
A 40-Year Cohort Study of Evolving Hypothalamic Dysfunction in Infants and Young Children (<3 years) with Optic Pathway Gliomas

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Demographic, Tumour Location, and Clinical Data at Presentation
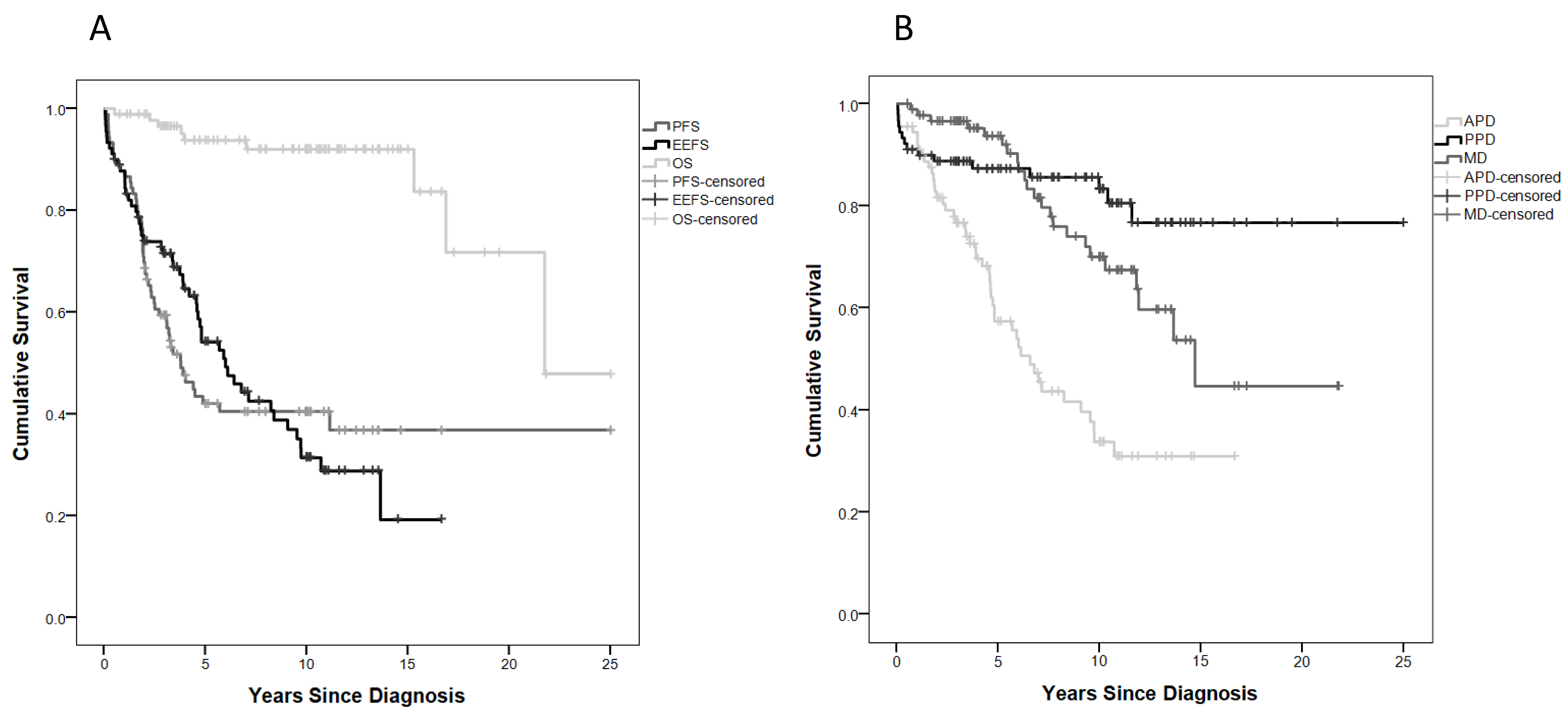
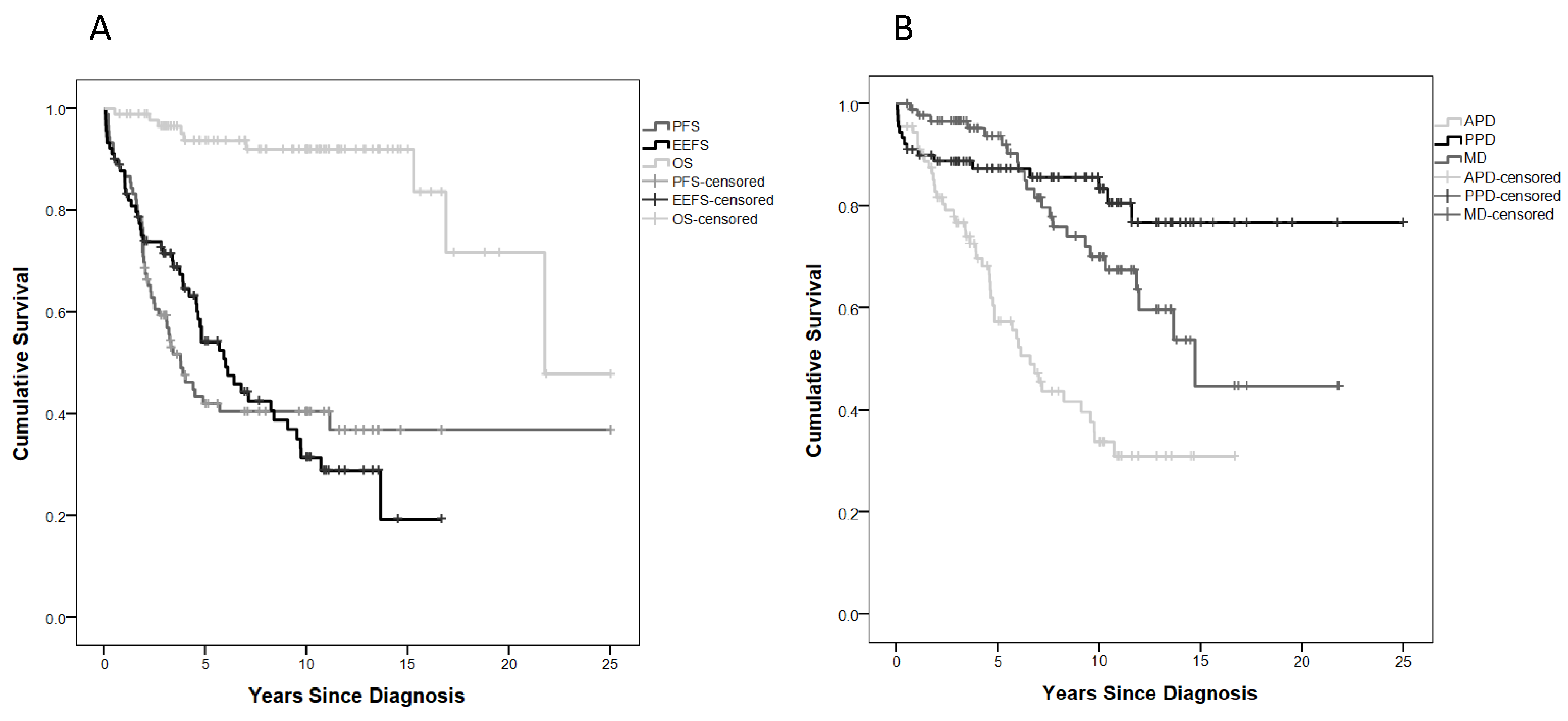
2.2. Treatment Burden and Survival Outcome
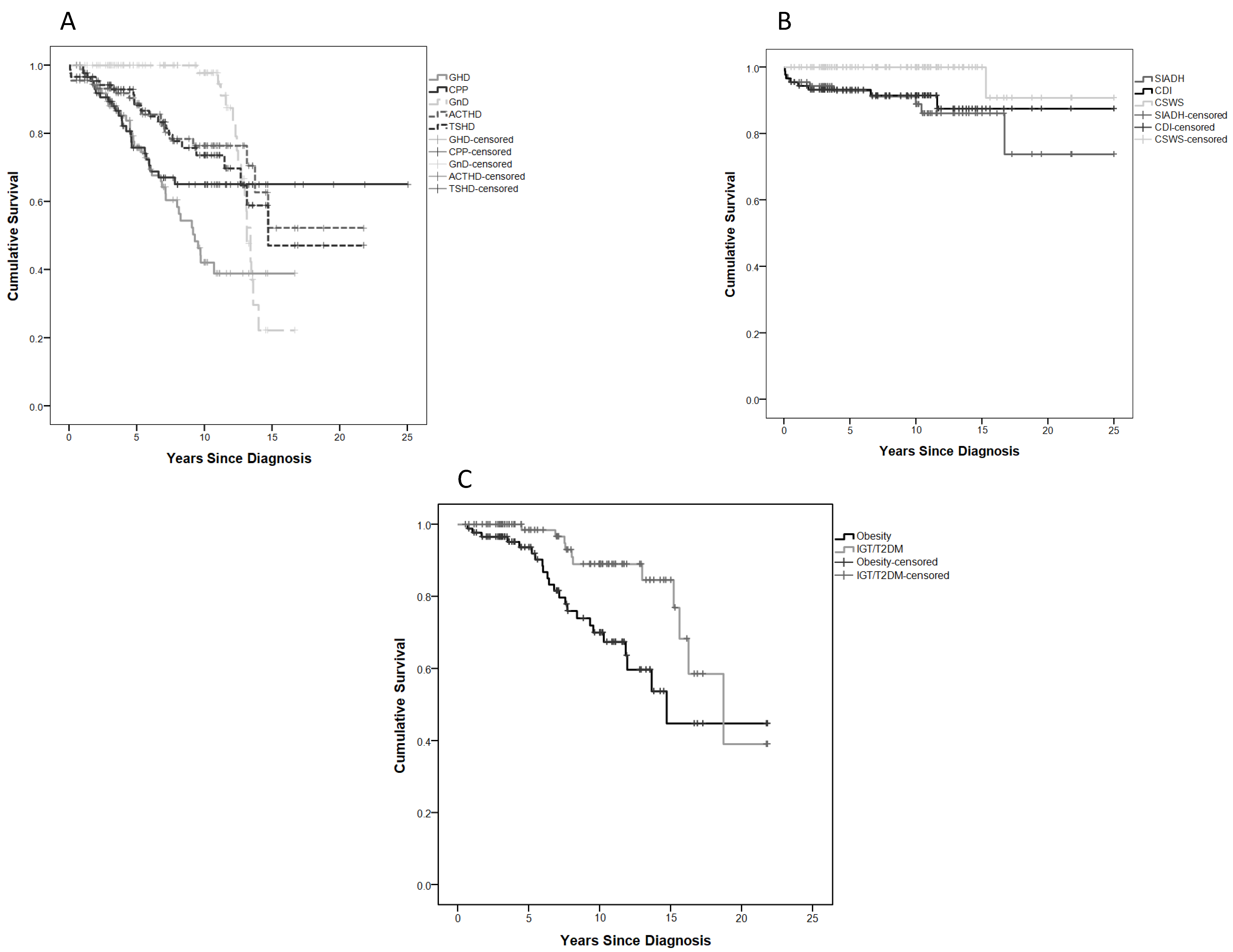
2.3. Endo-Metabolic Dysfunction
2.4. The Effect of Diencephalic Syndrome on Endo-Metabolic Disorders
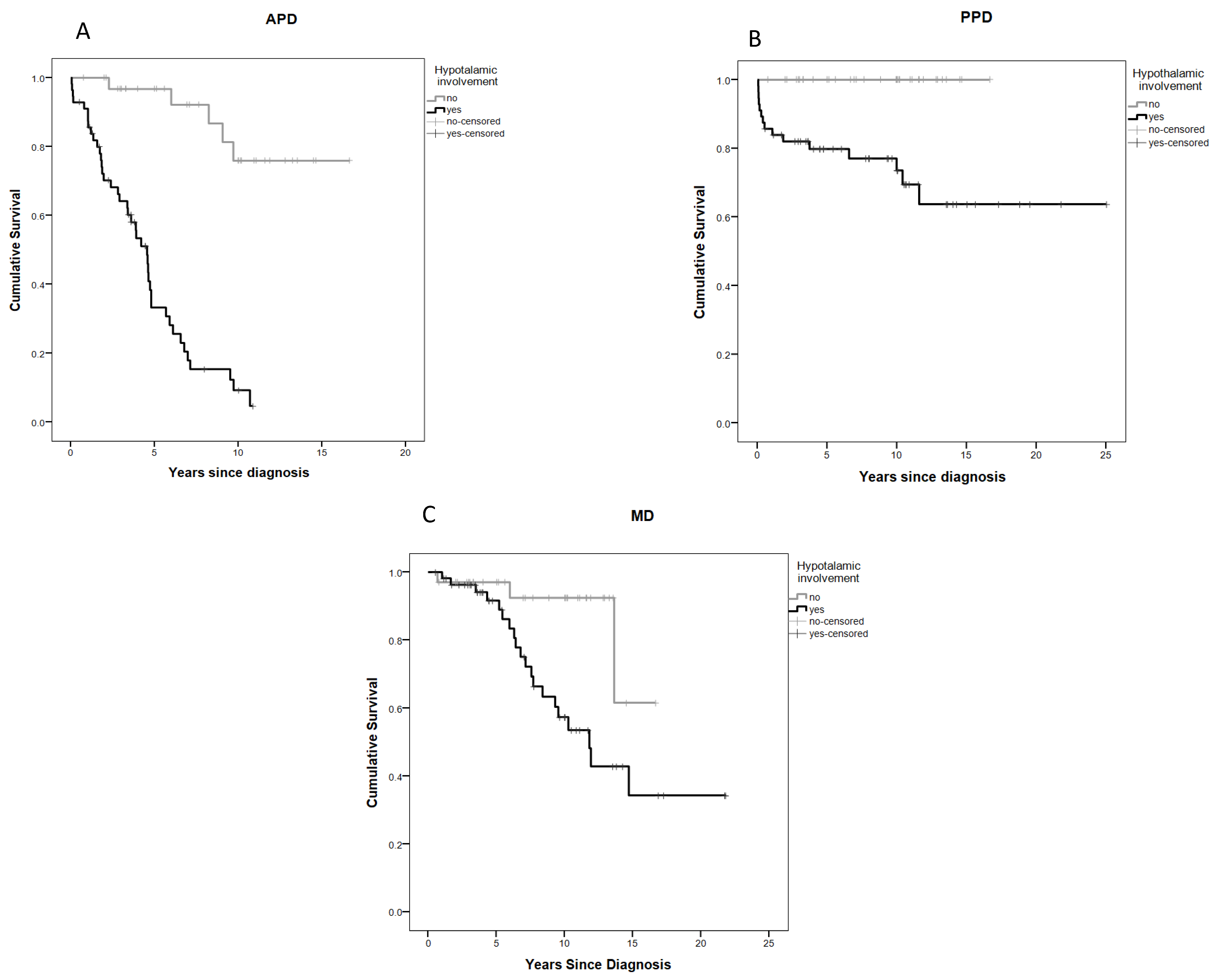
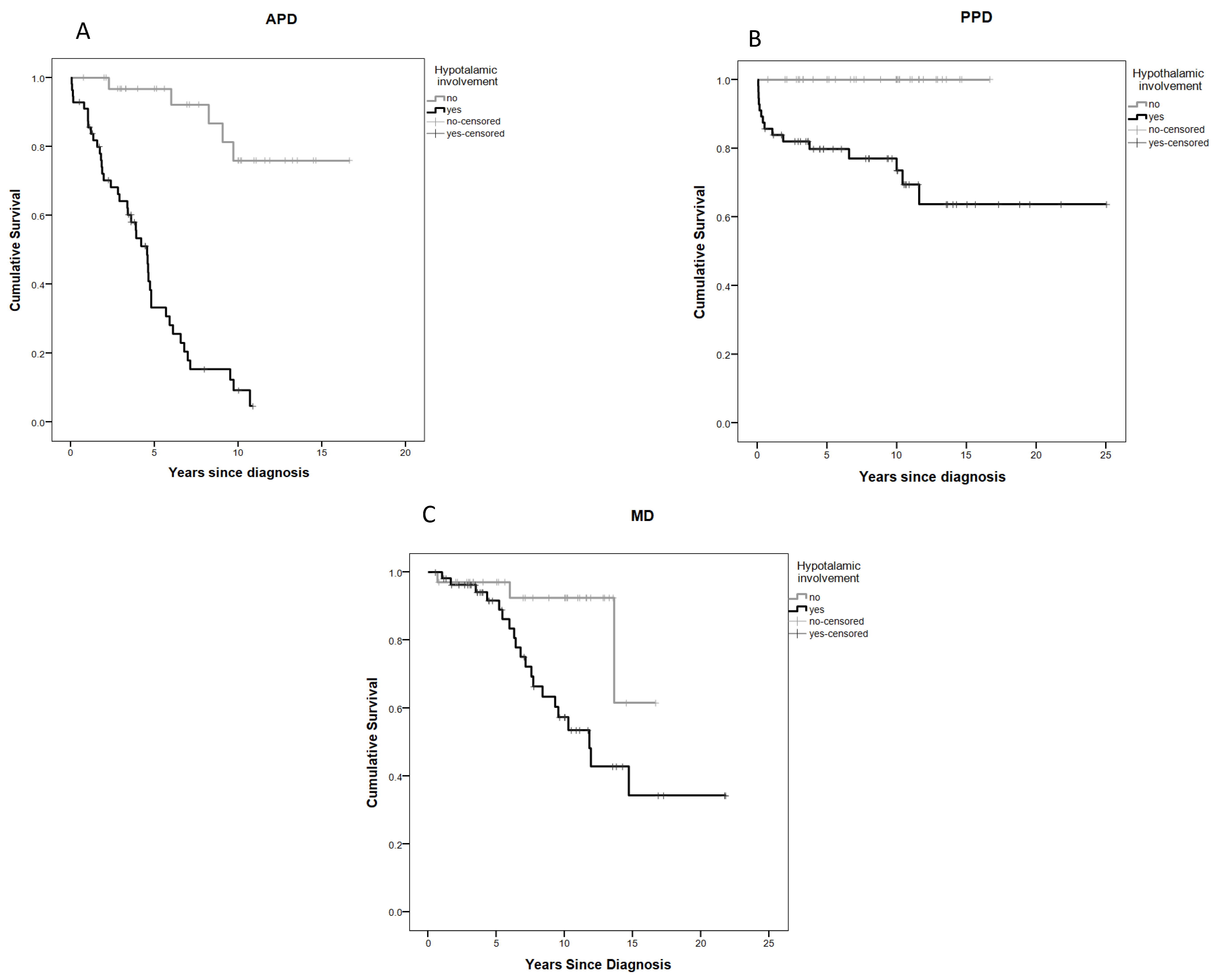
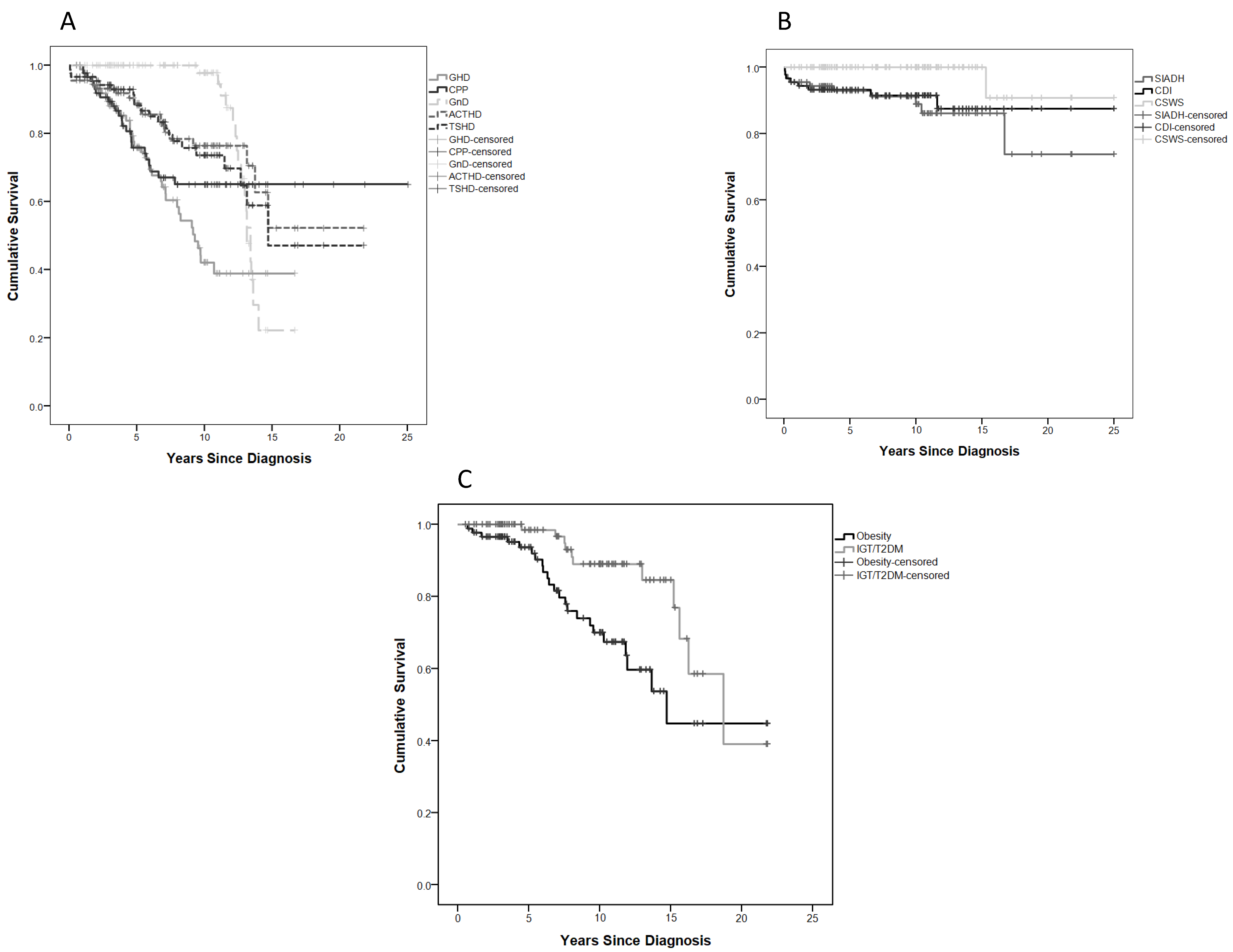
2.5. Anterior Pituitary Disorders (APDs)
2.6. Posterior Pituitary Disorders (PPDs)
2.7. Metabolic Disorders (MDs)
2.8. Neurobehavioural Morbidity
3. Discussion
4. Materials and Methods
Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- CBTRUS. Statistical Report: Primary Brain Tumors in the United States, 2000–2004; Central Brain Tumor Registry of the United States: Hinsdale, IL, USA, 2008. [Google Scholar]
- Pritchard-Jones, K. Childhood cancer in Britain: Incidence, survival and mortality. Br. J. Cancer 2007, 96, 1927. [Google Scholar] [CrossRef] [Green Version]
- De Blank, P.M.K.; Fisher, M.J.; Liu, G.T.; Gutmann, D.H.; Listernick, R.; Ferner, R.E.; Avery, R.A. Optic Pathway Gliomas in Neurofibromatosis Type 1: An Update: Surveillance, Treatment Indications, and Biomarkers of Vision. J. Neuroophthalmol. 2017, 37 (Suppl. 1), S23–S32. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, G.T.; Conklin, H.M.; Huang, S.; Srivastava, D.; Sanford, R.; Ellison, D.W.; Merchant, T.E.; Hudson, M.M.; Hoehn, M.E.; Robison, L.L.; et al. Survival and long-term health and cognitive outcomes after low-grade glioma. Neuro-Oncology 2011, 13, 223–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falzon, K.; Drimtzias, E.; Picton, S.; Simmons, I. Visual outcomes after chemotherapy for optic pathway glioma in children with and without neurofibromatosis type 1: Results of the International Society of Paediatric Oncology (SIOP) Low-Grade Glioma 2004 trial UK cohort. Br. J. Ophthalmol. 2018, 102, 1367–1371. [Google Scholar] [CrossRef] [PubMed]
