
Regulation of ZEB1 Function and Molecular Associations in Tumor Progression and Metastasis


{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Biological Function of ZEB1
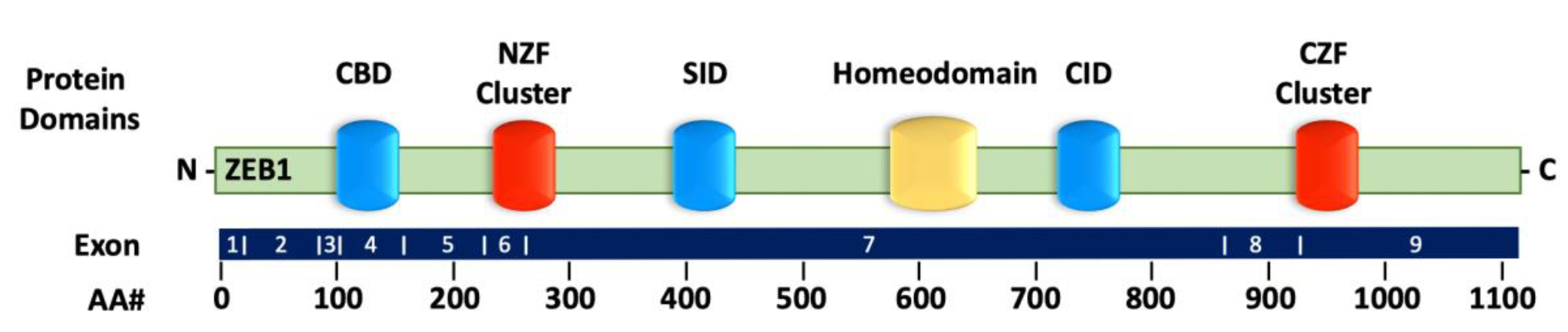
2.1. Structure of ZEB1
2.2. ZEB1 in Tumor Progression
2.3. ZEB1 in Metastasis
2.4. ZEB1 in Therapy Resistance
3. Regulation of ZEB1 Expression
3.1. Transcriptional Regulation of ZEB1
3.2. Feedback Loop-Mediated Regulation of ZEB1 Expression
3.3. Epigenetic Regulation of ZEB1
3.4. Translational Regulation of ZEB1
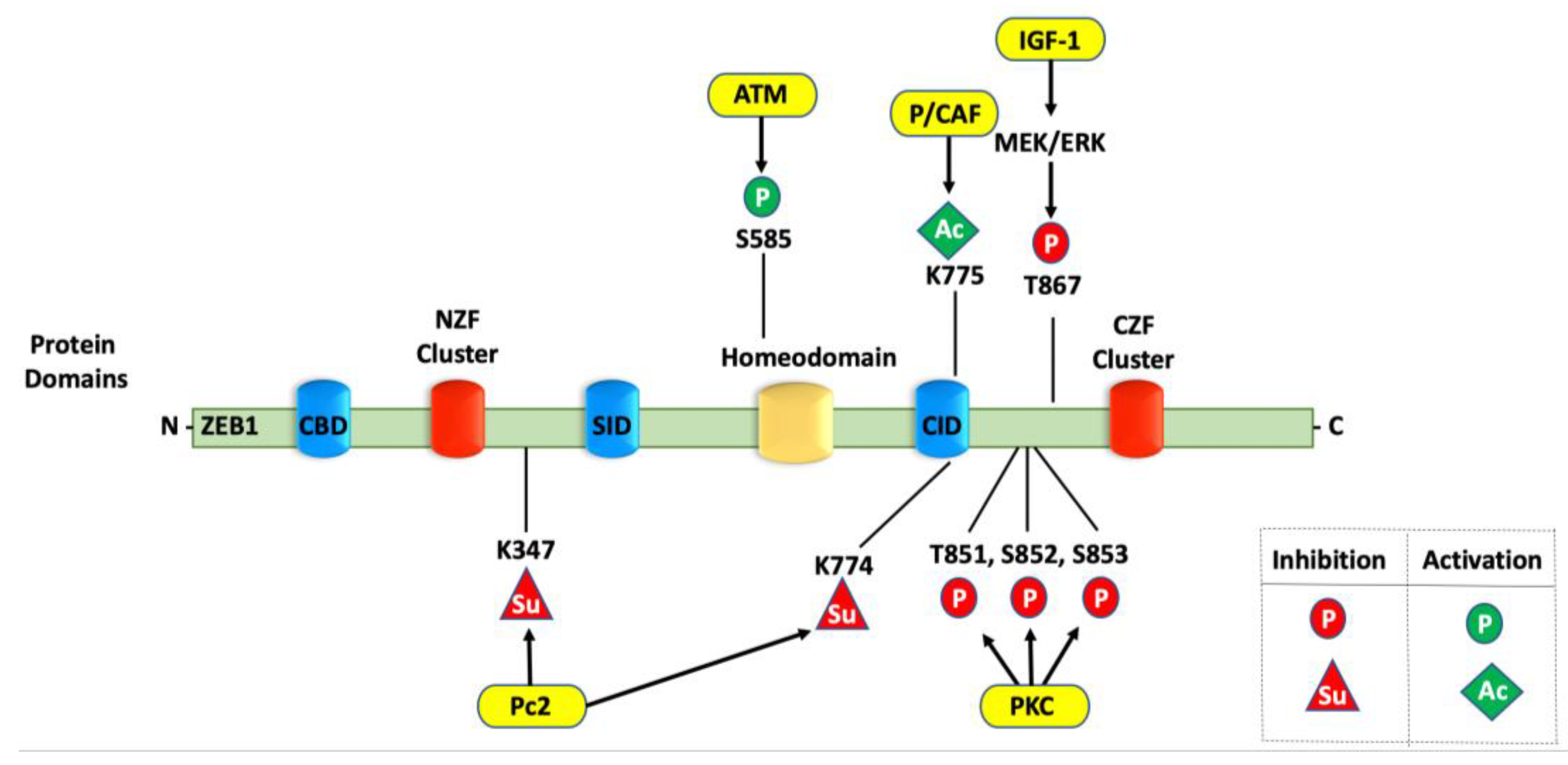
4. Post-Translational Modifications of ZEB1
4.1. ZEB1 Protein Stability
4.2. Phosphorylation of ZEB1
4.3. Ubiquitination of ZEB1
4.4. Sumoylation of ZEB1
4.5. ZEB1 Interactions with Histone Acetyltransferases
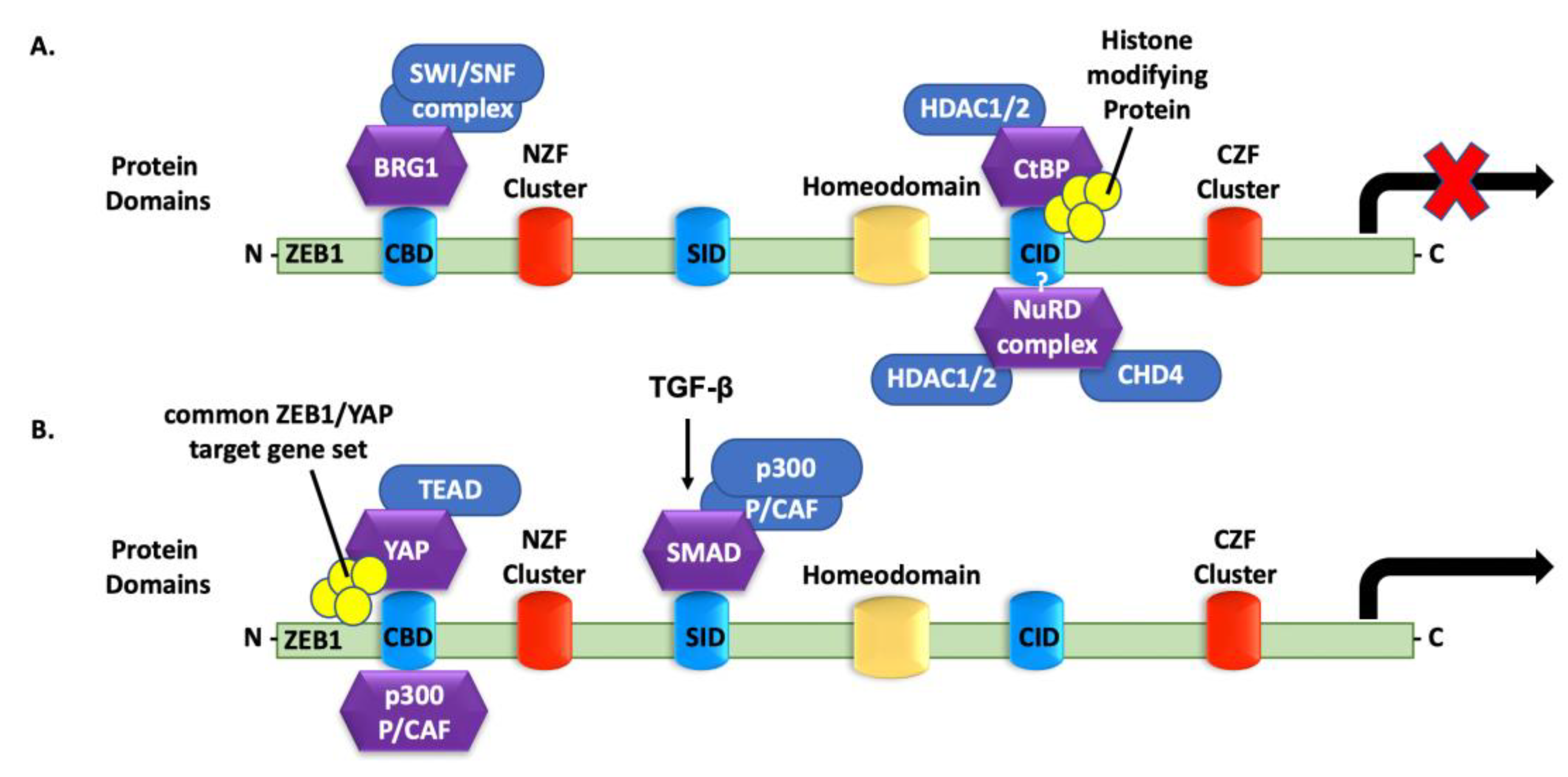
4.6. ZEB1 Binding to Co-Repressors
4.7. ZEB1 Binding to Transcriptional Activators
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Huysentruyt, L.C. On the origin of cancer metastasis. Crit. Rev. Oncog. 2013, 18, 43–73. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, Y.; Lin, Y.; Wang, X.; Cui, X.; Zhang, Z.; Xian, G.; Qin, C. Novel crosstalk between KLF4 and ZEB1 regulates gemcitabine resistance in pancreatic ductal adenocarcinoma. Int. J. Oncol. 2017, 51, 1239–1248. [Google Scholar] [CrossRef]
- Smita, S.; Ahad, A.; Ghosh, A.; Biswas, V.K.; Koga, M.M.; Gupta, B.; Acha-Orbea, H.; Raghav, S.K. Importance of EMT Factor ZEB1 in cDC1 “MutuDC Line” Mediated Induction of Th1 Immune Response. Front Immunol. 2018, 9, 2604. [Google Scholar] [CrossRef]
- Spaderna, S.; Schmalhofer, O.; Wahlbuhl, M.; Dimmler, A.; Bauer, K.; Sultabn, A.; Hlubek, F.; Jung, A.; Strand, D.; Eger, A.; et al. The Transcriptional Repressor ZEB1 Promotes Metastasis and Loss of Cell Polarity Cancer. Cancer Res. 2008, 68, 537–544. [Google Scholar] [CrossRef]
- Brabletz, S.; Brabletz, T. The ZEB/miR-200 feedback loop—A motor of cellular plasticity in development and cancer? EMBO Rep. 2010, 11, 670–677. [Google Scholar] [CrossRef]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.H.; Kundu, S.T.; Fradette, J.J.; Diao, L.; Tong, P.; Byers, L.A.; Wang, J.; Rodriguez, J.; Villalobos, P.; Mino, B.; et al. ZEB1 suppression sensitizes KRAS mutant cancers to MEK inhibition by an IL17RD-dependent mechanism. Sci. Transl. Med. 2019, 11, eaaq1238. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, V.; Postigo, A.; Sánchez-Tilló, E.; Andrew, S. ZEB1 and CtBP form a repressive complex at a distal promoter element of the BCL6 locus. Biochem. J. 2010, 427, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Tilló, E.; Lázaro, A.; Torrent, R.; Cuatrecasas, M.; Vaquero, E.C.; Castells, A.; Engel, P.; Postigo, A. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG. Oncogene 2010, 29, 3490–3500. [Google Scholar] [CrossRef]
- Manshouri, R.; Coyaud, E.; Kundu, S.T.; Peng, D.H.; Stratton, S.A.; Alton, K.; Bajaj, R.; Fradette, J.J.; Minelli, R.; Peoples, M.; et al. ZEB1/NuRD complex suppresses TBC1D2b to stimulate E-cadherin internalization and promote metastasis in lung cancer. Nat. Commun. 2019, 10, 5125. [Google Scholar] [CrossRef]
- Lehmann, W.; Mossmann, D.; Kleemann, J.; Mock, K.; Meiinger, C.; Brummer, T.; Herr, R.; Brabletz, S.; Stemmler, M.P.; Brabletz, T. ZEB1 turns into a transcriptional activator by interacting with YAP1 in aggressive cancer types. Nat. Commun. 2016, 7, 10498. [Google Scholar] [CrossRef]
