
Targeting High-Risk Neuroblastoma Patient-Derived Xenografts with Oncolytic Virotherapy

 , , , , , , , and
, , , , , , , and 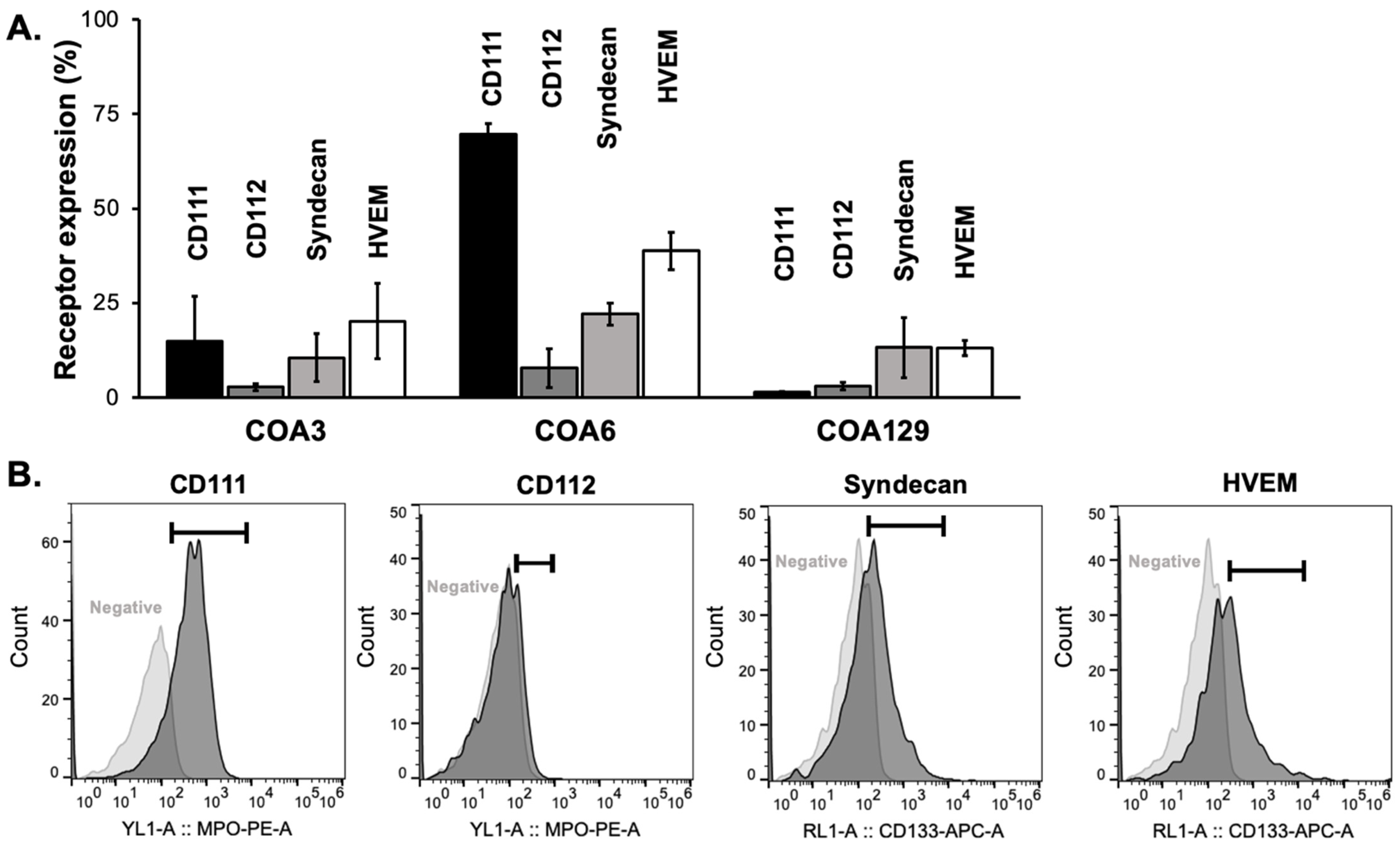{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture
2.2. Establishing Patient-Derived Xenografts
2.3. Virus
2.4. Flow Cytometry
2.5. Viral Replication
2.6. ELISA
2.7. Cytotoxicity
2.8. Antibodies
2.9. Immunoblotting
2.10. RNA Sequencing
2.11. Bioprinted Microtumors
2.12. Immunohistochemistry
2.13. Genomic Analyses
2.14. Data Analysis
3. Results
3.1. Expression of Viral Entry Mediated Receptors
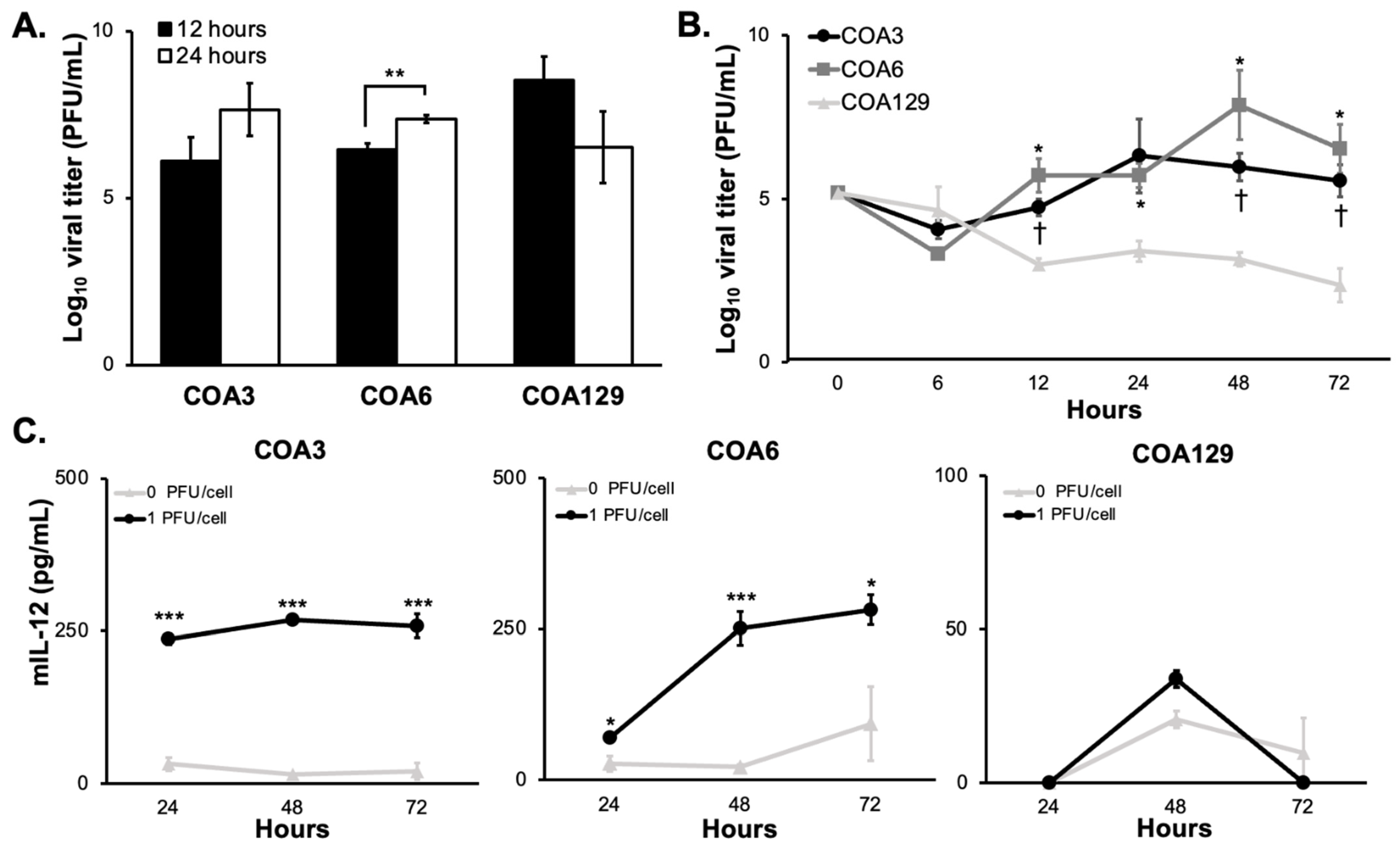
3.2. M002 Replication
3.3. mIL-12
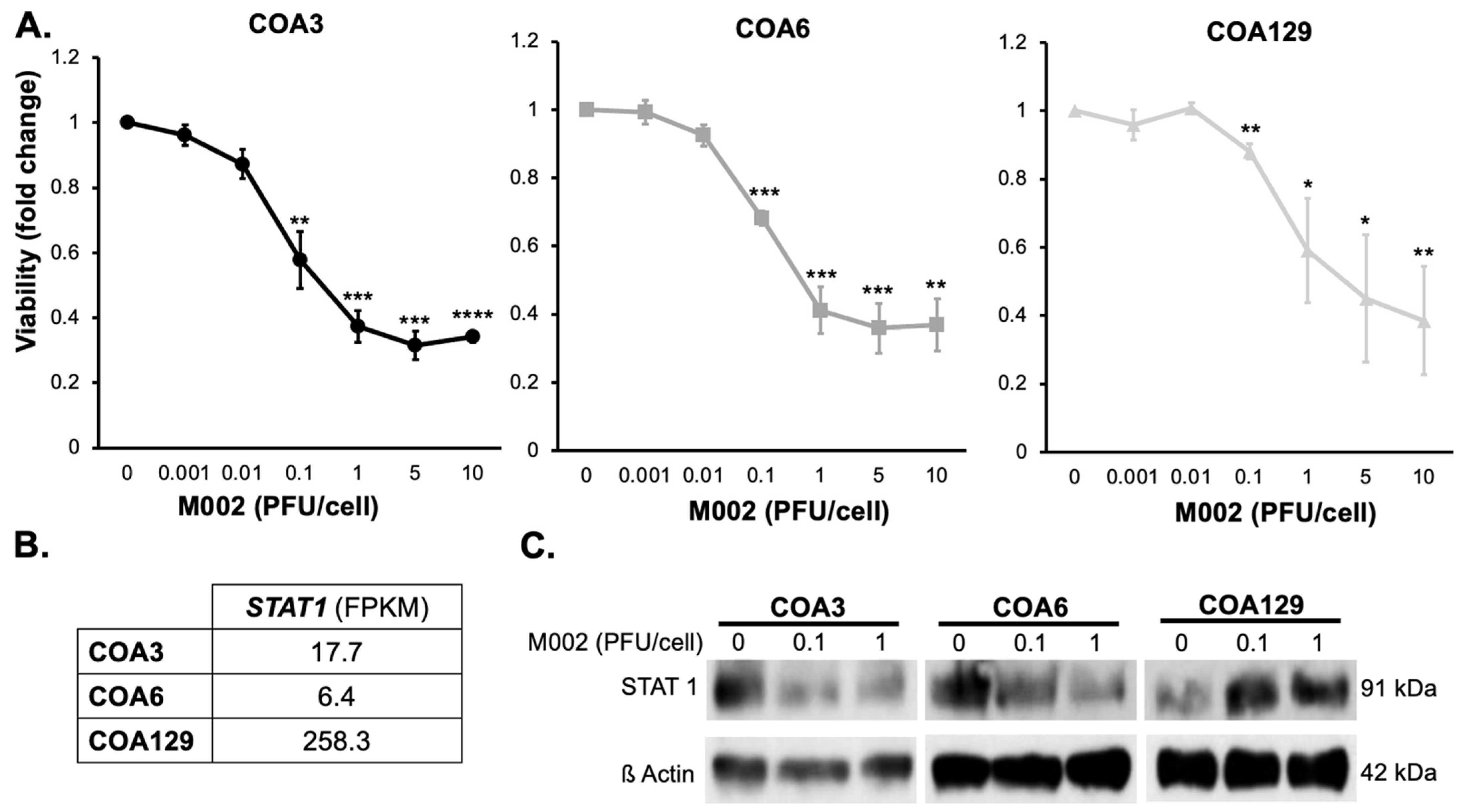
3.4. Cytotoxicity
3.5. M002 Effects STAT1 Expression
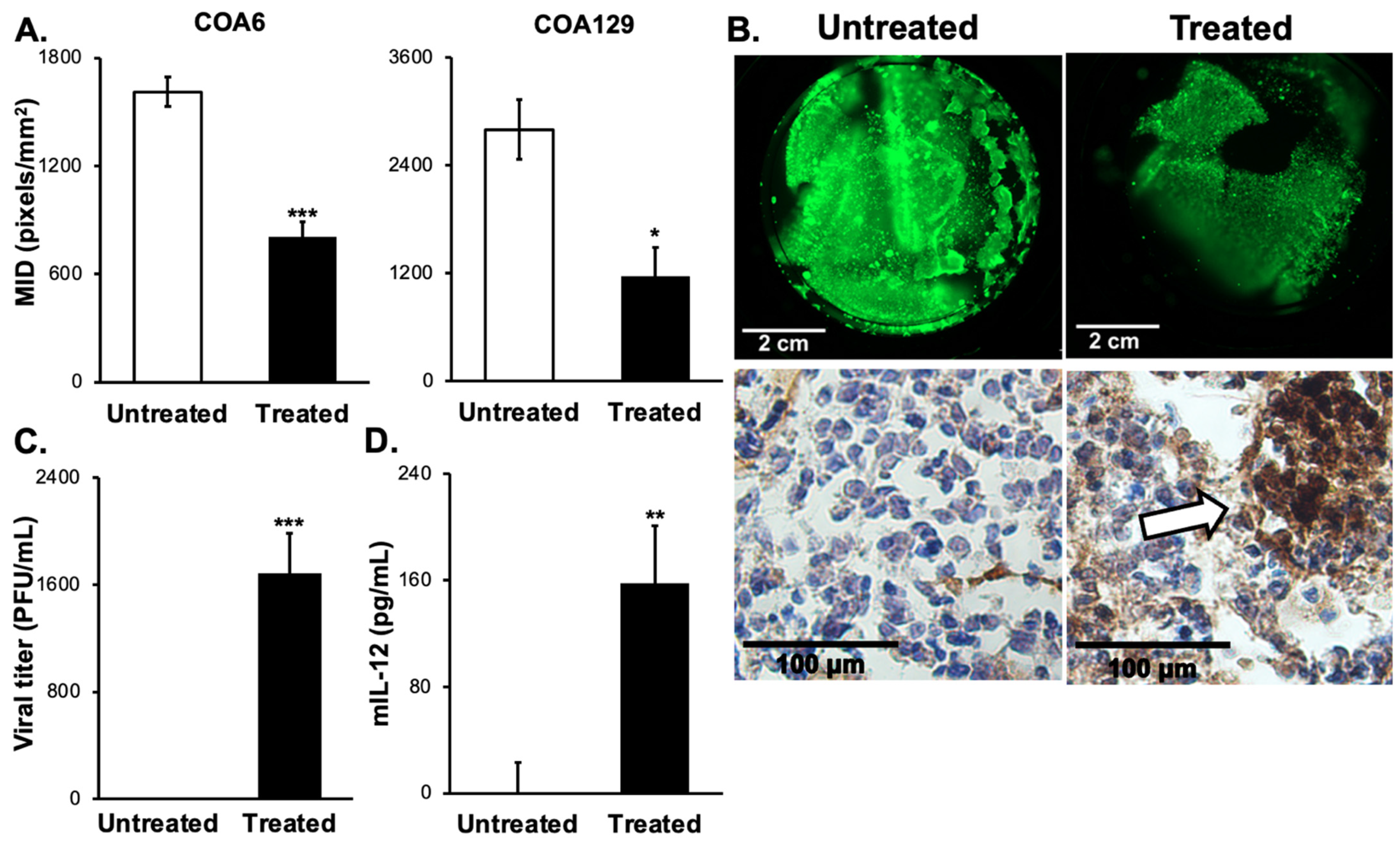
3.6. D Bioprinted PDX Tumors Respond to M002 Infection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Colon, N.C.; Chung, D.H. Neuroblastoma. Adv. Pediatr. 2011, 58, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Marayati, R.; Quinn, C.H.; Beierle, E.A. Immunotherapy in Pediatric Solid Tumors-A Systematic Review. Cancers 2019, 11, 2022. [Google Scholar] [CrossRef]
- Pistoia, V.; Morandi, F.; Bianchi, G.; Pezzolo, A.; Prigione, I.; Raffaghello, L. Immunosuppressive microenvironment in neuroblastoma. Front Oncol. 2013, 3, 167. [Google Scholar] [CrossRef]
- Friedman, G.K.; Johnston, J.M.; Bag, A.K.; Bernstock, J.D.; Li, R.; Aban, I.; Kachurak, K.; Nan, L.; Kang, K.D.; Totsch, S.; et al. Oncolytic HSV-1 G207 Immunovirotherapy for Pediatric High-Grade Gliomas. N. Engl. J. Med. 2021, 384, 1613–1622. [Google Scholar] [CrossRef]
- Ferrucci, P.F.; Pala, L.; Conforti, F.; Cocorocchio, E. Talimogene Laherparepvec (T-VEC): An Intralesional Cancer Immunotherapy for Advanced Melanoma. Cancers 2021, 13, 1383. [Google Scholar] [CrossRef]
- Streby, K.A.; Currier, M.A.; Triplet, M.; Ott, K.; Dishman, D.J.; Vaughan, M.R.; Ranalli, M.A.; Setty, B.; Skeens, M.A.; Whiteside, S.; et al. First-in-Human Intravenous Seprehvir in Young Cancer Patients: A Phase 1 Clinical Trial. Mol. Ther. 2019, 27, 1930–1938. [Google Scholar] [CrossRef]
