. J. Hematol. Oncol. 2021, 14, 186. [Google Scholar] [CrossRef]
- Lin, Z.P.; Zhu, Y.L.; Lo, Y.C.; Moscarelli, J.; Xiong, A.; Korayem, Y.; Huang, P.H.; Giri, S.; LoRusso, P.; Ratner, E.S. Combination of triapine, olaparib, and cediranib suppresses progression of BRCA-wild type and PARP inhibitor-resistant epithelial ovarian cancer. PLoS ONE 2018, 13, e0207399. [Google Scholar] [CrossRef]
- Pearce, O.M.T.; Delaine-Smith, R.M.; Maniati, E.; Nichols, S.; Wang, J.; Bohm, S.; Rajeeve, V.; Ullah, D.; Chakravarty, P.; Jones, R.R.; et al. Deconstruction of a Metastatic Tumor Microenvironment Reveals a Common Matrix Response in Human Cancers. Cancer Discov. 2018, 8, 304–319. [Google Scholar] [CrossRef]
- Diaz Osterman, C.J.; Ozmadenci, D.; Kleinschmidt, E.G.; Taylor, K.N.; Barrie, A.M.; Jiang, S.; Bean, L.M.; Sulzmaier, F.J.; Jean, C.; Tancioni, I.; et al. FAK activity sustains intrinsic and acquired ovarian cancer resistance to platinum chemotherapy. Elife 2019, 8, e47327. [Google Scholar] [CrossRef]
- Mundi, P.S.; Sachdev, J.; McCourt, C.; Kalinsky, K. AKT in cancer: New molecular insights and advances in drug development. Br. J. Clin. Pharmacol. 2016, 82, 943–956. [Google Scholar] [CrossRef]
- Clark, A.S.; West, K.; Streicher, S.; Dennis, P.A. Constitutive and inducible Akt activity promotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol. Cancer Ther. 2002, 1, 707–717. [Google Scholar]
- Hew, K.E.; Miller, P.C.; El-Ashry, D.; Sun, J.; Besser, A.H.; Ince, T.A.; Gu, M.; Wei, Z.; Zhang, G.; Brafford, P.; et al. MAPK Activation Predicts Poor Outcome and the MEK Inhibitor, Selumetinib, Reverses Antiestrogen Resistance in ER-Positive High-Grade Serous Ovarian Cancer. Clin. Cancer Res. 2016, 22, 935–947. [Google Scholar] [CrossRef]
- Wang, J.; Wu, G.S. Role of autophagy in cisplatin resistance in ovarian cancer cells. J. Biol. Chem. 2014, 289, 17163–17173. [Google Scholar] [CrossRef]
- Chefetz, I.; Grimley, E.; Yang, K.; Hong, L.; Vinogradova, E.V.; Suciu, R.; Kovalenko, I.; Karnak, D.; Morgan, C.A.; Chtcherbinine, M.; et al. A Pan-ALDH1A Inhibitor Induces Necroptosis in Ovarian Cancer Stem-like Cells. Cell Rep. 2019, 26, 3061–3075.e3066. [Google Scholar] [CrossRef]


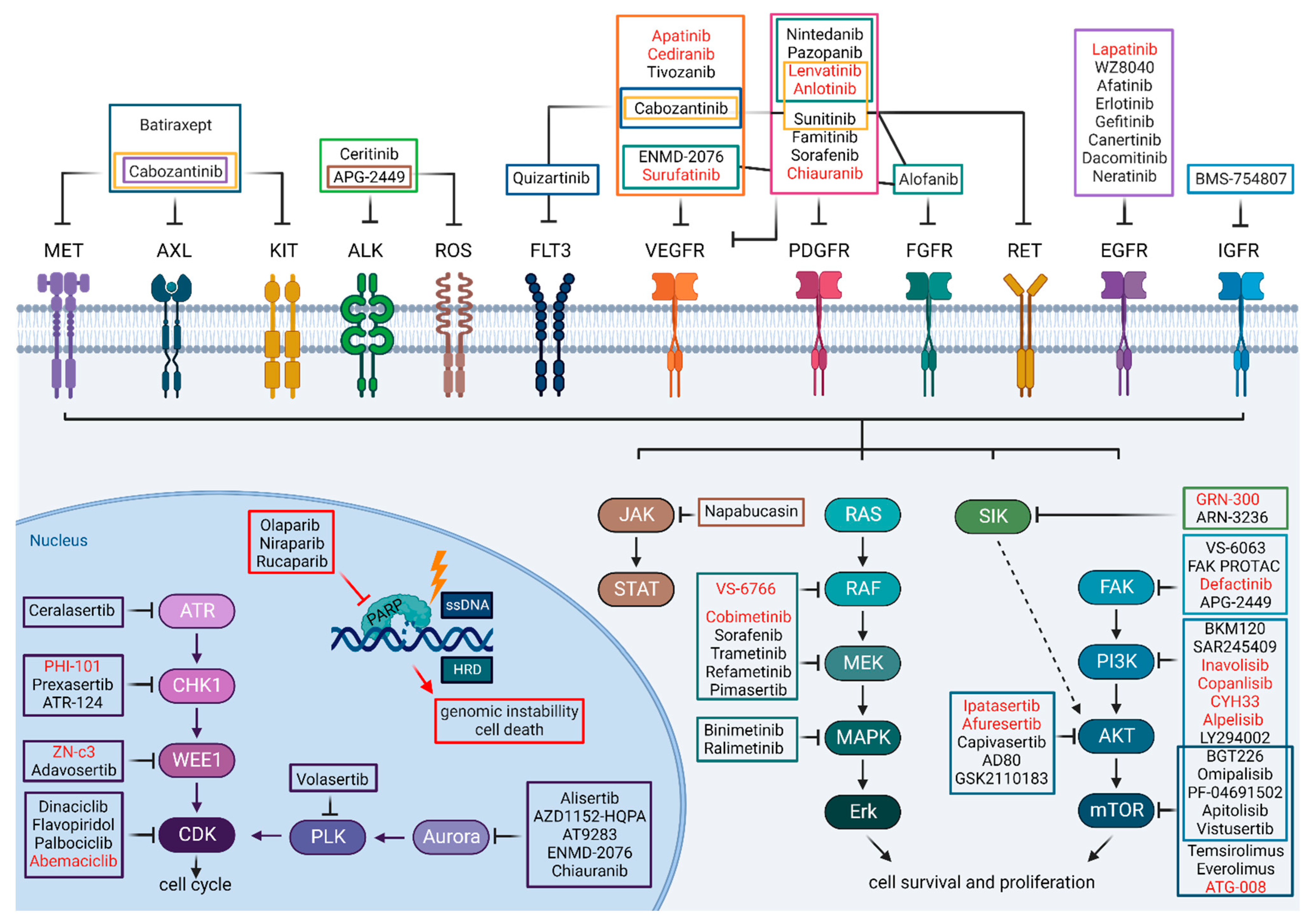{kind=link}
| Kinase Inhibitor | Target Kinase | Trial | Patient Group (ITT) | Study Design | Primary Outcomes | Secondary Outcomes | Conclusion | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| Tyrosine kinase inhibitors | Apatinib | VEGFR2 | Phase II RCT | Platinum-resistant, progressive, OVC. (n = 152) | 1:1 randomization to recieve pegylated liposomal doxyrubicin alone or in combination with apatinib. | PFS | OS, ORR, DCR, and safety | Apatinib plus pegylated liposomal doxorubicin showed promising efficacy and manageable toxic effects. | [83] |
| Single arm, phase II trial | Recurrent, platinum-resistant, OVC which failed available standard CTX. (n = 29) | Apatinib administered daily until progression or unacceptable toxicities. | ORR | PFS, OS DCR and toxicity | Apatinib may contribute to achieve clinical benefits with an acceptable safety profile. | [84] | |||
| Cediranib | VEGFRs | Phase III RCT | Platinum-sensitive, recurrent, high-grade serous or endometrioid OVC. (n = 565) | 1:1:1 randomization to platinum-based CTX, olaparib or olaparib + cediranib | PFS | Activity within gBRCAm or wt subgroups, and PROs | Cediranib + olaparib did not improve PFS and reduced PROs compared to CTX, but had significant clinical activity in patients with gBRCAm. | [85] | |
| Phase II RCT | Platinum-resistant, high-grade OVC. (n = 123) | 1:1:1 randomization to (1) weekly PAX, (2) olaparib + cediranib on a continuous schedule, or (3) olaparib + cediranib on intermittent schedule. | PFS and evacuations per day in first four weeks | Compliance, reasons for discontinuation, ORR, OS, and HRQoL. | Cediranib + olaparib showed clinical activity, but was not superior to CTX in terms of PFS. | [86] | |||
| Single-arm, phase II trial | Recurrent OVC with high-grade serous or high-grade endometrioid histology and disease progression on any PARPi. (n = 34) | 3 cohorts: platinum-sensitive, platinum-resistant, or progressive disease on PAPRi and subsequent CTX. Olaparib + cediranib on a continuous schedule. | ORR at 8 weeks and PFS at 16 weeks | DCR, safety, and mechanisms of resistance | Cediranib + olaparib was tolerable and showed some activity. | [87] | |||
| Phase I dose escalation trial | Recurrent, advanced breast or gynecologic malignancies. (n = 9, OVC = 7) | 3 + 3 design with cediranib + durvalumab + olaparib | RP2D | ORR, PKs, and correlative analyses | The RP2D was tolerable and showed preliminary activity in recurrent ovarian cancer. | [88] | |||