- Kikuchi, M.; Yamashita, K.; Waraya, M.; Minatani, N.; Ushiku, H.; Kojo, K.; Ema, A.; Kosaba, Y.; Katoh, H.; Sengoku, N.; et al. Epigenetic regulation of ZEB1-RAB25/ESRP1 axis plays a critical role in phenylbutyrate treatment-resistant breast cancer. Oncotarget 2016, 7, 1741–1753. [Google Scholar] [CrossRef]
- Gao, Y.; Zhao, Y.; Zhang, J.; Lu, Y.; Liu, X.; Geng, P.; Huang, B.; Zhang, Y.; Lu, J. The dual function of PRMT1 in modulating epithelial-mesenchymal transition and cellular senescence in breast cancer cells through regulation of ZEB. Sci. Rep. 2016, 6, 19874. [Google Scholar] [CrossRef]
- Chang, R.; Zhang, P.; You, J. Post-translational modifications of EMT transcriptional factors in cancer metastasis. Open Life Sci. 2016, 11, 237–243. [Google Scholar] [CrossRef]
- Funahashi, J.-I.; Kamachi, Y.; Goto, K.; Kondoh, H. Identification of nuclear factor δEF1 and its binding site essential for lens-specific activity of the δ1-crystallin enhancer. Nucleic Acids Res. 1991, 19, 3543–3547. [Google Scholar] [CrossRef]
- Williams, T.M.; Montoya, G.; Wu, Y.; Eddy, R.L.; Byers, M.G.; Shows, T.B. The TCF8 gene encoding a zinc finger protein (Nil-2-a) resides on human chromosome 10p11. Genomics 1992, 14, 194–196. [Google Scholar] [CrossRef]
- Braun, T.; Rudnicki, M.A.; Arnold, H.-H.; Jaenisch, R. Targeted inactivation of the muscle regulatory gene Myf-5 results in abnormal rib development and perinatal death. Cell 1992, 71, 369–382. [Google Scholar] [CrossRef]
- Comijn, J.; Berx, G.; Vermassen, P.; Verschueren, K.; Grunsven, L.V.; Bruyneel, E.; Mareel, M.; Huylebroeck, D.; Roy, F.V. The Two-Handed E Box Binding Zinc Finger Protein SIP1 Downregulates E-Cadherin and Induces Invasion. Mol. Cell 2001, 7, 1267–1278. [Google Scholar] [CrossRef]
- Genetta, T.; Ruezinsky, D.; Kadesch, T. Displacement of an E-box-binding repressor by basic helix-loop-helix proteins: Implications for B-cell specificity of the immunoglobulin heavy-chain enhancer. Mol. Cell. Biol. 1994, 14, 6153–6163. [Google Scholar] [PubMed]
- Postigo, A.A. Regulation of Smad signaling through a differential recruitment of coactivators and corepressors by ZEB proteins. EMBO J. 2003, 22, 2453–2462. [Google Scholar] [CrossRef]
- Fan, C.M.; Maniatis, T. A DNA-binding protein containing two widely separated zinc finger motifs that recognize the same DNA sequence. Genes Dev. 1990, 4, 29–42. [Google Scholar] [CrossRef]
- Shi, Y.; Sawada, J.-I.; Sui, G.; Affar, E.B.; Whetstine, J.R.; Lan, F.; Ogawa, H.; Luke, M. P-S.; Nakatani, Y.; Shi, Y. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature 2003, 422, 735–738. [Google Scholar] [CrossRef]
- Kenney, P.A.; Wszolek, M.F.; Rieger-Christ, K.M.; Silva-Neto, B.; Gould, J.J.; Harty, N.J.; Mosquera, J.M.; Zeheb, R.; Loda, M.; Darling, D.S.; et al. Novel ZEB1 expression in bladder tumorigenesis. BJU Int. 2011, 107, 656–663. [Google Scholar] [CrossRef]
- Wu, H.-T.; Zhong, H.-T.; Li, G.-W.; Shen, J.-X.; Ye, Q.-Q.; Zhang, M.-L.; Liu, J. Oncogenic functions of the EMT-related transcription factor ZEB1 in breast cancer. J. Transl. Med. 2020, 18, 51. [Google Scholar] [CrossRef]
- Wellner, U.; Brabletz, T.; Keck, T. ZEB1 in Pancreatic Cancer. Cancers 2010, 2, 1617–1628. [Google Scholar] [CrossRef]
- Singh, M.; Spoelstra, N.S.; Jean, A.; Howe, E.; Torkko, K.C.; Clarck, H.R.; Darling, D.S.; Shroyer, K.R.; Horwitz, K.B.; Broaddus, R.R.; et al. ZEB1 expression in type I vs type II endometrial cancers: A marker of aggressive disease. Mod. Pathol. 2008, 21, 912–923. [Google Scholar] [CrossRef] [PubMed]