- Streby, K.A.; Geller, J.I.; Currier, M.A.; Warren, P.S.; Racadio, J.M.; Towbin, A.J.; Vaughan, M.R.; Triplet, M.; Ott-Napier, K.; Dishman, D.J.; et al. Intratumoral Injection of HSV1716, an Oncolytic Herpes Virus, Is Safe and Shows Evidence of Immune Response and Viral Replication in Young Cancer Patients. Clin. Cancer Res. 2017, 23, 3566–3574. [Google Scholar] [CrossRef]
- Izumchenko, E.; Paz, K.; Ciznadija, D.; Sloma, I.; Katz, A.; Vasquez-Dunddel, D.; Ben-Zvi, I.; Stebbing, J.; McGuire, W.; Harris, W.; et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann. Oncol. 2017, 28, 2595–2605. [Google Scholar] [CrossRef] [PubMed]
- Quinn, C.H.; Beierle, A.M.; Beierle, E.A. Artificial Tumor Microenvironments in Neuroblastoma. Cancers 2021, 13, 1629. [Google Scholar] [CrossRef] [PubMed]
- Braekeveldt, N.; Bexell, D. Patient-derived xenografts as preclinical neuroblastoma models. Cell Tissue Res. 2018, 372, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Quinn, C.H.; Beierle, A.M.; Williams, A.P.; Marayati, R.; Bownes, L.V.; Markert, H.R.; Aye, J.M.; Stewart, J.E.; Mroczek-Musulman, E.; Crossman, D.K.; et al. Downregulation of PDGFRss Signaling Overcomes Crizotinib Resistance in a TYRO3 and ALK Mutated Neuroendocrine-Like Tumor. Transl. Oncol. 2021, 14, 101099. [Google Scholar] [CrossRef]
- Parker, J.N.; Gillespie, G.Y.; Love, C.E.; Randall, S.; Whitley, R.J.; Markert, J.M. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc. Natl. Acad. Sci. USA 2000, 97, 2208–2213. [Google Scholar] [CrossRef]
- Megison, M.L.; Gillory, L.A.; Stewart, J.E.; Nabers, H.C.; Mroczek-Musulman, E.; Waters, A.M.; Coleman, J.M.; Kelly, V.; Markert, J.M.; Gillespie, G.Y.; et al. Preclinical evaluation of engineered oncolytic herpes simplex virus for the treatment of pediatric solid tumors. PLoS ONE 2014, 9, e86843. [Google Scholar] [CrossRef]
- Friedman, G.K.; Moore, B.P.; Nan, L.; Kelly, V.M.; Etminan, T.; Langford, C.P.; Xu, H.; Han, X.; Markert, J.M.; Beierle, E.A.; et al. Pediatric medulloblastoma xenografts including molecular subgroup 3 and CD133+ and CD15+ cells are sensitive to killing by oncolytic herpes simplex viruses. Neuro-Oncology 2016, 18, 227–235. [Google Scholar] [CrossRef]
