
The Purinergic Landscape of Non-Small Cell Lung Cancer

 , ,
, ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
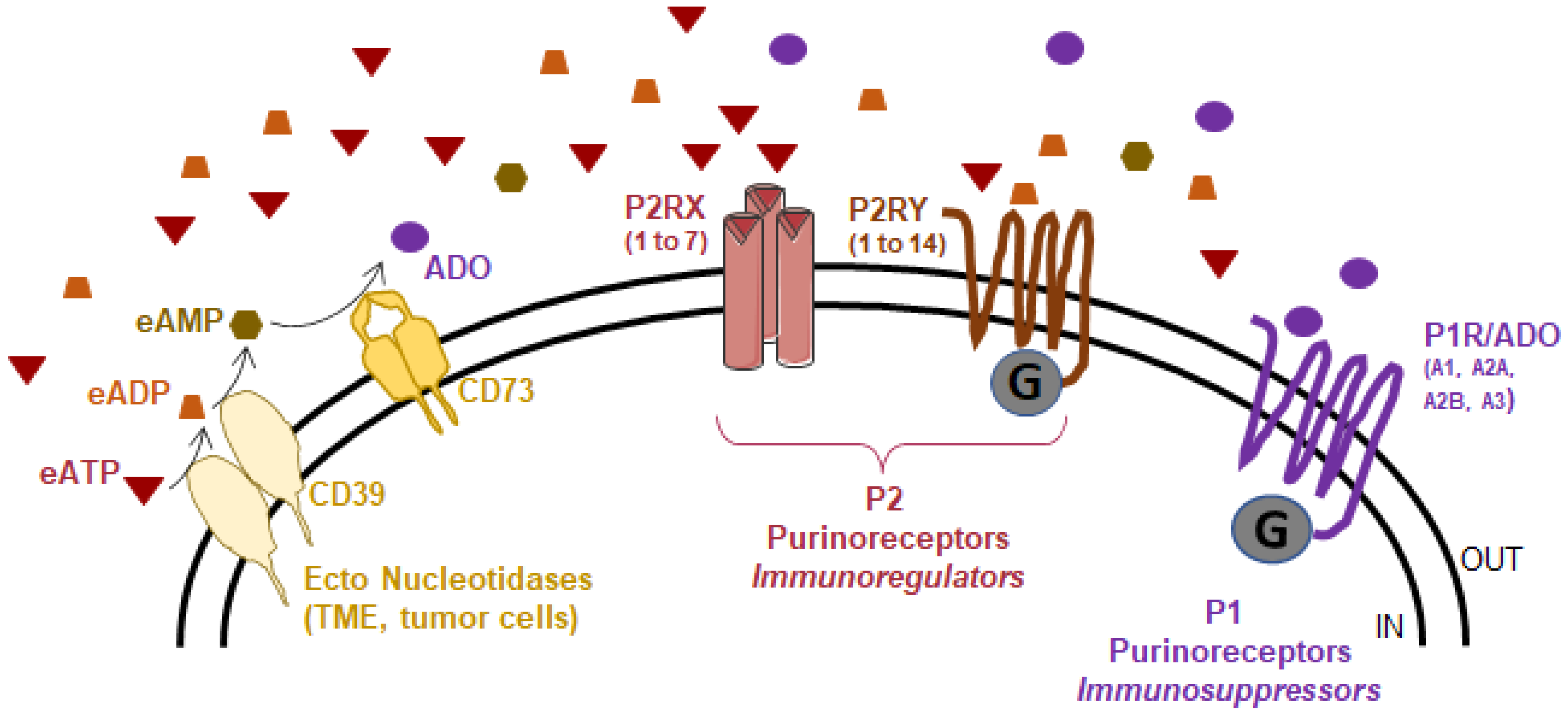
2. Source of Extracellular ATP
3. The Purinergic Landscape in NSCLC
3.1. Expression of Ectonucleotidases
3.2. Expresssion of the Purinergic Receptors
3.2.1. The Adenosine Receptors
3.2.2. The Purinergic Receptors
4. The Purinergic Landscape: A Therapeutically Targetable Circuitry to Treat NSCLC
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Shields, M.D.; Marin-Acevedo, J.A.; Pellini, B. Immunotherapy for Advanced Non–Small Cell Lung Cancer: A Decade of Progress. Am. Soc. Clin. Oncol. Educ. B 2021, 41, e105–e127. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Bezu, L.; Yamazaki, T.; Di Virgilio, F.; Smyth, M.J.; Kroemer, G.; Galluzzi, L. ATP and cancer immunosurveillance. EMBO J. 2021, 40, e108130. [Google Scholar] [CrossRef]
- Divirgilio, F. Liaisons dangereuses: P2X7 and the inflammasome. Trends Pharmacol. Sci. 2007, 28, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Pellegatti, P.; Raffaghello, L.; Bianchi, G.; Piccardi, F.; Pistoia, V.; Di Virgilio, F. Increased level of extracellular ATP at tumor sites: In vivo imaging with plasma membrane luciferase. PLoS ONE 2008, 3, e2599. [Google Scholar] [CrossRef] [PubMed]
- Stankovic, B.; Bjørhovde, H.A.K.; Skarshaug, R.; Aamodt, H.; Frafjord, A.; Müller, E.; Hammarström, C.; Beraki, K.; Bækkevold, E.S.; Woldbæk, P.R.; et al. Immune Cell Composition in Human Non-small Cell Lung Cancer. Front. Immunol. 2019, 9, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lennon, P.F.; Taylor, C.T.; Stahl, G.L.; Colgan, S.P. Neutrophil-derived 5′-Adenosine Monophosphate Promotes Endothelial Barrier Function via CD73-mediated Conversion to Adenosine and Endothelial A2B Receptor Activation. J. Exp. Med. 1998, 188, 1433–1443. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Ibla, J.C.; Furuta, G.T.; Leonard, M.O.; Jacobson, K.A.; Enjyoji, K.; Robson, S.C.; Colgan, S.P. Coordinated Adenine Nucleotide Phosphohydrolysis and Nucleoside Signaling in Posthypoxic Endothelium. J. Exp. Med. 2003, 198, 783–796. [Google Scholar] [CrossRef] [Green Version]
- Thompson, L.F.; Eltzschig, H.K.; Ibla, J.C.; Van De Wiele, C.J.; Resta, R.; Morote-Garcia, J.C.; Colgan, S.P. Crucial Role for Ecto-5′-Nucleotidase (CD73) in Vascular Leakage during Hypoxia. J. Exp. Med. 2004, 200, 1395–1405. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Eckle, T.; Mager, A.; Küper, N.; Karcher, C.; Weissmüller, T.; Boengler, K.; Schulz, R.; Robson, S.C.; Colgan, S.P. ATP Release From Activated Neutrophils Occurs via Connexin 43 and Modulates Adenosine-Dependent Endothelial Cell Function. Circ. Res. 2006, 99, 1100–1108. [Google Scholar] [CrossRef] [Green Version]
- Bao, Y.; Chen, Y.; Ledderose, C.; Li, L.; Junger, W.G. Pannexin 1 Channels Link Chemoattractant Receptor Signaling to Local Excitation and Global Inhibition Responses at the Front and Back of Polarized Neutrophils. J. Biol. Chem. 2013, 288, 22650–22657. [Google Scholar] [CrossRef] [Green Version]