- Bronsert, P.; Kohler, I.; Timme, S.; Kiefer, S.; Werner, M.; Schilling, O.; Vashist, Y.; Makoweic, F.; Brabletz, T.; Hopt, U.T.; et al. Prognostic significance of Zinc finger E-box binding homeobox 1 (ZEB1) expression in cancer cells and cancer-associated fibroblasts in pancreatic head cancer. Surgery 2014, 156, 97–108. [Google Scholar] [CrossRef]
- Wong, T.-S.; Gao, W.; Chan, J.Y.-W. Transcription Regulation of E-Cadherin by Zinc Finger E-Box Binding Homeobox Proteins in Solid Tumors. Biomed. Res. Int. 2014, 2014, 921564. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Liu, H.; Kong, Q.; Li, B. Overexpression of ZEB1 associated with metastasis and invasion in patients with gastric carcinoma. Mol. Cell. Biochem. 2012, 366, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Huang, B.; Li, B.M.; Cao, K.Y.; Mo, C.Q.; Jiang, S.J.; Pan, J.C.; Wang, Z.R.; Lin, H.Y.; Wang, D.H.; et al. ZEB1-mediated vasculogenic mimicry formation associates with epithelial-mesenchymal transition and cancer stem cell phenotypes in prostate cancer. J. Cell. Mol. Med. 2018, 22, 3768–3781. [Google Scholar] [CrossRef] [PubMed]
- Langer, E.M.; Kendsersky, N.D.; Daniel, C.J.; Kuziel, G.M.; Pelz, C.; Murphy, K.M.; Capecchi, M.R.; Sears, R.C. ZEB1-repressed microRNAs inhibit autocrine signaling that promotes vascular mimicry of breast cancer cells. Oncogene 2018, 37, 1005–1019. [Google Scholar] [CrossRef]
- Liu, L.; Tong, Q.; Liu, S.; Cui, J.; Zhang, Q.; Sun, W.; Yang, S. ZEB1 Upregulates VEGF Expression and Stimulates Angiogenesis in Breast Cancer. PLoS ONE 2016, 11, e0148774. [Google Scholar] [CrossRef]
- Fu, R.; Li, Y.; Jiang, N.; Ren, B.-X.; Zang, C.-Z.; Liu, L.-J.; Lv, W.-C.; Li, H.-M.; Weiss, S.; Li, Z.Y.; et al. Inactivation of endothelial ZEB1 impedes tumor progression and sensitizes tumors to conventional therapies. J. Clin. Investig. 2020, 130, 1252–1270. [Google Scholar] [CrossRef]
- Katsura, A.; Tamura, Y.; Hokari, S.; Harada, M.; Morikawa, M.; Sakurai, T.; Takahashi, K.; Mizutani, A.; Nishida, J.; Yokoyama, Y.; et al. ZEB1-regulated inflammatory phenotype in breast cancer cells. Mol. Oncol. 2017, 11, 1241–1262. [Google Scholar] [CrossRef]
- Fu, R.; Han, C.-F.; Ni, T.; Di, L.; Liu, L.-J.; Lv, W.-C.; Bi, Y.-R.; Jiang, N.; He, Y.; Li, H.-M.; et al. A ZEB1/p53 signaling axis in stromal fibroblasts promotes mammary epithelial tumours. Nat. Commun. 2019, 10, 3210. [Google Scholar] [CrossRef]
- Xu, R.; Won, J.-Y.; Kim, C.-H.; Kim, D.-E.; Yim, H. Roles of the Phosphorylation of Transcriptional Factors in Epithelial-Mesenchymal Transition. J. Oncol. 2019, 2019, 5810465. [Google Scholar] [CrossRef] [PubMed]
- Eger, A.; Aigner, K.; Sonderegger, S.; Dampier, B.; Oehler, S.; Schreiber, M.; Berx, G.; Cano, A.; Beug, H.; Foisner, R. DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene 2005, 24, 2375–2385. [Google Scholar] [CrossRef] [PubMed]
- Bindels, S.; Mestdagt, M.; Vandewalle, C.; Jacobs, N.; Volders, L.; Noel, A.; Roy, F.; Foidart, J.-M.; Giller, C. Regulation of vimentin by SIP1 in human epithelial breast tumor cells. Oncogene 2006, 25, 4975–4985. [Google Scholar] [CrossRef] [PubMed]
- Shirakihara, T.; Saitoh, M.; Miyazono, K. Differential Regulation of Epithelial and Mesenchymal Markers by EF1 Proteins in Epithelial Mesenchymal Transition Induced by TGF-beta. Mol. Biol. Cell 2007, 18, 3533–3544. [Google Scholar] [CrossRef] [PubMed]
