
Targeting Oncogenic Pathways in the Era of Personalized Oncology: A Systemic Analysis Reveals Highly Mutated Signaling Pathways in Cancer Patients and Potential Therapeutic Targets

 , ,
, , 
Simple Summary
Abstract
1. Personalized Therapeutic Strategies Require Understanding of the Underlying Signaling Pathways’ Mechanisms
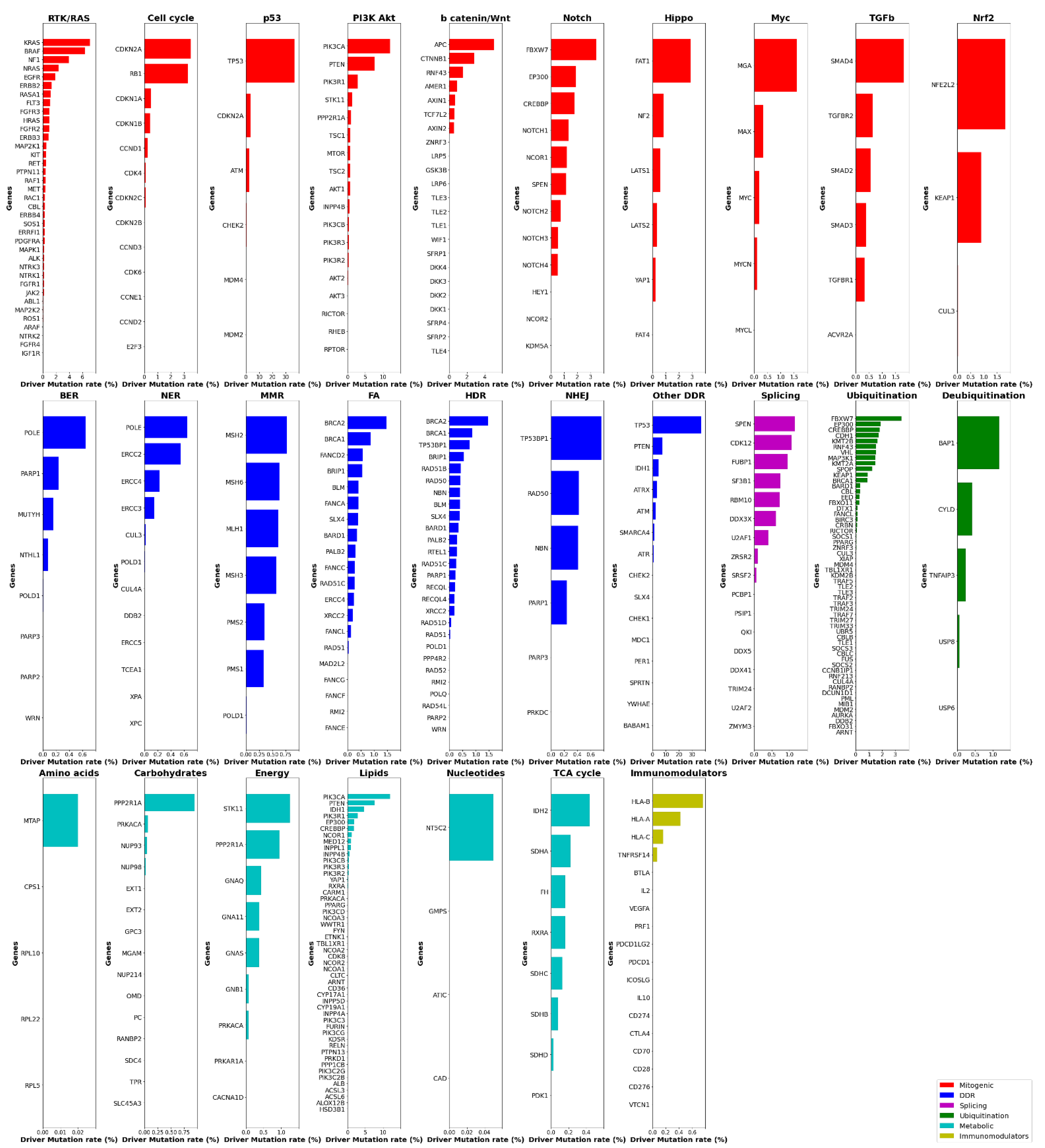
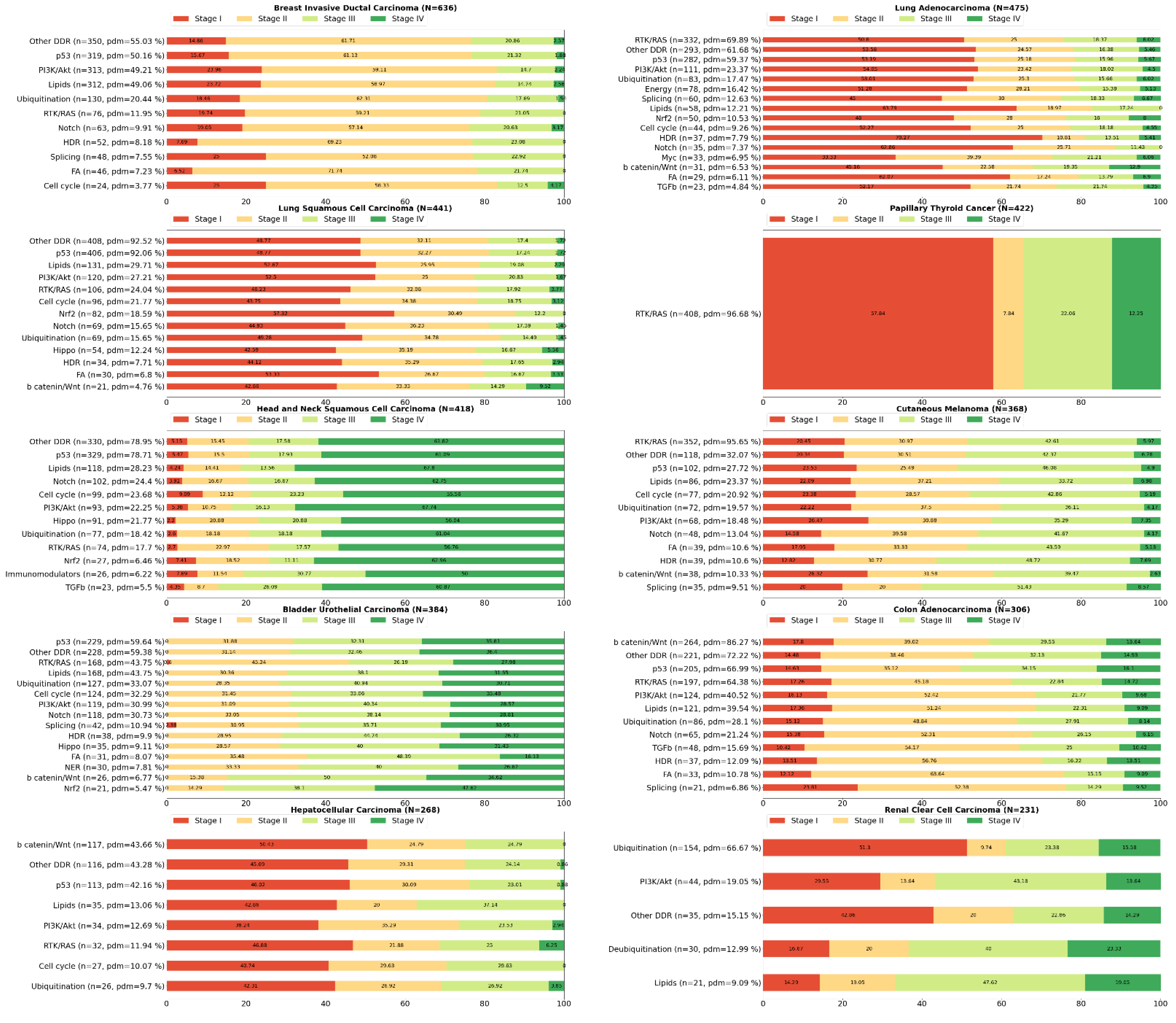
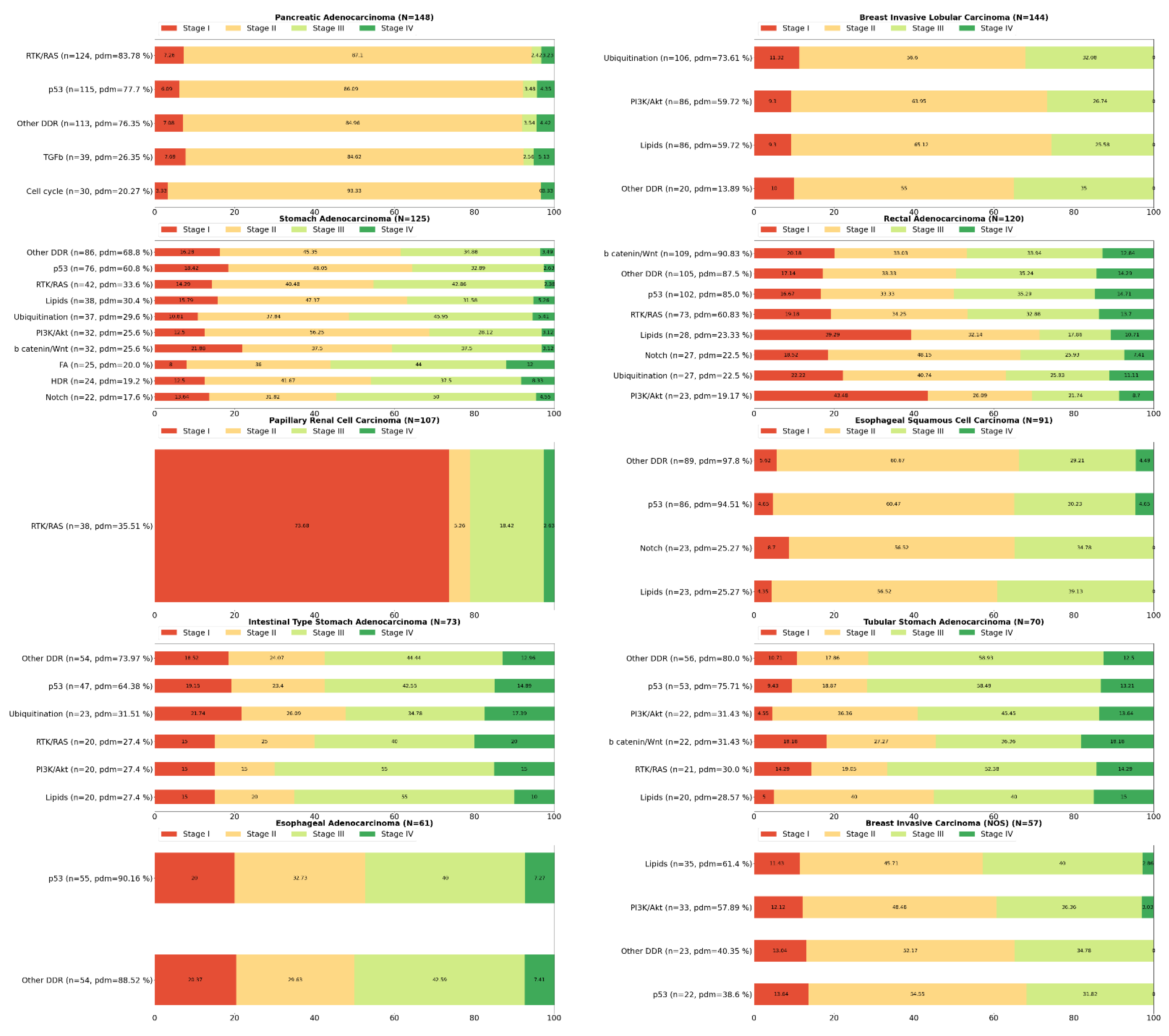
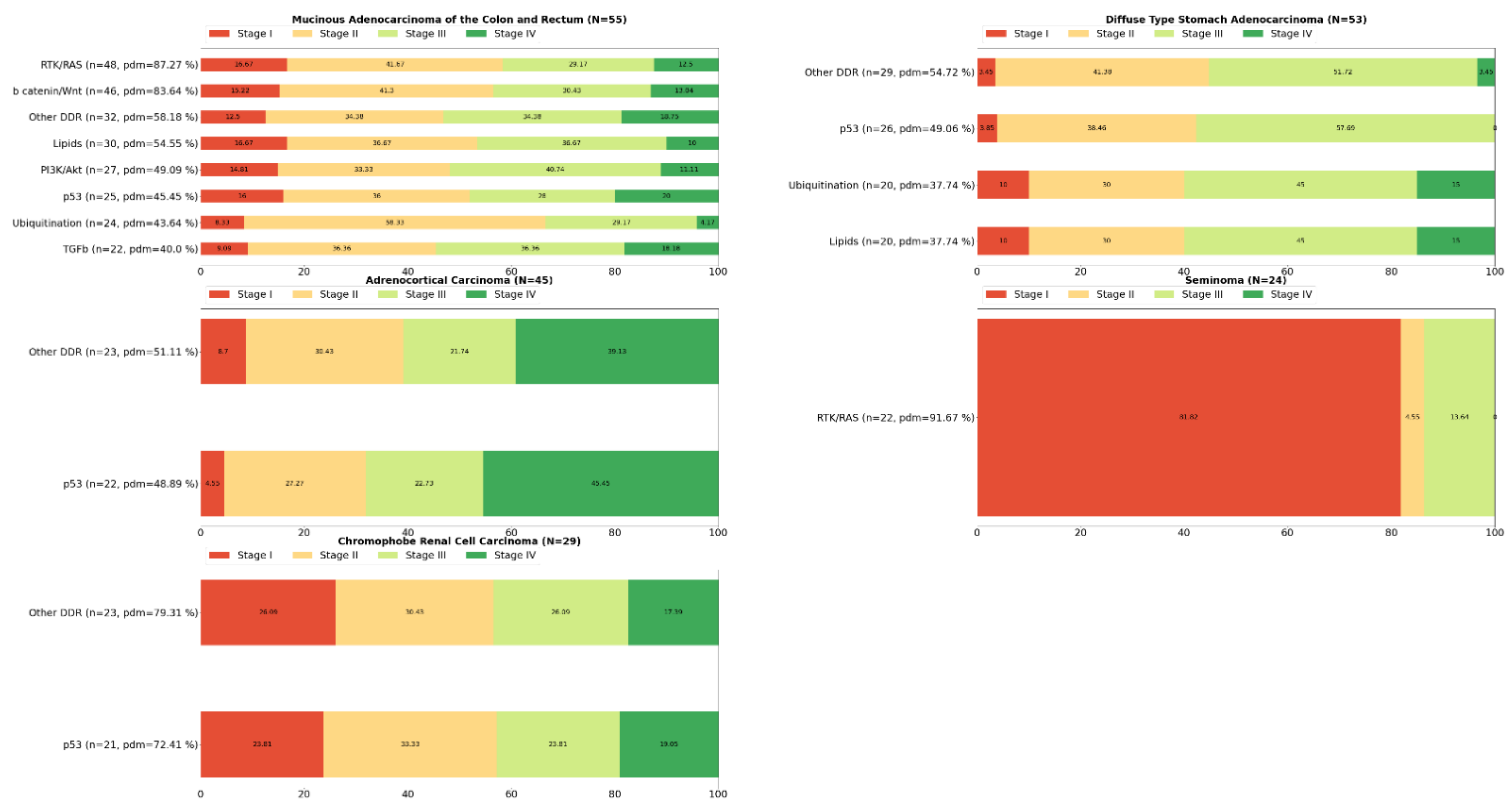
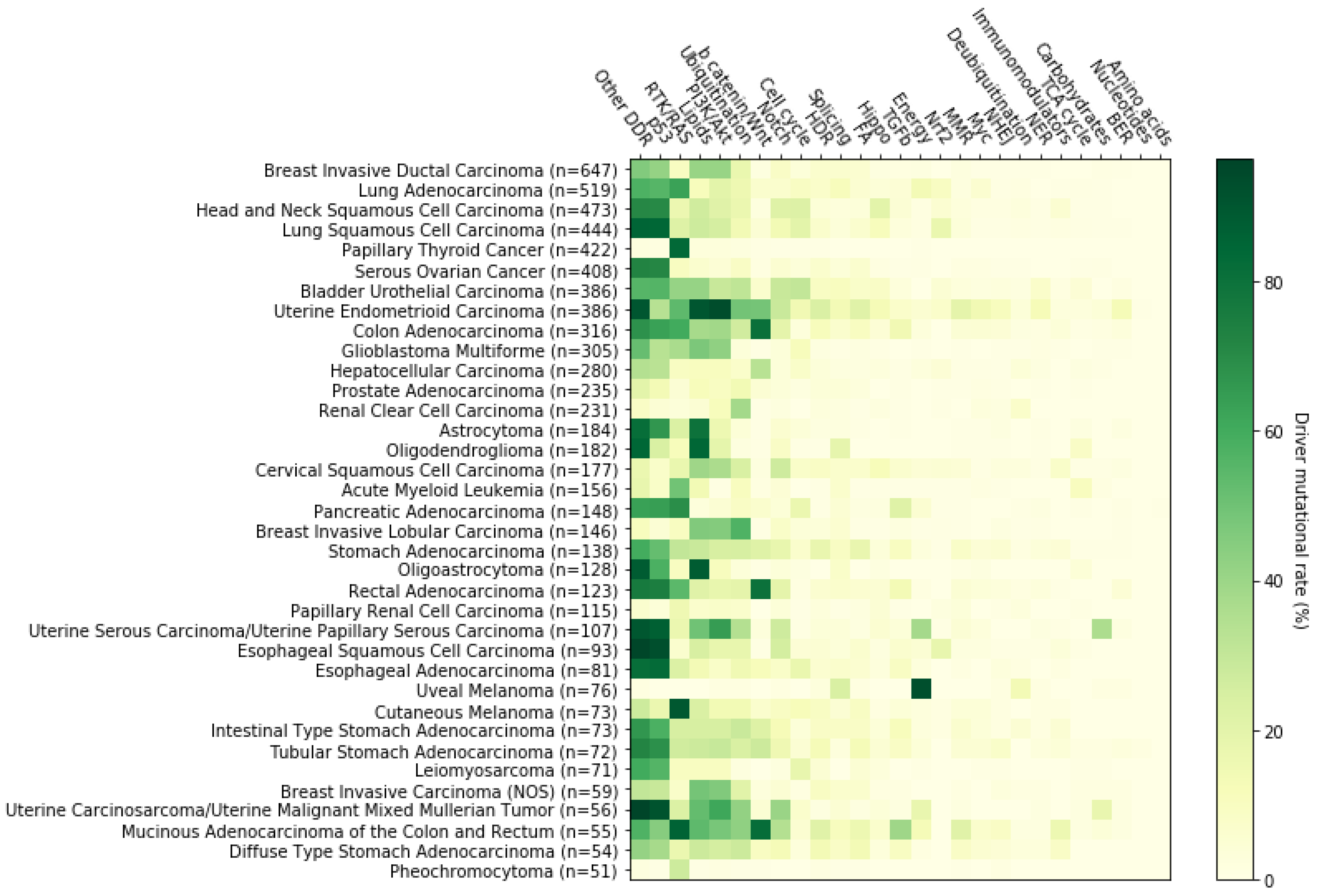
2. Systemic Analysis Determines Prevalent Mutated Carcinogenic Pathways
3. The p53 Pathway
4. The RTK-RAS Pathway
5. Lipid Metabolism
6. The PI3K/AKT Pathway
7. Ubiquitination and Acetylation Pathways
8. The WNT/b Catenin Pathway
9. The Notch Pathway
10. The Cell Cycle Pathway
11. The HDR Pathway
12. The Splicing Pathway
13. Conclusive Remarks
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-Ramiro, F.; Peiro-Pastor, R.; Aguado, B. Human genomics projects and precision medicine. Gene Ther. 2017, 24, 551–561. [Google Scholar] [CrossRef]
- Imai, K.; Takaoka, A. Comparing antibody and small-molecule therapies for cancer. Nat. Rev. Cancer 2006, 6, 714–727. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Parui, A.L.; Bose, K. Cancer Biology and Its Treatment Modalities: A Brief Historical Perspective. In Unravelling Cancer Signaling Pathways: A Multidisciplinary Approach; Bose, K., Chaudhari, P., Eds.; Springer: Singapore, 2019; pp. 1–11. [Google Scholar] [CrossRef]
- Martincorena, I.; Campbell, P.J. Somatic mutation in cancer and normal cells. Science 2016, 349, 1483–1489, Erratum in Science 2016, 351, aaf5401. [Google Scholar] [CrossRef]
- Okugawa, Y.; Grady, W.M.; Goel, A. Epigenetic Alterations in Colorectal Cancer: Emerging Biomarkers. Gastroenterology 2015, 149, 1204–1225.e1212. [Google Scholar] [CrossRef]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Koldobskiy, M.A.; Gondor, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e310. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Dempke, W.C.M.; Fenchel, K.; Uciechowski, P.; Chevassut, T. Targeting Developmental Pathways: The Achilles Heel of Cancer? Oncology 2017, 93, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Pon, J.R.; Marra, M.A. Driver and passenger mutations in cancer. Annu. Rev. Pathol. 2015, 10, 25–50. [Google Scholar] [CrossRef]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef]
- Johnson, A.; Zeng, J.; Bailey, A.M.; Holla, V.; Litzenburger, B.; Lara-Guerra, H.; Mills, G.B.; Mendelsohn, J.; Shaw, K.R.; Meric-Bernstam, F. The right drugs at the right time for the right patient: The MD Anderson precision oncology decision support platform. Drug Discov. Today 2015, 20, 1433–1438. [Google Scholar] [CrossRef]
- Saltz, L.B.; Meropol, N.J.; Loehrer, P.J., Sr.; Needle, M.N.; Kopit, J.; Mayer, R.J. Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J. Clin. Oncol. 2004, 22, 1201–1208. [Google Scholar] [CrossRef]
- Shaw, A.T.; Kim, D.W.; Nakagawa, K.; Seto, T.; Crino, L.; Ahn, M.J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Betge, J.; Ebert, M.P.; Boutros, M. CRISPR/Cas9 for cancer research and therapy. Semin. Cancer Biol. 2019, 55, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, K.; Janowicz, K.; Dyszkiewicz-Konwinska, M.; Hutchings, G.; Dompe, C.; Moncrieff, L.; Jankowski, M.; Machnik, M.; Oleksiewicz, U.; Kocherova, I.; et al. CRISPR/Cas9 in Cancer Immunotherapy: Animal Models and Human Clinical Trials. Genes 2020, 11, 921. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Leighton, J.S.; Wang, Y.; Peng, X.; Chen, Z.; Chen, H.; Sun, Y.; Yao, F.; Li, J.; Zhang, H.; et al. Integrated Genomic Analysis of the Ubiquitin Pathway across Cancer Types. Cell Rep. 2018, 23, 213–226.e213. [Google Scholar] [CrossRef] [PubMed]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254.e236. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Chen, Z.; Farshidfar, F.; Xu, X.; Lorenzi, P.L.; Wang, Y.; Cheng, F.; Tan, L.; Mojumdar, K.; Du, D.; et al. Molecular Characterization and Clinical Relevance of Metabolic Expression Subtypes in Human Cancers. Cell Rep. 2018, 23, 255–269.e254. [Google Scholar] [CrossRef]
- Seiler, M.; Peng, S.; Agrawal, A.A.; Palacino, J.; Teng, T.; Zhu, P.; Smith, P.G.; Cancer Genome Atlas Research, N.; Buonamici, S.; Yu, L. Somatic Mutational Landscape of Splicing Factor Genes and Their Functional Consequences across 33 Cancer Types. Cell Rep. 2018, 23, 282–296.e284. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e814. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Oren, M. Decision making by p53: Life, death and cancer. Cell Death Differ. 2003, 10, 431–442. [Google Scholar] [CrossRef]
- Harris, S.L.; Levine, A.J. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Labuschagne, C.F.; Zani, F.; Vousden, K.H. Control of metabolism by p53–Cancer and beyond. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Aubrey, B.J.; Strasser, A.; Kelly, G.L. Tumor-Suppressor Functions of the TP53 Pathway. Cold Spring Harb. Perspect. Med. 2016, 6, a026062. [Google Scholar] [CrossRef] [PubMed]
- Haronikova, L.; Olivares-Illana, V.; Wang, L.; Karakostis, K.; Chen, S.; Fahraeus, R. The p53 mRNA: An integral part of the cellular stress response. Nucleic Acids Res. 2019, 47, 3257–3271. [Google Scholar] [CrossRef]
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef]
- Li, J.; Yang, L.; Gaur, S.; Zhang, K.; Wu, X.; Yuan, Y.C.; Li, H.; Hu, S.; Weng, Y.; Yen, Y. Mutants TP53 p.R273H and p.R273C but not p.R273G enhance cancer cell malignancy. Hum. Mutat. 2014, 35, 575–584. [Google Scholar] [CrossRef]
- Sun, S.; Chen, H.; Sun, L.; Wang, M.; Wu, X.; Xiao, Z.J. Hotspot mutant p53-R273H inhibits KLF6 expression to promote cell migration and tumor metastasis. Cell Death Dis. 2020, 11, 595. [Google Scholar] [CrossRef]