- Waters, A.M.; Stafman, L.L.; Garner, E.F.; Mruthyunjayappa, S.; Stewart, J.E.; Friedman, G.K.; Coleman, J.M.; Markert, J.M.; Gillespie, G.Y.; Beierle, E.A. Effect of Repeat Dosing of Engineered Oncolytic Herpes Simplex Virus on Preclinical Models of Rhabdomyosarcoma. Transl. Oncol. 2016, 9, 419–430. [Google Scholar] [CrossRef]
- Ring, E.K.; Li, R.; Moore, B.P.; Nan, L.; Kelly, V.M.; Han, X.; Beierle, E.A.; Markert, J.M.; Leavenworth, J.W.; Gillespie, G.Y.; et al. Newly Characterized Murine Undifferentiated Sarcoma Models Sensitive to Virotherapy with Oncolytic HSV-1 M002. Mol. Ther. Oncolytics 2017, 7, 27–36. [Google Scholar] [CrossRef]
- Friedman, G.K.; Bernstock, J.D.; Chen, D.; Nan, L.; Moore, B.P.; Kelly, V.M.; Youngblood, S.L.; Langford, C.P.; Han, X.; Ring, E.K.; et al. Enhanced Sensitivity of Patient-Derived Pediatric High-Grade Brain Tumor Xenografts to Oncolytic HSV-1 Virotherapy Correlates with Nectin-1 Expression. Sci. Rep. 2018, 8, 13930. [Google Scholar] [CrossRef]
- Pressey, J.G.; Haas, M.C.; Pressey, C.S.; Kelly, V.M.; Parker, J.N.; Gillespie, G.Y.; Friedman, G.K. CD133 marks a myogenically primitive subpopulation in rhabdomyosarcoma cell lines that are relatively chemoresistant but sensitive to mutant HSV. Pediatr. Blood Cancer 2013, 60, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Gillory, L.A.; Megison, M.L.; Stewart, J.E.; Mroczek-Musulman, E.; Nabers, H.C.; Waters, A.M.; Kelly, V.; Coleman, J.M.; Markert, J.M.; Gillespie, G.Y.; et al. Preclinical evaluation of engineered oncolytic herpes simplex virus for the treatment of neuroblastoma. PLoS ONE 2013, 8, e77753. [Google Scholar] [CrossRef]
- Stafman, L.L.; Williams, A.P.; Marayati, R.; Aye, J.M.; Markert, H.R.; Garner, E.F.; Quinn, C.H.; Lallani, S.B.; Stewart, J.E.; Yoon, K.J.; et al. Focal Adhesion Kinase Inhibition Contributes to Tumor Cell Survival and Motility in Neuroblastoma Patient-Derived Xenografts. Sci. Rep. 2019, 9, 13259. [Google Scholar] [CrossRef] [PubMed]