- Bae, G.-Y.; Choi, S.-J.; Lee, J.-S.; Jo, J.; Lee, J.; Kim, J.; Cha, H.-J. Loss of E-cadherin activates EGFR-MEK/ERK signaling, which promotes invasion via the ZEB1/MMP2 axis in non-small cell lung cancer. Oncotarget 2013, 4, 2512–2522. [Google Scholar] [CrossRef] [PubMed]
- Aigner, K.; Dampier, B.; Descovich, L.; Mikula, M.; Sultan, A.; Schreiber, M.; Mikulits, W.; Brabletz, T.; Strand, D.; Obrist, P.; et al. The transcription factor ZEB1 (δEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene 2007, 26, 6979–6988. [Google Scholar] [CrossRef]
- Gibbons, D.L.; Lin, W.; Creighton, C.J.; Rizvi, Z.H.; Gregory, P.A.; Goodall, G.J.; Thilaganathan, N.; Du, L.; Zhang, Y.; Pertsemlidis, A.; et al. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev. 2009, 23, 2140–2151. [Google Scholar] [CrossRef]
- Scott, L.E.; Weinberg, S.H.; Lemmon, C.A. Mechanochemical Signaling of the Extracellular Matrix in Epithelial-Mesenchymal Transition. Front. Cell Dev. Biol. 2019, 7, 135. [Google Scholar] [CrossRef]
- Peng, D.H.; Ungewiss, C.; Tong, P.; Byers, L.A.; Wang, J.; Canales, J.R.; Villalobos, P.A.; Uraoka, N.; Mino, B.; Behrens, C.; et al. ZEB1 induces LOXL2-mediated collagen stabilization and deposition in the extracellular matrix to drive lung cancer invasion and metastasis. Oncogene 2017, 36, 1925–1938. [Google Scholar] [CrossRef]
- Ungewiss, C.; Rizvi, Z.H.; Roybal, J.D.; Peng, D.H.; Gold, K.A.; Shin, D.-H.; Creighton, C.J.; Gibbons, D.L. The microRNA-200/Zeb1 axis regulates ECM-dependent β1-integrin/FAK signaling, cancer cell invasion and metastasis through CRKL. Sci. Rep. 2016, 6, 18652. [Google Scholar] [CrossRef]
- Sánchez-Tilló, E.; Fanlo, L.; Siles, L.; Montes-Moreno, S.; Moros, A.; Chiva-Blanch, G.; Estruch, R.; Martinez, A.; Colomer, D.; Gyorffy, B.; et al. The EMT activator ZEB1 promotes tumor growth and determines differential response to chemotherapy in mantle cell lymphoma. Cell Death Differ. 2014, 21, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Z.; Zhang, Q.; Zhang, Q.; Sun, P.; Xiang, R.; Ren, G.; Yang, S. ZEB1 confers chemotherapeutic resistance to breast cancer by activating ATM. Cell Death Dis. 2018, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Santamaría, P.G.; Moreno-Bueno, G.; Cano, A. Contribution of Epithelial Plasticity to Therapy Resistance. J. Clin. Med. 2019, 8, 676. [Google Scholar] [CrossRef] [PubMed]
- Caramel, J.; Ligier, M.; Puisieux, A. Pleiotropic Roles for ZEB1 in Cancer. Cancer Res. 2018, 78, 30–35. [Google Scholar] [CrossRef]
- Zhang, P.; Wei, Y.; Wang, L.; Debeb, B.G.; Yuan, Y.; Zhang, J.; Yuan, J.; Wang, M.; Chen, D.; Sun, Y.; et al. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK. Nat. Cell Biol. 2014, 16, 864–875. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, L.; Rodriguez-Aguayo, C.; Yuan, Y.; Debeb, B.G.; Chen, D.; Sun, Y.; You, M.J.; Liu, Y.; Dean, D.C.; et al. miR-205 acts as a tumour radiosensitizer by targeting ZEB1 and Ubc. Nat. Commun. 2014, 5, 5671. [Google Scholar] [CrossRef]
- Tong, R. Transcriptional Regulation and Its Misregulation in Disease. Cell 2013, 152, 1237–1251. [Google Scholar]
- Gregory, P.A.; Bracken, C.P.; Smith, E.; Bert, A.G.; Wright, J.A.; Roslan, S.; Morris, M.; Wyatt, L.; Farshid, G.; Lin, Y.-Y.; et al. An autocrine TGF-/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol. Biol. Cell 2011, 22, 1686–1698. [Google Scholar] [CrossRef]