- Azizi, A.A.; Walker, D.A.; Liu, J.-F.; Sehested, A.; Jaspan, T.; Pemp, B.; Simmons, I.; Ferner, R.; Grill, J.; Hargrave, D.; et al. NF1 optic pathway glioma: Analyzing risk factors for visual outcome and indications to treat. Neuro-Oncology 2021, 23, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Acharya, S.; Quesada, S.; Coca, K.; Richardson, C.; Hoehn, M.E.; Chiang, J.; Qaddoumi, I.; Boop, F.A.; Gajjar, A.; Merchant, T.E. Long-term visual acuity outcomes after radiation therapy for sporadic optic pathway glioma. J. Neurooncol. 2019, 144, 603–610. [Google Scholar] [CrossRef]
- Dodgshun, A.J.; Elder, J.E.; Hansford, J.R.; Sullivan, M.J. Long-term visual outcome after chemotherapy for optic pathway glioma in children: Site and age are strongly predictive. Cancer 2015, 121, 4190–4196. [Google Scholar] [CrossRef] [Green Version]
- Gan, H.W.; Phipps, K.; Aquilina, K.; Gaze, M.N.; Hayward, R.; Spoudeas, H.A. Neuroendocrine Morbidity After Pediatric Optic Gliomas: A Longitudinal Analysis of 166 Children Over 30 Years. J. Clin. Endocrinol. Metab. 2015, 100, 3787–3799. [Google Scholar] [CrossRef]
- Rodriguez, L.A.; Edwards, M.S.; Levin, V.A. Management of hypothalamic gliomas in children: An analysis of 33 cases. Neurosurgery 1990, 26, 242–246. [Google Scholar] [CrossRef]
- Benesch, M.; Lackner, H.; Sovinz, P.; Suppan, E.; Schwinger, W.; Eder, H.G.; Dornbusch, H.J.; Moser, A.; Triebl-Roth, K.; Urban, C. Late sequela after treatment of childhood low-grade gliomas: A retrospective analysis of 69 long-term survivors treated between 1983 and 2003. J. Neurooncol. 2006, 78, 199–205. [Google Scholar] [CrossRef]
- Collet-Solberg, P.F.; Sernyak, H.; Satin-Smith, M.; Katz, L.L.; Sutton, L.; Molloy, P.; Moshang, T., Jr. Endocrine outcome in long-term survivors of low-grade hypothalamic/chiasmatic glioma. Clin. Endocrinol. 1997, 47, 79–85. [Google Scholar] [CrossRef]