- Friedman, G.K.; Langford, C.P.; Coleman, J.M.; Cassady, K.A.; Parker, J.N.; Markert, J.M.; Yancey Gillespie, G. Engineered herpes simplex viruses efficiently infect and kill CD133+ human glioma xenograft cells that express CD111. J. Neuro-Oncol. 2009, 95, 199–209. [Google Scholar] [CrossRef]
- Martinez, W.M.; Spear, P.G. Structural features of nectin-2 (HveB) required for herpes simplex virus entry. J. Virol. 2001, 75, 11185–11195. [Google Scholar] [CrossRef]
- Bacsa, S.; Karasneh, G.; Dosa, S.; Liu, J.; Valyi-Nagy, T.; Shukla, D. Syndecan-1 and syndecan-2 play key roles in herpes simplex virus type-1 infection. J. Gen. Virol. 2011, 92, 733–743. [Google Scholar] [CrossRef]
- Fu, X.; Tao, L.; Wang, P.Y.; Cripe, T.P.; Zhang, X. Comparison of infectivity and spread between HSV-1 and HSV-2 based oncolytic viruses on tumor cells with different receptor expression profiles. Oncotarget 2018, 9, 21348–21358. [Google Scholar] [CrossRef]
- Krummenacher, C.; Baribaud, F.; Ponce de Leon, M.; Baribaud, I.; Whitbeck, J.C.; Xu, R.; Cohen, G.H.; Eisenberg, R.J. Comparative usage of herpesvirus entry mediator A and nectin-1 by laboratory strains and clinical isolates of herpes simplex virus. Virology 2004, 322, 286–299. [Google Scholar] [CrossRef]
- Thomas, E.D.; Meza-Perez, S.; Bevis, K.S.; Randall, T.D.; Gillespie, G.Y.; Langford, C.; Alvarez, R.D. IL-12 Expressing oncolytic herpes simplex virus promotes anti-tumor activity and immunologic control of metastatic ovarian cancer in mice. J. Ovarian Res. 2016, 9, 70. [Google Scholar] [CrossRef]
- Nastala, C.L.; Edington, H.D.; McKinney, T.G.; Tahara, H.; Nalesnik, M.A.; Brunda, M.J.; Gately, M.K.; Wolf, S.F.; Schreiber, R.D.; Storkus, W.J.; et al. Recombinant IL-12 administration induces tumor regression in association with IFN-gamma production. J. Immunol. 1994, 153, 1697–1706. [Google Scholar]