- Wu, K.; Fan, J.; Zhang, L.; Ning, Z.; Zeng, J.; Zhou, J.; Li, L.; Chen, Y.; Zhang, T.; Wang, X.; et al. PI3K/Akt to GSK3β/β-catenin signaling cascade coordinates cell colonization for bladder cancer bone metastasis through regulating ZEB1 transcription. Cell. Signal. 2012, 24, 2273–2282. [Google Scholar] [CrossRef]
- Chua, H.L.; Bhat-Nakshatri, P.; Clare, S.E.; Morimiya, A.; Badve, S.; Nakshatri, H. NF-κB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: Potential involvement of ZEB-1 and ZEB. Oncogene 2007, 26, 711–724. [Google Scholar] [CrossRef]
- Han, Y.; Luo, Y.; Wang, Y.; Chen, Y.; Li, M.; Jiang, Y. Hepatocyte growth factor increases the invasive potential of PC-3 human prostate cancer cells via an ERK/MAPK and Zeb-1 signaling pathway. Oncol. Lett. 2015, 1, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Zheng, L.; Shen, J.; Zhang, D.; Xiong, M.; Zhang, Y.; He, X.; Tanyi, J.L.; Yang, F.; Montone, K.T.; et al. Suppression of MicroRNA 200 Family Expression by Oncogenic KRAS Activation Promotes Cell Survival and Epithelial-Mesenchymal Transition in KRAS-Driven Cancer. Mol. Cell. Biol. 2016, 36, 2742–2754. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.V.; Conroy, S.; Tomar, T.; Eggens-Meijer, E.; Bhat, K.; Copray, S.; Walenkamp, A.; Boddeke, E.; Balasubramanyian, V.; Wagemakers, M.; et al. TGF-β is an inducer of ZEB1-dependent mesenchymal transdifferentiation in glioblastoma that is associated with tumor invasion. Cell Death Dis. 2014, 5, e1443. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Jiang, T.; Cheng, N.; Wang, Q.; Ren, S.; Li, X.; Zhao, C.; Zhang, L.; Cai, W.; Zhou, C. Long non-coding RNA BC087858 induces non-T790M mutation acquired resistance to EGFR-TKIs by activating PI3K/AKT and MEK/ERK pathways and EMT in non-small-cell lung cancer. Oncotarget 2016, 7, 49948–49960. [Google Scholar] [CrossRef]
- Xu, X.M.; Liu, W.; Cao, Z.H.; Liu, M.X. Effects of ZEB1 on regulating osteosarcoma cells via NF-κB/iNOS. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 1184–1190. [Google Scholar]
- Nomura, A.; Majumder, K.; Giri, B.; Dauer, P.; Dudeja, V.; Roy, S.; Banerjee, S.; Saluja, A.K. Inhibition of NF-kappa B pathway leads to deregulation of epithelial-mesenchymal transition and neural invasion in pancreatic cancer. Lab. Investig. 2016, 96, 1268–1278. [Google Scholar] [CrossRef]
- Guenther, M.G.; Levine, S.S.; Boyer, L.A.; Jaenisch, R.; Young, R.A. A Chromatin Landmark and Transcription Initiation at Most Promoters in Human Cells. Cell 2007, 130, 77–88. [Google Scholar] [CrossRef]
- Mueller, D.; Bach, C.; Zeisig, D.; Garcia-Cuellar, M.-P.; Monroe, S.; Sreekumar, A.; Zhou, R.; Nesvizhskii, A.; Chinnaiyan, A.; Hess, J.L.; et al. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood 2007, 110, 4445–4454. [Google Scholar] [CrossRef]
- Khan, A.A.; Lee, A.J.; Roh, T.Y. Polycomb group protein-mediated histone modifications during cell differentiation. Epigenomics 2015, 7, 75–84. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A Bivalent Chromatin Structure Marks Key Developmental Genes in Embryonic Stem Cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef]
- Christine, N.; Lee, T.; Bell, G.; Kleer, C.G.; Reinhardt, F.; Alessio, A.; Young, R.A.; Weinberg, R.A. Poised Chromatin at the ZEB1 Promoter Enables Breast Cancer Cell Plasticity and Enhances Tumorigenicity. Cell 2013, 154, 61–74. [Google Scholar]
- Silveira, D.A.; Mombach, J.C.M. Dynamics of the feedback loops required for the phenotypic stabilization in the epithelial-mesenchymal transition. FEBS J. 2019, 287, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIPN1. Cell Biol. 2008, 10, 593–601. [Google Scholar]
