
Shikonin Inhibits Cell Growth of Sunitinib-Resistant Renal Cell Carcinoma by Activating the Necrosome Complex and Inhibiting the AKT/mTOR Signaling Pathway


 ,
,  ,
,
Abstract
:Simple Summary
Abstract
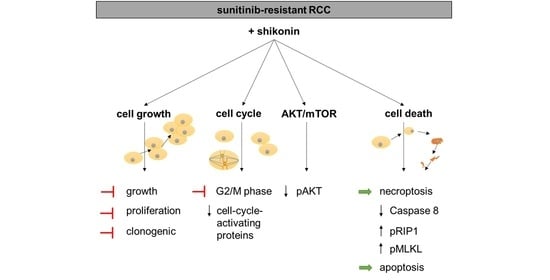
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Resistance Induction and Application of Sunitinib and Shikonin
2.3. RCC Cell Growth and Proliferation
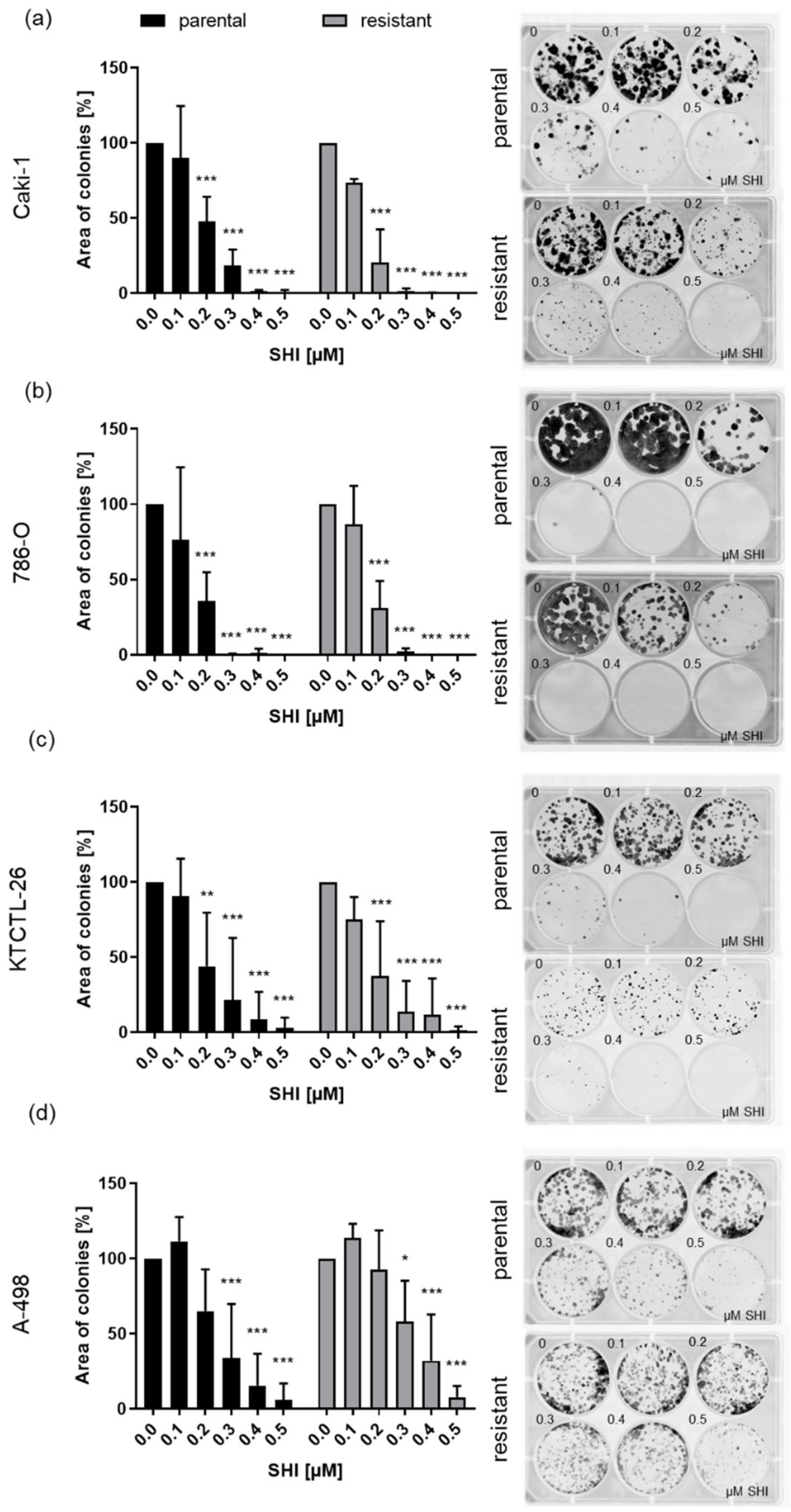
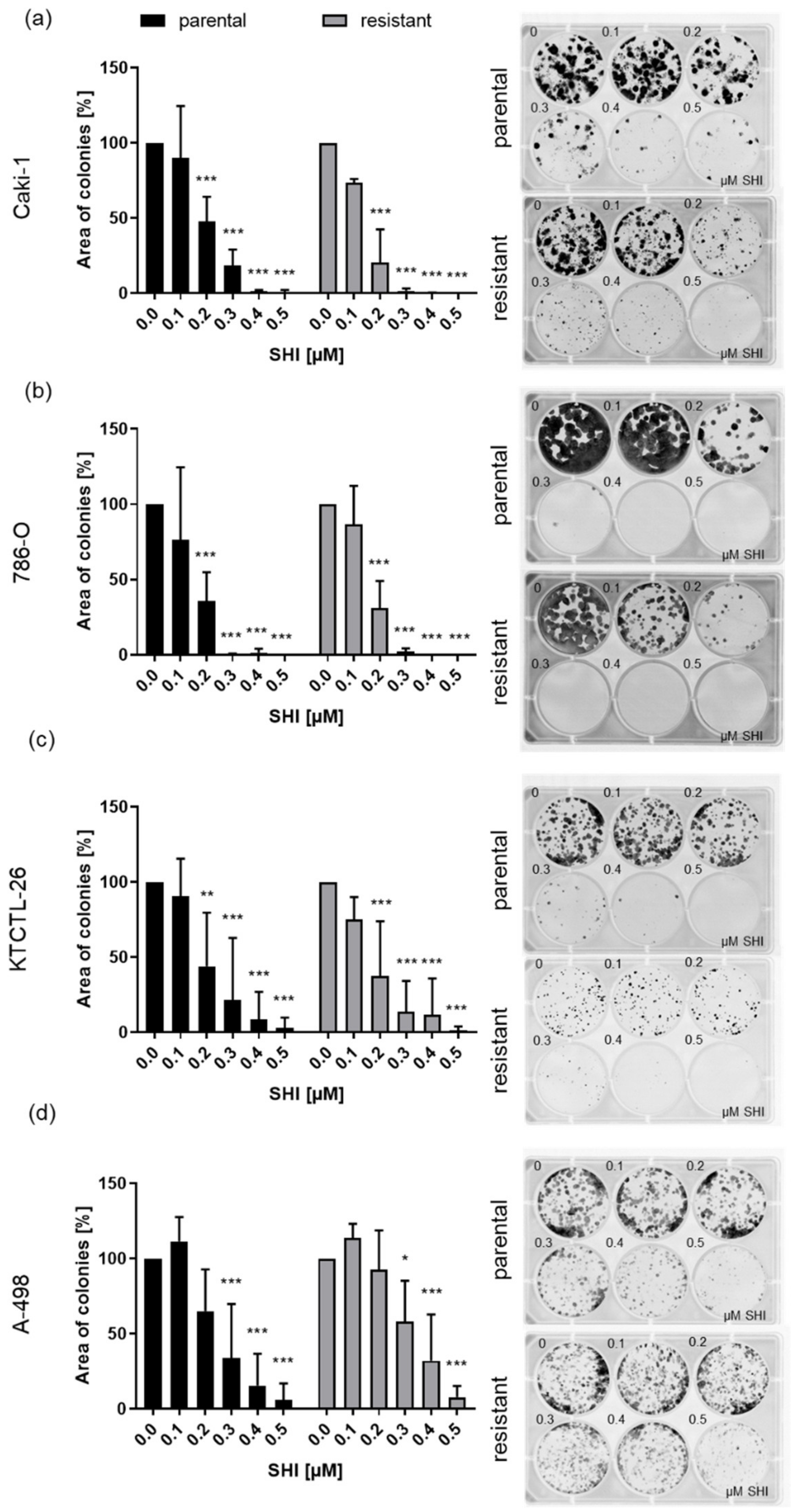
2.4. Clonogenic Growth Behavior
2.5. Cell Cycle Phase Distribution
2.6. RCC Cell Death
2.7. Protein Expression and Activity—Western Blot Analysis
2.8. AKT Blockade
2.9. Adhesion to Extracellular Matrix Proteins and Vascular Endothelium
2.10. Tumor Cell Motility
2.11. Statistical Analysis
3. Results
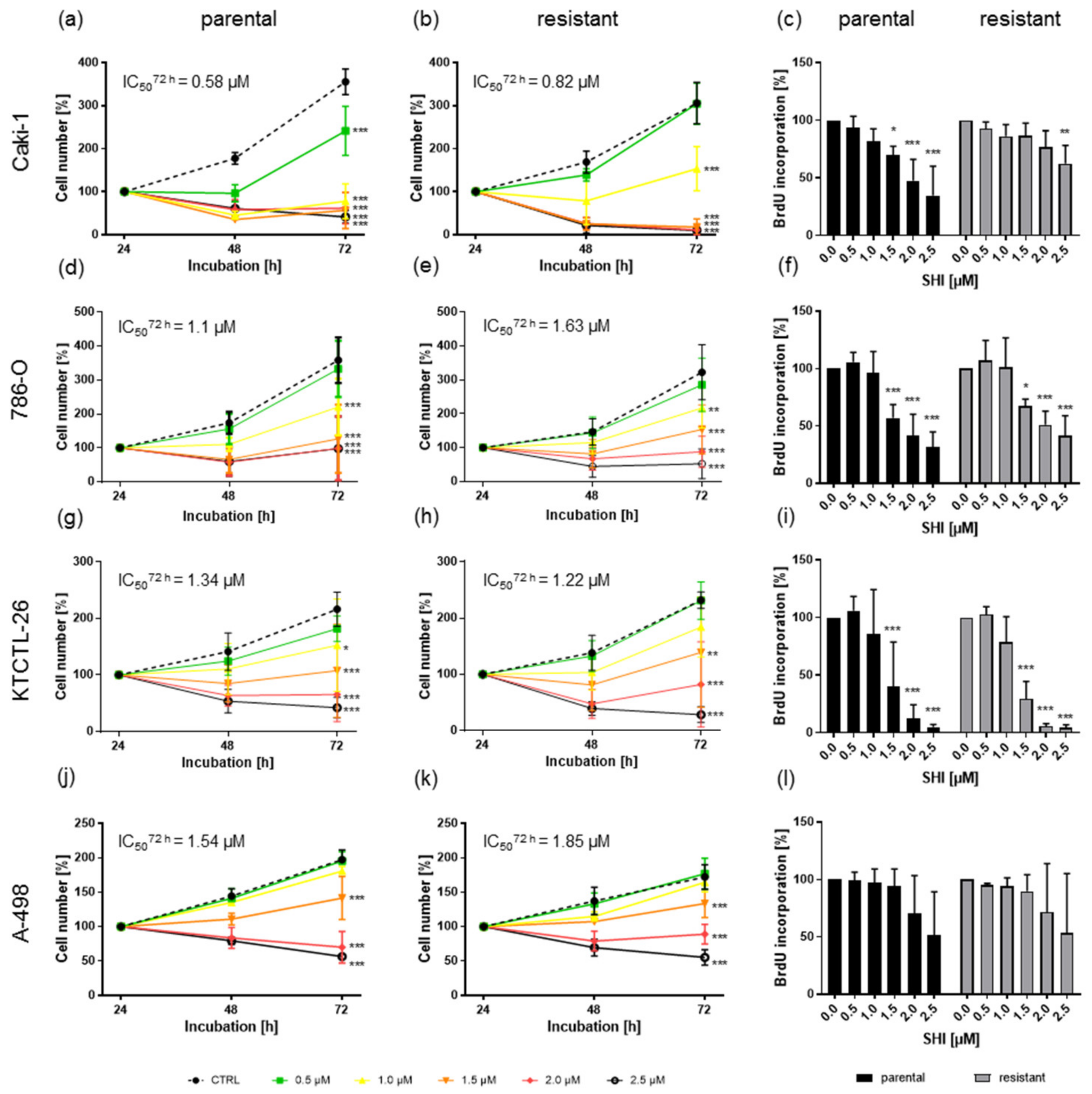
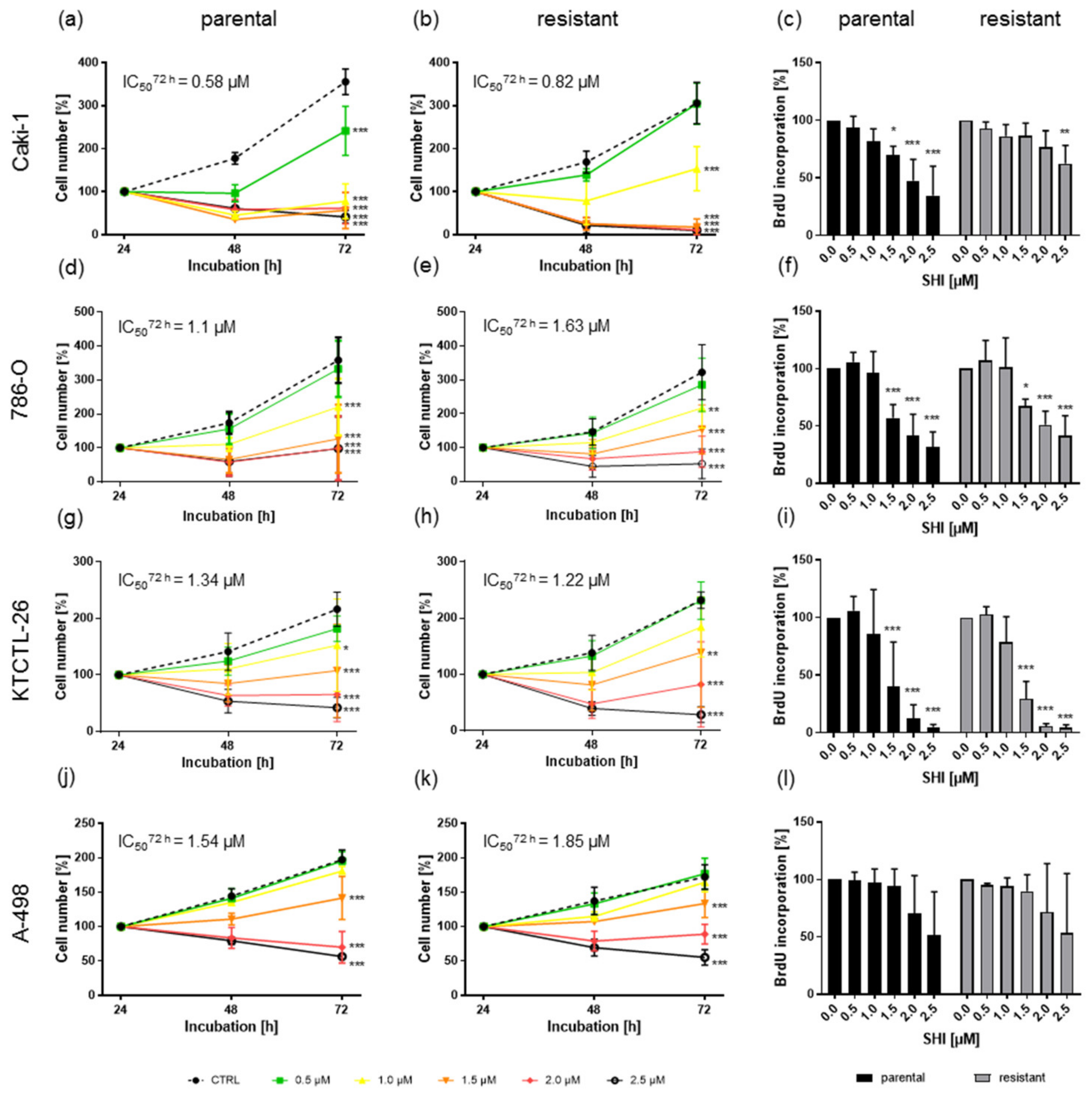
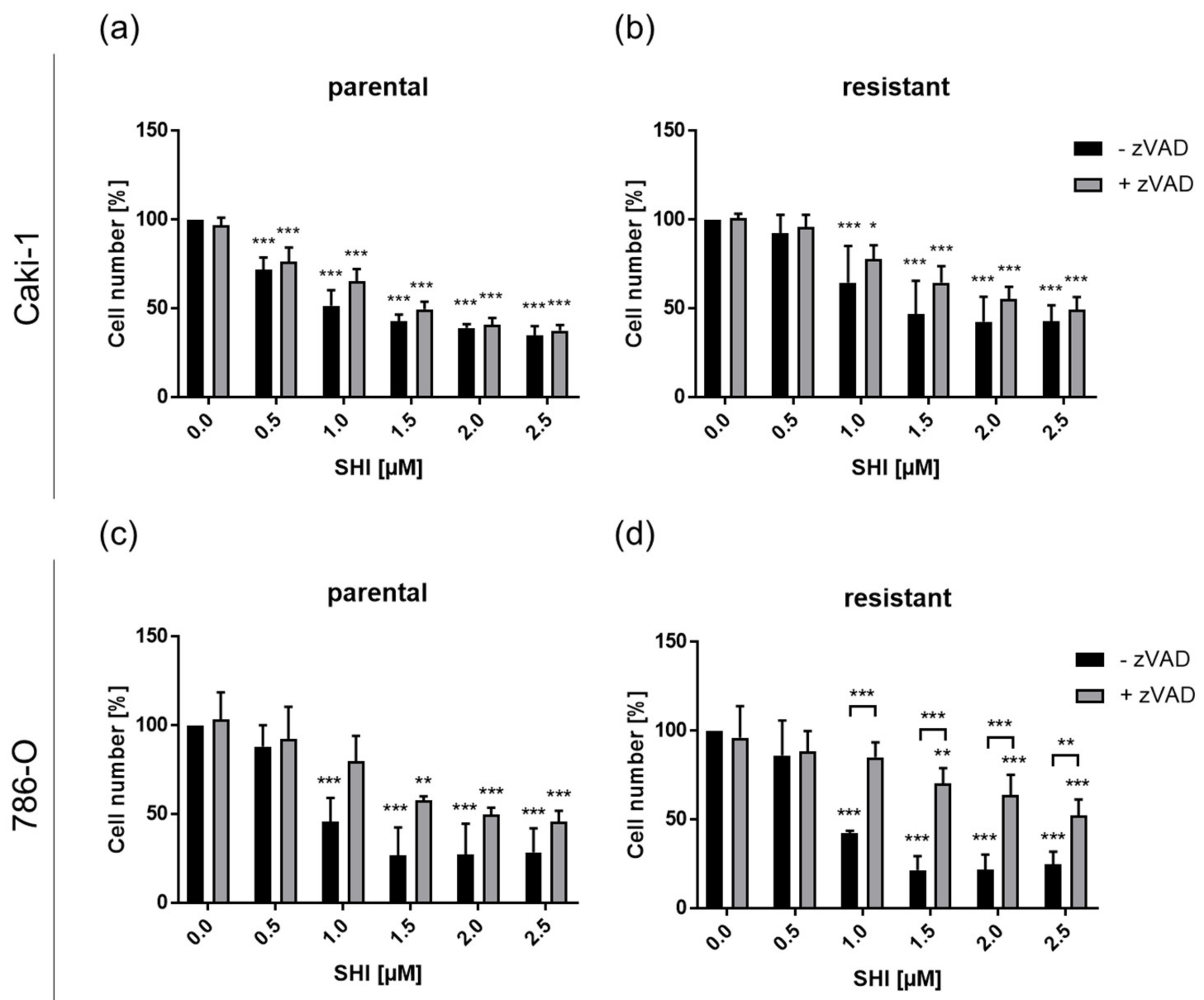
3.1. Shikonin Inhibited Progressive Growth Behavior of Parental and Sunitinib-Resistant RCC Cells
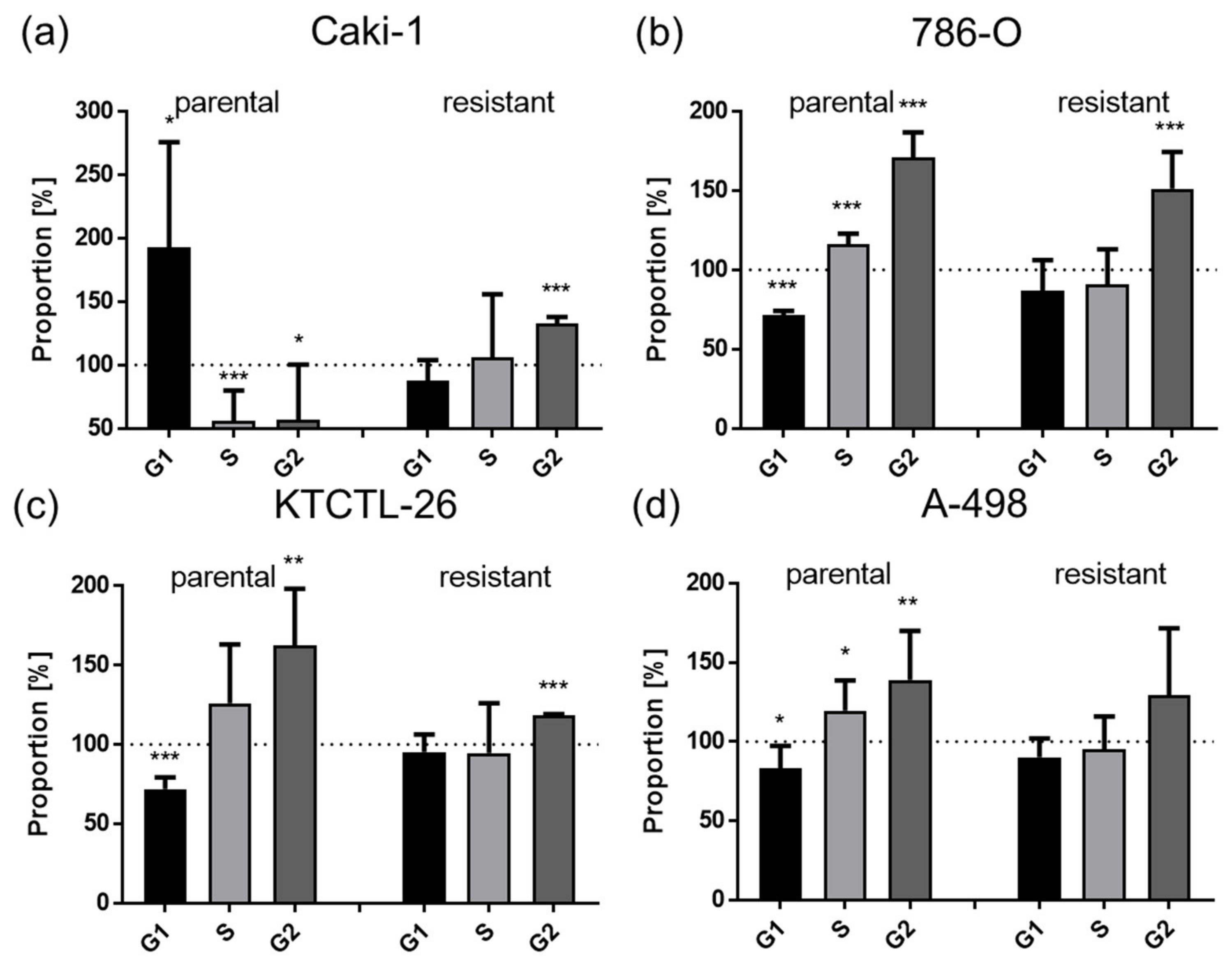
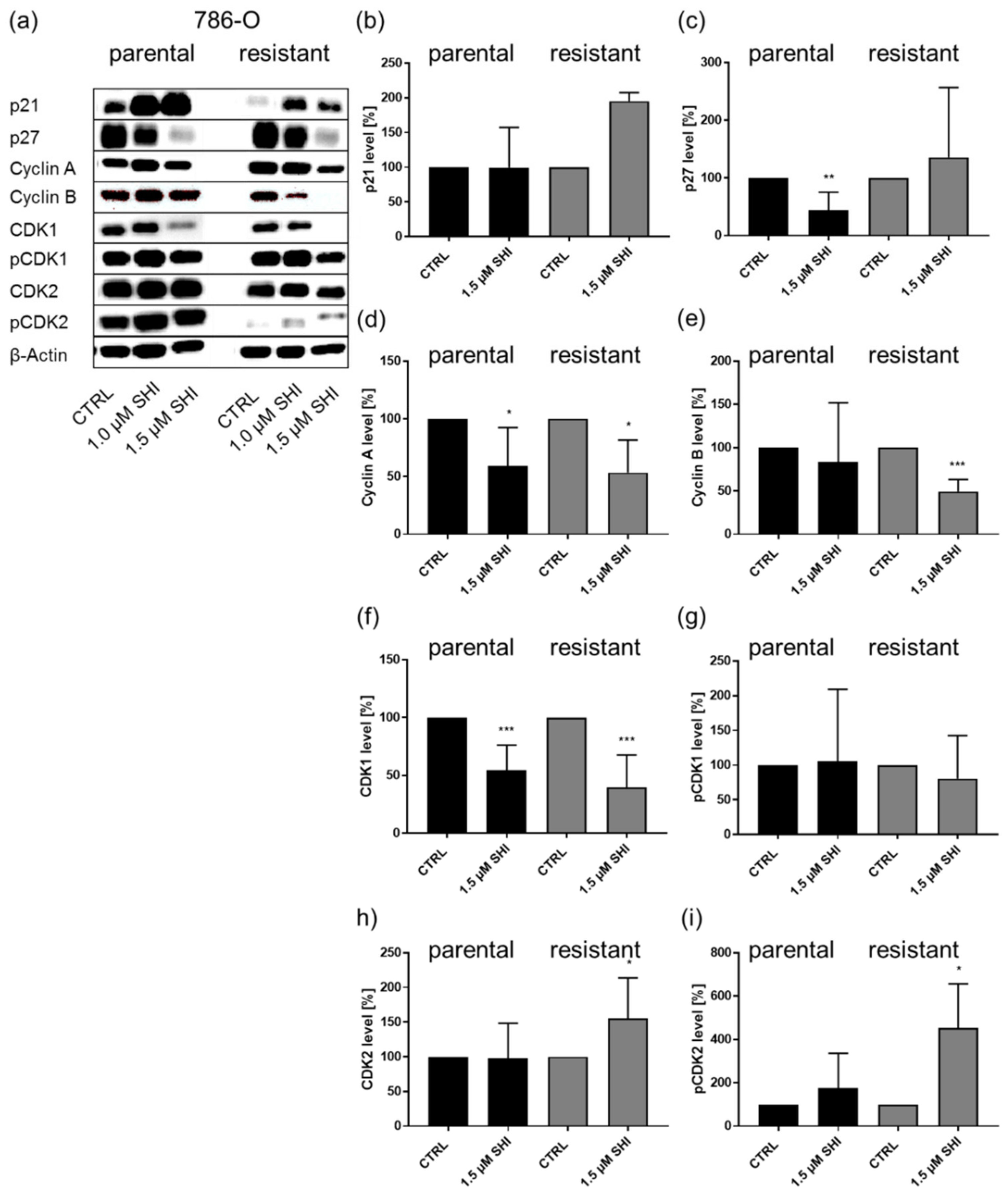
3.2. Shikonin-Induced Cell Cycle Arrest in the G2/M Phase
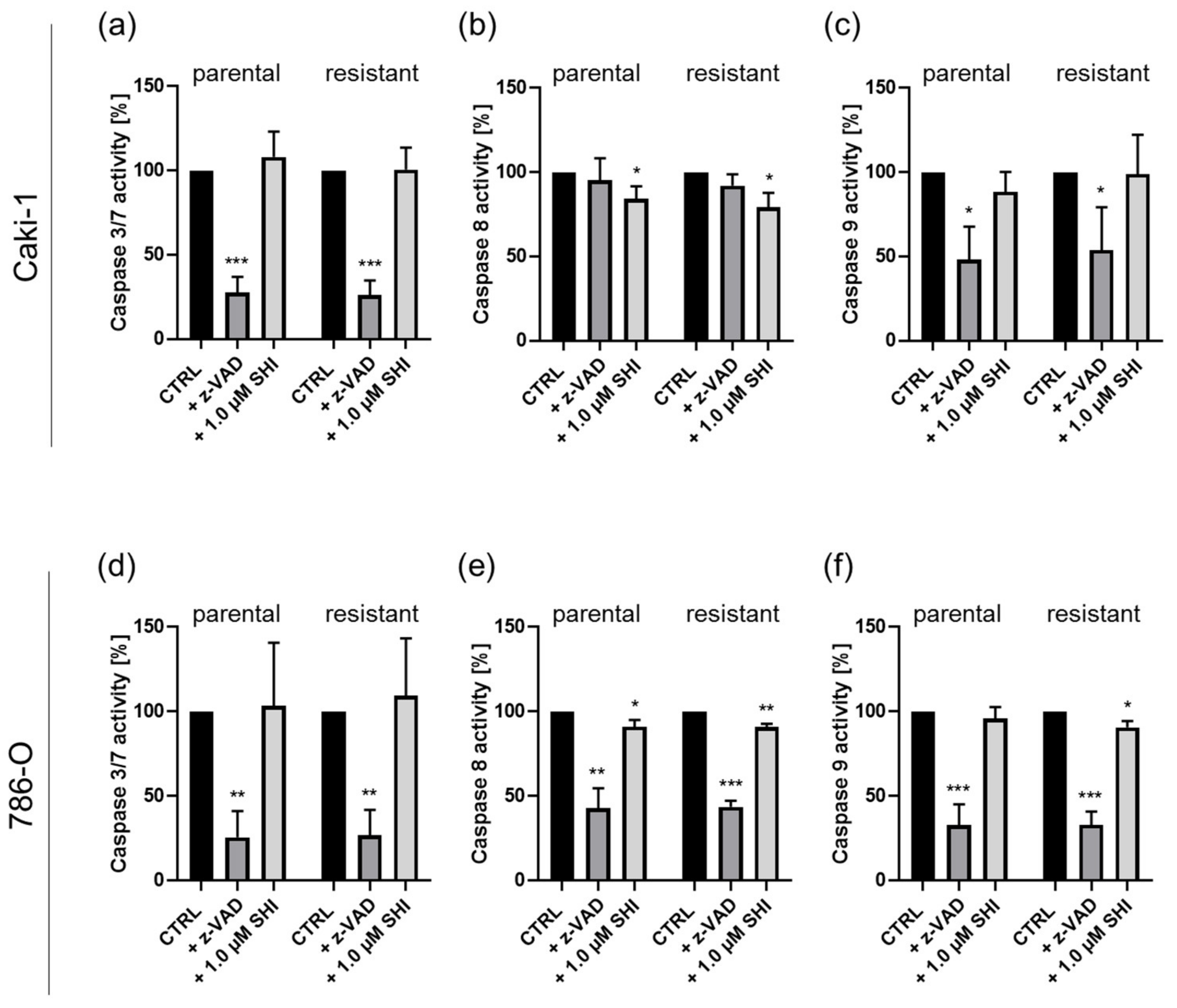
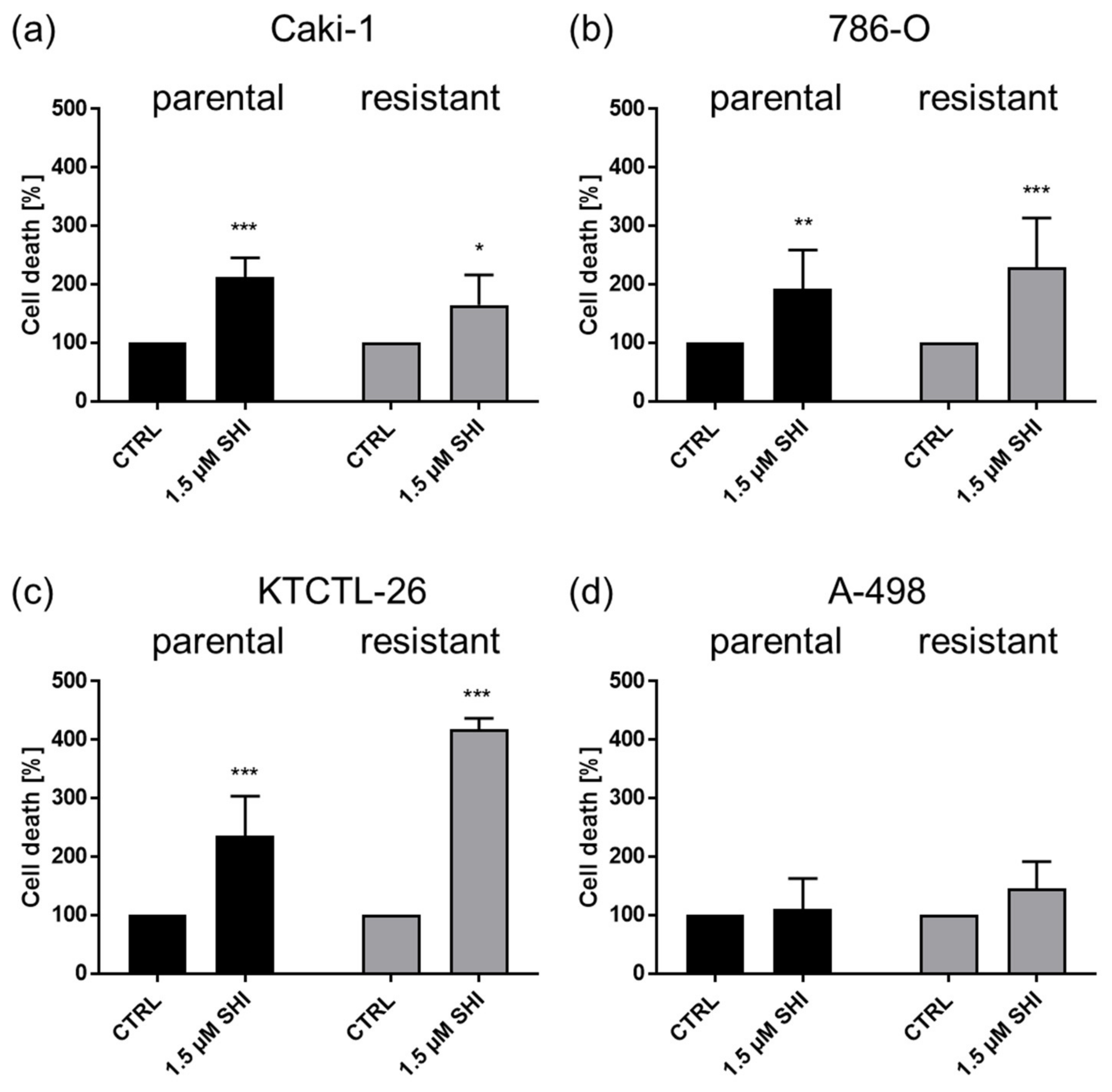
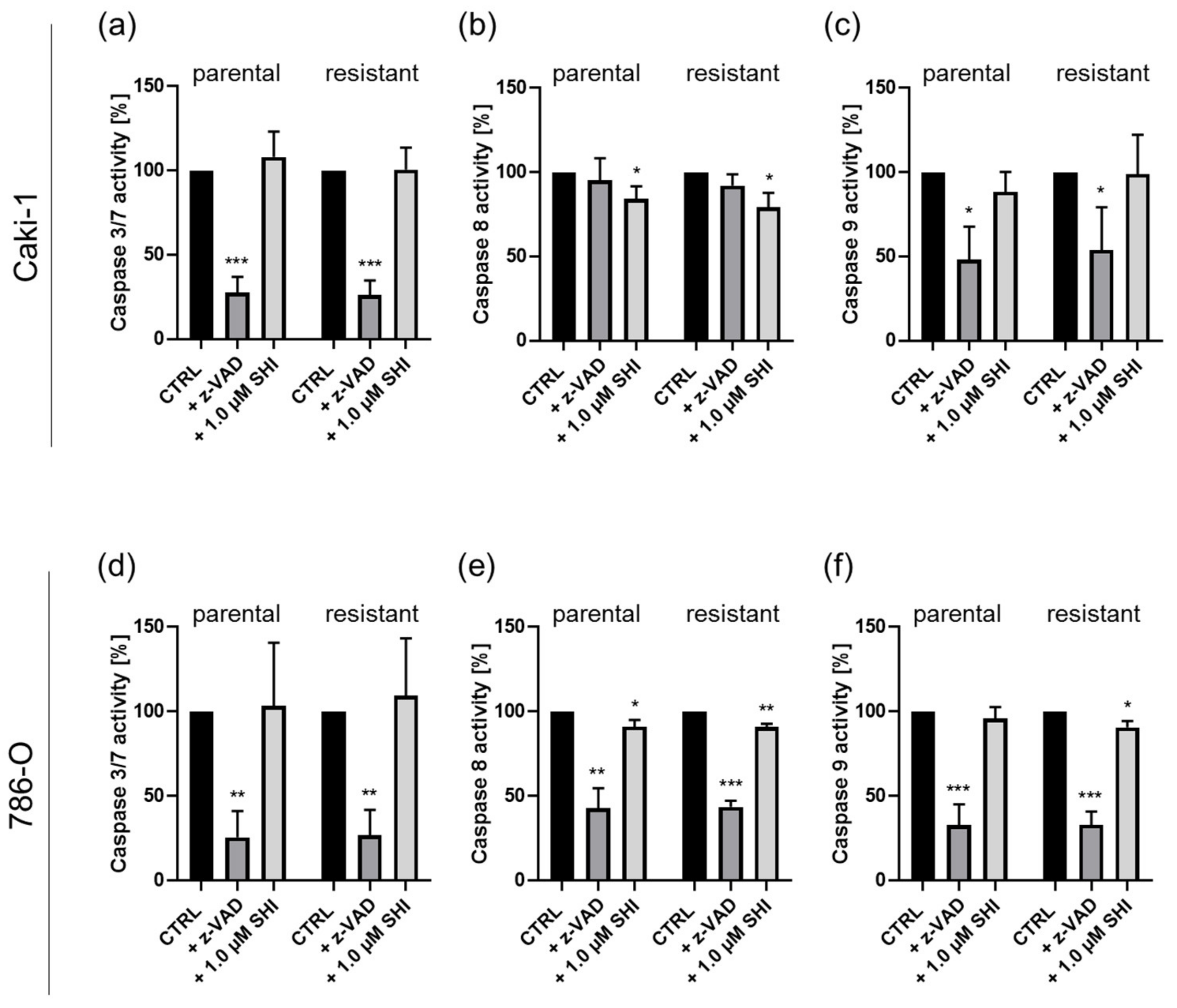
3.3. Shikonin-Induced Apoptosis
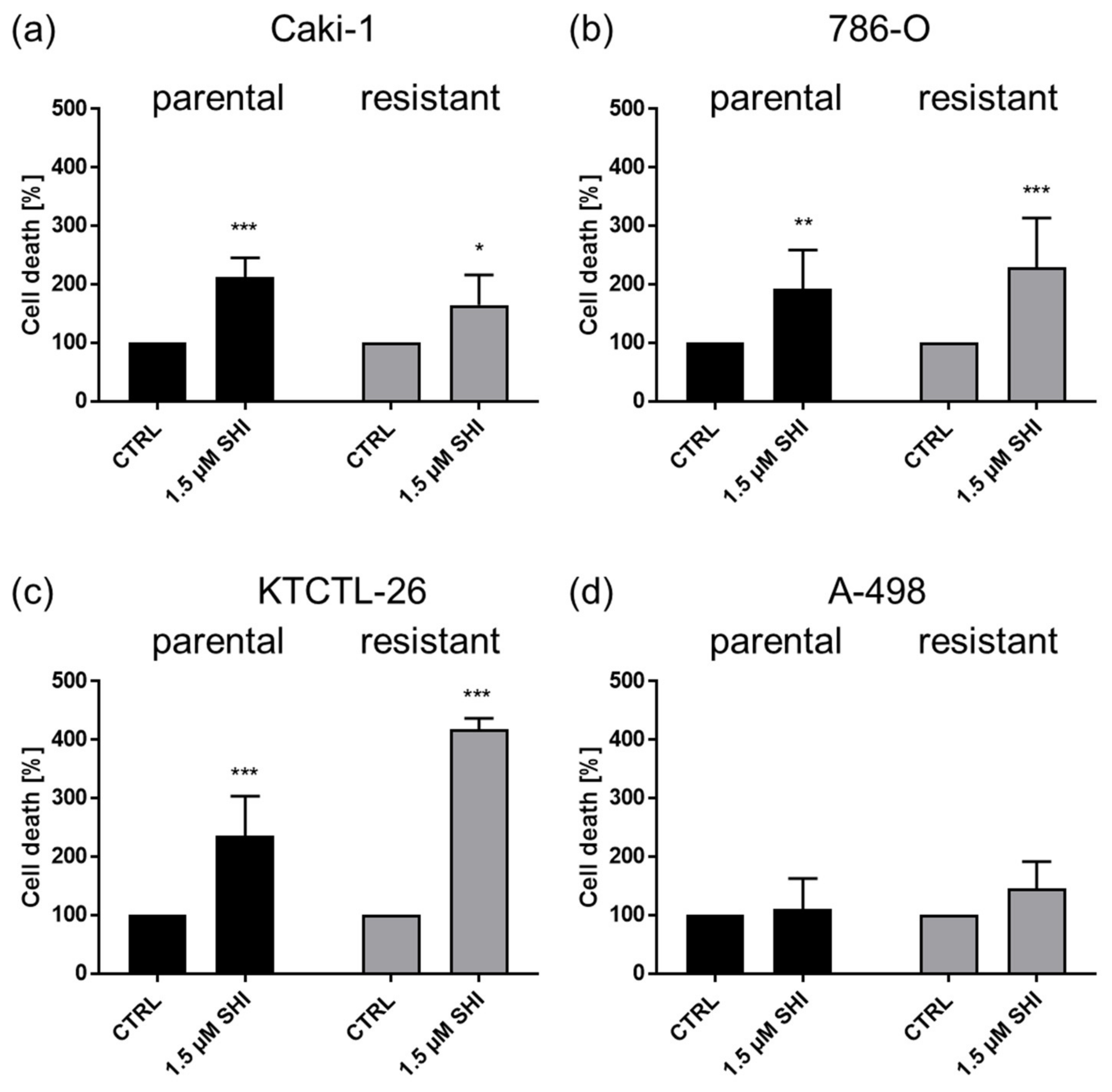
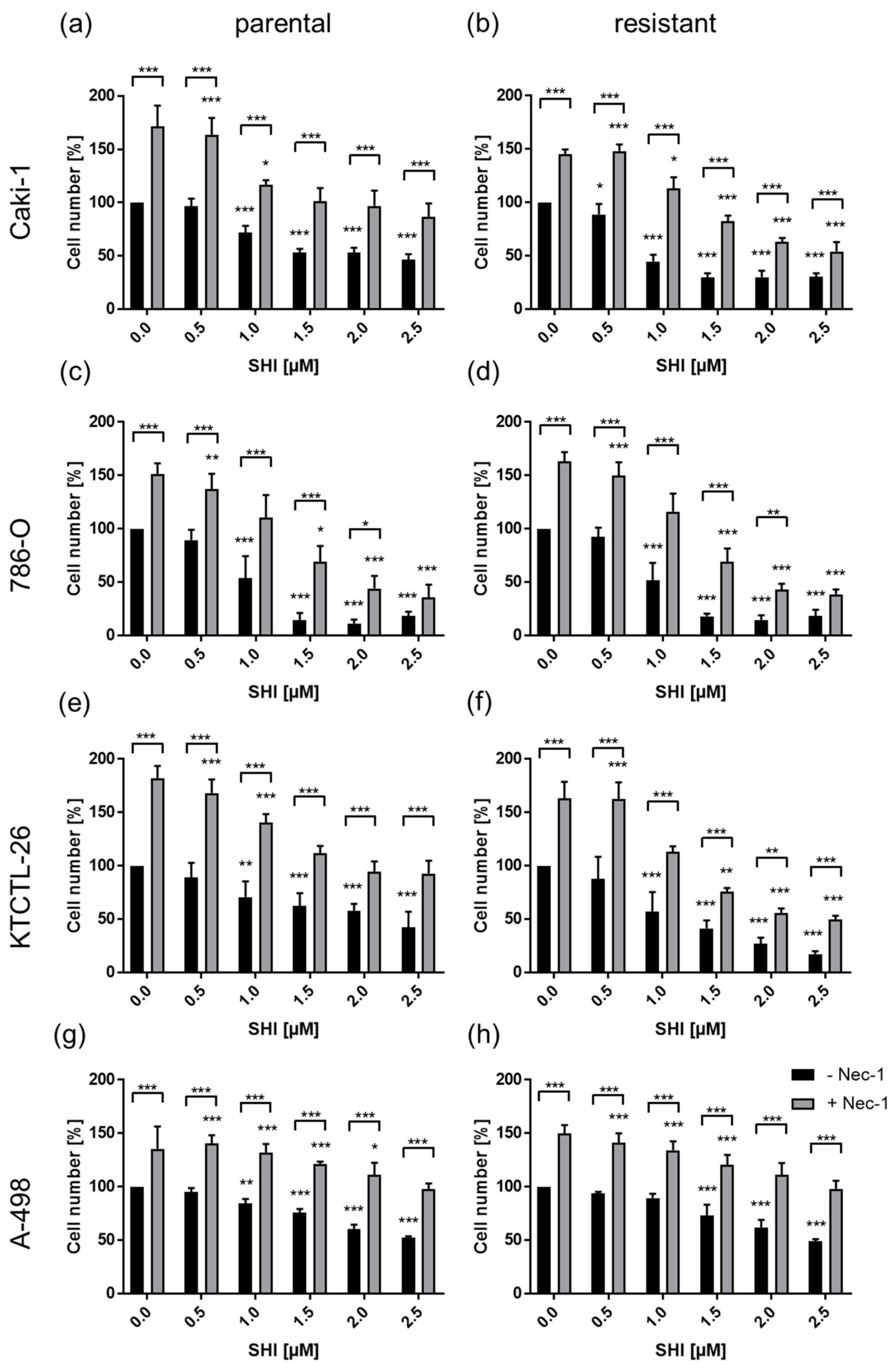
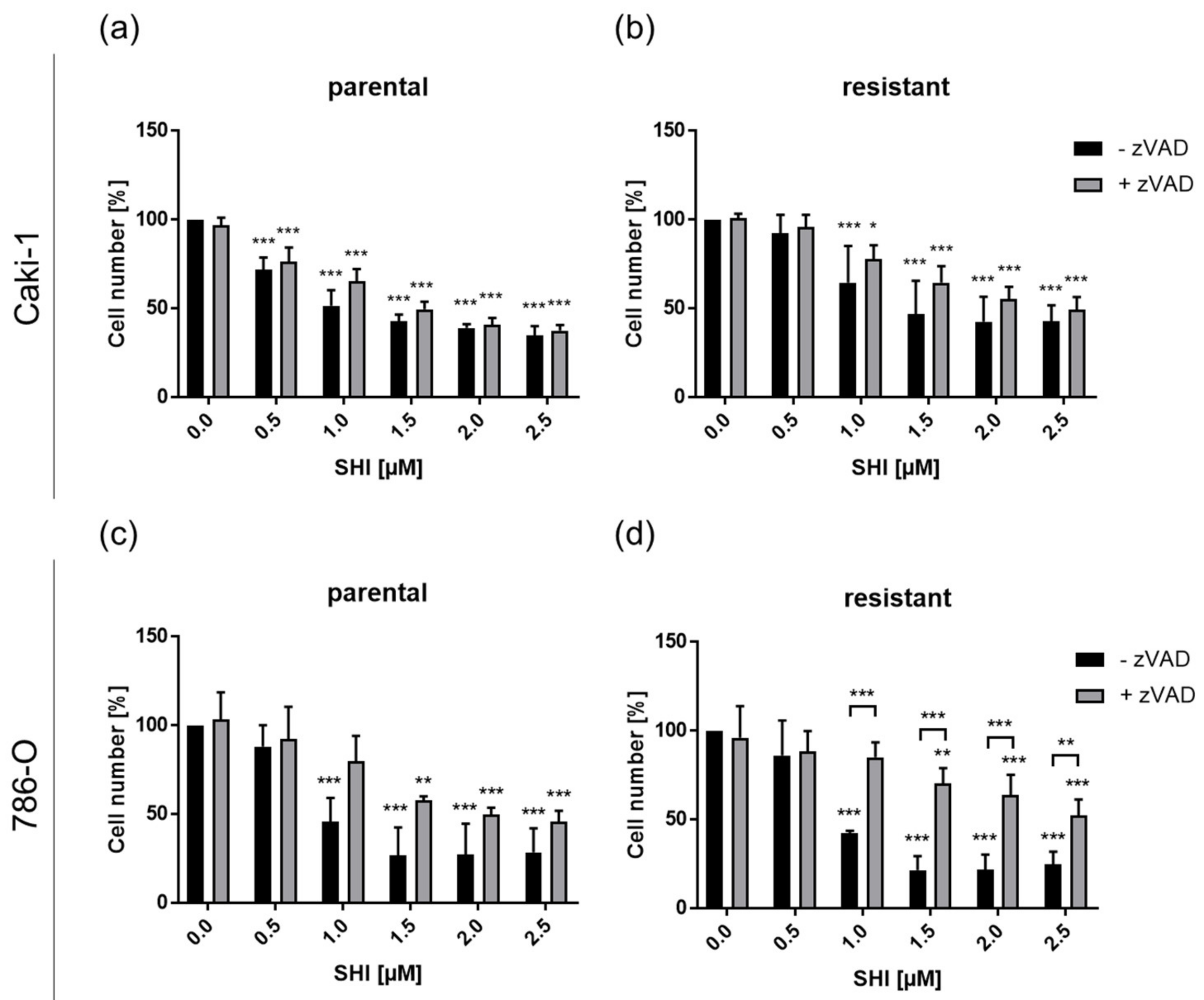
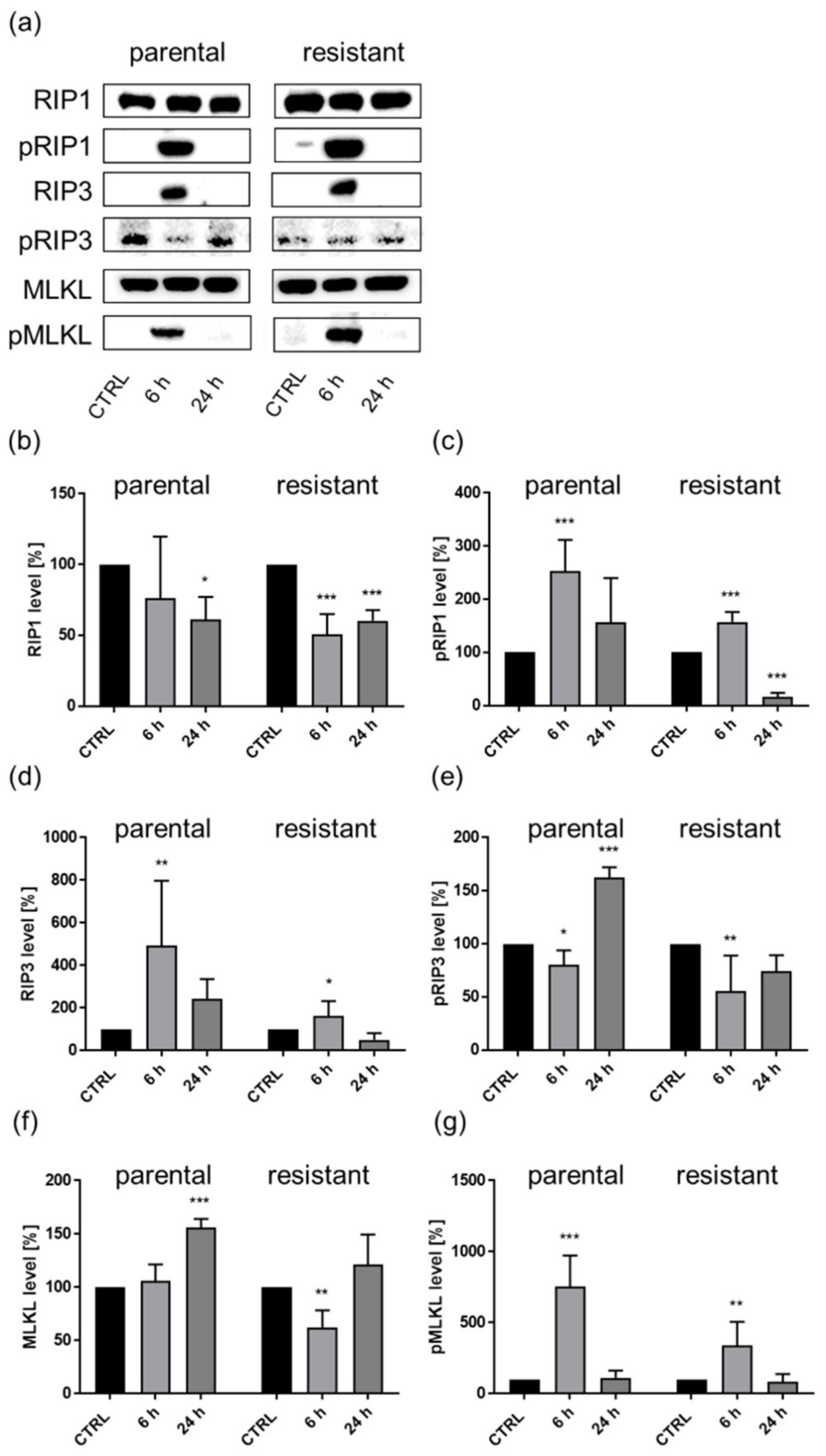
3.4. Shikonin-Induced Necroptosis in Parental and Sunitinib-Resistant RCC Cells
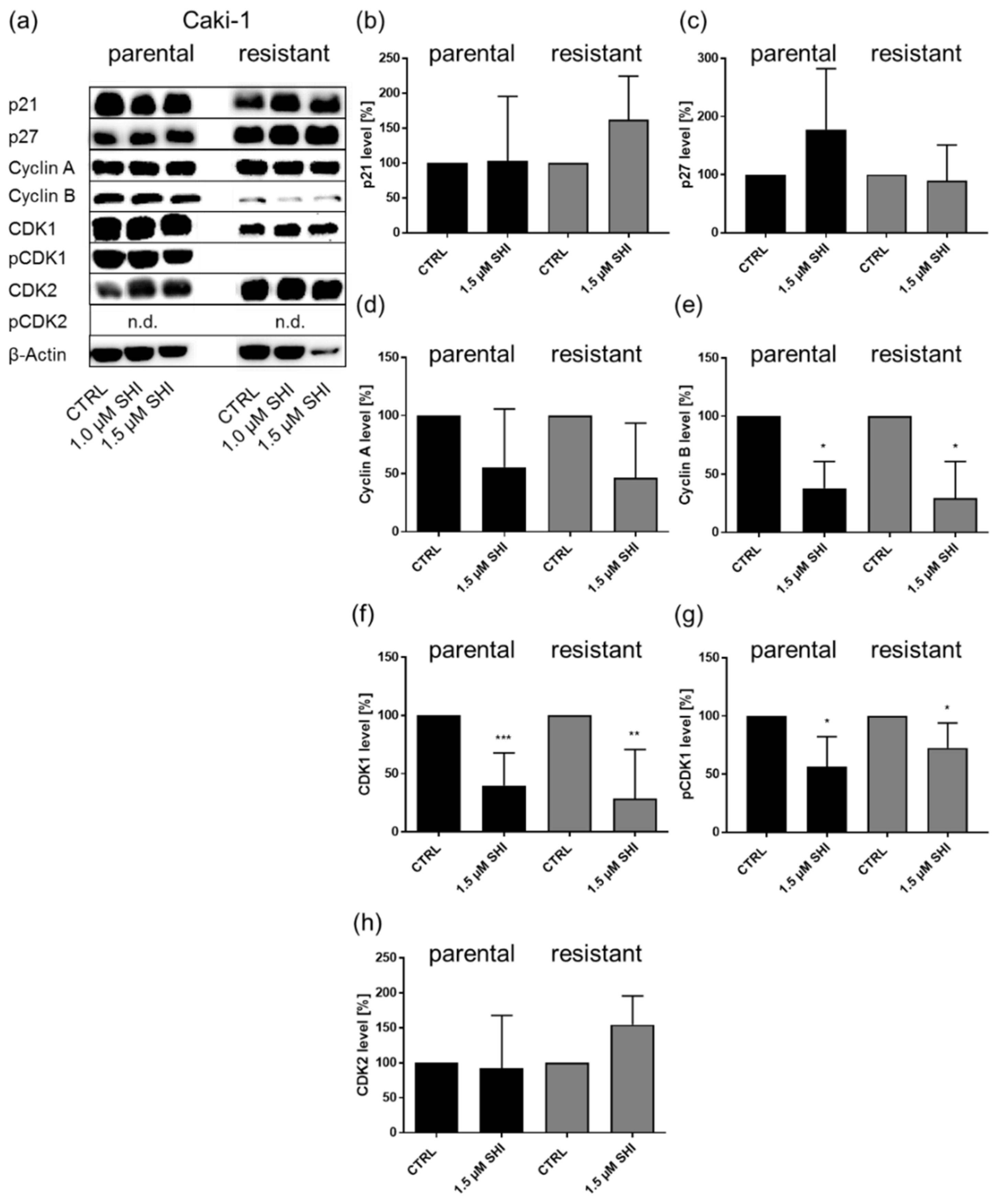
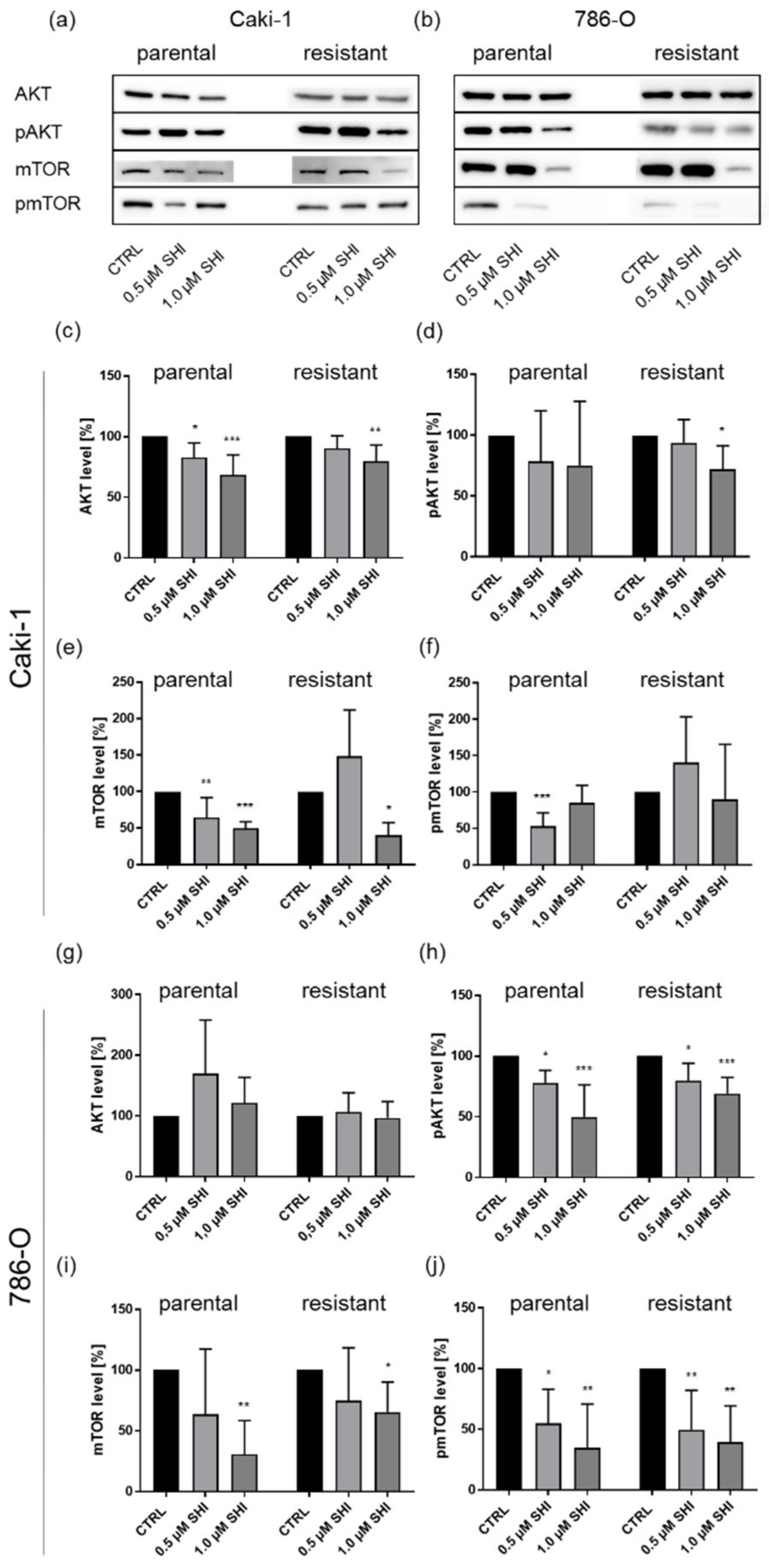
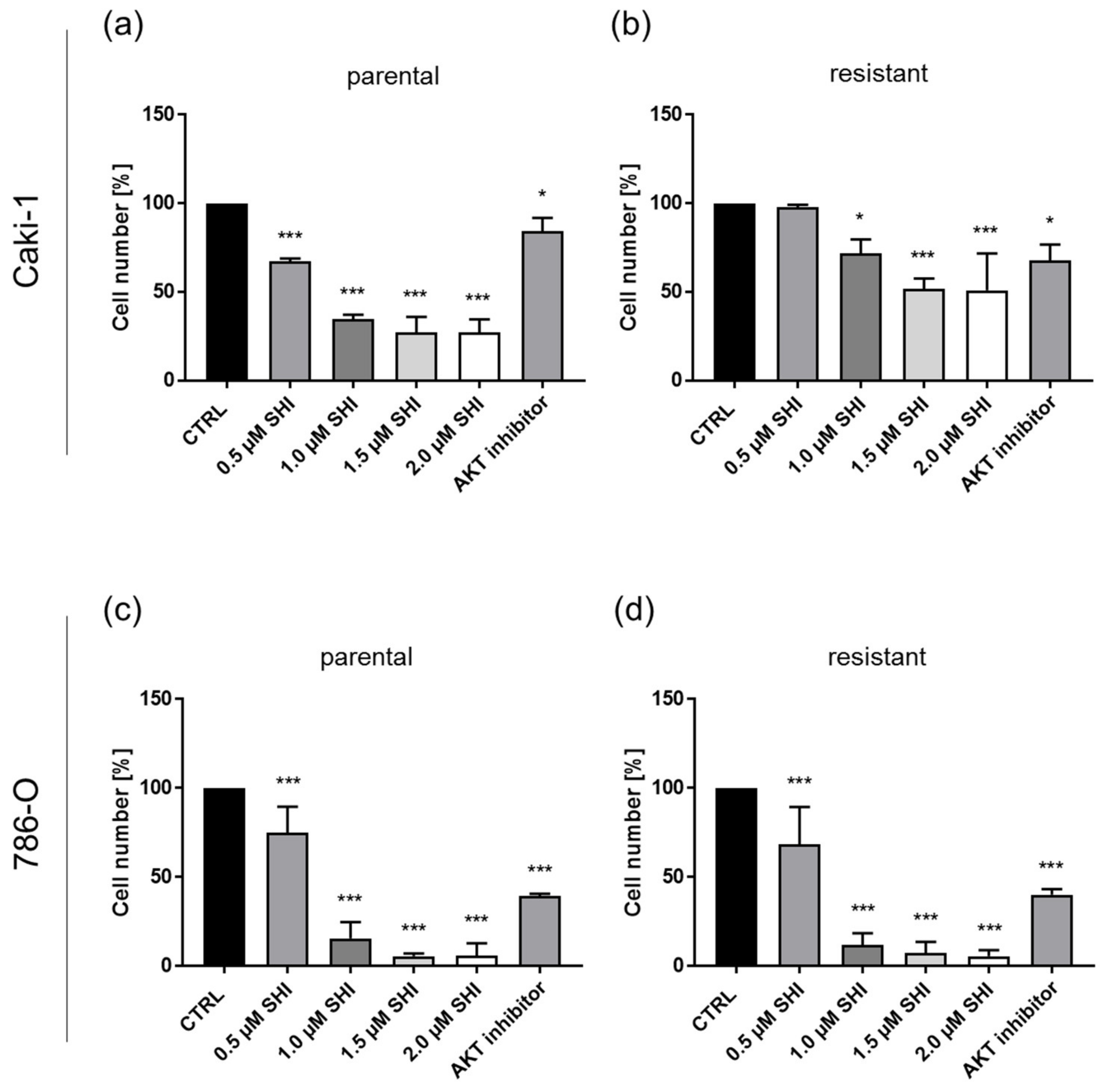
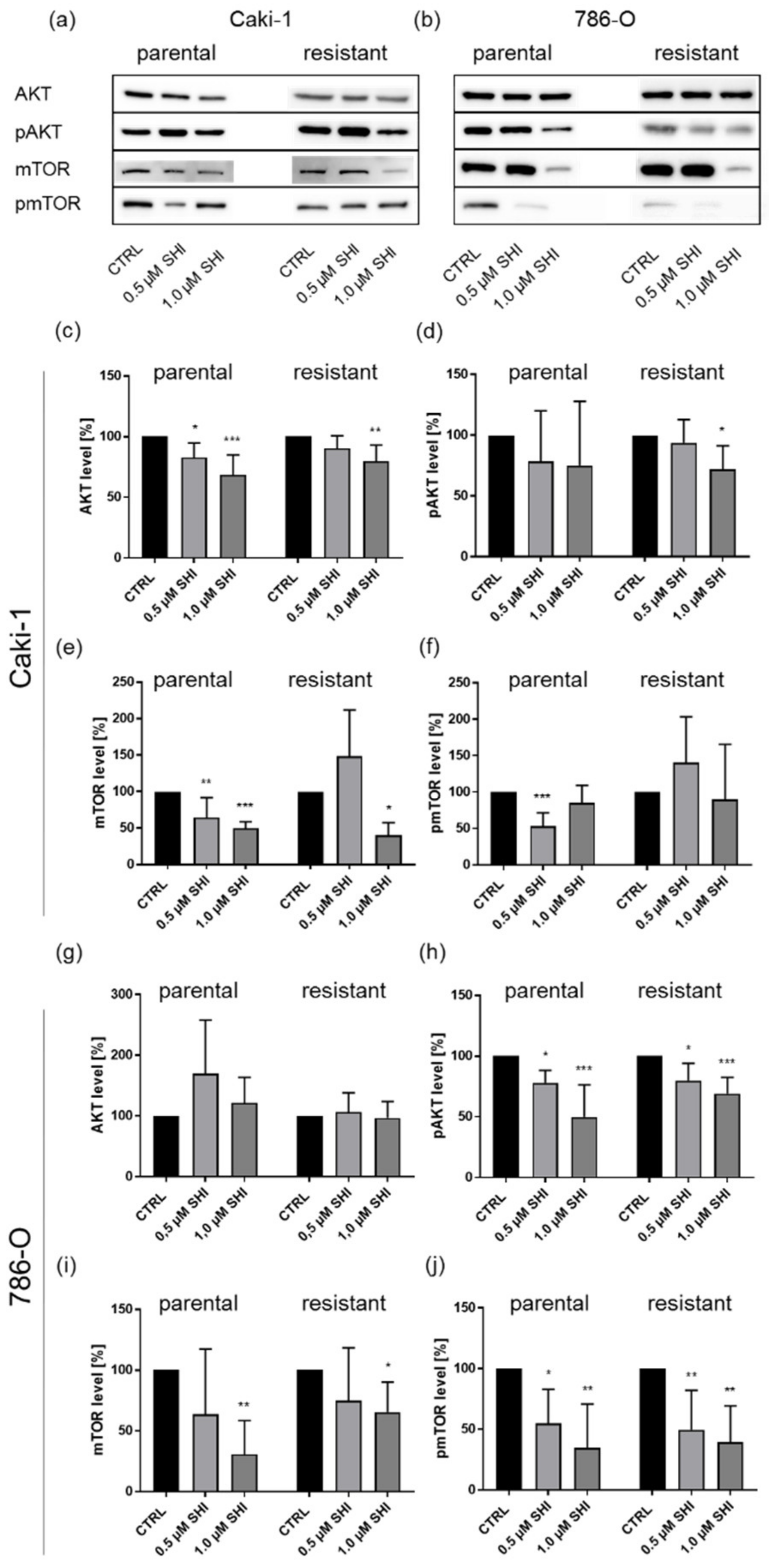
3.5. Shikonin Impaired AKT/mTOR Signaling
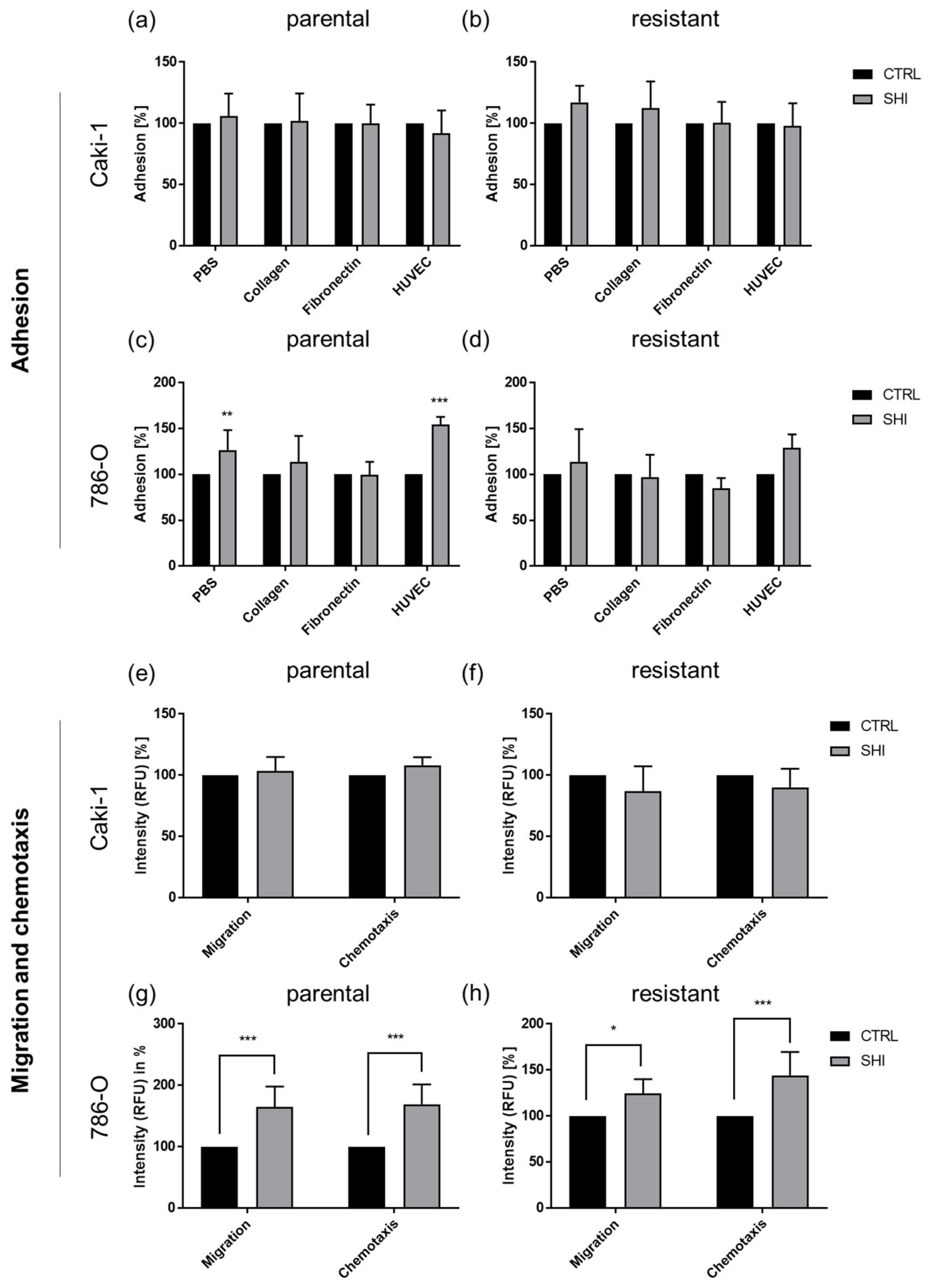
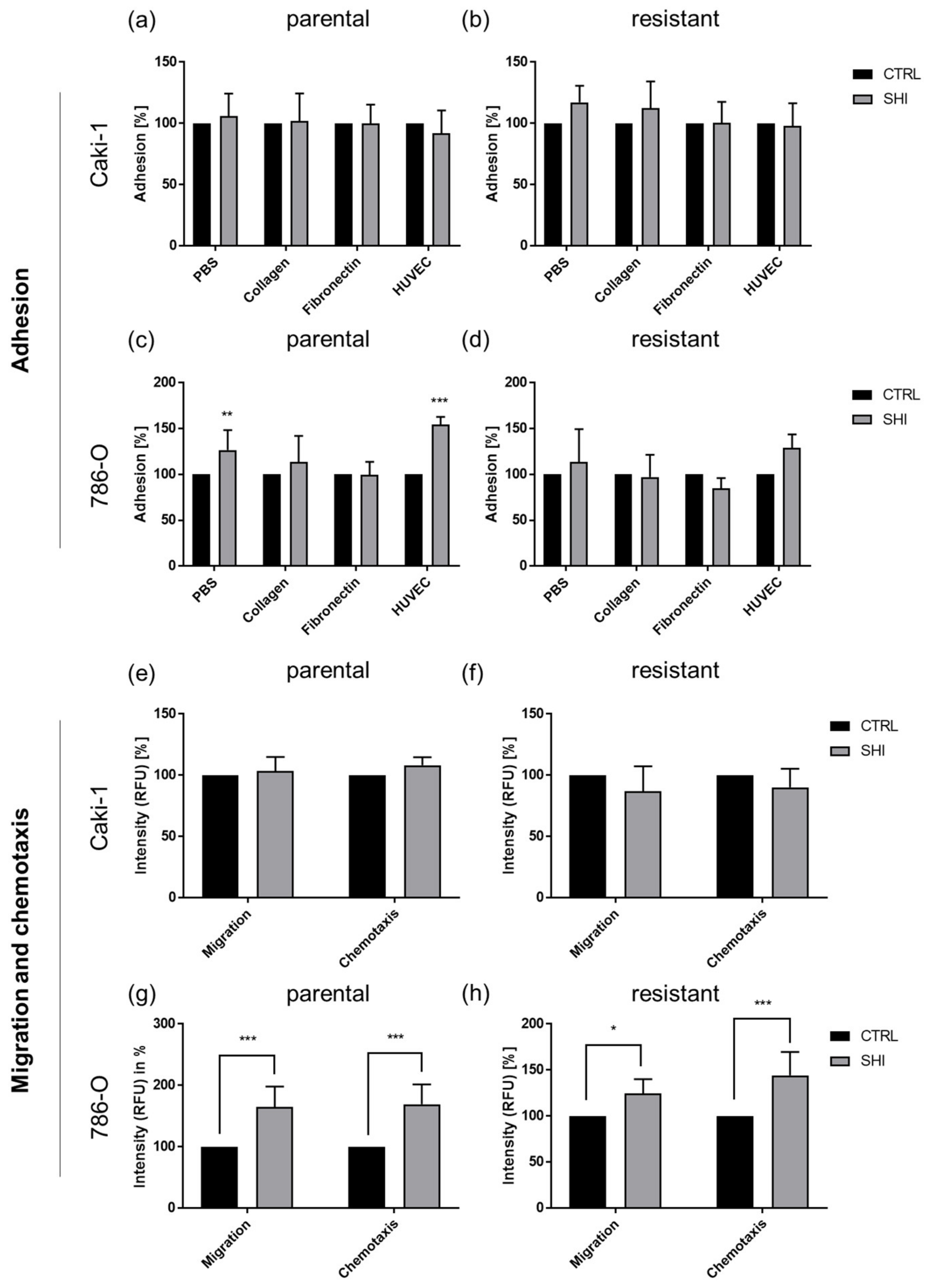
3.6. Shikonin Cell Line Specifically Influenced Adhesion and Motility
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Escudier, B.; Porta, C.; Schmidinger, M.; Rioux-Leclercq, N.; Bex, A.; Khoo, V.; Grünwald, V.; Gillessen, S.; Horwich, A.; ESMO Guidelines Committee. Renal cell carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019, 30, 706–720. [Google Scholar] [CrossRef] [Green Version]
- Ljungberg, B.; Bensalah, K.; Canfield, S.; Dabestani, S.; Hofmann, F.; Hora, M.; Kuczyk, M.A.; Lam, T.; Marconi, L.; Merseburger, A.S.; et al. EAU Guidelines on Renal Cell Carcinoma: 2014 Update. Eur. Urol. 2015, 67, 913–924. [Google Scholar] [CrossRef]
- Fornara, P.; Hoda, M.R. Das Nierenzellkarzinom. Der Urol. 2011, 50, 219–222. [Google Scholar] [CrossRef]
- Usher-Smith, J.; Simmons, R.K.; Rossi, S.H.; Stewart, G.D. Current evidence on screening for renal cancer. Nat. Rev. Urol. 2020, 17, 637–642. [Google Scholar] [CrossRef]
- Ljungberg, B.; Albiges, L.; Abu-Ghanem, Y.; Bensalah, K.; Dabestani, S.; Fernandez-Pello, S.; Giles, R.H.; Hofmann, F.; Hora, M.; Kuczyk, M.A.; et al. European Association of Urology Guidelines on Renal Cell Carcinoma: The 2019 Update. Eur. Urol. 2019, 75, 799–810. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Miller, K.; Bergmann, L.; Doehn, C.; Grünwald, V.; Gschwend, J.E.; Ivanyi, P.; Keilholz, U.; Kuczyk, M.A. Interdisziplinäre Empfehlungen zur Behandlung des fortgeschrittenen Nierenzellkarzinoms. Aktuel.-Urol. 2020, 51, 572–581. [Google Scholar] [CrossRef]