- Lester, S.N.; Li, K. Toll-like receptors in antiviral innate immunity. J. Mol. Biol. 2014, 426, 1246–1264. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Darnell, J.E., Jr. Antiviral response in cells containing Stat1 with heterologous transactivation domains. J. Virol. 2001, 75, 2627–2633. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Huo, C.; Xiao, J.; Fan, T.; Zou, S.; Qi, P.; Sun, L.; Wang, M.; Hu, Y. p-STAT1 regulates the influenza A virus replication and inflammatory response in vitro and vivo. Virology 2019, 537, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Mahller, Y.Y.; Sakthivel, B.; Baird, W.H.; Aronow, B.J.; Hsu, Y.H.; Cripe, T.P.; Mehrian-Shai, R. Molecular analysis of human cancer cells infected by an oncolytic HSV-1 reveals multiple upregulated cellular genes and a role for SOCS1 in virus replication. Cancer Gene Ther. 2008, 15, 733–741. [Google Scholar] [CrossRef]
- Stebbing, J.; Paz, K.; Schwartz, G.K.; Wexler, L.H.; Maki, R.; Pollock, R.E.; Morris, R.; Cohen, R.; Shankar, A.; Blackman, G.; et al. Patient-derived xenografts for individualized care in advanced sarcoma. Cancer 2014, 120, 2006–2015. [Google Scholar] [CrossRef]
- Langer, E.M.; Allen-Petersen, B.L.; King, S.M.; Kendsersky, N.D.; Turnidge, M.A.; Kuziel, G.M.; Riggers, R.; Samatham, R.; Amery, T.S.; Jacques, S.L.; et al. Modeling Tumor Phenotypes In Vitro with Three-Dimensional Bioprinting. Cell Rep. 2019, 26, 608–623.e6. [Google Scholar] [CrossRef]
- Pol, J.G.; Levesque, S.; Workenhe, S.T.; Gujar, S.; Le Boeuf, F.; Clements, D.R.; Fahrner, J.E.; Fend, L.; Bell, J.C.; Mossman, K.L.; et al. Trial Watch: Oncolytic viro-immunotherapy of hematologic and solid tumors. Oncoimmunology 2018, 7, e1503032. [Google Scholar] [CrossRef]
- Waters, A.M.; Friedman, G.K.; Ring, E.K.; Beierle, E.A. Oncolytic virotherapy for pediatric malignancies: Future prospects. Oncolytic. Virother. 2016, 5, 73–80. [Google Scholar] [CrossRef]
- Totsch, S.K.; Schlappi, C.; Kang, K.D.; Ishizuka, A.S.; Lynn, G.M.; Fox, B.; Beierle, E.A.; Whitley, R.J.; Markert, J.M.; Gillespie, G.Y.; et al. Oncolytic herpes simplex virus immunotherapy for brain tumors: Current pitfalls and emerging strategies to overcome therapeutic resistance. Oncogene 2019, 38, 6159–6171. [Google Scholar] [CrossRef]