- Schulz-Heddergott, R.; Stark, N.; Edmunds, S.J.; Li, J.; Conradi, L.C.; Bohnenberger, H.; Ceteci, F.; Greten, F.R.; Dobbelstein, M.; Moll, U.M. Therapeutic Ablation of Gain-of-Function Mutant p53 in Colorectal Cancer Inhibits Stat3-Mediated Tumor Growth and Invasion. Cancer Cell 2018, 34, 298–314.e297. [Google Scholar] [CrossRef]
- Klemke, L.; Fehlau, C.F.; Winkler, N.; Toboll, F.; Singh, S.K.; Moll, U.M.; Schulz-Heddergott, R. The Gain-of-Function p53 R248W Mutant Promotes Migration by STAT3 Deregulation in Human Pancreatic Cancer Cells. Front. Oncol. 2021, 11, 642603. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Vazquez, A.; Levine, A.J.; Carpizo, D.R. Allele-specific p53 mutant reactivation. Cancer Cell 2012, 21, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Grugan, K.D.; Vega, M.E.; Wong, G.S.; Diehl, J.A.; Bass, A.J.; Wong, K.K.; Nakagawa, H.; Rustgi, A.K. A common p53 mutation (R175H) activates c-Met receptor tyrosine kinase to enhance tumor cell invasion. Cancer Biol. Ther. 2013, 14, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Parry, D.; Peters, G. Temperature-sensitive mutants of p16CDKN2 associated with familial melanoma. Mol. Cell Biol. 1996, 16, 3844–3852. [Google Scholar] [CrossRef]
- Rutter, J.L.; Goldstein, A.M.; Davila, M.R.; Tucker, M.A.; Struewing, J.P. CDKN2A point mutations D153spl(c.457G>T) and IVS2+1G>T result in aberrant splice products affecting both p16INK4a and p14ARF. Oncogene 2003, 22, 4444–4448. [Google Scholar] [CrossRef]
- Kannengiesser, C.; Brookes, S.; del Arroyo, A.G.; Pham, D.; Bombled, J.; Barrois, M.; Mauffret, O.; Avril, M.F.; Chompret, A.; Lenoir, G.M.; et al. Functional, structural, and genetic evaluation of 20 CDKN2A germ line mutations identified in melanoma-prone families or patients. Hum. Mutat. 2009, 30, 564–574. [Google Scholar] [CrossRef]
- Angele, S.; Jones, C.; Reis Filho, J.S.; Fulford, L.G.; Treilleux, I.; Lakhani, S.R.; Hall, J. Expression of ATM, p53, and the MRE11-Rad50-NBS1 complex in myoepithelial cells from benign and malignant proliferations of the breast. J. Clin. Pathol. 2004, 57, 1179–1184. [Google Scholar] [CrossRef]
- Bailey, S.L.; Gurley, K.E.; Hoon-Kim, K.; Kelly-Spratt, K.S.; Kemp, C.J. Tumor suppression by p53 in the absence of Atm. Mol. Cancer Res. 2008, 6, 1185–1192. [Google Scholar] [CrossRef]
- Biddlestone-Thorpe, L.; Sajjad, M.; Rosenberg, E.; Beckta, J.M.; Valerie, N.C.; Tokarz, M.; Adams, B.R.; Wagner, A.F.; Khalil, A.; Gilfor, D.; et al. ATM kinase inhibition preferentially sensitizes p53-mutant glioma to ionizing radiation. Clin. Cancer Res. 2013, 19, 3189–3200. [Google Scholar] [CrossRef]
- Malbert-Colas, L.; Ponnuswamy, A.; Olivares-Illana, V.; Tournillon, A.S.; Naski, N.; Fahraeus, R. HDMX folds the nascent p53 mRNA following activation by the ATM kinase. Mol. Cell 2014, 54, 500–511. [Google Scholar] [CrossRef]
- Chwastek, J.; Jantas, D.; Lason, W. The ATM kinase inhibitor KU-55933 provides neuroprotection against hydrogen peroxide-induced cell damage via a gammaH2AX/p-p53/caspase-3-independent mechanism: Inhibition of calpain and cathepsin D. Int. J. Biochem. Cell Biol. 2017, 87, 38–53. [Google Scholar] [CrossRef] [PubMed]
- Karakostis, K.; Vadivel Gnanasundram, S.; Lopez, I.; Thermou, A.; Wang, L.; Nylander, K.; Olivares-Illana, V.; Fahraeus, R. A single synonymous mutation determines the phosphorylation and stability of the nascent protein. J. Mol. Cell Biol. 2019, 11, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.Y.; Kuk, M.U.; Kim, J.W.; Lee, Y.H.; Lee, Y.S.; Choy, H.E.; Park, S.C.; Park, J.T. ATM mediated-p53 signaling pathway forms a novel axis for senescence control. Mitochondrion 2020, 55, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Easton, D.F.; Pharoah, P.D.; Antoniou, A.C.; Tischkowitz, M.; Tavtigian, S.V.; Nathanson, K.L.; Devilee, P.; Meindl, A.; Couch, F.J.; Southey, M.; et al. Gene-panel sequencing and the prediction of breast-cancer risk. N. Engl. J. Med. 2015, 372, 2243–2257. [Google Scholar] [CrossRef]
- Griffith, O.L.; Spies, N.C.; Anurag, M.; Griffith, M.; Luo, J.; Tu, D.; Yeo, B.; Kunisaki, J.; Miller, C.A.; Krysiak, K.; et al. The prognostic effects of somatic mutations in ER-positive breast cancer. Nat. Commun. 2018, 9, 3476. [Google Scholar] [CrossRef]
- Nahar, R.; Zhai, W.; Zhang, T.; Takano, A.; Khng, A.J.; Lee, Y.Y.; Liu, X.; Lim, C.H.; Koh, T.P.T.; Aung, Z.W.; et al. Elucidating the genomic architecture of Asian EGFR-mutant lung adenocarcinoma through multi-region exome sequencing. Nat. Commun. 2018, 9, 216. [Google Scholar] [CrossRef]
- Jette, N.R.; Kumar, M.; Radhamani, S.; Arthur, G.; Goutam, S.; Yip, S.; Kolinsky, M.; Williams, G.J.; Bose, P.; Lees-Miller, S.P. ATM-Deficient Cancers Provide New Opportunities for Precision Oncology. Cancers 2020, 12, 687. [Google Scholar] [CrossRef]
- Zhu, G.; Pan, C.; Bei, J.X.; Li, B.; Liang, C.; Xu, Y.; Fu, X. Mutant p53 in Cancer Progression and Targeted Therapies. Front. Oncol. 2020, 10, 595187. [Google Scholar] [CrossRef]
- Salomao, N.; Karakostis, K.; Hupp, T.; Vollrath, F.; Vojtesek, B.; Fahraeus, R. What do we need to know and understand about p53 to improve its clinical value? J. Pathol. 2021, 254, 443–453. [Google Scholar] [CrossRef]
- Duffy, M.J.; Synnott, N.C.; O’Grady, S.; Crown, J. Targeting p53 for the treatment of cancer. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Dhillon, S. Palbociclib: First global approval. Drugs 2015, 75, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S. Abemaciclib: First Global Approval. Drugs 2017, 77, 2063–2070. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Ribociclib: First Global Approval. Drugs 2017, 77, 799–807. [Google Scholar] [CrossRef]