- Moran, M.; Nickens, D.; Adcock, K.; Bennetts, M.; Desscan, A.; Charnley, N.; Fife, K. Sunitinib for Metastatic Renal Cell Carcinoma: A Systematic Review and Meta-Analysis of Real-World and Clinical Trials Data. Target. Oncol. 2019, 14, 405–416. [Google Scholar] [CrossRef] [Green Version]
- Fiebig, J.; Kraywinkel, K. Epidemiologie des Nierenzellkarzinoms in Deutschland. Der Onkol. 2019, 25, 483–487. [Google Scholar] [CrossRef]
- Mantia, C.M.; McDermott, D.F. Vascular endothelial growth factor and programmed death-1 pathway inhibitors in renal cell carcinoma. Cancer 2019, 125, 4148–4157. [Google Scholar] [CrossRef]
- Duran, I.; Lambea, J.; Maroto, P.; González-Larriba, J.L.; Flores, L.; Granados-Principal, S.; Graupera, M.; Sáez, B.; Vivancos, A.; Casanovas, O. Resistance to Targeted Therapies in Renal Cancer: The Importance of Changing the Mechanism of Action. Target. Oncol. 2016, 12, 19–35. [Google Scholar] [CrossRef]
- Kutschan, S.; Freuding, M.; Keinki, C.; Huebner, J. Recommendations on complementary and alternative medicine within S3 guidelines in oncology: Systematic quality assessment of underlying methodology. J. Cancer Res. Clin. Oncol. 2020, 146, 2419–2425. [Google Scholar] [CrossRef]
- Mani, J.; Juengel, E.; Arslan, I.; Bartsch, G.; Filmann, N.; Ackermann, H.; Nelson, K.; Haferkamp, A.; Engl, T.; Blaheta, R.A. Use of complementary and alternative medicine before and after organ removal due to urologic cancer. Patient Prefer. Adherence 2015, 9, 1407–1412. [Google Scholar] [CrossRef] [Green Version]
- Ebel, M.-D.; Rudolph, I.; Keinki, C.; Hoppe, A.; Muecke, R.; Micke, O.; Muenstedt, K.; Huebner, J. Perception of cancer patients of their disease, self-efficacy and locus of control and usage of complementary and alternative medicine. J. Cancer Res. Clin. Oncol. 2015, 141, 1449–1455. [Google Scholar] [CrossRef]
- Boulos, J.; Rahama, M.; Hegazy, M.E.; Efferth, T. Shikonin derivatives for cancer prevention and therapy. Cancer Lett. 2019, 459, 248–267. [Google Scholar] [CrossRef]
- Guo, C.; He, J.; Song, X.; Tan, L.; Wang, M.; Jiang, P.; Li, Y.; Cao, Z.; Peng, C. Pharmacological properties and derivatives of shikonin—A review in recent years. Pharmacol. Res. 2019, 149, 104463. [Google Scholar] [CrossRef]
- Andujar, I.; Recio, M.C.; Giner, R.M.; Ríos, J.L. Traditional chinese medicine remedy to jury: The pharmacological basis for the use of shikonin as an anticancer therapy. Curr. Med. Chem. 2013, 20, 2892–2898. [Google Scholar] [CrossRef]
- Song, J.; Zhao, Z.; Fan, X.; Chen, M.; Cheng, X.; Zhang, D.; Wu, F.; Ying, X.; Ji, J. Shikonin potentiates the effect of arsenic trioxide against human hepatocellular carcinoma in vitro and in vivo. Oncotarget 2016, 7, 70504–70515. [Google Scholar] [CrossRef] [Green Version]
- Zang, F.; Rao, Y.; Zhu, X.; Wu, Z.; Jiang, H. Shikonin suppresses NEAT1 and Akt signaling in treating paclitaxel-resistant non-small cell of lung cancer. Mol. Med. 2020, 26, 28. [Google Scholar] [CrossRef]