- Marayati, R.; Bownes, L.V.; Quinn, C.H.; Wadhwani, N.; Williams, A.P.; Markert, H.R.; Atigadda, V.; Aye, J.M.; Stewart, J.E.; Yoon, K.J.; et al. Novel second-generation rexinoid induces growth arrest and reduces cancer cell stemness in human neuroblastoma patient-derived xenografts. J. Pediatr. Surg. 2021, 56, 1165–1173. [Google Scholar] [CrossRef]
- Augustine, R.; Kalva, S.N.; Ahmad, R.; Zahid, A.A.; Hasan, S.; Nayeem, A.; McClements, L.; Hasan, A. 3D Bioprinted cancer models: Revolutionizing personalized cancer therapy. Transl. Oncol. 2021, 14, 101015. [Google Scholar] [CrossRef] [PubMed]
- Bordoni, M.; Karabulut, E.; Kuzmenko, V.; Fantini, V.; Pansarasa, O.; Cereda, C.; Gatenholm, P. 3D Printed Conductive Nanocellulose Scaffolds for the Differentiation of Human Neuroblastoma Cells. Cells 2020, 9, 682. [Google Scholar] [CrossRef]
- Grunewald, L.; Lam, T.; Andersch, L.; Klaus, A.; Schwiebert, S.; Winkler, A.; Gauert, A.; Heeren-Hagemann, A.I.; Astrahantseff, K.; Klironomos, F.; et al. A Reproducible Bioprinted 3D Tumor Model Serves as a Preselection Tool for CAR T Cell Therapy Optimization. Front Immunol. 2021, 12, 689697. [Google Scholar] [CrossRef] [PubMed]
- Pinto, B.; Henriques, A.C.; Silva, P.M.A.; Bousbaa, H. Three-Dimensional Spheroids as In Vitro Preclinical Models for Cancer Research. Pharmaceutics 2020, 12, 1186. [Google Scholar] [CrossRef]
- Kraft, R.M.; Nguyen, M.L.; Yang, X.H.; Thor, A.D.; Blaho, J.A. Caspase 3 activation during herpes simplex virus 1 infection. Virus Res. 2006, 120, 163–175. [Google Scholar] [CrossRef]
- Galvan, V.; Roizman, B. Herpes simplex virus 1 induces and blocks apoptosis at multiple steps during infection and protects cells from exogenous inducers in a cell-type-dependent manner. Proc. Natl. Acad. Sci. USA 1998, 95, 3931–3936. [Google Scholar] [CrossRef]
- Geraghty, R.J.; Krummenacher, C.; Cohen, G.H.; Eisenberg, R.J.; Spear, P.G. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 1998, 280, 1618–1620. [Google Scholar] [CrossRef]