- Dhillon, S. Trilaciclib: First Approval. Drugs 2021, 81, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Young, R.J.; Waldeck, K.; Martin, C.; Foo, J.H.; Cameron, D.P.; Kirby, L.; Do, H.; Mitchell, C.; Cullinane, C.; Liu, W.; et al. Loss of CDKN2A expression is a frequent event in primary invasive melanoma and correlates with sensitivity to the CDK4/6 inhibitor PD0332991 in melanoma cell lines. Pigment Cell Melanoma Res. 2014, 27, 590–600. [Google Scholar] [CrossRef]
- Su, D.; Zhang, D.; Jin, J.; Ying, L.; Han, M.; Chen, K.; Li, B.; Wu, J.; Xie, Z.; Zhang, F.; et al. Identification of predictors of drug sensitivity using patient-derived models of esophageal squamous cell carcinoma. Nat. Commun. 2019, 10, 5076. [Google Scholar] [CrossRef]
- DeMichele, A.; Clark, A.S.; Tan, K.S.; Heitjan, D.F.; Gramlich, K.; Gallagher, M.; Lal, P.; Feldman, M.; Zhang, P.; Colameco, C.; et al. CDK 4/6 inhibitor palbociclib (PD0332991) in Rb+ advanced breast cancer: Phase II activity, safety, and predictive biomarker assessment. Clin. Cancer Res. 2015, 21, 995–1001. [Google Scholar] [CrossRef]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2015, 16, 25–35. [Google Scholar] [CrossRef]
- Arnedos, M.; Bayar, M.A.; Cheaib, B.; Scott, V.; Bouakka, I.; Valent, A.; Adam, J.; Leroux-Kozal, V.; Marty, V.; Rapinat, A.; et al. Modulation of Rb phosphorylation and antiproliferative response to palbociclib: The preoperative-palbociclib (POP) randomized clinical trial. Ann. Oncol. 2018, 29, 1755–1762. [Google Scholar] [CrossRef]
- Rose, T.L.; Chism, D.D.; Alva, A.S.; Deal, A.M.; Maygarden, S.J.; Whang, Y.E.; Kardos, J.; Drier, A.; Basch, E.; Godley, P.A.; et al. Phase II trial of palbociclib in patients with metastatic urothelial cancer after failure of first-line chemotherapy. Br. J. Cancer 2018, 119, 801–807. [Google Scholar] [CrossRef]
- Toulmonde, M.; Blay, J.Y.; Bouche, O.; Mir, O.; Penel, N.; Isambert, N.; Duffaud, F.; Bompas, E.; Esnaud, T.; Boidot, R.; et al. Activity and Safety of Palbociclib in Patients with Advanced Gastrointestinal Stromal Tumors Refractory to Imatinib and Sunitinib: A Biomarker-driven Phase II Study. Clin. Cancer Res. 2019, 25, 4611–4615. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.; Knittel, G.; Welcker, D.; Yang, T.P.; George, J.; Nowak, M.; Leeser, U.; Buttner, R.; Perner, S.; Peifer, M.; et al. ATM Deficiency Is Associated with Sensitivity to PARP1- and ATR Inhibitors in Lung Adenocarcinoma. Cancer Res. 2017, 77, 3040–3056. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, R.L.; Wijnhoven, P.W.G.; Ramos-Montoya, A.; Wilson, Z.; Illuzzi, G.; Falenta, K.; Jones, G.N.; James, N.; Chabbert, C.D.; Stott, J.; et al. Combined PARP and ATR inhibition potentiates genome instability and cell death in ATM-deficient cancer cells. Oncogene 2020, 39, 4869–4883. [Google Scholar] [CrossRef] [PubMed]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef]
- Castellano, E.; Molina-Arcas, M.; Krygowska, A.A.; East, P.; Warne, P.; Nicol, A.; Downward, J. RAS signalling through PI3-Kinase controls cell migration via modulation of Reelin expression. Nat. Commun. 2016, 7, 11245. [Google Scholar] [CrossRef]
- Imperial, R.; Toor, O.M.; Hussain, A.; Subramanian, J.; Masood, A. Comprehensive pancancer genomic analysis reveals (RTK)-RAS-RAF-MEK as a key dysregulated pathway in cancer: Its clinical implications. Semin. Cancer Biol. 2019, 54, 14–28. [Google Scholar] [CrossRef]
- Smith, M.J.; Neel, B.G.; Ikura, M. NMR-based functional profiling of RASopathies and oncogenic RAS mutations. Proc. Natl. Acad. Sci. USA 2013, 110, 4574–4579. [Google Scholar] [CrossRef]
- Cook, J.H.; Melloni, G.E.M.; Gulhan, D.C.; Park, P.J.; Haigis, K.M. The origins and genetic interactions of KRAS mutations are allele- and tissue-specific. Nat. Commun. 2021, 12, 1808. [Google Scholar] [CrossRef]
- Feig, L.A.; Cooper, G.M. Relationship among guanine nucleotide exchange, GTP hydrolysis, and transforming potential of mutated ras proteins. Mol. Cell Biol. 1988, 8, 2472–2478. [Google Scholar] [CrossRef]
- Poulin, E.J.; Bera, A.K.; Lu, J.; Lin, Y.J.; Strasser, S.D.; Paulo, J.A.; Huang, T.Q.; Morales, C.; Yan, W.; Cook, J.; et al. Tissue-Specific Oncogenic Activity of KRAS(A146T). Cancer Discov. 2019, 9, 738–755. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Cantwell-Dorris, E.R.; O’Leary, J.J.; Sheils, O.M. BRAFV600E: Implications for carcinogenesis and molecular therapy. Mol. Cancer Ther. 2011, 10, 385–394. [Google Scholar] [CrossRef]
- Falini, B.; Martelli, M.P.; Tiacci, E. BRAF V600E mutation in hairy cell leukemia: From bench to bedside. Blood 2016, 128, 1918–1927. [Google Scholar] [CrossRef]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef]
- Upadhyaya, M.; Kluwe, L.; Spurlock, G.; Monem, B.; Majounie, E.; Mantripragada, K.; Ruggieri, M.; Chuzhanova, N.; Evans, D.G.; Ferner, R.; et al. Germline and somatic NF1 gene mutation spectrum in NF1-associated malignant peripheral nerve sheath tumors (MPNSTs). Hum. Mutat. 2008, 29, 74–82. [Google Scholar] [CrossRef]
- Brems, H.; Beert, E.; de Ravel, T.; Legius, E. Mechanisms in the pathogenesis of malignant tumours in neurofibromatosis type 1. Lancet Oncol. 2009, 10, 508–515. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef]
- Dumbrava, E.I.; Meric-Bernstam, F. Personalized cancer therapy-leveraging a knowledge base for clinical decision-making. Cold Spring Harb. Mol. Case Stud. 2018, 4, a001578. [Google Scholar] [CrossRef]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [PubMed]