- Li, X.; Fan, X.-X.; Jiang, Z.-B.; Loo, W.T.; Yao, X.-J.; Leung, E.L.-H.; Chow, L.W.; Liu, L. Shikonin inhibits gefitinib-resistant non-small cell lung cancer by inhibiting TrxR and activating the EGFR proteasomal degradation pathway. Pharmacol. Res. 2017, 115, 45–55. [Google Scholar] [CrossRef]
- Liang, W.; Cai, A.; Chen, G.; Xi, H.; Wu, X.; Cui, J.; Zhang, K.; Zhao, X.; Yu, J.; Wei, B.; et al. Shikonin induces mitochondria-mediated apoptosis and enhances chemotherapeutic sensitivity of gastric cancer through reactive oxygen species. Sci. Rep. 2016, 6, 38267. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hao, F.; Nan, Y.; Qu, L.; Na, W.; Jia, C.; Chen, X. PKM2 Inhibitor Shikonin Overcomes the Cisplatin Resistance in Bladder Cancer by Inducing Necroptosis. Int. J. Biol. Sci. 2018, 14, 1883–1891. [Google Scholar] [CrossRef] [Green Version]
- Markowitsch, S.D.; Juetter, K.M.; Schupp, P.; Hauschulte, K.; Vakhrusheva, O.; Slade, K.S.; Thomas, A.; Tsaur, I.; Cinatl, J.; Michaelis, M.; et al. Shikonin Reduces Growth of Docetaxel-Resistant Prostate Cancer Cells Mainly through Necroptosis. Cancers 2021, 13, 882. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Chen, Z.; Ni, F.; Ye, X.; Qian, W. Shikonin overcomes drug resistance and induces necroptosis by regulating the miR-92a-1-5p/MLKL axis in chronic myeloid leukemia. Aging 2020, 12, 17662–17680. [Google Scholar] [CrossRef]
- Wiench, B.; Eichhorn, T.; Paulsen, M.; Efferth, T. Shikonin Directly Targets Mitochondria and Causes Mitochondrial Dysfunction in Cancer Cells. Evid.-Based Complement. Altern. Med. 2012, 2012, 726025. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Pozo, F.M.; Tian, D.; Geng, X.; Yao, X.; Zhang, Y.; Tang, J. Shikonin Inhibits Cancer Through P21 Upregulation and Apoptosis Induction. Front. Pharmacol. 2020, 11, 861. [Google Scholar] [CrossRef]
- Ding, Y.; He, C.; Lu, S.; Wang, X.; Wang, C.; Wang, L.; Zhang, J.; Piao, M.; Chi, G.; Luo, Y.; et al. MLKL contributes to shikonin-induced glioma cell necroptosis via promotion of chromatinolysis. Cancer Lett. 2019, 467, 58–71. [Google Scholar] [CrossRef]
- Liu, T.; Sun, X.; Cao, Z. Shikonin-induced necroptosis in nasopharyngeal carcinoma cells via ROS overproduction and upregulation of RIPK1/RIPK3/MLKL expression. OncoTargets Ther. 2019, 12, 2605–2614. [Google Scholar] [CrossRef] [Green Version]
- Bao, C.; Liu, T.; Qian, L.; Xiao, C.; Zhou, X.; Ai, H.; Wang, J.; Fan, W.; Pan, J. Shikonin inhibits migration and invasion of triple-negative breast cancer cells by suppressing epithelial-mesenchymal transition via miR-17-5p/PTEN/Akt pathway. J. Cancer 2021, 12, 76–88. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, J.; Xiao, S. Shikonin inhibits growth, invasion and glycolysis of nasopharyngeal carcinoma cells through inactivating the phosphatidylinositol 3 kinase/AKT signal pathway. Anti-Cancer Drugs 2020, 31, 932–941. [Google Scholar] [CrossRef]