- Merchant, T.E.; Conklin, H.M.; Wu, S.; Lustig, R.H.; Xiong, X. Late effects of conformal radiation therapy for pediatric patients with low-grade glioma: Prospective evaluation of cognitive, endocrine, and hearing deficits. J. Clin. Oncol. 2009, 27, 3691–3697. [Google Scholar] [CrossRef] [Green Version]
- Cappelli, C.; Grill, J.; Raquin, M.; Pierre-Kahn, A.; Lellouch-Tubiana, A.; Terrier-Lacombe, M.J.; Habrand, J.L.; Couanet, D.; Brauner, R.; Rodriguez, D.; et al. Long-term follow up of 69 patients treated for optic pathway tumours before the chemotherapy era. Arch. Dis. Child. 1998, 79, 334–338. [Google Scholar] [CrossRef]
- Huguenin, M.; Trivin, C.; Zerah, M.; Doz, F.; Brugieres, L.; Brauner, R. Adult height after cranial irradiation for optic pathway tumors: Relationship with neurofibromatosis. J. Pediatrics 2003, 142, 699–703. [Google Scholar] [CrossRef]
- Ahn, Y.; Cho, B.K.; Kim, S.K.; Chung, Y.N.; Lee, C.S.; Kim, I.H.; Yang, S.W.; Kim, H.S.; Kim, H.J.; Jung, H.W.; et al. Optic pathway glioma: Outcome and prognostic factors in a surgical series. Childs Nerv. Syst. 2006, 22, 1136–1142. [Google Scholar] [CrossRef]
- Silva, M.M.; Goldman, S.; Keating, G.; Marymont, M.A.; Kalapurakal, J.; Tomita, T. Optic pathway hypothalamic gliomas in children under three years of age: The role of chemotherapy. Pediatr. Neurosurg. 2000, 33, 151–158. [Google Scholar] [CrossRef]
- Kilday, J.P.; Bartels, U.; Huang, A.; Barron, M.; Shago, M.; Mistry, M.; Zhukova, N.; Laperriere, N.; Dirks, P.; Hawkins, C.; et al. Favorable survival and metabolic outcome for children with diencephalic syndrome using a radiation-sparing approach. J. Neurooncol. 2014, 116, 195–204. [Google Scholar] [CrossRef]
- Rakotonjanahary, J.; De Carli, E.; Delion, M.; Kalifa, C.; Grill, J.; Doz, F.; Leblond, P.; Bertozzi, A.I.; Rialland, X. Mortality in Children with Optic Pathway Glioma Treated with Up-Front BB-SFOP Chemotherapy. PLoS ONE 2015, 10, e0127676. [Google Scholar]
- Gnekow, A.K.; Kandels, D.; Tilburg, C.V.; Azizi, A.A.; Opocher, E.; Stokland, T.; Driever, P.H.; Schouten-van Meeteren, A.Y.N.; Thomale, U.W.; Schuhmann, M.U.; et al. SIOP-E-BTG and GPOH Guidelines for Diagnosis and Treatment of Children and Adolescents with Low Grade Glioma. Klin. Padiatr. 2019, 231, 107–135. [Google Scholar]