- Keeton, A.B.; Salter, E.A.; Piazza, G.A. The RAS-Effector Interaction as a Drug Target. Cancer Res. 2017, 77, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wang, Y.; Li, X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm. Sin. B 2019, 9, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, C. Grail of RAS cancer drugs within reach. Nat. Biotechnol. 2020, 38, 6–8. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A. Sotorasib: First Approval. Drugs 2021, 81, 1573–1579. [Google Scholar] [CrossRef]
- Riely, G.J.; Ou, S.H.I.; Rybkin, I.; Spira, A.; Papadopoulos, K.; Sabari, J.K.; Johnson, M.; Heist, R.S.; Bazhenova, L.; Barve, M.; et al. 99O_PR KRYSTAL-1: Activity and preliminary pharmacodynamic (PD) analysis of adagrasib (MRTX849) in patients (Pts) with advanced non–small cell lung cancer (NSCLC) harboring KRASG12C mutation. J. Thorac. Oncol. 2021, 16, S751–S752. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Yasothan, U.; Kirkpatrick, P. Vemurafenib. Nat. Rev. Drug Discov. 2011, 10, 811–812. [Google Scholar] [CrossRef]
- Ballantyne, A.D.; Garnock-Jones, K.P. Dabrafenib: First global approval. Drugs 2013, 73, 1367–1376. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef]
- Zhou, D.; Mattner, J.; Cantu, C., 3rd; Schrantz, N.; Yin, N.; Gao, Y.; Sagiv, Y.; Hudspeth, K.; Wu, Y.P.; Yamashita, T.; et al. Lysosomal glycosphingolipid recognition by NKT cells. Science 2004, 306, 1786–1789. [Google Scholar] [CrossRef]
- Groux-Degroote, S.; van Dijk, S.M.; Wolthoorn, J.; Neumann, S.; Theos, A.C.; De Maziere, A.M.; Klumperman, J.; van Meer, G.; Sprong, H. Glycolipid-dependent sorting of melanosomal from lysosomal membrane proteins by lumenal determinants. Traffic 2008, 9, 951–963. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Hishikawa, D.; Hashidate, T.; Shimizu, T.; Shindou, H. Diversity and function of membrane glycerophospholipids generated by the remodeling pathway in mammalian cells. J. Lipid Res. 2014, 55, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Gyamfi, D.; Ofori Awuah, E.; Owusu, S. Chapter 2—Lipid Metabolism: An Overview. In The Molecular Nutrition of Fats; Patel, V.B., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 17–32. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Zhao, L.; Vogt, P.K. Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc. Natl. Acad. Sci. USA 2008, 105, 2652–2657. [Google Scholar] [CrossRef]
- Leontiadou, H.; Galdadas, I.; Athanasiou, C.; Cournia, Z. Insights into the mechanism of the PIK3CA E545K activating mutation using MD simulations. Sci. Rep. 2018, 8, 15544. [Google Scholar] [CrossRef]
- Gkeka, P.; Evangelidis, T.; Pavlaki, M.; Lazani, V.; Christoforidis, S.; Agianian, B.; Cournia, Z. Investigating the structure and dynamics of the PIK3CA wild-type and H1047R oncogenic mutant. PLoS Comput. Biol. 2014, 10, e1003895. [Google Scholar] [CrossRef]
- Ikenoue, T.; Kanai, F.; Hikiba, Y.; Obata, T.; Tanaka, Y.; Imamura, J.; Ohta, M.; Jazag, A.; Guleng, B.; Tateishi, K.; et al. Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res. 2005, 65, 4562–4567. [Google Scholar] [CrossRef]
- Kang, S.; Bader, A.G.; Vogt, P.K. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc. Natl. Acad. Sci. USA 2005, 102, 802–807. [Google Scholar] [CrossRef]
- Bader, A.G.; Kang, S.; Vogt, P.K. Cancer-specific mutations in PIK3CA are oncogenic in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 1475–1479. [Google Scholar] [CrossRef] [PubMed]
- Dogruluk, T.; Tsang, Y.H.; Espitia, M.; Chen, F.; Chen, T.; Chong, Z.; Appadurai, V.; Dogruluk, A.; Eterovic, A.K.; Bonnen, P.E.; et al. Identification of Variant-Specific Functions of PIK3CA by Rapid Phenotyping of Rare Mutations. Cancer Res. 2015, 75, 5341–5354. [Google Scholar] [CrossRef] [PubMed]
- Rohrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef] [PubMed]
- Snaebjornsson, M.T.; Janaki-Raman, S.; Schulze, A. Greasing the Wheels of the Cancer Machine: The Role of Lipid Metabolism in Cancer. Cell Metab. 2020, 31, 62–76. [Google Scholar] [CrossRef]
- Georgescu, M.M.; Kirsch, K.H.; Akagi, T.; Shishido, T.; Hanafusa, H. The tumor-suppressor activity of PTEN is regulated by its carboxyl-terminal region. Proc. Natl. Acad. Sci. USA 1999, 96, 10182–10187. [Google Scholar] [CrossRef] [PubMed]
- Papa, A.; Wan, L.; Bonora, M.; Salmena, L.; Song, M.S.; Hobbs, R.M.; Lunardi, A.; Webster, K.; Ng, C.; Newton, R.H.; et al. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell 2014, 157, 595–610. [Google Scholar] [CrossRef]
- Rashmi, R.; DeSelm, C.; Helms, C.; Bowcock, A.; Rogers, B.E.; Rader, J.L.; Rader, J.; Grigsby, P.W.; Schwarz, J.K. AKT inhibitors promote cell death in cervical cancer through disruption of mTOR signaling and glucose uptake. PLoS ONE 2014, 9, e92948. [Google Scholar] [CrossRef]
- Leslie, N.R.; den Hertog, J. Mutant PTEN in Cancer: Worse Than Nothing. Cell 2014, 157, 527–529. [Google Scholar] [CrossRef][Green Version]
- Salmena, L.; Carracedo, A.; Pandolfi, P.P. Tenets of PTEN tumor suppression. Cell 2008, 133, 403–414. [Google Scholar] [CrossRef]
- Choi, S.W.; Lee, Y.; Shin, K.; Koo, H.; Kim, D.; Sa, J.K.; Cho, H.J.; Shin, H.M.; Lee, S.J.; Kim, H.; et al. Mutation-specific non-canonical pathway of PTEN as a distinct therapeutic target for glioblastoma. Cell Death Dis. 2021, 12, 374. [Google Scholar] [CrossRef]
- Kloosterhof, N.K.; Bralten, L.B.; Dubbink, H.J.; French, P.J.; van den Bent, M.J. Isocitrate dehydrogenase-1 mutations: A fundamentally new understanding of diffuse glioma? Lancet Oncol. 2011, 12, 83–91. [Google Scholar] [CrossRef]
- Clark, O.; Yen, K.; Mellinghoff, I.K. Molecular Pathways: Isocitrate Dehydrogenase Mutations in Cancer. Clin. Cancer Res. 2016, 22, 1837–1842. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2010, 465, 966. [Google Scholar] [CrossRef]
