
The Role of Macroautophagy and Chaperone-Mediated Autophagy in the Pathogenesis and Management of Hepatocellular Carcinoma

 ,
,  and
and {kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
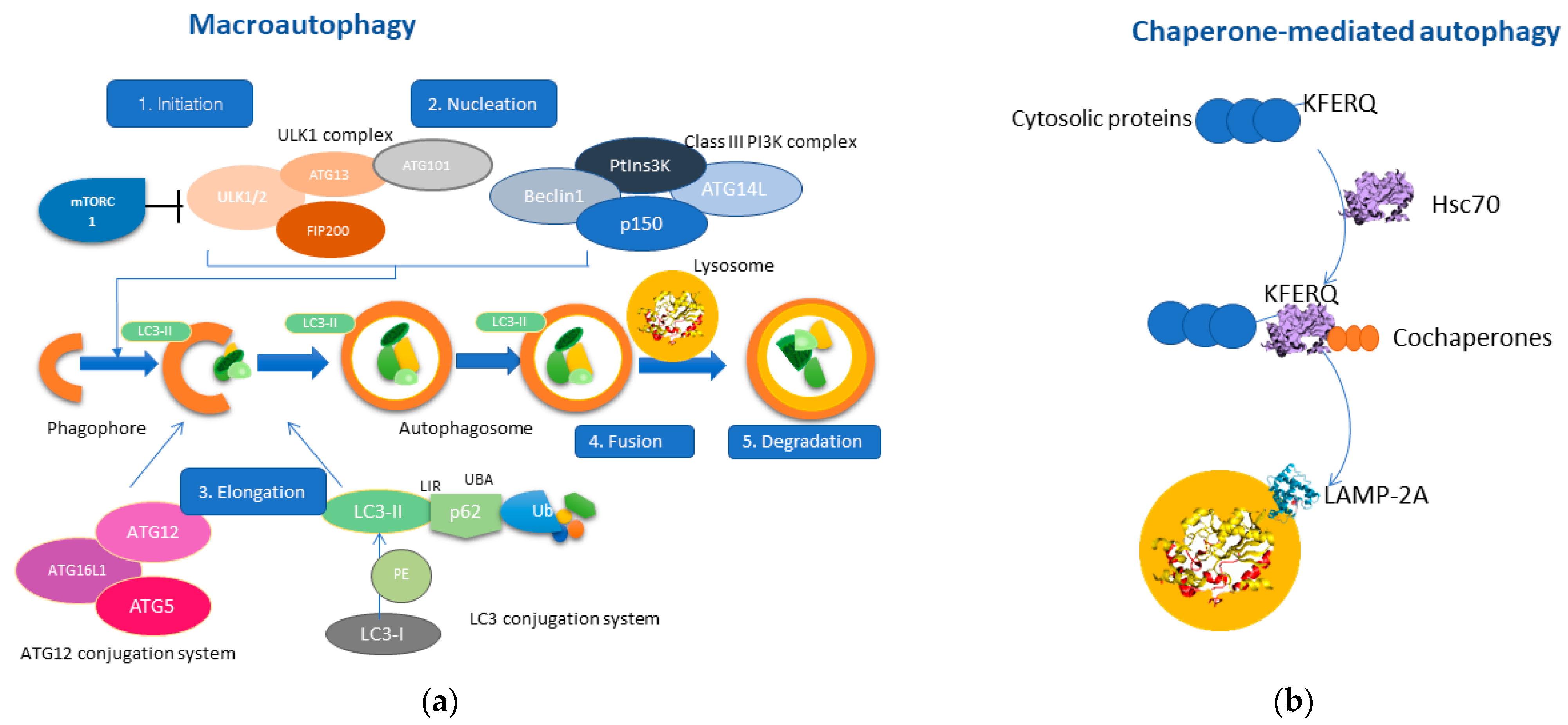
1. Overview of Autophagy
2. Autophagy Pathways Involved in Liver Pathophysiology
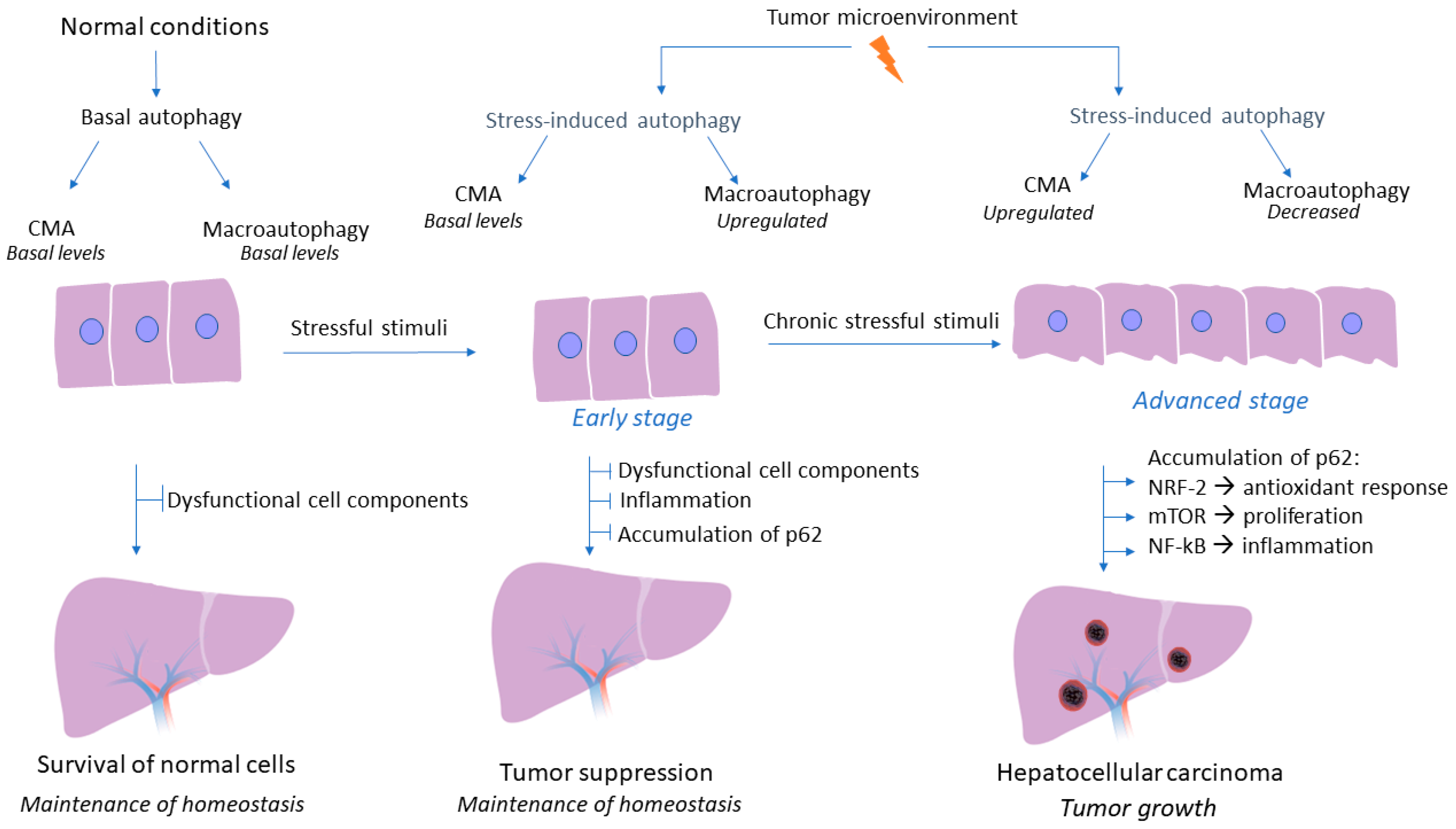
3. Role of Macroautophagy and CMA in HCC Development
3.1. Upregulation of Macroautophagy Acts as a Tumor-Suppressing Mechanism in Early Stages of HCC
3.2. Upregulation of the CMA Pathway Promotes Tumor Cell Survival in Later Stages of HCC
4. Autophagy and HCC Metastasis-Still a Matter of Discussion
5. Autophagy Mechanism Involved in HCC Therapy Resistance
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mijaljica, D.; Prescott, M.; Devenish, R.J. The intriguing life of autophagosomes. Int. J. Mol. Sci. 2012, 13, 3618–3635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox. Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.X.; Ni, H.M.; Gao, W.; Yoshimori, T.; Stolz, D.B.; Ron, D.; Yin, X.M. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am. J. Pathol. 2007, 171, 513–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Cuervo, A.M. Autophagy in the cellular energetic balance. Cell. Metab. 2011, 13, 495–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell. Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Tekirdag, K.; Cuervo, A.M. Chaperone-mediated autophagy and endosomal microautophagy: Joint by a chaperone. J. Biol. Chem. 2018, 293, 5414–5424. [Google Scholar] [CrossRef] [Green Version]
- Rautou, P.E.; Mansouri, A.; Lebrec, D.; Durand, F.; Valla, D.; Moreau, R. Autophagy in liver diseases. J. Hepatol. 2010, 53, 1123–1134. [Google Scholar] [CrossRef] [Green Version]
- Schneider, J.L.; Cuervo, A.M. Liver autophagy: Much more than just taking out the trash. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 187–200. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N. Physiological functions of autophagy. Curr. Top. Microbiol. Immunol. 2009, 335, 71–84. [Google Scholar] [CrossRef]
- Wang, L.; Ye, X.; Zhao, T. The physiological roles of autophagy in the mammalian life cycle. Biol. Rev. Camb. Philos. Soc. 2019, 94, 503–516. [Google Scholar] [CrossRef] [Green Version]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [Green Version]
- Malhi, H.; Gores, G.J. Cellular and molecular mechanisms of liver injury. Gastroenterology 2008, 134, 1641–1654. [Google Scholar] [CrossRef] [Green Version]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Khambu, B.; Yan, S.; Huda, N.; Liu, G.; Yin, X.M. Homeostatic Role of Autophagy in Hepatocytes. Semin. Liver. Dis. 2018, 38, 308–319. [Google Scholar] [CrossRef] [Green Version]