- Jing, X.; Cerveny, M.; Yang, K.; He, B. Replication of herpes simplex virus 1 depends on the gamma 134.5 functions that facilitate virus response to interferon and egress in the different stages of productive infection. J. Virol. 2004, 78, 7653–7666. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Liu, X.; Ma, Y.; Cao, Y.; He, B. Herpes Simplex Virus 1 gamma134.5 Protein Inhibits STING Activation That Restricts Viral Replication. J. Virol. 2018, 92, e01015-18. [Google Scholar] [CrossRef] [PubMed]
- Ghonime, M.G.; Jackson, J.; Shah, A.; Roth, J.; Li, M.; Saunders, U.; Coleman, J.; Gillespie, G.Y.; Markert, J.M.; Cassady, K.A. Chimeric HCMV/HSV-1 and Deltagamma134.5 oncolytic herpes simplex virus elicit immune mediated antigliomal effect and antitumor memory. Transl. Oncol. 2018, 11, 86–93. [Google Scholar] [CrossRef]
- Kurokawa, C.; Iankov, I.D.; Anderson, S.K.; Aderca, I.; Leontovich, A.A.; Maurer, M.J.; Oberg, A.L.; Schroeder, M.A.; Giannini, C.; Greiner, S.M.; et al. Constitutive Interferon Pathway Activation in Tumors as an Efficacy Determinant Following Oncolytic Virotherapy. J. Natl. Cancer Inst. 2018, 110, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F. Molecular mechanisms of IFN-gamma to up-regulate MHC class I antigen processing and presentation. Int. Rev. Immunol. 2009, 28, 239–260. [Google Scholar] [CrossRef] [PubMed]
- Squire, R.; Fowler, C.L.; Brooks, S.P.; Rich, G.A.; Cooney, D.R. The relationship of class I MHC antigen expression to stage IV-S disease and survival in neuroblastoma. J. Pediatr. Surg. 1990, 25, 381–386. [Google Scholar] [CrossRef]
- Raffaghello, L.; Prigione, I.; Bocca, P.; Morandi, F.; Camoriano, M.; Gambini, C.; Wang, X.; Ferrone, S.; Pistoia, V. Multiple defects of the antigen-processing machinery components in human neuroblastoma: Immunotherapeutic implications. Oncogene 2005, 24, 4634–4644. [Google Scholar] [CrossRef] [PubMed]