- Shim, E.H.; Livi, C.B.; Rakheja, D.; Tan, J.; Benson, D.; Parekh, V.; Kho, E.Y.; Ghosh, A.P.; Kirkman, R.; Velu, S.; et al. L-2-Hydroxyglutarate: An epigenetic modifier and putative oncometabolite in renal cancer. Cancer Discov. 2014, 4, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.J.; Metallo, C.M. Metabolic consequences of oncogenic IDH mutations. Pharmacol. Ther. 2015, 152, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ye, D.; Guan, K.L.; Xiong, Y. IDH1 and IDH2 mutations in tumorigenesis: Mechanistic insights and clinical perspectives. Clin. Cancer Res. 2012, 18, 5562–5571. [Google Scholar] [CrossRef]
- Reitman, Z.J.; Jin, G.; Karoly, E.D.; Spasojevic, I.; Yang, J.; Kinzler, K.W.; He, Y.; Bigner, D.D.; Vogelstein, B.; Yan, H. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc. Natl. Acad. Sci. USA 2011, 108, 3270–3275. [Google Scholar] [CrossRef] [PubMed]
- Esmaeili, M.; Hamans, B.C.; Navis, A.C.; van Horssen, R.; Bathen, T.F.; Gribbestad, I.S.; Leenders, W.P.; Heerschap, A. IDH1 R132H mutation generates a distinct phospholipid metabolite profile in glioma. Cancer Res. 2014, 74, 4898–4907. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, Z.; Hu, C.; Zhang, C.; Kovatcheva-Datchary, P.; Yu, D.; Liu, S.; Ren, F.; Wang, X.; Li, Y.; et al. Integrated Metabolomics and Lipidomics Analyses Reveal Metabolic Reprogramming in Human Glioma with IDH1 Mutation. J. Proteome Res. 2019, 18, 960–969. [Google Scholar] [CrossRef]
- Hudes, G.; Carducci, M.; Tomczak, P.; Dutcher, J.; Figlin, R.; Kapoor, A.; Staroslawska, E.; Sosman, J.; McDermott, D.; Bodrogi, I.; et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 2271–2281. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; Oudard, S.; Hutson, T.E.; Porta, C.; Bracarda, S.; Grunwald, V.; Thompson, J.A.; Figlin, R.A.; Hollaender, N.; et al. Efficacy of everolimus in advanced renal cell carcinoma: A double-blind, randomised, placebo-controlled phase III trial. Lancet 2008, 372, 449–456. [Google Scholar] [CrossRef]
- Benjamin, D.; Colombi, M.; Moroni, C.; Hall, M.N. Rapamycin passes the torch: A new generation of mTOR inhibitors. Nat. Rev. Drug Discov. 2011, 10, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A. Duvelisib: First Global Approval. Drugs 2018, 78, 1847–1853. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Alpelisib: First Global Approval. Drugs 2019, 79, 1249–1253. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Ivosidenib: First Global Approval. Drugs 2018, 78, 1509–1516. [Google Scholar] [CrossRef]
- Katso, R.; Okkenhaug, K.; Ahmadi, K.; White, S.; Timms, J.; Waterfield, M.D. Cellular function of phosphoinositide 3-kinases: Implications for development, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 2001, 17, 615–675. [Google Scholar] [CrossRef]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef]
- Courtney, K.D.; Corcoran, R.B.; Engelman, J.A. The PI3K pathway as drug target in human cancer. J. Clin. Oncol. 2010, 28, 1075–1083. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef]
- Cheung, L.W.; Yu, S.; Zhang, D.; Li, J.; Ng, P.K.; Panupinthu, N.; Mitra, S.; Ju, Z.; Yu, Q.; Liang, H.; et al. Naturally occurring neomorphic PIK3R1 mutations activate the MAPK pathway, dictating therapeutic response to MAPK pathway inhibitors. Cancer Cell 2014, 26, 479–494. [Google Scholar] [CrossRef]
- Seton-Rogers, S. Signalling: Uncovering new functions of PI3K mutations. Nat. Rev. Cancer 2014, 14, 766–767. [Google Scholar] [CrossRef] [PubMed]
- Moukarzel, L.A.; Da Cruz Paula, A.; Ferrando, L.; Hoang, T.; Sebastiao, A.P.M.; Pareja, F.; Park, K.J.; Jungbluth, A.A.; Capella, G.; Pineda, M.; et al. Clonal relationship and directionality of progression of synchronous endometrial and ovarian carcinomas in patients with DNA mismatch repair-deficiency associated syndromes. Mod. Pathol. 2021, 34, 994–1007. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Hillmann, P.; Hofmann, B.T.; Hart, J.R.; Vogt, P.K. Cancer-derived mutations in the regulatory subunit p85alpha of phosphoinositide 3-kinase function through the catalytic subunit p110alpha. Proc. Natl. Acad. Sci. USA 2010, 107, 15547–15552. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lau, A.Y.T.; Ng, A.S.N.; Aldehaiman, A.; Zhou, Y.; Ng, P.K.S.; Arold, S.T.; Cheung, L.W.T. Cancer-associated mutations in the p85alpha N-terminal SH2 domain activate a spectrum of receptor tyrosine kinases. Proc. Natl. Acad. Sci. USA 2021, 118, e2101751118. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Bode, A.M.; Dong, Z.; Lee, M.H. AKT as a Therapeutic Target for Cancer. Cancer Res. 2019, 79, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Martorana, F.; Motta, G.; Pavone, G.; Motta, L.; Stella, S.; Vitale, S.R.; Manzella, L.; Vigneri, P. AKT Inhibitors: New Weapons in the Fight Against Breast Cancer? Front. Pharmacol. 2021, 12, 662232. [Google Scholar] [CrossRef]
- Adamaki, M.; Zoumpourlis, V. Prostate Cancer Biomarkers: From diagnosis to prognosis and precision-guided therapeutics. Pharmacol. Ther. 2021, 228, 107932. [Google Scholar] [CrossRef]
- Deng, L.; Wang, C.; Spencer, E.; Yang, L.; Braun, A.; You, J.; Slaughter, C.; Pickart, C.; Chen, Z.J. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 2000, 103, 351–361. [Google Scholar] [CrossRef]
- Pickart, C.M. Ubiquitin in chains. Trends Biochem. Sci. 2000, 25, 544–548. [Google Scholar] [CrossRef]
- Spence, J.; Gali, R.R.; Dittmar, G.; Sherman, F.; Karin, M.; Finley, D. Cell cycle-regulated modification of the ribosome by a variant multiubiquitin chain. Cell 2000, 102, 67–76. [Google Scholar] [CrossRef]