- Hansen, T.E.; Johansen, T. Following autophagy step by step. BMC Biol. 2011, 9, 39. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Invest. 2015, 125, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Ganley, I.G.; Lam, d.H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell. Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Weidberg, H.; Shvets, E.; Elazar, Z. Biogenesis and cargo selectivity of autophagosomes. Annu. Rev. Biochem. 2011, 80, 125–156. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Eskelinen, E.L. To be or not to be? Examples of incorrect identification of autophagic compartments in conventional transmission electron microscopy of mammalian cells. Autophagy 2008, 4, 257–260. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. An overview of the molecular mechanism of autophagy. Curr. Top. Microbiol. Immunol. 2009, 335, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Yorimitsu, T.; Klionsky, D.J. Autophagy: Molecular machinery for self-eating. Cell Death Differ. 2005, 12 Suppl. 2, 1542–1552. [Google Scholar] [CrossRef] [Green Version]
- Bao, L.; Chandra, P.K.; Moroz, K.; Zhang, X.; Thung, S.N.; Wu, T.; Dash, S. Impaired autophagy response in human hepatocellular carcinoma. Exp. Mol. Pathol. 2014, 96, 149–154. [Google Scholar] [CrossRef] [Green Version]
- Chava, S.; Lee, C.; Aydin, Y.; Chandra, P.K.; Dash, A.; Chedid, M.; Thung, S.N.; Moroz, K.; Wu, T.; Nayak, N.C.; et al. Chaperone-mediated autophagy compensates for impaired macroautophagy in the cirrhotic liver to promote hepatocellular carcinoma. Oncotarget 2017, 8, 40019–40036. [Google Scholar] [CrossRef] [Green Version]
- Kon, M.; Kiffin, R.; Koga, H.; Chapochnick, J.; Macian, F.; Varticovski, L.; Cuervo, A.M. Chaperone-mediated autophagy is required for tumor growth. Sci. Transl. Med. 2011, 3, 109ra117. [Google Scholar] [CrossRef] [Green Version]
- Ding, Z.B.; Fu, X.T.; Shi, Y.H.; Zhou, J.; Peng, Y.F.; Liu, W.R.; Shi, G.M.; Gao, Q.; Wang, X.Y.; Song, K.; et al. Lamp2a is required for tumor growth and promotes tumor recurrence of hepatocellular carcinoma. Int. J. Oncol. 2016, 49, 2367–2376. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell. Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Kirchner, P.; Bourdenx, M.; Madrigal-Matute, J.; Tiano, S.; Diaz, A.; Bartholdy, B.A.; Will, B.; Cuervo, A.M. Proteome-wide analysis of chaperone-mediated autophagy targeting motifs. PLoS Biol. 2019, 17, e3000301. [Google Scholar] [CrossRef]
- Agarraberes, F.A.; Dice, J.F. A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J. Cell. Sci. 2001, 114, 2491–2499. [Google Scholar] [CrossRef]
- Juste, Y.R.; Cuervo, A.M. Analysis of Chaperone-Mediated Autophagy. Methods Mol. Biol. 2019, 1880, 703–727. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell. Res. 2014, 24, 92–104. [Google Scholar] [CrossRef] [Green Version]