| Double-blind, phase III RCT | Recurrent, platinum-sensitive OVC. (n = 456) | 2:3:3 randomization to A: placebo + CTX with placebo maintenance, B: Cediranib + CTX with placebo maintenance, or C: Cediranib + CTX with cediranib maintenance. | PFS comparing arms A and C | OS, toxicity, HRQoL, PFS | Cediranib + CTX with cediranib maintenance improved PFS but had added toxic effects compared to standard treatment. | [89] | |||
| Single-arm, two-stage phase II trial | Recurrent or persistent OVC. (n = 74) | Stratification into platinum-sensitive and platinum-resistant. Both groups recieved oral daily cediranib. | ORR at 16 weeks | PFS, OS and toxicity | Cediranib demonstrated activity. Toxicities were manageable at a reduced dose. | [90] | |||
| Updated analysis of phase II RCT | Recurrent, high-grade serous or high-grade endometrioid OVC or a high-grade histology with a known gBRCAm. (n = 90) | 1:1 randomization to cediranib + olaparib or olaparib alone with gBRCAm and previous anti-angiogenic therapy as stratification factors. | PFS | OS, AEs | Cediranib + olaparib increased PFS versus olaparib alone. OS was increased in patients without gBRCAm. | [91] | |||
| Tivozanib | VEGFRs | Single-arm phase II trial | Recurrent, platinum-resistant OVC. (n = 31) | Tivozanib as monotherapy until disease progression or withdrawal. | ORR | PFS, OS, and toxicity assessment. | Tivozanib was effective with moderate toxicity and no treatment-related deaths. | [92] | |
| Gefitinib | EGFR | Dose escalation phase Ib/II trial | Recurrent or persitent OVC with positive EGFR expression. (n = 19) | Phase Ib: 3 + 3 design with standard dose gefitinib and increasing doses of topotecan. Phase II: 10 patients treated with MTD from phase Ib. | Safety and tolerability | ORR and DOR. | The drug combination was relatively well tolerated, but did not show sufficient clinical activity. | [24] | |
| Nintedanib | VEGFRs, FGFRs and PDGFRs | Double-blind, phase II RCT | Recurrent OVC. (n = 117) | 1:1 randomization to cyclophosphamide + nintefanib or cyclophosphamide + placebo | OS | PFS, ORR, toxicity and HRQoL | Nintedanib + cyclophosphamide did not improve outcomes. More patients than expected remained on treatment for ≥6months. | [93] | |
| Double-blind, phase III RCT | Newly diagnosed, advanced (FIGO stage IIB–IV) OVC after initial debulking surgery. (n = 1366) | 2:1 randomization to CARB + PAX + nintedanib or CARB + PAX + placebo. | PFS and CA125 | OS | Nintedanib therapy did not affect final OS results. | [94] | |||
| Double-blind, phase III RCT | Newly diagnosed, advanced (FIGO stage IIB–IV) OVC after initial debulking surgery. (n = 1366) | 2:1 randomization to CARB + PAX + nintedanib or CARB + PAX + placebo. | PFS and CA125 | OS, time to CA125 progression, AEs, and HRQoL | CARB + PAX + nintedanib significantly increased PFS, but was associated with more gastrointestinal AEs. | [95] | |||
| Famitinib | VEGFR, PDGFR, and KIT | Single-arm phase II trial. | Platinum-resistant, recurrent OVC. (n = 37) | Camrelizumab + famitinib until disease progression or unacceptable toxicities | ORR | DCR, DOR, TTR, PFS, OS, OS at 12 months, and safety | Famitinib + camrelizumab showed antitumor activity with an acceptable safety profile. | [96] | |
| Pazopanib | VEGFRs, PDGFRs, KIT and FGFRs | Double-blind, phase III RCT | Advanced OVC, after surgical debulking, without progression after first-line platinum-taxane treatment. (n = 940) | 1:1 randomization to pazopanib or placebo as maintenance therapy for up to 24 months. | PFS | OS and safety | Pazopanib prolonged PFS, but was not associated with improved median OS. | [97] | |
| Double-blind phase IIb RCT | Recurrent or persistent OVC. (n = 106) | 1:1 randomization to PAX + pazopanib or PAX + placebo | PFS | AEs, ORR and OS. | Pazopanib + PAX was not superior to PAX alone. | [98] | |||
| Combination of two double-blind, phase III RCTs | East asian patients with advanced OVC without progression after first-line platinum-PAX treatment. (n = 354) | 1:1 randomization to pazopanib or placebo as maintenance therapy for up to 24 months. | PFS | OS, safety, and AEs | Pazopanib maintenance showed disadvantage in OS and AEs versus placebo. | [99] | |||
| Sorafenib | VEGFR2/3, BRAF, KIT, and PDGFRs | Single arm phase II trial | Recurrent or metastatic OVC. (n = 54) | Stratification by prior or no prior treatment with bevacizumab. Treatment with bevacizumab + sorafenib. | ORR | PFS and toxicity | Bevacizumab + sorafenib did not meet the primary endpoint, but did show some activity in the bevacizumab-naïve group. | [100] | |