- Korpal, M.; Lee, E.S.; Hu, G.; Kang, Y. The miR-200 Family Inhibits Epithelial-Mesenchymal Transition and Cancer Cell Migration by Direct Targeting of E-cadherin Transcriptional Repressors ZEB1 and ZEB. J. Biol. Chem. 2008, 283, 14910–14914. [Google Scholar] [CrossRef]
- Hugo, H.J.; Pereira, L.; Suryadinata, R.; Drabsch, Y.; Gonda, T.J.; Gunasinghe, N.; Pinto, C.; Soo, E.; Denderen, B.; Hill, P.; et al. Direct repression of MYB by ZEB1 suppresses proliferation and epithelial gene expression during epithelial-to-mesenchymal transition of breast cancer cells. Breast Cancer Res. 2013, 15, R113. [Google Scholar] [CrossRef]
- Chung, V.Y.; Tan, T.Z.; Tan, M.; Wong, M.K.; Kuay, K.T.; Yang, Z.; Ye, J.; Muller, J.; Koh, C.M.; Guccione, E.; et al. GRHL2-miR-200-ZEB1 maintains the epithelial status of ovarian cancer through transcriptional regulation and histone modification. Sci. Rep. 2016, 6, 19943. [Google Scholar] [CrossRef]
- Sommerova, L.; Ondrouskova, E.; Martisova, A.; Zoumpourlis, V.; Galtsidis, S.; Hrstka, R. ZEB1/miR-200c/AGR2, A New Regulatory Loop Modulating the Epithelial-Mesenchymal Transition in Lung Adenocarcinomas. Cancers 2020, 12, 1614. [Google Scholar] [CrossRef]
- Preca, B.-T.; Bajdak, K.; Mock, K.; Sundararajan, V.; Pfannstiel, J.; Maurer, J.; Wellner, U.; Hopt, U.T.; Brummer, T.; Brabletz, S.; et al. A self-enforcing CD44s/ZEB1 feedback loop maintains EMT and stemness properties in cancer cells. Int. J. Cancer 2015, 137, 2566–2577. [Google Scholar] [CrossRef]
- Kim, J.; Moon, M.; Kim, D.; Heo, S.; Jeong, Y. Hyaluronic Acid-Based Nanomaterials for Cancer Therapy. Polymers 2018, 10, 1133. [Google Scholar] [CrossRef]
- Preca, B.-T.; Bajdak, K.; Mock, K.; Lehmann, W.; Sundararajan, V.; Bronsert, P.; Matzge-Ogi, A.; Orian-Rousseau, V.; Brabletz, S.; Maurer, J.; et al. A novel ZEB1/HAS2 positive feedback loop promotes EMT in breast cancer. Oncotarget 2017, 8, 1133. [Google Scholar] [CrossRef]
- Jolly, M.K.; Preca, B.-T.; Tripathi, S.C.; Jia, D.; George, J.T.; Hanash, S.M.; Brabletz, T.; Stemmler, M.P.; Maurer, J.; Levine, H. Interconnected feedback loops among ESRP1, HAS2, and CD44 regulate epithelial-mesenchymal plasticity in cancer. APL Bioeng. 2018, 2, 031908. [Google Scholar] [CrossRef] [PubMed]
- Gibney, E.R.; Nolan, C.M. Epigenetics and gene expression. Heredity 2010, 105, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Stein, G.S.; Stein, J.L.; van Wijnen, A.J.; Lian, J.B.; Montecino, M.; Croce, C.M.; Choi, J.Y.; Ali, S.A.; Pande, S.; Hassan, M.Q.; et al. Transcription factor-mediated epigenetic regulation of cell growth and phenotype for biological control and cancer. Adv. Enzym. Regul. 2010, 50, 160–167. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vaklavas, C.; Blume, S.W.; Grizzle, W.E. Translational Dysregulation in Cancer: Molecular Insights and Potential Clinical Applications in Biomarker Development. Front. Oncol. 2017, 7, 158. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Chow, C.R.; Ebine, K.; Arslan, A.D.; Kwok, B.; Bentrem, D.J.; Eckerdt, F.D.; Platanias, L.C.; Munshi, H.G. Differential Regulation of ZEB1 and EMT by MAPK-Interacting Protein Kinases (MNK) and eIF4E in Pancreatic Cancer. Mol. Cancer Res. 2016, 14, 216–227. [Google Scholar] [CrossRef]