- Mishra, M.V.; Andrews, D.W.; Glass, J.; Evans, J.J.; Dicker, A.P.; Shen, X.; Lawrence, Y.R. Characterization and outcomes of optic nerve gliomas: A population-based analysis. J. Neurooncol. 2012, 107, 591–597. [Google Scholar] [CrossRef]
- Stokland, T.; Liu, J.F.; Ironside, J.W.; Ellison, D.W.; Taylor, R.; Robinson, K.J.; Picton, S.V.; Walker, D.A. A multivariate analysis of factors determining tumor progression in childhood low-grade glioma: A population-based cohort study (CCLG CNS9702). Neuro-Oncology 2010, 12, 1257–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brauner, R.; Malandry, F.; Rappaport, R.; Zucker, J.M.; Kalifa, C.; Pierre-Kahn, A.; Bataini, P.; Dufier, J.L. Growth and endocrine disorders in optic glioma. Eur. J. Pediatrics 1990, 149, 825–828. [Google Scholar] [CrossRef] [PubMed]
- Binning, M.J.; Liu, J.K.; Kestle, J.R.; Brockmeyer, D.L.; Walker, M.L. Optic pathway gliomas: A review. Neurosurg. Focus 2007, 23, E2. [Google Scholar] [CrossRef] [PubMed]
- Poussaint, T.Y.; Barnes, P.D.; Nichols, K.; Anthony, D.C.; Cohen, L.; Tarbell, N.J.; Goumnerova, L. Diencephalic syndrome: Clinical features and imaging findings. AJNR Am. J. Neuroradiol. 1997, 18, 1499–1505. [Google Scholar]
- Santoro, C.; Perrotta, S.; Picariello, S.; Scilipoti, M.; Cirillo, M.; Quaglietta, L.; Cinalli, G.; Cioffi, D.; Di Iorgi, N.; Maghnie, M.; et al. Pretreatment Endocrine Disorders Due to Optic Pathway Gliomas in Pediatric Neurofibromatosis Type 1: Multicenter Study. J. Clin. Endocrinol. Metab. 2020, 105, dgaa138. [Google Scholar] [CrossRef]
- Swerdlow, A.J.; Cooke, R.; Beckers, D.; Borgström, B.; Butler, G.; Carel, J.C.; Cianfarani, S.; Clayton, P.; Coste, J.; Deodati, A.; et al. Cancer Risks in Patients Treated with Growth Hormone in Childhood: The SAGhE European Cohort Study. J. Clin. Endocrinol. Metab. 2017, 102, 1661–1672. [Google Scholar] [CrossRef]