- Juengel, E.; Kim, D.; Makarević, J.; Reiter, M.; Tsaur, I.; Bartsch, G.; Haferkamp, A.; Blaheta, R.A. Molecular analysis of sunitinib resistant renal cell carcinoma cells after sequential treatment with RAD001 (everolimus) or sorafenib. J. Cell. Mol. Med. 2014, 19, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Juengel, E.; Nowaz, S.; Makarevi, J.; Natsheh, I.; Werner, I.; Nelson, K.; Reiter, M.; Tsaur, I.; Mani, J.; Harder, S.; et al. HDAC-inhibition counteracts everolimus resistance in renal cell carcinoma in vitro by diminishing cdk2 and cyclin A. Mol. Cancer 2014, 13, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juengel, E.; Makarević, J.; Tsaur, I.; Bartsch, G.; Nelson, K.; Haferkamp, A.; Blaheta, R.A. Resistance after Chronic Application of the HDAC-Inhibitor Valproic Acid Is Associated with Elevated Akt Activation in Renal Cell Carcinoma In Vivo. PLoS ONE 2013, 8, e53100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juengel, E.; Makarevic, J.; Reiter, M.; Mani, J.; Tsaur, I.; Bartsch, G.; Haferkamp, A.; Blaheta, R.A. Resistance to the mTOR Inhibitor Temsirolimus Alters Adhesion and Migration Behavior of Renal Cell Carcinoma Cells through an Integrin α5–and Integrin β3–Dependent Mechanism. Neoplasia 2014, 16, 291–300. [Google Scholar] [CrossRef] [Green Version]
- Juengel, E.; Dauselt, A.; Makarević, J.; Wiesner, C.; Tsaur, I.; Bartsch, G.; Haferkamp, A.; Blaheta, R.A. Acetylation of histone H3 prevents resistance development caused by chronic mTOR inhibition in renal cell carcinoma cells. Cancer Lett. 2012, 324, 83–90. [Google Scholar] [CrossRef]
- Zhao, F.; Vakhrusheva, O.; Markowitsch, S.D.; Slade, K.S.; Tsaur, I.; Cinatl, J.J.; Michaelis, M.; Efferth, T.; Haferkamp, A.; Juengel, E. Artesunate Impairs Growth in Cisplatin-Resistant Bladder Cancer Cells by Cell Cycle Arrest, Apoptosis and Autophagy Induction. Cells 2020, 9, 2643. [Google Scholar] [CrossRef]
- Juengel, E.; Euler, S.; Maxeiner, S.; Rutz, J.; Justin, S.; Roos, F.; Khoder, W.; Nelson, K.; Bechstein, W.; Blaheta, R.A. Sulforaphane as an adjunctive to everolimus counteracts everolimus resistance in renal cancer cell lines. Phytomedicine 2017, 27, 1–7. [Google Scholar] [CrossRef]
- Juengel, E.; Maxeiner, S.; Rutz, J.; Justin, S.; Roos, F.; Khoder, W.; Tsaur, I.; Nelson, K.; Bechstein, W.; Haferkamp, A.; et al. Sulforaphane inhibits proliferation and invasive activity of everolimus-resistant kidney cancer cells in vitro. Oncotarget 2016, 7, 85208–85219. [Google Scholar] [CrossRef]
- Justin, S.; Rutz, J.; Maxeiner, S.; Chun, F.K.-H.; Juengel, E.; Blaheta, R.A. Bladder Cancer Metastasis Induced by Chronic Everolimus Application Can Be Counteracted by Sulforaphane In Vitro. Int. J. Mol. Sci. 2020, 21, 5582. [Google Scholar] [CrossRef]
- Justin, S.; Rutz, J.; Maxeiner, S.; Chun, F.K.-H.; Juengel, E.; Blaheta, R.A. Chronic Sulforaphane Administration Inhibits Resistance to the mTOR-Inhibitor Everolimus in Bladder Cancer Cells. Int. J. Mol. Sci. 2020, 21, 4026. [Google Scholar] [CrossRef]