| Cabozantinib | MET, VEGFR2, AXL, KIT, FLT3 and RET | Phase II RCT | Persistent or recurrent OVC. (n = 111) | 1:1 randomization to daily cabozantinib versus weekly PAX. | PFS at week 16 and week 32 | Toxicities, ORR, OS and EFS | No difference in PFS between cabozantinib and weekly PAX. OS, EFS, and ORR were worse in the cabozantinib group. | [101] | |
| Double-blind, phase II discontinuation RCT | Progressive OVC. (n = 70) | Patients with SD after 12-week open-label lead-in phase were randomized 1:1 to daily carbozantinib or placebo. | ORR at week 12 and PFS | CA125 response and AEs. | Cabozantinib showed clinical activity. Toxicities were acceptable. | [102] | |||
| Lenvatinib | VEGFRs, FGFRs, PDGFRβ, RET, and KIT. | Phase I dose escalation trial. | Recurrent endometrial, OVC. (n = 26) | 5 dose cohorts with an accelerated titration design until DLT. Then accural transitioned to 3 + 3 design for further dose levels. | AEs | OR, PFS, and duration of response. | Lenvatinib + PAX showed tolerable side effects and clinical activity. | [103] | |
| Sunitinib | VEGFRs, PDGFRs, RET, KIT, CD114, and CD135. | Single arm phase II trial | Recurrent or persistent clear cell ovarian cancer. (n = 30) | Sunitinib every day for 4 weeks in 6-week cycles until disease progression or prohibitive toxicity. | PFS at 6 months and clinical response | OS | Sunitinib showed minimal activity as second- and thrid-line treatment. | [104] | |
| Tyrosine and serine/threonine kinase inhibitors | Binimetinib | MEK1/2 | Phase III RCT | Recurrent LGSC. (n = 303) | 2:1 randomized study of binimetinib versus CTX. | PFS | OS, ORR, DOR, CBR, biomarkers and safety | Binimetinib did not show difference in PFS versus CTX. | [105] |
| Dose-escalation, phase Ib trial | Platinum-resistant- or refractory OVC. (n = 34) | 3 + 3 design for dose escalation of binimetinib on continuous or intermittent schedule. Additionally 12 patients were enrolled in each group after RP2D determination. | RP2D | Predictive biomarkers of clinical activity (by NGS), CR, PR, ORR and SD. | Binimetinib + PAX was tolerable and RP2D was determined. ORR was modest, but higher in patients with genetic alterations affecting the MAPK pathway. | [106] | |||
| ENMD-2076 | VEGFRs, FGFRs, FLT3, KIT, and Aurora A | Single-arm, phase II trial | Platinum-resistant or recurrent OCCC. (n = 40) | ENMD-2076 on contiuous schedule until disease progression or unacceptable toxicity. | ORR and PFS at 6 months | Duration of response | ENMD-2076 did not meet the pre-determined bar for efficacy. | [107] | |
| Serine/threonine kinase inhibitors | Ralimetinib | p38 MAPK | Double-blind, phase Ib/II RCT | Platinum-sensitive, recurrent OVC. (n = 118) | Phase Ib: open-label 3 + 3 dose escalation design. Phase II: 1:1 randomization to ralimetinib + gemcitabene + CARB or placebo + gemcitabine + CARB followed by ralimeinib or placebo maintenance. | Phase 1b: RP2D, phase 2: PFS | OS, ORR, CA125, safety and tolerability. | Addition of ralimetinib to gemcitabene + CTX resulted in a modest improvement in PFS. | [72] |
| Capivasertib | AKT | Dose expansion phase Ib trial | Recurrent endometrial, triple negative breast, and OVC. (n = 38, OVC = 16) | Olaparib + capivasertib on an intermittent schedule until progression or toxicity. | MTD and RP2D | ORR, SD, and duration of response. | Olaparib + capivasertib showed no serious AEs, and demonstrated durable activity. | [108] | |
| Afuresertib | Dose escalation phase Ib trial. | Progressive serous OVC following prior platinum-based treatment. (n = 29 for part I and n = 30 for part II) | Afuresertib + CARB + PAX. Part I was a 3 + 3 dose escalation study and part II was a single-arm evaluation of the clinical activity. | Safety and tolerability (part I) and ORR (part II) | CA125 response and PFS | Afuresertib + CARB + PAX showed clinical activity with the MTD of afuresertib defined as 125 mg/ml. | [109] | ||
| BKM120 | PI3K | Dose escalation phase I trial | Recurrent HGSOC or TNBC, or other histology of OVC or BC but with gBRCAm. (n = 69; 45 OVC and 24 BC) | 3 + 3 design dose escalation study of olaparib + BKM120 with expansion cohorts of 12 patients per cancer type. | MTD and RP2D | AEs | Clinical benefit was observed for both gBRCAm and gBRCAwt. BKM120 and olaparib can be co-administered with attenuated BKM120 dose. | [110] | |