- Darendeliler, F.; Karagiannis, G.; Wilton, P.; Ranke, M.B.; Albertsson-Wikland, K.; Anthony Price, D.; on Behalf of The Kigs International Board. Recurrence of brain tumours in patients treated with growth hormone: Analysis of KIGS (Pfizer International Growth Database). Acta Paediatr. 2006, 95, 1284–1290. [Google Scholar] [CrossRef]
- Müller, H.L.; Gebhardt, U.; Schröder, S.; Pohl, F.; Kortmann, R.D.; Faldum, A.; Zwiener, I.; Warmuth-Metz, M.; Pietsch, T.; Calaminus, G.; et al. Analyses of treatment variables for patients with childhood craniopharyngioma—Results of the multicenter prospective trial KRANIOPHARYNGEOM 2000 after three years of follow-up. Horm. Res. Paediatr. 2010, 73, 175–180. [Google Scholar] [CrossRef]
- Packer, R.J.; Boyett, J.M.; Janss, A.J.; Stavrou, T.; Kun, L.; Wisoff, J.; Russo, C.; Geyer, R.; Phillips, P.; Kieran, M.; et al. Growth hormone replacement therapy in children with medulloblastoma: Use and effect on tumor control. J. Clin. Oncol. 2001, 19, 480–487. [Google Scholar] [CrossRef]
- Sklar, C.A.; Antal, Z.; Chemaitilly, W.; Cohen, L.E.; Follin, C.; Meacham, L.R.; Murad, M.H. Hypothalamic-Pituitary and Growth Disorders in Survivors of Childhood Cancer: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2018, 103, 2761–2784. [Google Scholar] [CrossRef] [Green Version]
- Sani, I.; Albanese, A. Endocrine Long-Term Follow-Up of Children with Neurofibromatosis Type 1 and Optic Pathway Gliomas. Horm. Res. Paediatr. 2017, 87, 179–188. [Google Scholar] [CrossRef]
- Shofty, B.; Ben Sira, L.; Constantini, S. Neurofibromatosis 1-associated optic pathway gliomas. Childs Nerv. Syst. 2020, 36, 2351–2361. [Google Scholar] [CrossRef]
- Papini, C.; Dineen, R.A.; Walker, D.A.; Thomas, S.; Pitchford, N.J. Neuropsychological outcomes of children with Optic Pathway Glioma. Sci. Rep. 2020, 10, 3344. [Google Scholar] [CrossRef]
- Liu, A.P.Y.; Hastings, C.; Wu, S.; Bass, J.K.; Heitzer, A.M.; Ashford, J.; Vestal, R.; Hoehn, M.E.; Ghazwani, Y.; Acharya, S.; et al. Treatment burden and long-term health deficits of patients with low-grade gliomas or glioneuronal tumors diagnosed during the first year of life. Cancer 2019, 125, 1163–1175. [Google Scholar] [CrossRef]