- Dosch, M.; Gerber, J.; Jebbawi, F.; Beldi, G. Mechanisms of ATP Release by Inflammatory Cells. Int. J. Mol. Sci. 2018, 19, 1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawada, K.; Echigo, N.; Juge, N.; Miyaji, T.; Otsuka, M.; Omote, H.; Yamamoto, A.; Moriyama, Y. Identification of a vesicular nucleotide transporter. Proc. Natl. Acad. Sci. USA 2008, 105, 5683–5686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Südhof, T.C.; Rothman, J.E. Membrane Fusion: Grappling with SNARE and SM Proteins. Science 2009, 323, 474–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [Green Version]
- Martins, I.; Wang, Y.; Michaud, M.; Ma, Y.; Sukkurwala, A.Q.; Shen, S.; Kepp, O.; Métivier, D.; Galluzzi, L.; Perfettini, J.-L.; et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ. 2014, 21, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Robinson, N.; Ganesan, R.; Hegedűs, C.; Kovács, K.; Kufer, T.A.; Virág, L. Programmed necrotic cell death of macrophages: Focus on pyroptosis, necroptosis, and parthanatos. Redox Biol. 2019, 26, 101239. [Google Scholar] [CrossRef]
- Santos, S.A.C.S.; Persechini, P.M.; Henriques-Santos, B.M.; Bello-Santos, V.G.; Castro, N.G.; Costa de Sousa, J.; Genta, F.A.; Santiago, M.F.; Coutinho-Silva, R.; Savio, L.E.B.; et al. P2X7 Receptor Triggers Lysosomal Leakage Through Calcium Mobilization in a Mechanism Dependent on Pannexin-1 Hemichannels. Front. Immunol. 2022, 13, 75105. [Google Scholar] [CrossRef]
- Brock, V.J.; Wolf, I.M.A.; Er-Lukowiak, M.; Lory, N.; Stähler, T.; Woelk, L.-M.; Mittrücker, H.-W.; Müller, C.E.; Koch-Nolte, F.; Rissiek, B.; et al. P2X4 and P2X7 are essential players in basal T cell activity and Ca 2+ signaling milliseconds after T cell activation. Sci. Adv. 2022, 8, abl9770. [Google Scholar] [CrossRef]
- Grassi, F. The P2X7 Receptor as Regulator of T Cell Development and Function. Front. Immunol. 2020, 11, 1179. [Google Scholar] [CrossRef]
- Fucikova, J.; Kepp, O.; Kasikova, L.; Petroni, G.; Yamazaki, T.; Liu, P.; Zhao, L.; Spisek, R.; Kroemer, G.; Galluzzi, L. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 2020, 11, 1013. [Google Scholar] [CrossRef]
- Golden, E.B.; Apetoh, L. Radiotherapy and Immunogenic Cell Death. Semin. Radiat. Oncol. 2015, 25, 11–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, G.; Glaser, T.; Lameu, C.; Abdelbaset-Ismail, A.; Sellers, Z.P.; Moniuszko, M.; Ulrich, H.; Ratajczak, M.Z. Extracellular nucleotides as novel, underappreciated pro-metastatic factors that stimulate purinergic signaling in human lung cancer cells. Mol. Cancer 2015, 14, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ide, S.; Nishimaki, N.; Tsukimoto, M.; Kojima, S. Purine receptor P2Y6 mediates cellular response to γ-ray-induced DNA damage. J. Toxicol. Sci. 2014, 39, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimaki, N.; Tsukimoto, M.; Kitami, A.; Kojima, S. Autocrine regulation of γ-irradiation-induced DNA damage response via extracellular nucleotides-mediated activation of P2Y6 and P2Y12 receptors. DNA Repair (Amst.) 2012, 11, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Adjemian, S.; Yang, H.; Catani, J.P.P.; Hannani, D.; Martins, I.; Michaud, M.; Kepp, O.; Sukkurwala, A.Q.; Vacchelli, E.; et