- Ghonime, M.G.; Cassady, K.A. Combination Therapy Using Ruxolitinib and Oncolytic HSV Renders Resistant MPNSTs Susceptible to Virotherapy. Cancer Immunol. Res. 2018, 6, 1499–1510. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Espitia, C.M.; Persky, D.O.; Carew, J.S.; Nawrocki, S.T. Targeting JAK/STAT Signaling Antagonizes Resistance to Oncolytic Reovirus Therapy Driven by Prior Infection with HTLV-1 in Models of T-Cell Lymphoma. Viruses 2021, 13, 1406. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.R.; Dash, A.; Jacobson, B.A.; Ji, Y.; Baumann, D.; Ismail, K.; Kratzke, R.A. JAK/STAT inhibition with ruxolitinib enhances oncolytic virotherapy in non-small cell lung cancer models. Cancer Gene Ther. 2019, 26, 411–418. [Google Scholar] [CrossRef]
- Tang, M.; Xie, Q.; Gimple, R.C.; Zhong, Z.; Tam, T.; Tian, J.; Kidwell, R.L.; Wu, Q.; Prager, B.C.; Qiu, Z.; et al. Three-dimensional bioprinted glioblastoma microenvironments model cellular dependencies and immune interactions. Cell Res. 2020, 30, 833–853. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quinn, C.H.; Beierle, A.M.; Hutchins, S.C.; Marayati, R.; Bownes, L.V.; Stewart, J.E.; Markert, H.R.; Erwin, M.H.; Aye, J.M.; Yoon, K.J.; et al. Targeting High-Risk Neuroblastoma Patient-Derived Xenografts with Oncolytic Virotherapy. Cancers 2022, 14, 762. https://doi.org/10.3390/cancers14030762
Quinn CH, Beierle AM, Hutchins SC, Marayati R, Bownes LV, Stewart JE, Markert HR, Erwin MH, Aye JM, Yoon KJ, et al. Targeting High-Risk Neuroblastoma Patient-Derived Xenografts with Oncolytic Virotherapy. Cancers. 2022; 14(3):762. https://doi.org/10.3390/cancers14030762
Chicago/Turabian StyleQuinn, Colin H., Andee M. Beierle, Sara Claire Hutchins, Raoud Marayati, Laura V. Bownes, Jerry E. Stewart, Hooper R. Markert, Michael H. Erwin, Jamie M. Aye, Karina J. Yoon, and et al. 2022. "Targeting High-Risk Neuroblastoma Patient-Derived Xenografts with Oncolytic Virotherapy" Cancers 14, no. 3: 762. https://doi.org/10.3390/cancers14030762
APA StyleQuinn, C. H., Beierle, A. M., Hutchins, S. C., Marayati, R., Bownes, L. V., Stewart, J. E., Markert, H. R., Erwin, M. H., Aye, J. M., Yoon, K. J., Friedman, G. K., Willey, C. D., Markert, J. M., & Beierle, E. A. (2022). Targeting High-Risk Neuroblastoma Patient-Derived Xenografts with Oncolytic Virotherapy. Cancers, 14(3), 762. https://doi.org/10.3390/cancers14030762