- Barnard, R.A.; Regan, D.P.; Hansen, R.J.; Maycotte, P.; Thorburn, A.; Gustafson, D.L. Autophagy Inhibition Delays Early but Not Late-Stage Metastatic Disease. J. Pharmacol. Exp. Ther. 2016, 358, 282–293. [Google Scholar] [CrossRef] [Green Version]
- Ichimura, Y.; Komatsu, M. Pathophysiological role of autophagy: Lesson from autophagy-deficient mouse models. Exp. Anim. 2011, 60, 329–345. [Google Scholar] [CrossRef] [Green Version]
- Gewirtz, D.A. When cytoprotective autophagy isn’t and even when it is. Autophagy 2014, 10, 391–392. [Google Scholar] [CrossRef] [Green Version]
- Alfaro, I.E.; Albornoz, A.; Molina, A.; Moreno, J.; Cordero, K.; Criollo, A.; Budini, M. Chaperone Mediated Autophagy in the Crosstalk of Neurodegenerative Diseases and Metabolic Disorders. Front. Endocrinol. 2018, 9, 778. [Google Scholar] [CrossRef]
- Aydin, Y.; Stephens, C.M.; Chava, S.; Heidari, Z.; Panigrahi, R.; Williams, D.D.; Wiltz, K.; Bell, A.; Wilson, W.; Reiss, K.; et al. Chaperone-Mediated Autophagy Promotes Beclin1 Degradation in Persistently Infected Hepatitis C Virus Cell Culture. Am. J. Pathol. 2018, 188, 2339–2355. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.A.; Ding, W.X. Role of autophagy in alcohol and drug-induced liver injury. Food Chem. Toxicol. 2020, 136, 111075. [Google Scholar] [CrossRef]
- Chao, X.; Ding, W.X. Role and mechanisms of autophagy in alcohol-induced liver injury. Adv. Pharmacol. 2019, 85, 109–131. [Google Scholar] [CrossRef]
- Sun, K.; Guo, X.L.; Zhao, Q.D.; Jing, Y.Y.; Kou, X.R.; Xie, X.Q.; Zhou, Y.; Cai, N.; Gao, L.; Zhao, X.; et al. Paradoxical role of autophagy in the dysplastic and tumor-forming stages of hepatocarcinoma development in rats. Cell Death Dis. 2013, 4, e501. [Google Scholar] [CrossRef] [Green Version]
- Ding, Z.B.; Shi, Y.H.; Zhou, J.; Qiu, S.J.; Xu, Y.; Dai, Z.; Shi, G.M.; Wang, X.Y.; Ke, A.W.; Wu, B.; et al. Association of autophagy defect with a malignant phenotype and poor prognosis of hepatocellular carcinoma. Cancer Res. 2008, 68, 9167–9175. [Google Scholar] [CrossRef] [Green Version]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Invest. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [Green Version]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Yu, J.; Wen, Z.; Wang, M.; Chen, W. Decreased expression of Beclin-1 in patients with hepatocellular carcinoma. J. BUON 2019, 24, 634–641. [Google Scholar]