al. ATP-dependent recruitment, survival and differentiation of dendritic cell precursors in the tumor bed after anticancer chemotherapy. Oncoimmunology 2013, 2, e24568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Adjemian, S.; Mattarollo, S.R.; Yamazaki, T.; Aymeric, L.; Yang, H.; Portela Catani, J.P.; Hannani, D.; Duret, H.; Steegh, K.; et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 2013, 38, 729–741. [Google Scholar] [CrossRef] [Green Version]
- Schetinger, M.R.C.; Morsch, V.M.; Bonan, C.D.; Wyse, A.T.S. NTPDase and 5’-nucleotidase activities in physiological and disease conditions: New perspectives for human health. BioFactors 2007, 31, 77–98. [Google Scholar] [CrossRef]
- Chini, E.N.; Chini, C.C.S.; Espindola Netto, J.M.; de Oliveira, G.C.; van Schooten, W. The Pharmacology of CD38/NADase: An Emerging Target in Cancer and Diseases of Aging. Trends Pharmacol. Sci. 2018, 39, 424–436. [Google Scholar] [CrossRef]
- Gupta, P.K.; Godec, J.; Wolski, D.; Adland, E.; Yates, K.; Pauken, K.E.; Cosgrove, C.; Ledderose, C.; Junger, W.G.; Robson, S.C.; et al. CD39 Expression Identifies Terminally Exhausted CD8+ T Cells. PLoS Pathog. 2015, 11, e1005177. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Bo, C.; Kang, Y.; Li, H. What Else Can CD39 Tell Us? Front. Immunol. 2017, 8, 727. [Google Scholar] [CrossRef] [Green Version]
- Bastid, J.; Regairaz, A.; Bonnefoy, N.; Déjou, C.; Giustiniani, J.; Laheurte, C.; Cochaud, S.; Laprevotte, E.; Funck-Brentano, E.; Hemon, P.; et al. Inhibition of CD39 Enzymatic Function at the Surface of Tumor Cells Alleviates Their Immunosuppressive Activity. Cancer Immunol. Res. 2015, 3, 254–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tøndell, A.; Wahl, S.G.F.; Sponaas, A.-M.; Sørhaug, S.; Børset, M.; Haug, M. Ectonucleotidase CD39 and Checkpoint Signalling Receptor Programmed Death 1 are Highly Elevated in Intratumoral Immune Cells in Non–small-cell Lung Cancer. Transl. Oncol. 2020, 13, 17–24. [Google Scholar] [CrossRef]
- Yeong, J.; Suteja, L.; Simoni, Y.; Lau, K.W.; Tan, A.C.; Li, H.H.; Lim, S.; Loh, J.H.; Wee, F.Y.T.; Nerurkar, S.N.; et al. Intratumoral CD39+CD8+ T Cells Predict Response to Programmed Cell Death Protein-1 or Programmed Death Ligand-1 Blockade in Patients With NSCLC. J. Thorac. Oncol. 2021, 16, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Horenstein, A.L.; Chillemi, A.; Zaccarello, G.; Bruzzone, S.; Quarona, V.; Zito, A.; Serra, S.; Malavasi, F. A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. Oncoimmunology 2013, 2, e26246. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Wainwright, D.A.; Wu, J.D.; Wan, Y.; Matei, D.E.; Zhang, Y.; Zhang, B. CD73: An emerging checkpoint for cancer immunotherapy. Immunotherapy 2019, 11, 983–997. [Google Scholar] [CrossRef]
- Giatromanolaki, A.; Kouroupi, M.; Pouliliou, S.; Mitrakas, A.; Hasan, F.; Pappa, A.; Koukourakis, M.I. Ectonucleotidase CD73 and CD39 expression in non-small cell lung cancer relates to hypoxia and immunosuppressive pathways. Life Sci. 2020, 259, 118389. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Yoshimura, K.; Kurabe, N.; Kahyo, T.; Kawase, A.; Tanahashi, M.; Ogawa, H.; Inui, N.; Funai, K.; Shinmura, K.; et al. Prognostic impact of CD73 and A2A adenosine receptor expression in non-small-cell lung cancer. Oncotarget 2017, 8, 8738–8751. [Google Scholar] [CrossRef] [Green Version]