- Markowitsch, S.; Schupp, P.; Lauckner, J.; Vakhrusheva, O.; Slade, K.; Mager, R.; Efferth, T.; Haferkamp, A.; Juengel, E. Artesunate Inhibits Growth of Sunitinib-Resistant Renal Cell Carcinoma Cells through Cell Cycle Arrest and Induction of Ferroptosis. Cancers 2020, 12, 3150. [Google Scholar] [CrossRef] [PubMed]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Motzer, R.J. Systemic Therapy for Metastatic Renal-Cell Carcinoma. N. Engl. J. Med. 2017, 376, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Zhai, T.; Hei, Z.; Ma, Q.; Liang, H.; Xu, Y.; Zhang, Y.; Jin, L.; Han, C.; Wang, J. Shikonin induces apoptosis and G0/G1�phase arrest of gallbladder cancer cells via the JNK signaling pathway. Oncol. Rep. 2017, 38, 3473–3480. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Gao, W.; Tao, S.; Wang, Y.; Niu, J.; Zhao, P.; Rao, C.; Yang, L. ER-mediated anti-tumor effects of shikonin on breast cancer. Eur. J. Pharmacol. 2019, 863, 172667. [Google Scholar] [CrossRef]
- Liang, W.; Cui, J.; Zhang, K.; Xi, H.; Cai, A.; Li, J.; Gao, Y.; Hu, C.; Liu, Y.; Lu, Y.; et al. Shikonin induces ROS-based mitochondria-mediated apoptosis in colon cancer. Oncotarget 2017, 8, 109094–109106. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Luo, H.-S.; Li, M.; Tan, S.-Y. Artesunate inhibits the growth and induces apoptosis of human gastric cancer cells by downregulating COX-2. OncoTargets Ther. 2015, 8, 845–854. [Google Scholar] [CrossRef] [Green Version]
- Wen, L.; Liu, L.; Wen, L.; Yu, T.; Wei, F. Artesunate promotes G2/M cell cycle arrest in MCF7 breast cancer cells through ATM activation. Breast Cancer 2018, 25, 681–686. [Google Scholar] [CrossRef]
- Greenshields, A.L.; Shepherd, T.G.; Hoskin, D.W. Contribution of reactive oxygen species to ovarian cancer cell growth arrest and killing by the anti-malarial drug artesunate. Mol. Carcinog. 2016, 56, 75–93. [Google Scholar] [CrossRef]
- Makhov, P.B.; Golovine, K.; Kutikov, A.; Teper, E.; Canter, D.J.; Simhan, J.; Uzzo, R.G.; Kolenko, V.M. Modulation of Akt/mTOR Signaling Overcomes Sunitinib Resistance in Renal and Prostate Cancer Cells. Mol. Cancer Ther. 2012, 11, 1510–1517. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Lu, S.; Li, J.; Deng, L. Shikonin-mediated up-regulation of miR-34a and miR-202 inhibits retinoblastoma proliferation. Toxicol. Res. 2018, 7, 907–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandimali, N.; Sun, H.-N.; Kong, L.-Z.; Zhen, X.; Liu, R.; Kwon, T.; Lee, D.-S. Shikonin-induced Apoptosis of Colon Cancer Cells Is Reduced by Peroxiredoxin V Expression. Anticancer Res. 2019, 39, 6115–6123. [Google Scholar] [CrossRef]
- Zhang, N.; Peng, F.; Wang, Y.; Yang, L.; Wu, F.; Wang, X.; Ye, C.; Han, B.; He, G. Shikonin induces colorectal carcinoma cells apoptosis and autophagy by targeting galectin-1/JNK signaling axis. Int. J. Biol. Sci. 2020, 16, 147–161. [Google Scholar] [CrossRef]
- Gara, R.K.; Srivastava, V.K.; Duggal, S.; Bagga, J.K.; Bhatt, M.; Sanyal, S.; Mishra, D.P. Shikonin selectively induces apoptosis in human prostate cancer cells through the endoplasmic reticulum stress and mitochondrial apoptotic pathway. J. Biomed. Sci. 2015, 22, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.-J.; Hwang, K.-E.; Park, D.-S.; Oh, S.-H.; Jun, H.Y.; Yoon, K.-H.; Jeong, E.-T.; Kim, H.-R.; Kim, Y.-S. Shikonin-induced necroptosis is enhanced by the inhibition of autophagy in non-small cell lung cancer cells. J. Transl. Med. 2017, 15, 123. [Google Scholar] [CrossRef] [PubMed]