- Ris, M.D.; Leisenring, W.M.; Goodman, P.; Di, C.; Noll, J.; Levy, W.; Robison, L.L.; Armstrong, G.T. Neuropsychological and socioeconomic outcomes in adult survivors of pediatric low-grade glioma. Cancer 2019, 125, 3050–3058. [Google Scholar] [CrossRef]
- Taylor, T.; Jaspan, T.; Milano, G.; Gregson, R.; Parker, T.; Ritzmann, T.; Benson, C.; Walker, D. Radiological classification of optic pathway gliomas: Experience of a modified functional classification system. Br. J. Radiol. 2008, 81, 761–766. [Google Scholar] [CrossRef]
- Hoffmann, A.; Gebhardt, U.; Sterkenburg, A.S.; Warmuth-Metz, M.; Müller, H.L. Diencephalic syndrome in childhood craniopharyngioma--results of German multicenter studies on 485 long-term survivors of childhood craniopharyngioma. J. Clin. Endocrinol. Metab. 2014, 99, 3972–3977. [Google Scholar] [CrossRef] [Green Version]
- Brauner, R.; Trivin, C.; Zerah, M.; Souberbielle, J.C.; Doz, F.; Kalifa, C.; Sainte-Rose, C. Diencephalic syndrome due to hypothalamic tumor: A model of the relationship between weight and puberty onset. J. Clin. Endocrinol. Metab. 2006, 91, 2467–2473. [Google Scholar] [CrossRef] [Green Version]
- DeVile, C.J.; Grant, D.B.; Hayward, R.D.; Stanhope, R. Growth and endocrine sequelae of craniopharyngioma. Arch. Dis. Child. 1996, 75, 108–114. [Google Scholar] [CrossRef] [Green Version]
- Hill, C.S.; Khan, M.; Phipps, K.; Green, K.; Hargrave, D.; Aquilina, K. Neurosurgical experience of managing optic pathway gliomas. Child’s Nerv. Syst. 2021, 37, 1917–1929. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Study Population (n = 90) | |
|---|---|
| Male | 42 (46.7) |
| Age at diagnosis (years) | 1.84 (0.83–2.51) (0.06–3.00) |
| ≤1 year | 30 (33.3) |
| NF1 positive | 27 (30.0) |
| Symptoms at diagnosis | |
| 41 (45.6) |
| 27 (30.0) |
| 22 (24.4) |
| 14 (15.5) |
| Radiological Classification | |
| 19 (21.1) |
| 25 (27.8) |
| 46 (51.1) |
| Hypothalamic involvement | 56 (62.2) |
| Leptomeningeal metastases | 11 (12.2) |
| Follow-up (years) | 9.49 (4.00–12.53) (0.52–25.00) |
| Deceased | 9 (10) |
| Death age (years) | 5.6 (2.76–18.03) (0.58–23.66) |