- Huen, M.S.; Grant, R.; Manke, I.; Minn, K.; Yu, X.; Yaffe, M.B.; Chen, J. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 2007, 131, 901–914. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, D.; Riezman, H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 2007, 315, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Yumimoto, K.; Akiyoshi, S.; Ueo, H.; Sagara, Y.; Onoyama, I.; Ueo, H.; Ohno, S.; Mori, M.; Mimori, K.; Nakayama, K.I. F-box protein FBXW7 inhibits cancer metastasis in a non-cell-autonomous manner. J. Clin. Investig. 2015, 125, 621–635. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.H.; Bellon, M.; Nicot, C. FBXW7: A critical tumor suppressor of human cancers. Mol. Cancer 2018, 17, 115. [Google Scholar] [CrossRef]
- Close, V.; Close, W.; Kugler, S.J.; Reichenzeller, M.; Yosifov, D.Y.; Bloehdorn, J.; Pan, L.; Tausch, E.; Westhoff, M.A.; Dohner, H.; et al. FBXW7 mutations reduce binding of NOTCH1, leading to cleaved NOTCH1 accumulation and target gene activation in CLL. Blood 2019, 133, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Koepp, D.M.; Schaefer, L.K.; Ye, X.; Keyomarsi, K.; Chu, C.; Harper, J.W.; Elledge, S.J. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 2001, 294, 173–177. [Google Scholar] [CrossRef]
- Oberg, C.; Li, J.; Pauley, A.; Wolf, E.; Gurney, M.; Lendahl, U. The Notch intracellular domain is ubiquitinated and negatively regulated by the mammalian Sel-10 homolog. J. Biol. Chem. 2001, 276, 35847–35853. [Google Scholar] [CrossRef]
- Yada, M.; Hatakeyama, S.; Kamura, T.; Nishiyama, M.; Tsunematsu, R.; Imaki, H.; Ishida, N.; Okumura, F.; Nakayama, K.; Nakayama, K.I. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 2004, 23, 2116–2125. [Google Scholar] [CrossRef]
- Wei, W.; Jin, J.; Schlisio, S.; Harper, J.W.; Kaelin, W.G., Jr. The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell 2005, 8, 25–33. [Google Scholar] [CrossRef]
- Mao, J.H.; Kim, I.J.; Wu, D.; Climent, J.; Kang, H.C.; DelRosario, R.; Balmain, A. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science 2008, 321, 1499–1502. [Google Scholar] [CrossRef]
- Inuzuka, H.; Shaik, S.; Onoyama, I.; Gao, D.; Tseng, A.; Maser, R.S.; Zhai, B.; Wan, L.; Gutierrez, A.; Lau, A.W.; et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 2011, 471, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, L.; Zhao, K.; Thompson, P.R.; Hwang, Y.; Marmorstein, R.; Cole, P.A. The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature 2008, 451, 846–850. [Google Scholar] [CrossRef] [PubMed]
- Delvecchio, M.; Gaucher, J.; Aguilar-Gurrieri, C.; Ortega, E.; Panne, D. Structure of the p300 catalytic core and implications for chromatin targeting and HAT regulation. Nat. Struct. Mol. Biol. 2013, 20, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- Pao, G.M.; Janknecht, R.; Ruffner, H.; Hunter, T.; Verma, I.M. CBP/p300 interact with and function as transcriptional coactivators of BRCA1. Proc. Natl. Acad. Sci. USA 2000, 97, 1020–1025. [Google Scholar] [CrossRef]
- Chan, H.M.; Krstic-Demonacos, M.; Smith, L.; Demonacos, C.; La Thangue, N.B. Acetylation control of the retinoblastoma tumour-suppressor protein. Nat. Cell Biol. 2001, 3, 667–674. [Google Scholar] [CrossRef]
- Grossman, S.R. p300/CBP/p53 interaction and regulation of the p53 response. Eur. J. Biochem. 2001, 268, 2773–2778. [Google Scholar] [CrossRef]
- Friedrichs, N.; Jager, R.; Paggen, E.; Rudlowski, C.; Merkelbach-Bruse, S.; Schorle, H.; Buettner, R. Distinct spatial expression patterns of AP-2alpha and AP-2gamma in non-neoplastic human breast and breast cancer. Mod. Pathol. 2005, 18, 431–438. [Google Scholar] [CrossRef][Green Version]
- Salloum, R.; McConechy, M.K.; Mikael, L.G.; Fuller, C.; Drissi, R.; DeWire, M.; Nikbakht, H.; De Jay, N.; Yang, X.; Boue, D.; et al. Characterizing temporal genomic heterogeneity in pediatric high-grade gliomas. Acta Neuropathol. Commun. 2017, 5, 78. [Google Scholar] [CrossRef]
- Shi, D.; Pop, M.S.; Kulikov, R.; Love, I.M.; Kung, A.L.; Grossman, S.R. CBP and p300 are cytoplasmic E4 polyubiquitin ligases for p53. Proc. Natl. Acad. Sci. USA 2009, 106, 16275–16280. [Google Scholar] [CrossRef]
- Fahraeus, R.; Olivares-Illana, V. MDM2’s social network. Oncogene 2014, 33, 4365–4376. [Google Scholar] [CrossRef]
- Klein, A.M.; de Queiroz, R.M.; Venkatesh, D.; Prives, C. The roles and regulation of MDM2 and MDMX: It is not just about p53. Genes Dev. 2021, 35, 575–601. [Google Scholar] [CrossRef] [PubMed]
- Attar, N.; Kurdistani, S.K. Exploitation of EP300 and CREBBP Lysine Acetyltransferases by Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a026534. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Dominguez-Sola, D.; Chiarenza, A.; Fabbri, G.; Grunn, A.; Trifonov, V.; Kasper, L.H.; Lerach, S.; Tang, H.; Ma, J.; et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 2011, 471, 189–195. [Google Scholar] [CrossRef]
- Peifer, M.; Fernandez-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Merk, D.J.; Ohli, J.; Merk, N.D.; Thatikonda, V.; Morrissy, S.; Schoof, M.; Schmid, S.N.; Harrison, L.; Filser, S.; Ahlfeld, J.; et al. Opposing Effects of CREBBP Mutations Govern the Phenotype of Rubinstein-Taybi Syndrome and Adult SHH Medulloblastoma. Dev. Cell 2018, 44, 709–724.e706. [Google Scholar] [CrossRef] [PubMed]
- Mondello, P.; Tadros, S.; Teater, M.; Fontan, L.; Chang, A.Y.; Jain, N.; Yang, H.; Singh, S.; Ying, H.Y.; Chu, C.S.; et al. Selective Inhibition of HDAC3 Targets Synthetic Vulnerabilities and Activates Immune Surveillance in Lymphoma. Cancer Discov. 2020, 10, 440–459. [Google Scholar] [CrossRef]
- Deng, L.; Meng, T.; Chen, L.; Wei, W.; Wang, P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct. Target. Ther. 2020, 5, 11. [Google Scholar] [CrossRef]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.K.; Bradner, J.E.; Kaelin, W.G., Jr. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef]
- Stewart, A.K. Medicine. How thalidomide works against cancer. Science 2014, 343, 256–257. [Google Scholar] [CrossRef]