- Qiu, D.M.; Wang, G.L.; Chen, L.; Xu, Y.Y.; He, S.; Cao, X.L.; Qin, J.; Zhou, J.M.; Zhang, Y.X.; Qun, E. The expression of beclin-1, an autophagic gene, in hepatocellular carcinoma associated with clinical pathological and prognostic significance. BMC Cancer 2014, 14, 327. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Li, W.; Ge, H.; Zhang, K.; Li, G.; Wu, J. Role of Beclin1 expression in patients with hepatocellular carcinoma: A meta-analysis. Onco. Targets Ther. 2018, 11, 2387–2397. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell. Biol. 2005, 169, 425–434. [Google Scholar] [CrossRef]
- An, C.H.; Kim, M.S.; Yoo, N.J.; Park, S.W.; Lee, S.H. Mutational and expressional analyses of ATG5, an autophagy-related gene, in gastrointestinal cancers. Pathol. Res. Pract. 2011, 207, 433–437. [Google Scholar] [CrossRef]
- Chao, X.; Qian, H.; Wang, S.; Fulte, S.; Ding, W.X. Autophagy and liver cancer. Clin. Mol. Hepatol. 2020, 26, 606–617. [Google Scholar] [CrossRef]
- Chen, N.; Debnath, J. Autophagy and tumorigenesis. FEBS Lett. 2010, 584, 1427–1435. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, M.A.; Bowman, J.W.; Liang, Q.; Jung, J.U. Regulation where autophagy intersects the inflammasome. Antioxid Redox Signal. 2014, 20, 495–506. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Liu, Y.; Varley, P.; Chang, Y.; He, X.X.; Huang, H.; Tang, D.; Lotze, M.T.; Lin, J.; Tsung, A. High-Mobility Group Box 1 Promotes Hepatocellular Carcinoma Progression through miR-21-Mediated Matrix Metalloproteinase Activity. Cancer Res. 2015, 75, 1645–1656. [Google Scholar] [CrossRef] [Green Version]
- Elliott, E.I.; Sutterwala, F.S. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol. Rev. 2015, 265, 35–52. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Wang, Y.; Jing, L.; Claret, F.X.; Li, Q.; Tian, T.; Liang, X.; Ruan, Z.; Jiang, L.; Yao, Y.; et al. Autophagy in the “inflammation-carcinogenesis” pathway of liver and HCC immunotherapy. Cancer Lett. 2017, 411, 82–89. [Google Scholar] [CrossRef]
- White, E.; Karp, C.; Strohecker, A.M.; Guo, Y.; Mathew, R. Role of autophagy in suppression of inflammation and cancer. Curr. Opin. Cell. Biol. 2010, 22, 212–217. [Google Scholar] [CrossRef] [Green Version]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Yang, L.; Xu, S.; Zhang, Z.; Cao, Y. The receptor proteins: Pivotal roles in selective autophagy. Acta. Biochim. Biophys. Sin. 2015, 47, 571–580. [Google Scholar] [CrossRef] [Green Version]
- Akkoç, Y.; Gözüaçık, D. Autophagy and liver cancer. Turk. J. Gastroenterol. 2018, 29, 270–282. [Google Scholar] [CrossRef]
- Umemura, A.; He, F.; Taniguchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandan, S.; Duran, A.; et al. p62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell. 2016, 29, 935–948. [Google Scholar] [CrossRef] [Green Version]
- Denk, H.; Stumptner, C.; Abuja, P.M.; Zatloukal, K. Sequestosome 1/p62-related pathways as therapeutic targets in hepatocellular carcinoma. Expert. Opin. Ther. Targets 2019, 23, 393–406. [Google Scholar] [CrossRef]