| Number of Cases | 5-Year EFS | 10-Year EFS | Time Since Diagnosis (Years) | Age at Diagnosis (Years) | |
|---|---|---|---|---|---|
| APD | |||||
| GHD | 37 (41.1) | 76.1 | 42.2 | 5.3 (2.8–8) (0.79–10.7) | 7 (4.9–9.2) (1.5–12.5) |
| CPP | 24 (26.7) | 75.8 | 65.1 | 3.8 (1.9–5.3) (1.02–7.8) | 5.6 (3–6.5) (2.1–8.9) |
| GnD | 17 (18.9) | 100 | 97.8 | 12.7 (11.8–13.3) (9.4–14) | 14 (14–14.3) (13.4–15.2) |
| ACTHD | 19 (22.2) | 88.3 | 76.3 | 5.3 (1.7–7.4) (0.05–14.7) | 7 (3.5–9.2) (0.3–15) |
| TSHD | 21 (23.3) | 88.8 | 76.4 | 5.9 (2.7–8.9) (0.05–14.7) | 7.8 (4.9–10.8) (0.3–15) |
| PPD | |||||
| SIADH | 10 (11.1) | 92.9 | 88.9 | 2.8 (0.2–10.1) (0–16.7) | 3.9 (1.1–11.3) (0.8–18.8) |
| CDI | 8 (8.9) | 93.3 | 91.5 | 0.8 (0.1–5.4) (0.05–11.6) | 2.9 (2.3–7.6) (0.3–13.46) |
| CSWS | 1 (1.1) | 100 | 100 | 15.28 | 17.07 |
| MD | |||||
| Obesity | 23 (25.5) | 93.6 | 70 | 6.8 (5.2–9.5) (0.7–14.7) | 8.9 (6.9–10.8) (2.8–16) |
| IGT/T2D | 11 (12.2) | 98.4 | 89 | 8.1 (7.5–15.6) (4.5–18.7) | 11.1 (9.2–17) (6.8–19) |
| Univariate | Multivariate | |||
|---|---|---|---|---|
| HR (95% CI) | p | HR (95% CI) | p | |
| Absence of NF1 | 5.3 (2.2–12.5) | <0.0001 | 1.5 (0.5–4.4) | 0.414 |
| Age ≤ 1 year | 2.3 (1.3–4.2) | 0.007 | 0.7 (0.3–1.4) | 0.317 |
| Diencephalic syndrome | 3.2 (1.8– 5.9) | <0.0001 | 1.6 (0.8–3.2) | 0.137 |
| Hypothalamic involvement | 11.7 (4.5–30.2) | <0.0001 | 4.2 (1.3–13.8) | 0.018 |
| MDC | 3.1 (1.9–5.1) | <0.0001 | 1.9 (0.9–3.7) | 0.065 |
| Hydrocephalus | 2.7 (1.4–4.9) | 0.002 | 1.1 (0.6–2.1) | 0.775 |
| OR (95% CI) | p | OR (95% CI) | p | |
| Radiotherapy | 31.1 (3.9–245.5) | 0.001 | 15.7 (1.2–211.8) | 0.038 |
| Chemotherapy | 4 (1.4–11.6) | 0.009 | 5.3 (0.9–28.7) | 0.057 |
| Surgery | 12.8 (4.4–36.9) | <0.0001 | 9.9 (2.6–37.3) | 0.001 |
| Number of progressions | 1.9 (1.4–2.6) | <0.0001 | 1.1 (0.8–1.6) | 0.567 |
| Years of follow-up | 1.2 (1.1–1.3) | 0.003 | 2 (1.1–3.8) | 0.034 |
| Univariate | Multivariate | |||
|---|---|---|---|---|
| HR (95% CI) | p | HR (95% CI) | p | |
| Absence of NF1 | 2.1 (0.7–6.3) | 0.169 | NI | NI |