- Fricker, L.D. Proteasome Inhibitor Drugs. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 457–476. [Google Scholar] [CrossRef]
- Dick, L.R.; Fleming, P.E. Building on bortezomib: Second-generation proteasome inhibitors as anti-cancer therapy. Drug Discov. Today 2010, 15, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Nelson, W.J.; Nusse, R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science 2004, 303, 1483–1487. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Zhong, X.S.; Palmer, D.C.; Ji, Y.; Hinrichs, C.S.; Yu, Z.; Wrzesinski, C.; Boni, A.; Cassard, L.; Garvin, L.M.; et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat. Med. 2009, 15, 808–813. [Google Scholar] [CrossRef]
- Petersen, C.P.; Reddien, P.W. Wnt signaling and the polarity of the primary body axis. Cell 2009, 139, 1056–1068. [Google Scholar] [CrossRef]
- Spranger, S.; Gajewski, T.F. A new paradigm for tumor immune escape: Beta-catenin-driven immune exclusion. J. Immunother. Cancer 2015, 3, 43. [Google Scholar] [CrossRef] [PubMed]
- Junge, H.J. Ligand-Selective Wnt Receptor Complexes in CNS Blood Vessels: RECK and GPR124 Plugged In. Neuron 2017, 95, 983–985. [Google Scholar] [CrossRef]
- Miyoshi, Y.; Nagase, H.; Ando, H.; Horii, A.; Ichii, S.; Nakatsuru, S.; Aoki, T.; Miki, Y.; Mori, T.; Nakamura, Y. Somatic mutations of the APC gene in colorectal tumors: Mutation cluster region in the APC gene. Hum. Mol. Genet. 1992, 1, 229–233. [Google Scholar] [CrossRef]
- Fearnhead, N.S.; Britton, M.P.; Bodmer, W.F. The ABC of APC. Hum. Mol. Genet. 2001, 10, 721–733. [Google Scholar] [CrossRef]
- Azzopardi, D.; Dallosso, A.R.; Eliason, K.; Hendrickson, B.C.; Jones, N.; Rawstorne, E.; Colley, J.; Moskvina, V.; Frye, C.; Sampson, J.R.; et al. Multiple rare nonsynonymous variants in the adenomatous polyposis coli gene predispose to colorectal adenomas. Cancer Res. 2008, 68, 358–363. [Google Scholar] [CrossRef]
- Pai, S.G.; Carneiro, B.A.; Mota, J.M.; Costa, R.; Leite, C.A.; Barroso-Sousa, R.; Kaplan, J.B.; Chae, Y.K.; Giles, F.J. Wnt/beta-catenin pathway: Modulating anticancer immune response. J. Hematol. Oncol. 2017, 10, 101. [Google Scholar] [CrossRef]
- Imperial, R.; Ahmed, Z.; Toor, O.M.; Erdogan, C.; Khaliq, A.; Case, P.; Case, J.; Kennedy, K.; Cummings, L.S.; Melton, N.; et al. Comparative proteogenomic analysis of right-sided colon cancer, left-sided colon cancer and rectal cancer reveals distinct mutational profiles. Mol. Cancer 2018, 17, 177. [Google Scholar] [CrossRef] [PubMed]
- Ficari, F.; Cama, A.; Valanzano, R.; Curia, M.C.; Palmirotta, R.; Aceto, G.; Esposito, D.L.; Crognale, S.; Lombardi, A.; Messerini, L.; et al. APC gene mutations and colorectal adenomatosis in familial adenomatous polyposis. Br. J. Cancer 2000, 82, 348–353. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mihalatos, M.; Danielides, I.; Belogianni, J.; Harokopos, E.; Papadopoulou, E.; Kalimanis, G.; Tsiava, M.; Triantafillidis, J.K.; Kosmidis, P.A.; Fountzilas, G.; et al. Novel mutations of the APC gene in familial adenomatous polyposis in Greek patients. Cancer Genet. Cytogenet. 2003, 141, 65–70. [Google Scholar] [CrossRef]
- Kitagawa, M.; Hatakeyama, S.; Shirane, M.; Matsumoto, M.; Ishida, N.; Hattori, K.; Nakamichi, I.; Kikuchi, A.; Nakayama, K.; Nakayama, K. An F-box protein, FWD1, mediates ubiquitin-dependent proteolysis of beta-catenin. EMBO J. 1999, 18, 2401–2410. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, A. Tumor formation by genetic mutations in the components of the Wnt signaling pathway. Cancer Sci. 2003, 94, 225–229. [Google Scholar] [CrossRef]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef]
- Rubinfeld, B.; Robbins, P.; El-Gamil, M.; Albert, I.; Porfiri, E.; Polakis, P. Stabilization of beta-catenin by genetic defects in melanoma cell lines. Science 1997, 275, 1790–1792. [Google Scholar] [CrossRef]
- Liu, C.; Li, Y.; Semenov, M.; Han, C.; Baeg, G.H.; Tan, Y.; Zhang, Z.; Lin, X.; He, X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 2002, 108, 837–847. [Google Scholar] [CrossRef]
- Rebouissou, S.; Franconi, A.; Calderaro, J.; Letouze, E.; Imbeaud, S.; Pilati, C.; Nault, J.C.; Couchy, G.; Laurent, A.; Balabaud, C.; et al. Genotype-phenotype correlation of CTNNB1 mutations reveals different ss-catenin activity associated with liver tumor progression. Hepatology 2016, 64, 2047–2061. [Google Scholar] [CrossRef]
- Tu, J.; Park, S.; Yu, W.; Zhang, S.; Wu, L.; Carmon, K.; Liu, Q.J. The most common RNF43 mutant G659Vfs*41 is fully functional in inhibiting Wnt signaling and unlikely to play a role in tumorigenesis. Sci. Rep. 2019, 9, 18557. [Google Scholar] [CrossRef]
- Hao, H.X.; Xie, Y.; Zhang, Y.; Charlat, O.; Oster, E.; Avello, M.; Lei, H.; Mickanin, C.; Liu, D.; Ruffner, H.; et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature 2012, 485, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.K.; Spit, M.; Jordens, I.; Low, T.Y.; Stange, D.E.; van de Wetering, M.; van Es, J.H.; Mohammed, S.; Heck, A.J.; Maurice, M.M.; et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature 2012, 488, 665–669. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Spit, M.; Fenderico, N.; Jordens, I.; Radaszkiewicz, T.; Lindeboom, R.G.; Bugter, J.M.; Cristobal, A.; Ootes, L.; van Osch, M.; Janssen, E.; et al. RNF43 truncations trap CK1 to drive niche-independent self-renewal in cancer. EMBO J. 2020, 39, e103932. [Google Scholar] [CrossRef]
- Anastas, J.N.; Moon, R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef]
- Jung, Y.S.; Park, J.I. Wnt signaling in cancer: Therapeutic targeting of Wnt signaling beyond beta-catenin and the destruction complex. Exp. Mol. Med. 2020, 52, 183–191. [Google Scholar] [CrossRef]