| Pimasertib and SAR245409 | MEK and PI3K, respectively | Double-blind, phase II RCT | Recurrent LMP or LGSC. (n = 65) | 1:1 randomization to pimaserib + SAR245409 or pimasertib + placebo, stratified by tumor histology (LGSOC or LMP/borderline). | ORR | PFS, DCR and AEs. | Pimasertib as single treatment can be alternative to CTX. Pimasertib + SER245409 was not more effective than pimasertib alone. | [111] | |
| Temsirolimus | mTOR | Two single-arm, single-stage phase II trials | Primary stage III or IV OCCC. (n = 90) | 1 cohort form the US and Korea and 1 cohort from Japan recieved CARB + PAX + temsirolimus for 6 cycles or until progression followed by temsirolimus consolidation therapy. | PFS at 12 months | OS, PFS and AEs. | PFS at 12 months, was not increased compared to historical controls. The treatment was well tolerated. | [112] | |
| Single arm phase II trial | Progressive OVC following platinum-based CTX. (n = 22) | Temsirolimus every seven days until disease progression, inacceptable toxicities, or withdrawal. | PFS | AEs and OS | Temsirolimus treatment was well tolerated, but did not meet the predefined efficacy criteria. Few patients had long lasting SD. | [113] | |||
| Everolimus | Single-arm phase II trial | Recurrent OVC. (n= 50) | Everolimus + bevacizumab until disease progression or unacceptabel toxicities. | PFS at 6 months | Molecular profiling and AEs. | Everolimus + beavcizumab did not show added clinical activity compared to studies of bevacizumab alone. | [114] | ||
| Double-blind phase II RCT | Persistent or recurrent OVC. (n = 150) | 1:1 randomization to bevacizumab + everolimus or bevecizumab+ placebo until progression or toxicity. | PFS | Safety and ORR | Bevacizumab + everolimus did not increase PFS compared to bevacizumab alone, and was associated with higer AE rate and discontinuation. | [115] | |||
| Ceralasertib | ATR | Single-arm phase II trial | Recurrent, high-grade serous OVC. (n = 14) | Ceralasertib + olaparib until progression or toxicity. | Toxicity and ORR. | PFS | Olaparib + ceralasertib was well-tolerated, but ORR was unaffected. Some activity was associated with gBRCA1m. | [116] | |
| Berzosertib | Phase II RCT | Recurrent, platinum-resistant HGSC. (n = 70) | 1:1 randomization to gemcitabene alone or gemcitabene + berzosertib. | PFS | OS, ORR, CBR, CR, PR, SD, DOR, CA125, and safety | Gemcitabene + berzosertib increased PFS. No added toxic efects were observed. | [117] | ||
| Prexasertib | CHK1 | Phase I trial | gBRCAm patients with HGSC, who have previously progressed on PARP-inhibitor. (n = 29) | 3 + 3 design with a 7-day lead-in of olaparib followed by intermittent prexasertib + attenuated dose of olaparib. | Safety and tolerability | Preliminary anti-tumor activity and PDs. | Prexasertib + olaparib showed preliminary clinical activity in this patient group. | [118] | |
| Volasertib | PLK1 | Phase II RCT. | Recurrent, platinum-resistant- or refractory OVC. (n = 109) | 1:1 randomization to volasertib or non-platinum CTX. Two-step design for early saftey analysis. | DCR at 24 weeks | ORR, OS, PFS, HRQoL, safety, PK and biomarker analysis. | Volasertib demonstrated antitumor activity, and AEs were manageable. | [119] | |
| Adavosertib | WEE1 | Double-blind phase II RCT | Platinum-resistant or- refractory, recurrent OVC. (n = 124) | Stratification into HGSOC and non-HGSOC. HGSOC randomized 2:1 to adavosertib + gemcitabine or adavosertib + placebo, and non-HGSOC recieved adavosertib + gemcitabine. | PFS | ORR, OS, safety and tolerability, TP53 mutations and p53 expression. | Adavosertib + gemcitabene extended PFS and OS. | [80] | |
| Double-blind, phase II RCT | Platinum-sensitive TP53 mutant OVC. (n = 121) | 1:1 randomization to adavosertib + CTX or placebo + CTX. | ePFS, safety and tolerability | PFS, ORR, and OS. | Adavosertib + CTX improved ePFS, clinical benefit was observed depending on TP53 mutation, and AEs were increased. | [81] |
| Kinase Inhibitor | Target Kinase | Trial | Patient Group (EE) | Study Design | Primary Outcomes | Secondary Outcomes | Clinical Trials ID | First Posted | |
|---|---|---|---|---|---|---|---|---|---|
| Tyrosine kinase inhibitors | Cediranib | VEGFRs | Phase II RCT | Recurrent platinum-resistant OVC with prior bevacizumab. (n = 164) | Comparison of durvalumab + olaparib + cediranib, durvalumab + cediranib, and olaparib + cediranib to CTX. | PFS | ORR, DOR, OS, AEs. | NCT04739800 | 2021 |
| Apatinib | VEGFR2 | Phase II RCT | Platinum-sensitive, relapsed, high-grade predominantly serous OVC. (n = 132) | Fluzoparib + apatinib versus fluzoparib monotherapy as maintenance treatment. | PFS in PARPi treated patients | PFS, PFS in gBRCAm patients, ORR, DCR, DOR, OS, and AEs. | NCT05479487 | 2022 | |