- Negrao, M.V.; Skoulidis, F.; Montesion, M.; Schulze, K.; Bara, I.; Shen, V.; Xu, H.; Hu, S.; Sui, D.; Elamin, Y.Y.; et al. Oncogene-specific differences in tumor mutational burden, PD-L1 expression, and outcomes from immunotherapy in non-small cell lung cancer. J. Immunother. Cancer 2021, 9, e002891. [Google Scholar] [CrossRef]
- Ricciuti, B.; Arbour, K.C.; Lin, J.J.; Vajdi, A.; Vokes, N.; Hong, L.; Zhang, J.; Tolstorukov, M.Y.; Li, Y.Y.; Spurr, L.F.; et al. Diminished Efficacy of Programmed Death-(Ligand)1 Inhibition in STK11- and KEAP1-Mutant Lung Adenocarcinoma Is Affected by KRAS Mutation Status. J. Thorac. Oncol. 2022, 17, 399–410. [Google Scholar] [CrossRef]
- Le, X.; Negrao, M.V.; Reuben, A.; Federico, L.; Diao, L.; McGrail, D.; Nilsson, M.; Robichaux, J.; Munoz, I.G.; Patel, S.; et al. Characterization of the Immune Landscape of EGFR-Mutant NSCLC Identifies CD73/Adenosine Pathway as a Potential Therapeutic Target. J. Thorac. Oncol. 2021, 16, 583–600. [Google Scholar] [CrossRef]
- Burnstock, G.; Di Virgilio, F. Purinergic signalling and cancer. Purinergic Signal. 2013, 9, 491–540. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Brouns, I.; Adriaensen, D.; Timmermans, J.-P. Purinergic Signaling in the Airways. Pharmacol. Rev. 2012, 64, 834–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sek, K.; Mølck, C.; Stewart, G.; Kats, L.; Darcy, P.; Beavis, P. Targeting Adenosine Receptor Signaling in Cancer Immunotherapy. Int. J. Mol. Sci. 2018, 19, 3837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazemi, M.H.; Raoofi Mohseni, S.; Hojjat-Farsangi, M.; Anvari, E.; Ghalamfarsa, G.; Mohammadi, H.; Jadidi-Niaragh, F. Adenosine and adenosine receptors in the immunopathogenesis and treatment of cancer. J. Cell. Physiol. 2018, 233, 2032–2057. [Google Scholar] [CrossRef] [Green Version]
- MacKenzie, W.M.; Hoskin, D.W.; Blay, J. Adenosine Suppresses α4β7 Integrin-Mediated Adhesion of T Lymphocytes to Colon Adenocarcinoma Cells. Exp. Cell Res. 2002, 276, 90–100. [Google Scholar] [CrossRef]
- Williams, B.A.; Manzer, A.; Blay, J.; Hoskin, D.W. Adenosine Acts through a Novel Extracellular Receptor to Inhibit Granule Exocytosis by Natural Killer Cells. Biochem. Biophys. Res. Commun. 1997, 231, 264–269. [Google Scholar] [CrossRef]
- Panther, E.; Idzko, M.; Herouy, Y.; Rheinen, H.; Gebicke-Haerter, P.J.; Mrowietz, U.; Dichmann, S.; Norgauer, J. Expression and function of adenosine receptors in human dendritic cells. FASEB J. 2001, 15, 1963–1970. [Google Scholar] [CrossRef]
- Zarek, P.E.; Huang, C.-T.; Lutz, E.R.; Kowalski, J.; Horton, M.R.; Linden, J.; Drake, C.G.; Powell, J.D. A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood 2008, 111, 251–259. [Google Scholar] [CrossRef]
- Sitkovsky, M.V.; Hatfield, S.; Abbott, R.; Belikoff, B.; Lukashev, D.; Ohta, A. Hostile, Hypoxia–A2-Adenosinergic Tumor Biology as the Next Barrier to Overcome for Tumor Immunologists. Cancer Immunol. Res. 2014, 2, 598–605. [Google Scholar] [CrossRef] [Green Version]