| Age ≤ 1 year | 1 (0.4–2.4) | 0.989 | NI | NI |
| Diencephalic Syndrome | 2.3 (1.1–5.4) | 0.043 | 1.2 (0.5–3) | 0.679 |
| Hypothalamic involvement | 4.4 (1.3–14.9) | 0.017 | 2.1 (0.4–10.2) | 0.359 |
| MDC | 2.2 (1.1–4.4) | 0.024 | 1.2 (0.5–3.1) | 0.623 |
| Hydrocephalus | 4.1 (1.8–9.4) | 0.001 | 2.3 (0.9–5.9) | 0.072 |
| OR (95% CI) | p | OR (95% CI) | p | |
| Radiotherapy | 5.3 (1.8–15.1) | 0.002 | 0.6 (0.1–3.9) | 0.625 |
| Chemotherapy | 1.2 (0.4–3.8) | 0.727 | NI | NI |
| Surgery | 2.7 (0.9–8.3) | 0.072 | NI | NI |
| Number of concomitant APD | 1.8 (1.1 -3) | 0.014 | 2 (1.1–3.8) | 0.034 |
| Number of progressions | 1.2 (0.9–1.6) | 0.095 | NI | |
| Years of follow-up | 1.2 (1.1–1.3) | 0.001 | 1 (0.8–1.2) | 0.888 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Picariello, S.; Cerbone, M.; D’Arco, F.; Gan, H.-W.; O’Hare, P.; Aquilina, K.; Opocher, E.; Hargrave, D.; Spoudeas, H.A. A 40-Year Cohort Study of Evolving Hypothalamic Dysfunction in Infants and Young Children (<3 years) with Optic Pathway Gliomas. Cancers 2022, 14, 747. https://doi.org/10.3390/cancers14030747
Picariello S, Cerbone M, D’Arco F, Gan H-W, O’Hare P, Aquilina K, Opocher E, Hargrave D, Spoudeas HA. A 40-Year Cohort Study of Evolving Hypothalamic Dysfunction in Infants and Young Children (<3 years) with Optic Pathway Gliomas. Cancers. 2022; 14(3):747. https://doi.org/10.3390/cancers14030747
Chicago/Turabian StylePicariello, Stefania, Manuela Cerbone, Felice D’Arco, Hoong-Wei Gan, Patricia O’Hare, Kristian Aquilina, Enrico Opocher, Darren Hargrave, and Helen A. Spoudeas. 2022. "A 40-Year Cohort Study of Evolving Hypothalamic Dysfunction in Infants and Young Children (<3 years) with Optic Pathway Gliomas" Cancers 14, no. 3: 747. https://doi.org/10.3390/cancers14030747
APA StylePicariello, S., Cerbone, M., D’Arco, F., Gan, H.-W., O’Hare, P., Aquilina, K., Opocher, E., Hargrave, D., & Spoudeas, H. A. (2022). A 40-Year Cohort Study of Evolving Hypothalamic Dysfunction in Infants and Young Children (<3 years) with Optic Pathway Gliomas. Cancers, 14(3), 747. https://doi.org/10.3390/cancers14030747