| Phase II RCT | High-grade serous or endometrioid recurrent OVC. (n = 142) | Safety-lead-in of fluzoparib + apatinib, exploratory cohort of fluzoparib + apatinib in patients with prior PARPi treatment, and fluzoparib monotherapy cohort as active comparator. | Safety lead-in: DLT and RP2D, phase II: ORR | AEs, PFS, DCR, DOR, RR, and CA125 | NCT04517357 | 2020 | |||
| Single-arm, exploratory phase II trial | Treatment-naïve stage II-IV OVC. (n = 58) | Apatinib + abraxane and carboplatin or cisplatinum as first-line treatment. | R0 resection rate and PFS | N/A | NCT04590625 | 2020 | |||
| Lapatinib | HER2/neu and EGFR | Dose-escalation phase I trial | Platinum-resistant OVC. (n = 15) | Lapatinib + PAX therapy tested with 4 different concentrations of lapatinib. | PFS and DLT | ∆plasma lapatinib, and ABCB1 expression | NCT04608409 | 2020 | |
| Surufatinib | VEGFR, FGFR, and CSF1R | Single-arm phase Ib/II trial | Platinum-resistant OVC. (n = 38) | Phase Ib: dose de-escalation schedule with 3 + 3 design administering surufatinib + pamiparib. Phase II: RP2D of surufatinib + pamiparib. | Phase Ib: MTD and RP2D, phase II: ORR | PFS, DCR, DOR, OS, PROs, and safety | NCT05494580 | 2022 | |
| Anlotinib | VEGFRs, FGFRs, PDGFRs, KIT and RET. | Phase III RCT | Platinum-resistant, recurrent, OVC. (n = 405) | TQB2450 + anlotinib versus PAX as weekly treatment | PFS and OS | PFS at 6 months, ORR, DOR, DCR, OS at 12 months, AEs | NCT05145218 | 2021 | |
| Single-arm, exploratory phase II trial | Newly diagnosed advanced (FIGO stage III-IV) OVC. (n = 56) | Anlotinib + CARB/PAX as first-line treatment. | PFS | ORR, DCR, OS, AEs | NCT04807166 | 2021 | |||
| Single-arm, exploratory phase II trial | Platinum-resistant, recurrent OVC. (n = 68) | Anlotinib + dose-reduced olaparib until disease progression. | PFS, AEs | ORR, DCR, OS, TFST, and QoL | NCT04566952 | 2020 | |||
| Lenvatinib | VEGFRs, FGFRs, PDGFRs, KIT, and RET | Single-arm phase II trial | Recurrent or persistent OCCC. (n = 31) | Lenvatinib + pembrolizumab until progression of disease or unacceptable toxicity. | ORR and 6-month PFS | PFS, AEs, CBR, OS, median PFS, and median OS | NCT05296512 | 2022 | |
| Single-arm phase II trial | Platinum-resistant, recurrent OVC. (n = 20) | Envafolimab + lenvatinib + VP-16 for 6 cycles, followed by envafolimab maintenance therapy. | ORR | OS, PFS, DCR, and AEs | NCT05422183 | 2022 | |||
| Randomized phase II trial | High-grade serous OVC. (n = 16) | Pembrolizumab or lenvatinib administered first as monotherapy and then as combination therapy. Cohort A: Lenvatinib as monothrapy, cohort B: pembrolizumab as monotherapy. | T-cell dysfunction and proliferation | ORR, T-cell effector function, and T-cell memory establishment | NCT05114421 | 2021 | |||
| Single-arm phase II trial | Platinum-sensitive, recurrent, OVC (except from low grade tumors and mucinous histology). (n = 24) | Pembrolizumab/lenvitanib for up to 35 21-day cycles. | PFS | ORR, time to next-line therapy, OS, HRQoL, AEs, safety and tolerability | NCT04519151 | 2020 | |||
| Tyrosine and serine/threonine kinase inhibitors | Ipatasertib, cobimetinib, abemaciclib, inavolisib, palbociclib | AKT, MEK, CDK4- and 6, PI3K, CDK4- and 6, respectively | Phase II platform study | Persistent or recurrent rare OVC. (n = 400) | Stratificatin into 8 arms depending on biomarker expression: (1) Ipatasertib + PAX, (2) cobimetinib, (3) trastuzumab emtansine, (4) atezolizumab + bevacizumab, (5) giredestrant + abemaciclib, (6) inavolisib + palbociclib, (7) inavolisib + palbociclib + letrozole, and (8) inavolisib + olaparib. | ORR | DOR, DCR, PFS, OS, and AEs. | NCT04931342 | 2021 |
| VS-6766 and Defactinib | BRAF/MEK and FAK, respectively | Single-stage exploratory, parallel cohort, phase II trial | Endometrioid, MOC, HGSC and cervical cancer patients with RAS/BRAF/NF1 mutations. (n = 55) | VS-6766 + defectanib for 3 weeks in 28-day cycles. | ORR | AEs, PFS, DCR, DOR, and OR | NCT05512208 | 2022 | |
| Phase II RCT | Molecularly profiled recurrent LGSC. (n = 100) | Randomization to either VS-6766 monotherapy or VS-6766 + defactinib combination therapy. | ORR | DOR, DCR, PFS and OS | NCT04625270 | 2020 | |||
| Serine/threonine kinase inhibitors | Copanlisib | PI3K | Phase II RCT | Patients with recurrent, platinum resistant OVC with progression on PARPi therapy. (n = 96) | Randomization to (1) Experimental arm: copanlisib + olaparib, or (2) Active comparator arm: PAX or liposomal doxorubicin or topotecan hydrochloride. | PFS | ORR, OS, and AEs | NCT05295589 | 2022 |