- Lukashev, D.; Ohta, A.; Sitkovsky, M. Hypoxia-dependent anti-inflammatory pathways in protection of cancerous tissues. Cancer Metastasis Rev. 2007, 26, 273–279. [Google Scholar] [CrossRef]
- Burnstock, G. Purinergic signalling. Br. J. Pharmacol. 2006, 147, S172–S181. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, R.; Sedehizade, F.; Welte, T.; Reiser, G. ATP- and UTP-activated P2Y receptors differently regulate proliferation of human lung epithelial tumor cells. Am. J. Physiol. Cell. Mol. Physiol. 2003, 285, L376–L385. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Knight, G.E. Cellular Distribution and Functions of P2 Receptor Subtypes in Different Systems. Int. Rev. Cytol. 2004, 240, 31–304. [Google Scholar] [PubMed]
- Song, S.; Jacobson, K.N.; McDermott, K.M.; Reddy, S.P.; Cress, A.E.; Tang, H.; Dudek, S.M.; Black, S.M.; Garcia, J.G.N.; Makino, A.; et al. ATP promotes cell survival via regulation of cytosolic [Ca2+] and Bcl-2/Bax ratio in lung cancer cells. Am. J. Physiol. Physiol. 2016, 310, C99–C114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, E.; Tsukimoto, M.; Harada, H.; Sawada, K.; Moriyama, Y.; Kojima, S. Autocrine regulation of TGF-β1-induced cell migration by exocytosis of ATP and activation of P2 receptors in human lung cancer cells. J. Cell Sci. 2012, 125, 5051–5060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, E.; Tsukimoto, M.; Harada, H.; Kojima, S. Autocrine signaling via release of ATP and activation of P2X7 receptor influences motile activity of human lung cancer cells. Purinergic Signal. 2014, 10, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Schmid, S.; Kübler, M.; Korcan Ayata, C.; Lazar, Z.; Haager, B.; Hoßfeld, M.; Meyer, A.; Cicko, S.; Elze, M.; Wiesemann, S.; et al. Altered purinergic signaling in the tumor associated immunologic microenvironment in metastasized non-small-cell lung cancer. Lung Cancer 2015, 90, 516–521. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, X.; Li, Y.; Evers, M.; Zhang, H.; Chen, X. Extracellular and macropinocytosis internalized ATP work together to induce epithelial–mesenchymal transition and other early metastatic activities in lung cancer. Cancer Cell Int. 2019, 19, 254. [Google Scholar] [CrossRef]
- Buell, G.; Chessell, I.P.; Michel, A.D.; Collo, G.; Salazzo, M.; Herren, S.; Gretener, D.; Grahames, C.; Kaur, R.; Kosco-Vilbois, M.H.; et al. Blockade of human P2X7 receptor function with a monoclonal antibody. Blood 1998, 92, 3521–3528. [Google Scholar] [CrossRef]
- Benzaquen, J.; Dit Hreich, S.J.; Heeke, S.; Juhel, T.; Lalvee, S.; Bauwens, S.; Saccani, S.; Lenormand, P.; Hofman, V.; Butori, M.; et al. P2RX7B is a new theranostic marker for lung adenocarcinoma patients. Theranostics 2020, 10, 10849–10860. [Google Scholar] [CrossRef]