- Jiang, T.; Harder, B.; Rojo de la Vega, M.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 links autophagy and Nrf2 signaling. Free Radic. Biol. Med. 2015, 88, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, K.; Dan, K.; Goto, F.; Tshuchihashi, N.; Nomura, Y.; Fujioka, M.; Kanzaki, S.; Ogawa, K. The autophagy pathway maintained signaling crosstalk with the Keap1-Nrf2 system through p62 in auditory cells under oxidative stress. Cell Signal. 2015, 27, 382–393. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Hennig, P.; Fenini, G.; Di Filippo, M.; Karakaya, T.; Beer, H.D. The Pathways Underlying the Multiple Roles of p62 in Inflammation and Cancer. Biomedicines 2021, 9, 707. [Google Scholar] [CrossRef]
- Arias, E. Methods to Study Chaperone-Mediated Autophagy. Methods Enzymol. 2017, 588, 283–305. [Google Scholar] [CrossRef]
- Massey, A.; Kiffin, R.; Cuervo, A.M. Pathophysiology of chaperone-mediated autophagy. Int. J. Biochem. Cell. Biol. 2004, 36, 2420–2434. [Google Scholar] [CrossRef]
- Dash, S.; Aydin, Y.; Moroz, K. Chaperone-Mediated Autophagy in the Liver: Good or Bad? Cells 2019, 8, 1308. [Google Scholar] [CrossRef] [Green Version]
- Lum, J.J.; Bauer, D.E.; Kong, M.; Harris, M.H.; Li, C.; Lindsten, T.; Thompson, C.B. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 2005, 120, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [Green Version]
- Eskelinen, E.L. The dual role of autophagy in cancer. Curr. Opin. Pharmacol. 2011, 11, 294–300. [Google Scholar] [CrossRef]
- Chen, S.; Rehman, S.K.; Zhang, W.; Wen, A.; Yao, L.; Zhang, J. Autophagy is a therapeutic target in anticancer drug resistance. Biochim. Biophys. Acta. 2010, 1806, 220–229. [Google Scholar] [CrossRef]
- Lee, Y.J.; Jang, B.K. The Role of Autophagy in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2015, 16, 26629–26643. [Google Scholar] [CrossRef] [Green Version]
- Dice, J.F. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem. Sci. 1990, 15, 305–309. [Google Scholar] [CrossRef]
- Zhou, Y.; Sun, K.; Ma, Y.; Yang, H.; Zhang, Y.; Kong, X.; Wei, L. Autophagy inhibits chemotherapy-induced apoptosis through downregulating Bad and Bim in hepatocellular carcinoma cells. Sci. Rep. 2014, 4, 5382. [Google Scholar] [CrossRef] [Green Version]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [Green Version]
- Yazdani, H.O.; Huang, H.; Tsung, A. Autophagy: Dual Response in the Development of Hepatocellular Carcinoma. Cells 2019, 8, 91. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Sun, Y.; Fei, M.; Tan, C.; Wu, J.; Zheng, J.; Tang, J.; Sun, W.; Lv, Z.; Bao, J.; et al. Disruption of chaperone-mediated autophagy-dependent degradation of MEF2A by oxidative stress-induced lysosome destabilization. Autophagy 2014, 10, 1015–1035. [Google Scholar] [CrossRef] [Green Version]