| CYH33 | Single-arm phase II study | Recurrent/persistent OVC with clear cell histology. (n = 86) | CYH33 monotherapy | ORR in patients with PI3KCA hotspot mutations | PFS, OS, biomarker alterations impacting PI3K pathway | NCT05043922 | 2021 | ||
| Alpelisib | Open-label phase III RCT | Platinum-resistant/refractory HGSC with no gBRCAm detected. (n = 358) | Randomization to (1) Experimental arm: alpelisib + olaparib, or (2)Active comparator arm: either PAX or liposomal doxorubicin. | PFS | OS, tolerability, PS, ORR, CBR, TTR, DOR, PKs, HRQoL | NCT04729387 | 2021 | ||
| Ipatasertib | AKT | Single-arm phase I/Ib trial | High grade serous OVC, and endometrioid adenocarcinoma. (n = 24) | CARB + PAX for up to 3 cycles + ipatasertib until 24 hours before surgery. | DLT in dose escalation and dose expansion phase | Tumor response | NCT05276973 | 2022 | |
| Afuresertib | Phase II RCT | High grade serous, endometroid, or clear cell OVC. (n = 141) | Randomization to (1) Experimental arm: afuresertib + PAX, or (2) Active comparator arm: PAX. | PFS | OS, ORR, DOR, DCR, BOR, CA125, PKs, and AEs | NCT04374630 | 2020 | ||
| ATG-008 | mTOR | Two-arm phase II trial | High grade relapsed or metastatic serous OVC, endometrial cancer, and cervical cancer. (n = 96) | Assigment to either ATG-008 + CTX or ATG010 + CTX. | ORR | TTR, DOR, DCR, OS, PFS, AEs, and safety and tolerability | NCT04998760 | 2021 | |
| GRN-300 | SIK2- and 3 | Single-arm phase I/Ib trial | Recurrent OVC. (n = 64) | Phase Ia: GRN-300 as monotherapy, phase Ib: GRN-300 + PAX | RP2D and AEs | PKs, CBR, PFS, PDs and biomarkers | NCT04711161 | 2021 | |
| PHI-101 | CHK2 | Phase I dose-finding trial | Platinum-resistance/refractory OVC. (n = 36) | Accelerated 3 + 3 design of PHI-101 to determine MTD | DLT and MTD | Dose interruption, reduction or termination, PKs, ORR, DCR, DOR, PFS, OS, TTP, genetic variation, AEs. | NCT04678102 | 2020 | |
| Abemaciclib | CDK4- and 6 | Single-arm phase II trial | Recurrent OVC, or recurrent endometrial cancer. (n = 32) | All patients receive abemaciclib. Patients with HR+ tumors also receive anastrozole or letrozole per standard of care. | PFS at 16 weeks | ORR, PFS (up to 1 year), AEs, and CBR | NCT04469764 | 2020 | |
| ZN-c3 | WEE1 | Single-arm phase I trial | Advanced ovarian cancer or triple-negative breast cancer. (n = 14) | ZN-c3 monotherapy for up to 12 cycles. | Decrease in pCDK1 and/or Ki67, or pHH3 or PCHK1 in tumor cells, and AEs. | CBR, CBR in ovarian cancer, PFS, OS and time to disease progression | NCT05368506 | 2022 | |
| Single-arm phase I/II trial | Recurrent, high grade OVC with histologic subtypes of serous, clear cell or endometrial. (n = 138) | ZN-c3 + niraparib combination therapy. | Phase I: DLT, phase II: PFS and ORR | DOR, CBR, ORR, OS, AEs, PROs, and PKs | NCT05198804 | 2022 | |||
| Phase Ib trial | Platinum-resistant OVC. (n = 140) | 4 cohorts receiving either ZN-c3 + PLD, ZN-c3 + CARB, ZN-c3 + PAX pr ZN-c3 + gemcitabene. | Safety and tolerability and MTD | ORR, DOR, PFS, CA125, and PKs | NCT04516447 | 2020 | |||
| Chiauranib | Aurora B, VEGFRs, KIT, PDGFRs | Double-blind phase III RCT | Platinum-refractory, resistant, OVC. (n = 376) | Chiauranib + PAX or placebo + PAX for 6 cycles followed by single agent therapy of chiauranib or placebo. | PFS and OS | ORR, DOR, DCR, HRQoL, and toxicity | NCT04921527 | 2021 |
| Kinase Inhibitor | Target Kinase | Combination Treatment | Patient-Derived Organoid Samples | Conclusion | Ref. | ||
|---|---|---|---|---|---|---|---|
| Tyrosine Kinase Inhibitors | Cediranib | VEGFR | Monotherapy | HGSOC short-term PDOs (n = 3) | Organoids sensitive to the drug. | [125,126] | |
| Pazopanib | VEGFR | Monotherapy | HGSOC short-term PDOs (n = 3) | Organoids display different sensitivity towards the drug. | |||
| Sunitinib | VEGFR | Monotherapy | HGSOC short-term PDOs (n = 3) | No drug sensitivity. | |||
| Gefitinib | EGFR | Monotherapy Monotherapy | HGSOC short-term PDOs (n = 3) PDOs from ascites or tumor tissue (n = 3). | Organoids display different sensitivity towards the drug. Effective response against cell growth. | |||
| Lapatinib | EGFR | Monotherapy Monotherapy | HGSOC short-term PDOs (n = 3) Platinium resistant HGSOC PDO (n = 1) | Organoids display different sensitivity towards the drug. Moderate response. | [125,127] | ||
| WZ8040 | EGFR | Monotherapy | Platinium resistant HGSOC PDO (n = 1) | Moderate response. | [127] | ||
| Afatinib | EGFR | Monotherapy Monotherapy | PDOs (n = 36) PDOs from ascites or tumor tissue (n = 3). | Low responsivness with intrapatient heterogeneity. Effective response against cell growth. | [126,128] | ||