- Adinolfi, E.; Raffaghello, L.; Giuliani, A.L.; Cavazzini, L.; Capece, M.; Chiozzi, P.; Bianchi, G.; Kroemer, G.; Pistoia, V.; Di Virgilio, F. Expression of P2X7 receptor increases in vivo tumor growth. Cancer Res. 2012, 72, 2957–2969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrot, I.; Michaud, H.-A.; Giraudon-Paoli, M.; Augier, S.; Docquier, A.; Gros, L.; Courtois, R.; Déjou, C.; Jecko, D.; Becquart, O.; et al. Blocking Antibodies Targeting the CD39/CD73 Immunosuppressive Pathway Unleash Immune Responses in Combination Cancer Therapies. Cell Rep. 2019, 27, 2411–2425.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.-Y.; Moesta, A.K.; Xiao, C.; Nakamura, K.; Casey, M.; Zhang, H.; Madore, J.; Lepletier, A.; Aguilera, A.R.; Sundarrajan, A.; et al. Targeting CD39 in Cancer Reveals an Extracellular ATP- and Inflammasome-Driven Tumor Immunity. Cancer Discov. 2019, 9, 1754–1773. [Google Scholar] [CrossRef] [Green Version]
- Mediavilla-Varela, M.; Castro, J.; Chiappori, A.; Noyes, D.; Hernandez, D.C.; Allard, B.; Stagg, J.; Antonia, S.J. A Novel Antagonist of the Immune Checkpoint Protein Adenosine A2a Receptor Restores Tumor-Infiltrating Lymphocyte Activity in the Context of the Tumor Microenvironment. Neoplasia 2017, 19, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Lara, R.; Adinolfi, E.; Harwood, C.A.; Philpott, M.; Barden, J.A.; Di Virgilio, F.; McNulty, S. P2X7 in Cancer: From Molecular Mechanisms to Therapeutics. Front. Pharmacol. 2020, 11, 793. [Google Scholar] [CrossRef] [PubMed]
- Amoroso, F.; Salaro, E.; Falzoni, S.; Chiozzi, P.; Giuliani, A.L.; Cavallesco, G.; Maniscalco, P.; Puozzo, A.; Bononi, I.; Martini, F.; et al. P2X7 targeting inhibits growth of human mesothelioma. Oncotarget 2016, 7, 49664–49676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adinolfi, E.; Capece, M.; Franceschini, A.; Falzoni, S.; Giuliani, A.L.; Rotondo, A.; Sarti, A.C.; Bonora, M.; Syberg, S.; Corigliano, D.; et al. Accelerated tumor progression in mice lacking the ATP receptor P2X7. Cancer Res. 2015, 75, 635–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofman, P.; Cherfils-Vicini, J.; Bazin, M.; Ilie, M.; Juhel, T.; Hébuterne, X.; Gilson, E.; Schmid-Alliana, A.; Boyer, O.; Adriouch, S.; et al. Genetic and pharmacological inactivation of the purinergic P2RX7 receptor dampens inflammation but increases tumor incidence in a mouse model of colitis-associated cancer. Cancer Res. 2015, 75, 835–845. [Google Scholar] [CrossRef] [Green Version]
- Douguet, L.; Janho dit Hreich, S.; Benzaquen, J.; Seguin, L.; Juhel, T.; Dezitter, X.; Duranton, C.; Ryffel, B.; Kanellopoulos, J.; Delarasse, C.; et al. A small-molecule P2RX7 activator promotes anti-tumor immune responses and sensitizes lung tumor to immunotherapy. Nat. Commun. 2021, 12, 653. [Google Scholar] [CrossRef]
- Janho dit Hreich, S.; Benzaquen, J.; Hofman, P.; Vouret-Craviari, V. To inhibit or to boost the ATP/P2RX7 pathway to fight cancer—that is the question. Purinergic Signal. 2021, 17, 619–631. [Google Scholar] [CrossRef]