- Guo, N.; Miao, R.; Gao, X.; Huang, D.; Hu, Z.; Ji, N.; Nan, Y.; Jiang, F. Shikonin inhibits proliferation and induces apoptosis in glioma cells via downregulation of CD147. Mol. Med. Rep. 2019, 19, 4335–4343. [Google Scholar] [CrossRef]
- Wei, Y.; Li, M.; Cui, S.; Wang, D.; Zhang, C.-Y.; Zen, K.; Li, L. Shikonin Inhibits the Proliferation of Human Breast Cancer Cells by Reducing Tumor-Derived Exosomes. Molecules 2016, 21, 777. [Google Scholar] [CrossRef] [PubMed]
- Shahsavari, Z.; Karami-Tehrani, F.; Salami, S.; Ghasemzadeh, M. RIP1K and RIP3K provoked by shikonin induce cell cycle arrest in the triple negative breast cancer cell line, MDA-MB-468: Necroptosis as a desperate programmed suicide pathway. Tumor Biol. 2015, 37, 4479–4491. [Google Scholar] [CrossRef]
- Chen, Y.; Zheng, L.; Liu, J.; Zhou, Z.; Cao, X.; Lv, X.; Chen, F. Shikonin inhibits prostate cancer cells metastasis by reducing matrix metalloproteinase-2/-9 expression via AKT/mTOR and ROS/ERK1/2 pathways. Int. Immunopharmacol. 2014, 21, 447–455. [Google Scholar] [CrossRef]
- Gavet, O.; Pines, J. Progressive Activation of CyclinB1-Cdk1 Coordinates Entry to Mitosis. Dev. Cell 2010, 18, 533–543. [Google Scholar] [CrossRef] [Green Version]
- Otto, T.; Sicinski, T.O.P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.-W.; Tsai, S.-C.; Peng, S.-F.; Lin, M.-W.; Chiang, J.-H.; Chiu, Y.-J.; Fushiya, S.; Tseng, M.T.; Yang, J.-S. Kaempferol induces autophagy through AMPK and AKT signaling molecules and causes G2/M arrest via downregulation of CDK1/cyclin B in SK-HEP-1 human hepatic cancer cells. Int. J. Oncol. 2013, 42, 2069–2077. [Google Scholar] [CrossRef] [PubMed]
- He, G.; He, G.; Zhou, R.; Pi, Z.; Zhu, T.; Jiang, L.; Xie, Y. Enhancement of cisplatin-induced colon cancer cells apoptosis by shikonin, a natural inducer of ROS in vitro and in vivo. Biochem. Biophys. Res. Commun. 2016, 469, 1075–1082. [Google Scholar] [CrossRef]
- Chen, C.; Xiao, W.; Huang, L.; Yu, G.; Ni, J.; Yang, L.; Wan, R.; Hu, G. Shikonin induces apoptosis and necroptosis in pancreatic cancer via regulating the expression of RIP1/RIP3 and synergizes the activity of gemcitabine. Am. J. Transl. Res. 2017, 9, 5507–5517. [Google Scholar] [PubMed]
- Jeung, Y.-J.; Kim, H.-G.; Ahn, J.; Lee, H.-J.; Lee, S.-B.; Won, M.; Jung, C.-R.; Im, J.-Y.; Kim, B.-K.; Park, S.-K.; et al. Shikonin induces apoptosis of lung cancer cells via activation of FOXO3a/EGR1/SIRT1 signaling antagonized by p300. Biochim. Biophys. Acta 2016, 1863, 2584–2593. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Han, H.-W.; Wang, Y.-S.; He, D.-L.; Sun, W.-X.; Feng, L.; Wen, Z.-L.; Yang, M.-K.; Lu, G.-H.; Wang, X.-M.; et al. Shikonin and 4-hydroxytamoxifen synergistically inhibit the proliferation of breast cancer cells through activating apoptosis signaling pathway in vitro and in vivo. Chin. Med. 2020, 15, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klöditz, K.; Fadeel, B. Three cell deaths and a funeral: Macrophage clearance of cells undergoing distinct modes of cell death. Cell Death Discov. 2019, 5, 65. [Google Scholar] [CrossRef]