| Erlotinib | EGFR | Monotherapy | PDOs from ascites or tumor tissue (n = 3). | Effective response against cell growth. | |||
| Canertinib | EGFR | Monotherapy | PDOs from ascites or tumor tissue (n = 3). | Effective response against cell growth, especially under 3D culture conditions. | |||
| Dacominitib | EGFR | Monotherapy | PDOs from ascites or tumor tissue (n = 3). | Effective response against cell growth, especially under 3D culture conditions. | |||
| Neratinib | EGFR | Monotherapy | PDOs from ascites or tumor tissue (n = 3). | Effective response against cell growth, especially under 3D culture conditions. | |||
| BMS-754807 | IGF1R/InsR | Monotherapy | PDOs from ascites or tumor tissue (n = 3). | Highly effective response against cell growth, irrespetive of 2D or 3D cuture conditions. | |||
| Sorafenib | MEK, ERK, VEGFR, Carboplatin/Paclitaxel PDGFR | HGSOC PDOs from ascites or pleural fluid speciments (n = 10) | Consistent inhibitory effects in low micromolar range. IC50 lower to Cmax acssociated with therapeutic dosage, but variability between subjects. | [129] | |||
| Batiraxcept (AVB- 500) | Chemoresistant POV71-hTERT cell AXL Carboplatin/Paclitaxel, Olaparib culture from ascites (n = 1) | Synergistic effect with chemotherapy. | [130] | ||||
| Quizartinib AC220 | FLT3 Monotherapy Platinium resistant HGSOC PDO (n = 1) | Moderate response. | [127] | ||||
| Monotherapy | HGSOC PDOs from ascites or pleural fluid No consistent sensitivity towards all samples speciments (n = 10) (n = 5). | [129] | |||||
| Serine/Threonine Kinase Inhibitors | LY294002 | PI3K | Cisplatin | MCW-OV-SL-3, established cell line from tumor tissue | Sensitization towards cisplatin. | [126,127,131] | |
| BGT226 | PI3K/mTOR | Monotherapy | Platinium resistant HGSOC PDO (n = 1) | Organoids sensitive to the drug. | |||
| Omipalisib | PI3K/mTOR | Monotherapy | PDOs from ascites or tumor tissue (n = 3). | Highly effective response against cell growth, irrespetive of 2D or 3D cuture conditions. | |||
| PF-04691502 | PI3K/mTOR | Monotherapy | PDOs from ascites or tumor tissue (n = 3). | Highly effective response against cell growth, irrespetive of 2D or 3D cuture conditions. | |||
| Apitolisib | PI3K/mTOR | Monotherapy | PDOs from ascites or tumor tissue (n = 3). | Effective response against cell growth. | |||
| Vistusertib (AZD1152) | PI3K/mTOR | Monotherapy | PDOs from ascites or tumor tissue (n = 3). | Highly effective response against cell growth, irrespetive of 2D or 3D cuture conditions. | |||
| Everolimus | mTOR | Monotherapy | HGSOC short-term organoid culture (n = 3) | Organoids sensitive to the drug. | [125] | ||
| Capivasertib (AZD5363) | AKT | Monotherapy | HGSOC PDOs from ascites or pleural fluid No consistent sensitivity towards all samples speciments (n = 10) (n = 4). | [125,126,129] | |||
| Trametinib | MEK1, MEK2 | Monotherapy Monotherapy Monotherapy | HGSOC PDOs from ascites or pleural fluid No inhibitory effects. speciments (n = 10) Organoids display different sensitivity towards HGSOC short-term organoid culture (n = 3) the drug. PDOs from ascites or tumor tissue (n = 3). Effective response against cell growth. | ||||
| Refametinib | MEK | Monotherapy | Highly effective response against cell growth, PDOs from ascites or tumor tissue (n = 3). irrespetive of 2D or 3D cuture conditions. | ||||
| Adavosertib (AZD1775) | Wee1 | Monotherapy Monotherapy Monotherapy | PDOs (n = 36) HGSOC PDOs from ascites or pleural fluid speciments (n = 10) Patient-ascites-derived established cell lines (n = 2) | Low responsivness with intrapatient heterogeneity. Consistent inhibitory effects in low micromolar range. IC50 lower to Cmax acssociated with therapeutic dosage, but variability between subjects. Induced apoptosis and reduced proliferation independently of the HR status of the patient. | [128,129,132] | ||
| Berzosertib (VE822) | ATR | Monotherapy | HGSC short-term organoid culture (n = 10) | Organoids display different sensitivity/resistance towards the drug. | [133] | ||
| Prexasertib | CHEK1 | Carboplatin, Gemcitabine | HGSC short-term organoid culture (n = 10) | Sesitive for fork-unstable organoids. Resistant for stable. But, combination with carboplatin or gemcitaine promotes instability. | |||
| CHIR-124 | CHEK1 | Monotherapy | Platinium resistant HGSOC PDO (n = 1) | Moderate response. | [127] | ||
| Alisertib | Aurora | Monotherapy | PDOs from ascites or tumor tissue (n = 3). | Effective response against cell growth. | [126] | ||
| AZD1152-HQPA | Aurora | Monotherapy | PDOs from ascites or tumor tissue (n = 3). | Effective response against cell growth. | |||