- Gebremeskel, S.; LeVatte, T.; Liwski, R.S.; Johnston, B.; Bezuhly, M. The reversible P2Y12 inhibitor ticagrelor inhibits metastasis and improves survival in mouse models of cancer. Int. J. Cancer 2015, 136, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Gareau, A.J.; Brien, C.; Gebremeskel, S.; Liwski, R.S.; Johnston, B.; Bezuhly, M. Ticagrelor inhibits platelet–tumor cell interactions and metastasis in human and murine breast cancer. Clin. Exp. Metastasis 2018, 35, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, C.C.; Lyon, A.R.; Wojta, J.; Huber, K. Is P2Y12 inhibitor therapy associated with an increased risk of cancer? Eur. Heart J. Cardiovasc. Pharmacother. 2019, 5, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.; Bachelot-Loza, C.; Nesseler, N.; Gaussem, P.; Gouin-Thibault, I. P2Y12 Inhibition beyond Thrombosis: Effects on Inflammation. Int. J. Mol. Sci. 2020, 21, 1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Gao, Y.; He, C.; Wei, H.; Zhang, J.; Zhang, H.; Hu, L.; Jiang, W. Purinergic Receptor P2Y 6 Is a Negative Regulator of NK Cell Maturation and Function. J. Immunol. 2021, 207, 1555–1565. [Google Scholar] [CrossRef]
- Chiappori, A.; Williams, C.C.; Creelan, B.C.; Tanvetyanon, T.; Gray, J.E.; Haura, E.B.; Thapa, R.; Chen, D.-T.; Beg, A.A.; Boyle, T.A.; et al. Phase I/II study of the A2AR antagonist NIR178 (PBF-509), an oral immunotherapy, in patients (pts) with advanced NSCLC. J. Clin. Oncol. 2018, 36, 9089. [Google Scholar] [CrossRef]
- Fong, L.; Forde, P.M.; Powderly, J.D.; Goldman, J.W.; Nemunaitis, J.J.; Luke, J.J.; Hellmann, M.D.; Kummar, S.; Doebele, R.C.; Mahadevan, D.; et al. Safety and clinical activity of adenosine A2a receptor (A2aR) antagonist, CPI-444, in anti-PD1/PDL1 treatment-refractory renal cell (RCC) and non-small cell lung cancer (NSCLC) patients. J. Clin. Oncol. 2017, 35, 3004. [Google Scholar] [CrossRef]
- Kamiya, H.; Kanno, T.; Fujita, Y.; Gotoh, A.; Nakano, T.; Nishizaki, T. Apoptosis-Related Gene Transcription in Human A549 Lung Cancer Cells via A 3 Adenosine Receptor. Cell. Physiol. Biochem. 2012, 29, 687–696. [Google Scholar] [CrossRef]
- Kanno, T.; Nakano, T.; Fujita, Y.; Gotoh, A.; Nishizaki, T. Adenosine Induces Apoptosis in SBC-3 Human Lung Cancer Cells through A 3 Adenosine Receptor-Dependent AMID Upregulation. Cell. Physiol. Biochem. 2012, 30, 666–676. [Google Scholar] [CrossRef]
- Otsuki, T.; Kanno, T.; Fujita, Y.; Tabata, C.; Fukuoka, K.; Nakano, T.; Gotoh, A.; Nishizaki, T. A 3 Adenosine Receptor-Mediated p53-Dependent Apoptosis in Lu-65 Human Lung Cancer Cells. Cell. Physiol. Biochem. 2012, 30, 210–220. [Google Scholar] [CrossRef]
- Nakamura, K.; Yoshikawa, N.; Yamaguchi, Y.; Kagota, S.; Shinozuka, K.; Kunitomo, M. Antitumor effect of cordycepin (3′-deoxyadenosine) on mouse melanoma and lung carcinoma cells involves adenosine A3 receptor stimulation. Anticancer Res. 2006, 26, 43–47. [Google Scholar] [PubMed]
- Lam, E.T.; Au, J.L.-S.; Otterson, G.A.; Guillaume Wientjes, M.; Chen, L.; Shen, T.; Wei, Y.; Li, X.; Bekaii-Saab, T.; Murgo, A.J.; et al. Phase I trial of non-cytotoxic suramin as a modulator of docetaxel and gemcitabine therapy in previously treated patients with non-small cell lung cancer. Cancer Chemother. Pharmacol. 2010, 66, 1019–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villalona-Calero, M.A.; Otterson, G.A.; Wientjes, M.G.; Weber, F.; Bekaii-Saab, T.; Young, D.; Murgo, A.J.; Jensen, R.; Yeh, T.