- Li, S.; Zhang, H.-Y.; Du, Z.-X.; Li, C.; An, M.X.; Zong, Z.-H.; Liu, B.-Q.; Wang, H.-Q. Induction of epithelial-mesenchymal transition (EMT) by Beclin 1 knockdown via posttranscriptional upregulation of ZEB1 in thyroid cancer cells. Oncotarget 2016, 7, 70364–70377. [Google Scholar] [CrossRef][Green Version]
- Zhao, X.; Wang, D.; Ding, Y.; Zhou, J.; Liu, G.; Ji, Z. lncRNA ZEB1-AS1 promotes migration and metastasis of bladder cancer cells by post-transcriptional activation of ZEB. Int. J. Mol. Med. 2019, 44, 196–206. [Google Scholar]
- Buchanan, B.W.; Lloyd, M.E.; Engle, S.M.; Rubenstein, E.M. Cycloheximide Chase Analysis of Protein Degradation in Saccharomyces cerevisiae. J. Vis. Exp. 2016, 110, 53975. [Google Scholar] [CrossRef]
- Chen, A.; Wong, C.S.F.; Liu, M.C.P.; House, C.M.; Sceneay, J.; Bowtell, D.D.; Thompson, E.W.; Moller, A. The ubiquitin ligase Siah is a novel regulator of Zeb1 in breast cancer. Oncotarget 2015, 6, 862–873. [Google Scholar] [CrossRef]
- Abshire, C.F.; Carroll, J.L.; Dragoi, A.M. FLASH protects ZEB1 from degradation and supports cancer cells’ epithelial-to-mesenchymal transition. Oncogenesis 2016, 5, e254. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhang, P.; Hu, X.; Kim, J.; Yao, F.; Xiao, Z.; Zeng, L.; Chang, L.; Sun, Y.; Ma, L. USP51 promotes deubiquitination and stabilization of ZEB1. J. Cancer Res. 2017, 7, 2020–2031. [Google Scholar]
- Llorens, M.C.; Lorenzatti, G.; Cavallo, N.L.; Vaglienti, M.V.; Perrone, A.P.; Carenbauer, A.L.; Darling, D.S.; Cabanillas, A.M. Phosphorylation Regulates Functions of ZEB1 Transcription Factor. J. Cell. Physiol. 2016, 7, 2020–2031. [Google Scholar] [CrossRef] [PubMed]
- Voutsadakis, I.A. Ubiquitination and the Ubiquitin-Proteasome System as regulators of transcription and transcription factors in epithelial mesenchymal transition of cancer. Tumor Biol. 2012, 33, 897–910. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lu, X.; Liu, Z.; Chen, L.; Xu, Y.; Wang, Y.; Wei, G.; Chen, Y. FBXW7 suppresses epithelial-mesenchymal transition, stemness and metastatic potential of cholangiocarcinoma cells. Oncotarget 2015, 6, 6310–6325. [Google Scholar] [CrossRef]
- Zhou, F.; Du, C.; Xu, D.; Lu, J.; Zhou, L.; Wu, C.; Wu, B.; Huang, J. Knockdown of ubiquitin-specific protease 51 attenuates cisplatin resistance in lung cancer through ubiquitination of zinc-finger E-box binding homeobox. Mol. Med. Rep. 2020, 22, 1382–1390. [Google Scholar] [CrossRef]
- Bogachek, M.V.; de Andrade, J.P.; Weigel, R.J. Regulation of Epithelial-Mesenchymal Transition through SUMOylation of Transcription Factors. Cancer Res. 2015, 75, 11–15. [Google Scholar] [CrossRef]
- Herranz, N.; Pasini, D.; Diaz, V.M.; Franci, C.; Gutierrez, A.; Dave, N.; Escriva, M.; Hernandez-Munoz, I.; Croce, L.D.; Helin, K.; et al. Polycomb Complex 2 Is Required for E-cadherin Repression by the Snail1 Transcription Factor. Mol. Cell. Biol. 2008, 28, 4772–4781. [Google Scholar] [CrossRef]
- Kuppuswamy, M.; Vijayalingam, S.; Zhao, L.J.; Zhou, Y.; Subramanian, T.; Ryerse, J.; Chinnadurai, G. Role of the PLDLS-Binding Cleft Region of CtBP1 in Recruitment of Core and Auxiliary Components of the Corepressor Complex. Mol. Cell. Biol. 2008, 28, 269–281. [Google Scholar] [CrossRef][Green Version]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol. Cancer 2016, 15, 18. [Google Scholar] [CrossRef]
- Kim, J.-H.; Cho, E.-J.; Kim, S.-T.; Youn, H.-D. CtBP represses p300-mediated transcriptional activation by direct association with its bromodomain. Nat. Struct. Mol. Biol. 2005, 12, 423–428. [Google Scholar] [CrossRef]