| AT9283 | Aurora | Monotherapy | PDOs from ascites or tumor tissue (n = 3). | Effective response against cell growth. | |||
| Volasertib | PLK1 | Monotherapy | PDOs from ascites or tumor tissue (n = 3). | Effective response against cell growth. | |||
| Napabucasin | STAT | Monotherapy | HGSOC PDOs from ascites or pleural fluid speciments (n = 10) | No consistent sensitivity towards all samples | [129] | ||
| Vemurafenib | B-raf | Monotherapy | PDOs (n = 36) | High Responsivness. | [128] | ||
| Flavopiridol | CDK | Monotherapy | PDOs (n = 36) | High Responsivness | |||
| Kinase Inhibitor | Target Kinase | Combination Patient-Derived Treatment Tissue or Cell Lines | Mice | Conclusion | Ref. | ||
|---|---|---|---|---|---|---|---|
| Olaparib PDX | − | Broad anti-tumor effect irrespective of HR tumor status. Combination treatment reduced tumor metastasis and prolonged overall survival. | [139] | ||||
| Tyrosine Kinase Inhibitors | Cediranib | VEGF | Triapine, Olaparib OVCAR3, SKOV3 HGSOC mouse orthrotropic cell anti-Il6 antibody, antilines; 30200, 60,577 expressing PD1 antibody Trp53-/-, BRCA1-/-,Rb, HGS2 | Nude, SCID FVB/NCrl, C57BL/6J | Anti-cancer effect regardlesss of HR status. Combination of anti-angiogenic agents with anti-Il6 or anti-PD1 result in prolonged mouse survival. | [140,141] | |
| Alofanib | FGFR2 | Carboplatin/Paclitaxel | SKOV3 | Nude | Delayed tumor growth and proliferation in combination treatment. | [142] | |
| VS6063, FAK PROTAC | FAK | Monotherapy | OVCAR8 | NOD/SCID gamma | FAK PROTAC is more effective than VS6063 in inhibiting tumor growth, migration and invasion. | [143] | |
| APG-2449 | ALK/ROS/FAK | Paclitaxel | PDX, OVCAR3 | Nude with NSCLC H3122 CDX, SCID with KARPAS-299 CDX | Adminestered alone or in combination SOC can overcome primary and secondary TKI resistance. | [144] | |
| Ceritinib | ALK | Olaparib | PDX | SCID | Induces more effective tumor regression in combination treatment with Olaparib. | [145] | |
| Batiraxcept (AVB500) | AXL | Carboplatin/Paclitaxel | PDX, OVCAR5, OVCAR8 | NOD/SCID gamma | Improves response to carboplatin, increased DNA damage | [130] | |
| Serine/Threonine Kinase Inhibitors | Uprosertib (LAE003) | AKT | Olaparib | PDX from platinium-resistant EOC patients with former PARPi treament (n = 5) | Balb/c nude | Combination treatment delays tumor growth with higher efficiency compared to monotherapy. | [146] |
| AD80 BMS777607 BKM120 GSK2110183 | AKT, S6K1 Ron PI3K pan-AKT | BMS777607 Monotherapy | PDX, OVCAR4 OVCAR3sfRon | NOD/SCID gamma | Superior inhibition of tumor growth and metastasis development than SOC. Anti-tumor activity is hindered after cessation of treatment. Anti-tumor activity is hindered after cessation of treatment. Anti-tumor activity is hindered after cessation of treatment. | [147] | |
| BAY1217389 CFI-402257 | TTK, mTOR | Cisplatin | CAOV3, OV90 | Nude | Inhibits tumor growth and increased cisplatin sensitivity via inhibiting autophagy in vitro and in vivo. | [148] | |
| Trametinib | MEK1/2 | Monotherapy | PEO4 | NOD/SCID gamma | Reduces the rate of tumor growth in vivo, but corellates with cancer stem-like features. | [149] | |
| Prexasertib | CHK1 | Olaparib | Patient-derived ascites (n = 14) | NOD/SCID gamma | As monotherapy or in combination kills tumors cells with de novo or acquired PARP resistance via DNA damage. | [150] | |
| Dinaciclib | CDK | MK-2206 | OVCAR3, CAOV3 | NOD/SCID gamma | Delayed tumor growth in CCNE-1-amplified HGSOC xenografts. Selectivly synergistic effect with MK2206. | [151] | |
| ARN-3236 | SIK2 | Paclitaxel | OVCAR8 | NOD/SCID gamma | Enhances paclitaxel sensitivity. | [152] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skorda, A.; Bay, M.L.; Hautaniemi, S.; Lahtinen, A.; Kallunki, T. Kinase Inhibitors in the Treatment of Ovarian Cancer: Current State and Future Promises. Cancers 2022, 14, 6257. https://doi.org/10.3390/cancers14246257
Skorda A, Bay ML, Hautaniemi S, Lahtinen A, Kallunki T. Kinase Inhibitors in the Treatment of Ovarian Cancer: Current State and Future Promises. Cancers. 2022; 14(24):6257. https://doi.org/10.3390/cancers14246257
Chicago/Turabian StyleSkorda, Aikaterini, Marie Lund Bay, Sampsa Hautaniemi, Alexandra Lahtinen, and Tuula Kallunki. 2022. "Kinase Inhibitors in the Treatment of Ovarian Cancer: Current State and Future Promises" Cancers 14, no. 24: 6257. https://doi.org/10.3390/cancers14246257
APA StyleSkorda, A., Bay, M. L., Hautaniemi, S., Lahtinen, A., & Kallunki, T. (2022). Kinase Inhibitors in the Treatment of Ovarian Cancer: Current State and Future Promises. Cancers, 14(24), 6257. https://doi.org/10.3390/cancers14246257