- Menegon, S.; Columbano, A.; Giordano, S. The Dual Roles of NRF2 in Cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Inpanathan, S.; Botelho, R.J. The Lysosome Signaling Platform: Adapting With the Times. Front. Cell Dev. Biol. 2019, 7, 113. [Google Scholar] [CrossRef] [Green Version]
- Maishman, T.; Cutress, R.I.; Hernandez, A.; Gerty, S.; Copson, E.R.; Durcan, L.; Eccles, D.M. Local Recurrence and Breast Oncological Surgery in Young Women With Breast Cancer: The POSH Observational Cohort Study. Ann. Surg. 2017, 266, 165–172. [Google Scholar] [CrossRef] [Green Version]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef]
- Kenific, C.M.; Thorburn, A.; Debnath, J. Autophagy and metastasis: Another double-edged sword. Curr. Opin. Cell. Biol. 2010, 22, 241–245. [Google Scholar] [CrossRef] [Green Version]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef]
- Peng, Y.F.; Shi, Y.H.; Ding, Z.B.; Ke, A.W.; Gu, C.Y.; Hui, B.; Zhou, J.; Qiu, S.J.; Dai, Z.; Fan, J. Autophagy inhibition suppresses pulmonary metastasis of HCC in mice via impairing anoikis resistance and colonization of HCC cells. Autophagy 2013, 9, 2056–2068. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Luo, R.Z.; Lu, Y.; Zhang, X.; Yu, Q.; Khare, S.; Kondo, S.; Kondo, Y.; Yu, Y.; Mills, G.B.; et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J. Clin. Invest. 2008, 118, 3917–3929. [Google Scholar] [CrossRef] [Green Version]
- Fung, C.; Lock, R.; Gao, S.; Salas, E.; Debnath, J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol. Biol. Cell 2008, 19, 797–806. [Google Scholar] [CrossRef] [Green Version]
- Dower, C.M.; Wills, C.A.; Frisch, S.M.; Wang, H.G. Mechanisms and context underlying the role of autophagy in cancer metastasis. Autophagy 2018, 14, 1110–1128. [Google Scholar] [CrossRef] [Green Version]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Yang, B.; Zhou, Q.; Wu, Y.; Shang, D.; Guo, Y.; Song, Z.; Zheng, Q.; Xiong, J. Autophagy promotes hepatocellular carcinoma cell invasion through activation of epithelial-mesenchymal transition. Carcinogenesis 2013, 34, 1343–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.F.; Shi, Y.H.; Shen, Y.H.; Ding, Z.B.; Ke, A.W.; Zhou, J.; Qiu, S.J.; Fan, J. Promoting colonization in metastatic HCC cells by modulation of autophagy. PLoS ONE 2013, 8, e74407. [Google Scholar] [CrossRef]
- Heimbach, J.K.; Kulik, L.M.; Finn, R.S.; Sirlin, C.B.; Abecassis, M.M.; Roberts, L.R.; Zhu, A.X.; Murad, M.H.; Marrero, J.A. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology 2018, 67, 358–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.W.; Talati, C.; Kim, R. Hepatocellular carcinoma (HCC): Beyond sorafenib-chemotherapy. J. Gastrointest. Oncol. 2017, 8, 256–265. [Google Scholar] [CrossRef] [Green Version]