- Al-Lamki, R.S.; Lu, W.; Manalo, P.; Wang, J.; Warren, A.Y.; Tolkovsky, A.M.; Pober, J.S.; Bradley, J. Tubular epithelial cells in renal clear cell carcinoma express high RIPK1/3 and show increased susceptibility to TNF receptor 1-induced necroptosis. Cell Death Dis. 2016, 7, e2287. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.-F.; Chen, S.-M.; Ong, A.-Z.; Chung, Y.-H.; Chen, P.-N.; Hsieh, Y.-H.; Kang, Y.-T.; Hsu, L.-S. Shikonin Induced Program Cell Death through Generation of Reactive Oxygen Species in Renal Cancer Cells. Antioxidants 2021, 10, 1831. [Google Scholar] [CrossRef]
- Huang, C.; Luo, Y.; Zhao, J.; Yang, F.; Zhao, H.; Fan, W.; Ge, P. Shikonin Kills Glioma Cells through Necroptosis Mediated by RIP-1. PLoS ONE 2013, 8, e66326. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Y.k.; Peng, F.h.; Guo, Y.q.; Ge, H.; Cai, S.y.; Fan, L.x.; Peng, Y.x.; Wen, H.; Wang, Q.; Tao, L. Connexin32 Activates Necroptosis via Src-Mediated Inhibition of Caspase 8 in Hepatocellular Carcinoma. Cancer Sci. 2021, 112, 3507. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S. Caspase function in programmed cell death. Cell Death Differ. 2006, 14, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Mandal, R.; Barrón, J.C.; Kostova, I.; Becker, S.; Strebhardt, K. Caspase-8: The double-edged sword. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2020, 1873, 188357. [Google Scholar] [CrossRef] [PubMed]
- Wada, N.; Kawano, Y.; Fujiwara, S.; Kikukawa, Y.; Okuno, Y.; Tasaki, M.; Ueda, M.; Ando, Y.; Yoshinaga, K.; Ri, M.; et al. Shikonin, dually functions as a proteasome inhibitor and a necroptosis inducer in multiple myeloma cells. Int. J. Oncol. 2014, 46, 963–972. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Zhang, T.; Xu, W.; Ding, J.; Yin, F.; Xu, J.; Sun, W.; Wang, H.; Sun, M.; Cai, Z.; et al. Sarcoma-Targeting Peptide-Decorated Polypeptide Nanogel Intracellularly Delivers Shikonin for Upregulated Osteosarcoma Necroptosis and Diminished Pulmonary Metastasis. Theranostics 2018, 8, 1361–1375. [Google Scholar] [CrossRef] [Green Version]
- Fresno Vara, J.Á.; Casado, E.; De Castro, J.; Cejas, P.; Belda-Iniesta, C.; González-Barón, M. PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef]
- Miricescu, D.; Balan, D.G.; Tulin, A.; Stiru, O.; Vacaroiu, I.A.; Mihai, D.A.; Popa, C.C.; Papacocea, R.I.; Enyedi, M.; Sorin, N.A.; et al. PI3K/AKT/mTOR signalling pathway involvement in renal cell carcinoma pathogenesis (Review). Exp. Ther. Med. 2021, 21, 540. [Google Scholar] [CrossRef]
- Lu, D.; Qian, J.; Li, W.; Feng, Q.; Pan, S.; Zhang, S. β-hydroxyisovaleryl-shikonin induces human cervical cancer cell apoptosis via PI3K/AKT/mTOR signaling Corrigendum in /10.3892/ol.2021.12456. Oncol. Lett. 2015, 10, 3434–3442. [Google Scholar] [CrossRef] [Green Version]
- Ni, F.; Huang, X.; Chen, Z.; Qian, W.; Tong, X. Shikonin exerts antitumor activity in Burkitt’s lymphoma by inhibiting C-MYC and PI3K/AKT/mTOR pathway and acts synergistically with doxorubicin. Sci. Rep. 2018, 8, 3317. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, G. Activation of CaMKKβ-AMPK-mTOR pathway is required for autophagy induction by β,β-dimethylacrylshikonin against lung adenocarcinoma cells. Biochem. Biophys. Res. Commun. 2019, 517, 477–483. [Google Scholar] [CrossRef]