-K.; Wei, Y.; et al. Noncytotoxic suramin as a chemosensitizer in patients with advanced non-small-cell lung cancer: A phase II study. Ann. Oncol. 2008, 19, 1903–1909. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, S.M.; Gidley Baird, A.; Glazer, S.; Barden, J.A.; Glazer, A.; Teh, L.C.; King, J. A phase I clinical trial demonstrates that nfP2X 7 -targeted antibodies provide a novel, safe and tolerable topical therapy for basal cell carcinoma. Br. J. Dermatol. 2017, 177, 117–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Clinical Trial NCT | Targeted Protein | Name of Compound | Treatment | Phase | Results |
|---|---|---|---|---|---|
| 04306900 | CD39 | TTX-30 | ±ICI, chemo | 1 | Active |
| 04336098 | CD39 | SRF-617 | ±ICI, chemo | 1 | Recruiting |
| 05143970 | CD73 | IPH-5301 (Ab) | ±ICI, chemo | 1 | Recruiting |
| 05001347 | CD73 | TJ004309 (Ab) | ±ICI | 2 | Recruiting |
| 04148937 | CD73 | LY3475070 | ±osinertinib | 1 | Active |
| 03381274 | CD73 | MEDI-9447 (Ab) | ±ICI | 1 | Recruiting |
| 04672434 | CD73 | Syn-024 (Ab) | ±ICI | 1 | Recruiting |
| 03549000 | CD73 | NZV-930 (Ab) | ±ICI | 1/1b | Recruiting |
| 03454451 | CD73/A2AR | CPI-006 (Ab) | ±ICI | 1/1b | Recruiting |
| 03549000 | CD73/A2AR | NZV-930/NIR178 | ±ICI | 1/1b | Recruiting |
| 02403193 | A2AR | PDF-509 (NIR178) | ±ICI | 1/1b | Completed with Clinical benefit [76,77] |
| 03207867 | A2AR | PDF-509 (NIR178) | ±ICI | 2 | Recruiting |
| 05060432 | A2AR | EOS-448 | ±ICI | 1/1b | Recruiting |
| 04969315 | A2AR | TT-10 | ±ICI | 1/2 | Not yet recruiting |
| 03629756 | A2AR | AB-298 (Etrumadenant) | ±ICI | 1 | Completed, no results |
| 03381274 | A2AR | AZD-4635 | ±ICI, targeted therapy | 1/2 | Active |
| 04262856 | A2AR/A2BR | AB-298 (Etrumadenant) | ±ICI | 2 | Recruiting |
| 03846310 | A2AR/A2BR | AB-298 (Etrumadenant) | ±ICI | 1/1b | Active |
| 03274479 | A2AR | PDF-1129 | 1 | Recruiting | |
| 05234307 | A2AR | PDF-1129 | ±ICI | 1 | Not yet recruiting |
| 03337698 | A2AR | CPI-444 | ±ICI, targeted therapy, chemo, radio | 1b/2 | Recruiting |
| 02080078 | A2AR/A2BR | Theophylline | ±TKI | 1 | Completed, no results |
| 01038752 | P2Rs | suramin | ±chemo | 2 | Completed |
| 00006929 | P2Rs | suramin | ±chemo | 2 | Completed |
| 00066768 | P2Rs | suramin | ±chemo | 1 | Completed |
| 01671332 | P2Rs | suramin | ±chemo | 2 | Completed |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janho dit Hreich, S.; Benzaquen, J.; Hofman, P.; Vouret-Craviari, V. The Purinergic Landscape of Non-Small Cell Lung Cancer. Cancers 2022, 14, 1926. https://doi.org/10.3390/cancers14081926
Janho dit Hreich S, Benzaquen J, Hofman P, Vouret-Craviari V. The Purinergic Landscape of Non-Small Cell Lung Cancer. Cancers. 2022; 14(8):1926. https://doi.org/10.3390/cancers14081926
Chicago/Turabian StyleJanho dit Hreich, Serena, Jonathan Benzaquen, Paul Hofman, and Valérie Vouret-Craviari. 2022. "The Purinergic Landscape of Non-Small Cell Lung Cancer" Cancers 14, no. 8: 1926. https://doi.org/10.3390/cancers14081926
APA StyleJanho dit Hreich, S., Benzaquen, J., Hofman, P., & Vouret-Craviari, V. (2022). The Purinergic Landscape of Non-Small Cell Lung Cancer. Cancers, 14(8), 1926. https://doi.org/10.3390/cancers14081926