- Glantzounis, G.K.; Karampa, A.; Peristeri, D.V.; Pappas-Gogos, G.; Tepelenis, K.; Tzimas, P.; Cyrochristos, D.J. Recent advances in the surgical management of hepatocellular carcinoma. Ann. Gastroenterol. 2021, 34, 453–465. [Google Scholar] [CrossRef]
- Glantzounis, G.K.; Paliouras, A.; Stylianidi, M.C.; Milionis, H.; Tzimas, P.; Roukos, D.; Pentheroudakis, G.; Felekouras, E. The role of liver resection in the management of intermediate and advanced stage hepatocellular carcinoma. A systematic review. Eur. J. Surg. Oncol. 2018, 44, 195–208. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Yang, S.; Yang, L.; Li, X.; Li, B.; Li, Y.; Zhang, X.; Ma, Y.; Peng, X.; Jin, H.; Li, H. New insights into autophagy in hepatocellular carcinoma: Mechanisms and therapeutic strategies. Am. J. Cancer Res. 2019, 9, 1329–1353. [Google Scholar]
- Liu, T.; Zhang, J.; Li, K.; Deng, L.; Wang, H. Combination of an Autophagy Inducer and an Autophagy Inhibitor: A Smarter Strategy Emerging in Cancer Therapy. Front. Pharmacol 2020, 11, 408. [Google Scholar] [CrossRef] [Green Version]
- Sheng, J.; Qin, H.; Zhang, K.; Li, B.; Zhang, X. Targeting autophagy in chemotherapy-resistant of hepatocellular carcinoma. Am. J. Cancer Res. 2018, 8, 354–365. [Google Scholar] [PubMed]
- Li, Y.; McGreal, S.; Zhao, J.; Huang, R.; Zhou, Y.; Zhong, H.; Xia, M.; Ding, W.X. A cell-based quantitative high-throughput image screening identified novel autophagy modulators. Pharmacol Res. 2016, 110, 35–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pursiheimo, J.P.; Rantanen, K.; Heikkinen, P.T.; Johansen, T.; Jaakkola, P.M. Hypoxia-activated autophagy accelerates degradation of SQSTM1/p62. Oncogene 2009, 28, 334–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Gao, Y.; Wang, D.; Xu, Z.; Sun, W.; Ren, P. Autophagy Inhibitor (LY294002) and 5-fluorouracil (5-FU) Combination-Based Nanoliposome for Enhanced Efficacy Against Esophageal Squamous Cell Carcinoma. Nanoscale. Res. Lett. 2018, 13, 325. [Google Scholar] [CrossRef] [Green Version]
- Wang, J. Beclin 1 bridges autophagy, apoptosis and differentiation. Autophagy 2008, 4, 947–948. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Li, M.; Wang, Y.; Chen, Y.; He, C.; Zhang, X.; Yang, T.; Lu, Y.; You, J.; Lee, R.J.; et al. Enhancing anti-tumor efficiency in hepatocellular carcinoma through the autophagy inhibition by miR-375/sorafenib in lipid-coated calcium carbonate nanoparticles. Acta. Biomater. 2018, 72, 248–255. [Google Scholar] [CrossRef]