- Hsieh, Y.; Liao, C.; Chen, W.; Pai, J.; Weng, M. Shikonin Inhibited Migration and Invasion of Human Lung Cancer Cells via Suppression of c-Met-Mediated Epithelial-to-Mesenchymal Transition. J. Cell. Biochem. 2017, 118, 4639–4651. [Google Scholar] [CrossRef] [PubMed]
- Tee, S.S.; Suster, I.; Truong, S.; Jeong, S.; Eskandari, R.; Digialleonardo, V.; Alvarez, J.A.; Aldeborgh, H.N.; Keshari, K.R. Targeted AKT Inhibition in Prostate Cancer Cells and Spheroids Reduces Aerobic Glycolysis and Generation of Hyperpolarized [1-13C] Lactate. Mol. Cancer Res. 2018, 16, 453–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.S.; Kim, J.E.; Kim, K.; Lee, J.; Park, J.O.; Lim, H.Y.; Park, Y.S.; Kang, W.K.; Kim, S.T. The Impact of Cetuximab Plus AKT- or mTOR- Inhibitor in a Patient-Derived Colon Cancer Cell Model with Wild-Type RAS and PIK3CA Mutation. J. Cancer 2017, 8, 2713–2719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.; Yu, E.J.; Ham, I.-H.; Hur, H.; Kim, Y.-S. AKT inhibition is an effective treatment strategy in ARID1A-deficient gastric cancer cells. OncoTargets Ther. 2017, 10, 4153–4159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, B.R.; Greenwood, H.; Dudley, P.; Crafter, C.; Yu, D.-H.; Zhang, J.; Li, J.; Gao, B.; Ji, Q.; Maynard, J.; et al. Preclinical Pharmacology of AZD5363, an Inhibitor of AKT: Pharmacodynamics, Antitumor Activity, and Correlation of Monotherapy Activity with Genetic Background. Mol. Cancer Ther. 2012, 11, 873–887. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markowitsch, S.D.; Vakhrusheva, O.; Schupp, P.; Akele, Y.; Kitanovic, J.; Slade, K.S.; Efferth, T.; Thomas, A.; Tsaur, I.; Mager, R.; et al. Shikonin Inhibits Cell Growth of Sunitinib-Resistant Renal Cell Carcinoma by Activating the Necrosome Complex and Inhibiting the AKT/mTOR Signaling Pathway. Cancers 2022, 14, 1114. https://doi.org/10.3390/cancers14051114
Markowitsch SD, Vakhrusheva O, Schupp P, Akele Y, Kitanovic J, Slade KS, Efferth T, Thomas A, Tsaur I, Mager R, et al. Shikonin Inhibits Cell Growth of Sunitinib-Resistant Renal Cell Carcinoma by Activating the Necrosome Complex and Inhibiting the AKT/mTOR Signaling Pathway. Cancers. 2022; 14(5):1114. https://doi.org/10.3390/cancers14051114
Chicago/Turabian StyleMarkowitsch, Sascha D., Olesya Vakhrusheva, Patricia Schupp, Yasminn Akele, Jovana Kitanovic, Kimberly S. Slade, Thomas Efferth, Anita Thomas, Igor Tsaur, René Mager, and et al. 2022. "Shikonin Inhibits Cell Growth of Sunitinib-Resistant Renal Cell Carcinoma by Activating the Necrosome Complex and Inhibiting the AKT/mTOR Signaling Pathway" Cancers 14, no. 5: 1114. https://doi.org/10.3390/cancers14051114
APA StyleMarkowitsch, S. D., Vakhrusheva, O., Schupp, P., Akele, Y., Kitanovic, J., Slade, K. S., Efferth, T., Thomas, A., Tsaur, I., Mager, R., Haferkamp, A., & Juengel, E. (2022). Shikonin Inhibits Cell Growth of Sunitinib-Resistant Renal Cell Carcinoma by Activating the Necrosome Complex and Inhibiting the AKT/mTOR Signaling Pathway. Cancers, 14(5), 1114. https://doi.org/10.3390/cancers14051114