- Mizuguchi, Y.; Specht, S.; Lunz, J.G.; Isse, K.; Corbitt, N.; Takizawa, T.; Demetris, A.J. Cooperation of p300 and PCAF in the Control of MicroRNA 200c/141 Transcription and Epithelial Characteristics. PLoS ONE 2012, 7, e32449. [Google Scholar]
- Fu, R.; Lv, W.-C.; Xu, Y.; Gong, M.-Y.; Chen, X.-J.; Jiang, N.; Xu, Y.; Yao, Q.-Q.; Di, L.; Lu, T.; et al. Endothelial ZEB1 promotes angiogenesis-dependent bone formation and reverses osteoporosis. Nat. Commun. 2020, 11, 460. [Google Scholar] [CrossRef] [PubMed]
- Siles, L.; Sanchez-Tillo, E.; Lim, J.W.; Darling, D.S.; Kroll, K.L.; Postigo, A. ZEB1 Imposes a Temporary Stage-Dependent Inhibition of Muscle Gene Expression and Differentiation via CtBP-Mediated Transcriptional Repression. Mol. Cell. Biol. 2013, 33, 1368–1382. [Google Scholar] [CrossRef] [PubMed]
- Postigo, A.A. Opposing functions of ZEB proteins in the regulation of the TGFbeta/BMP signaling pathway. EMBO J. 2003, 22, 2443–2452. [Google Scholar] [CrossRef]
- Peña, C.; García, J.M.; García, V.; Silva, J.; Dominguez, G.; Rodriguez, R.; Maximiano, C.; de Herreros, A.G.; Munoz, A.; Bonilla, F. The expression levels of the transcriptional regulators p300 and CtBP modulate the correlations between SNAIL, ZEB1, E-cadherin and vitamin D receptor in human colon carcinomas. Int. J. Cancer 2006, 119, 2098–2104. [Google Scholar] [CrossRef]
- Krubasik, D.; Iyer, N.G.; English, W.R.; Ahmed, A.A.; Vias, M.; Roskelley, C.; Brenton, J.D.; Caldas, C.; Murphy, G. Absence of p300 induces cellular phenotypic changes characteristic of epithelial to mesenchyme transition. Br. J. Cancer 2006, 95, 245. [Google Scholar] [CrossRef]
- Amoutzias, G.D.; Robertson, D.L.; van de Peer, Y.; Oliver, S.G. Choose your partners: Dimerization in eukaryotic transcription factors. Trends Biochem. Sci. 2008, 33, 220–229. [Google Scholar] [CrossRef]
- Moraitis, A.N.; Giguère, V. Transition from Monomeric to Homodimeric DNA Binding by Nuclear Receptors: Identification of RevErbAα Determinants Required for RORα Homodimer Complex Formation. Mol. Endocrinol. 1999, 13, 431–439. [Google Scholar] [CrossRef][Green Version]
- Zhuang, S. Regulation of STAT signaling by acetylation. Cell. Signal. 2013, 25, 1924–1931. [Google Scholar] [CrossRef]
- Sethi, G.; Chatterjee, S.; Rajendran, P.; Li, F.; Shanmugam, M.K.; Wong, K.F.; Kuman, A.P.; Senapati, P.; Behera, A.K.; Hui, K.M.; et al. Inhibition of STAT3 dimerization and acetylation by garcinol suppresses the growth of human hepatocellular carcinoma in vitro and in vivo. Mol. Cancer 2014, 13, 66. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perez-Oquendo, M.; Gibbons, D.L. Regulation of ZEB1 Function and Molecular Associations in Tumor Progression and Metastasis. Cancers 2022, 14, 1864. https://doi.org/10.3390/cancers14081864
Perez-Oquendo M, Gibbons DL. Regulation of ZEB1 Function and Molecular Associations in Tumor Progression and Metastasis. Cancers. 2022; 14(8):1864. https://doi.org/10.3390/cancers14081864
Chicago/Turabian StylePerez-Oquendo, Mabel, and Don L. Gibbons. 2022. "Regulation of ZEB1 Function and Molecular Associations in Tumor Progression and Metastasis" Cancers 14, no. 8: 1864. https://doi.org/10.3390/cancers14081864
APA StylePerez-Oquendo, M., & Gibbons, D. L. (2022). Regulation of ZEB1 Function and Molecular Associations in Tumor Progression and Metastasis. Cancers, 14(8), 1864. https://doi.org/10.3390/cancers14081864