- Brun, S.; Bestion, E.; Raymond, E.; Bassissi, F.; Jilkova, Z.M.; Mezouar, S.; Rachid, M.; Novello, M.; Tracz, J.; Hamaï, A.; et al. GNS561, a clinical-stage PPT1 inhibitor, is efficient against hepatocellular carcinoma. Autophagy 2021, 1–17. [Google Scholar] [CrossRef]
- Allaire, M.; Rautou, P.E.; Codogno, P.; Lotersztajn, S. Autophagy in liver diseases: Time for translation? J. Hepatol. 2019, 70, 985–998. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Sedano, S.; Allen, R.; Gong, J.; Cho, M.; Sharma, S. Current Treatment Landscape for Advanced Hepatocellular Carcinoma: Patient Outcomes and the Impact on Quality of Life. Cancers 2019, 11, 841. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, X.; Wu, H.; Ni, P.; Gu, Z.; Qiao, Y.; Chen, N.; Sun, F.; Fan, Q. CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucleic Acids. Res. 2010, 38, 5366–5383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, T. Long noncoding RNAs act as regulators of autophagy in cancer. Pharmacol. Res. 2018, 129, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Xiang, X.; Feng, B.; Zhou, H.; Wang, T.; Chu, X.; Wang, R. Targeting Long Non-Coding RNAs in Hepatocellular Carcinoma: Progress and Prospects. Front. Oncol. 2021, 11, 670838. [Google Scholar] [CrossRef] [PubMed]
- Ghafouri-Fard, S.; Gholipour, M.; Hussen, B.M.; Taheri, M. The Impact of Long Non-Coding RNAs in the Pathogenesis of Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 1150. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karampa, A.D.; Goussia, A.C.; Glantzounis, G.K.; Mastoridou, E.M.; Anastasopoulos, N.-A.T.; Charchanti, A.V. The Role of Macroautophagy and Chaperone-Mediated Autophagy in the Pathogenesis and Management of Hepatocellular Carcinoma. Cancers 2022, 14, 760. https://doi.org/10.3390/cancers14030760
Karampa AD, Goussia AC, Glantzounis GK, Mastoridou EM, Anastasopoulos N-AT, Charchanti AV. The Role of Macroautophagy and Chaperone-Mediated Autophagy in the Pathogenesis and Management of Hepatocellular Carcinoma. Cancers. 2022; 14(3):760. https://doi.org/10.3390/cancers14030760
Chicago/Turabian StyleKarampa, Anastasia D., Anna C. Goussia, Georgios K. Glantzounis, Eleftheria M. Mastoridou, Nikolaos-Andreas T. Anastasopoulos, and Antonia V. Charchanti. 2022. "The Role of Macroautophagy and Chaperone-Mediated Autophagy in the Pathogenesis and Management of Hepatocellular Carcinoma" Cancers 14, no. 3: 760. https://doi.org/10.3390/cancers14030760
APA StyleKarampa, A. D., Goussia, A. C., Glantzounis, G. K., Mastoridou, E. M., Anastasopoulos, N.-A. T., & Charchanti, A. V. (2022). The Role of Macroautophagy and Chaperone-Mediated Autophagy in the Pathogenesis and Management of Hepatocellular Carcinoma. Cancers, 14(3), 760. https://doi.org/10.3390/cancers14030760