- Hori, K.; Sen, A.; Artavanis-Tsakonas, S. Notch signaling at a glance. J. Cell Sci. 2013, 126, 2135–2140. [Google Scholar] [CrossRef]
- Bray, S.J. Notch signalling in context. Nat. Rev. Mol. Cell Biol. 2016, 17, 722–735. [Google Scholar] [CrossRef]
- Siebel, C.; Lendahl, U. Notch Signaling in Development, Tissue Homeostasis, and Disease. Physiol. Rev. 2017, 97, 1235–1294. [Google Scholar] [CrossRef]
- Pancewicz, J.; Taylor, J.M.; Datta, A.; Baydoun, H.H.; Waldmann, T.A.; Hermine, O.; Nicot, C. Notch signaling contributes to proliferation and tumor formation of human T-cell leukemia virus type 1-associated adult T-cell leukemia. Proc. Natl. Acad. Sci. USA 2010, 107, 16619–16624. [Google Scholar] [CrossRef]
- Yeh, C.H.; Bellon, M.; Pancewicz-Wojtkiewicz, J.; Nicot, C. Oncogenic mutations in the FBXW7 gene of adult T-cell leukemia patients. Proc. Natl. Acad. Sci. USA 2016, 113, 6731–6736. [Google Scholar] [CrossRef] [PubMed]
- Oswald, F.; Tauber, B.; Dobner, T.; Bourteele, S.; Kostezka, U.; Adler, G.; Liptay, S.; Schmid, R.M. p300 acts as a transcriptional coactivator for mammalian Notch-1. Mol. Cell Biol. 2001, 21, 7761–7774. [Google Scholar] [CrossRef] [PubMed]
- Wallberg, A.E.; Pedersen, K.; Lendahl, U.; Roeder, R.G. p300 and PCAF act cooperatively to mediate transcriptional activation from chromatin templates by notch intracellular domains in vitro. Mol. Cell Biol. 2002, 22, 7812–7819. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Cai, K.; Xu, P.P.; Wang, L.; Huang, C.X.; Fang, Y.; Cheng, S.; Sun, X.J.; Liu, F.; Huang, J.Y.; et al. CREBBP/EP300 mutations promoted tumor progression in diffuse large B-cell lymphoma through altering tumor-associated macrophage polarization via FBXW7-NOTCH-CCL2/CSF1 axis. Signal Transduct. Target. Ther. 2021, 6, 10. [Google Scholar] [CrossRef]
- Majumder, S.; Crabtree, J.S.; Golde, T.E.; Minter, L.M.; Osborne, B.A.; Miele, L. Targeting Notch in oncology: The path forward. Nat. Rev. Drug Discov. 2021, 20, 125–144. [Google Scholar] [CrossRef]
- Pannuti, A.; Foreman, K.; Rizzo, P.; Osipo, C.; Golde, T.; Osborne, B.; Miele, L. Targeting Notch to target cancer stem cells. Clin. Cancer Res. 2010, 16, 3141–3152. [Google Scholar] [CrossRef]
- Takebe, N.; Nguyen, D.; Yang, S.X. Targeting notch signaling pathway in cancer: Clinical development advances and challenges. Pharmacol. Ther. 2014, 141, 140–149. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Matthews, H.K.; Bertoli, C.; de Bruin, R.A.M. Cell cycle control in cancer. Nat. Rev. Mol. Cell Biol. 2022, 23, 74–88. [Google Scholar] [CrossRef]
- Cowell, J.K.; Smith, T.; Bia, B. Frequent constitutional C to T mutations in CGA-arginine codons in the RB1 gene produce premature stop codons in patients with bilateral (hereditary) retinoblastoma. Eur. J. Hum. Genet. 1994, 2, 281–290. [Google Scholar] [CrossRef]
- Richter, S.; Vandezande, K.; Chen, N.; Zhang, K.; Sutherland, J.; Anderson, J.; Han, L.; Panton, R.; Branco, P.; Gallie, B. Sensitive and efficient detection of RB1 gene mutations enhances care for families with retinoblastoma. Am. J. Hum. Genet. 2003, 72, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Ayari Jeridi, H.; Bouguila, H.; Ansperger-Rescher, B.; Baroudi, O.; Mdimegh, I.; Omran, I.; Charradi, K.; Bouzayene, H.; Benammar-Elgaaied, A.; Lohmann, D.R. Genetic testing in Tunisian families with heritable retinoblastoma using a low cost approach permits accurate risk prediction in relatives and reveals incomplete penetrance in adults. Exp. Eye Res. 2014, 124, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.A.; Suzawa, K.; Jordan, E.; Zehir, A.; Ni, A.; Kim, R.; Kris, M.G.; Hellmann, M.D.; Li, B.T.; Somwar, R.; et al. Concurrent Alterations in EGFR-Mutant Lung Cancers Associated with Resistance to EGFR Kinase Inhibitors and Characterization of MTOR as a Mediator of Resistance. Clin. Cancer Res. 2018, 24, 3108–3118. [Google Scholar] [CrossRef] [PubMed]
- Trimarchi, J.M.; Lees, J.A. Sibling rivalry in the E2F family. Nat. Rev. Mol. Cell Biol. 2002, 3, 11–20. [Google Scholar] [CrossRef]
- Burkhart, D.L.; Sage, J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 2008, 8, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Hannon, G.J.; Zhang, H.; Casso, D.; Kobayashi, R.; Beach, D. p21 is a universal inhibitor of cyclin kinases. Nature 1993, 366, 701–704. [Google Scholar] [CrossRef]
- Waga, S.; Hannon, G.J.; Beach, D.; Stillman, B. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature 1994, 369, 574–578. [Google Scholar] [CrossRef]
- Jackson, R.J.; Adnane, J.; Coppola, D.; Cantor, A.; Sebti, S.M.; Pledger, W.J. Loss of the cell cycle inhibitors p21(Cip1) and p27(Kip1) enhances tumorigenesis in knockout mouse models. Oncogene 2002, 21, 8486–8497. [Google Scholar] [CrossRef]
- Poole, A.J.; Heap, D.; Carroll, R.E.; Tyner, A.L. Tumor suppressor functions for the Cdk inhibitor p21 in the mouse colon. Oncogene 2004, 23, 8128–8134. [Google Scholar] [CrossRef]
- Forster, K.; Obermeier, A.; Mitina, O.; Simon, N.; Warmuth, M.; Krause, G.; Hallek, M. Role of p21(WAF1/CIP1) as an attenuator of both proliferative and drug-induced apoptotic signals in BCR-ABL-transformed hematopoietic cells. Ann. Hematol. 2008, 87, 183–193. [Google Scholar] [CrossRef]
- Suzuki, A.; Tsutomi, Y.; Miura, M.; Akahane, K. Caspase 3 inactivation to suppress Fas-mediated apoptosis: Identification of binding domain with p21 and ILP and inactivation machinery by p21. Oncogene 1999, 18, 1239–1244. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Megyesi, J.; Safirstein, R.L.; Price, P.M. Identification of the functional domain of p21(WAF1/CIP1) that protects cells from cisplatin cytotoxicity. Am. J. Physiol. Renal Physiol. 2005, 289, F514–F520. [Google Scholar] [CrossRef] [PubMed]
- Suski, J.M.; Braun, M.; Strmiska, V.; Sicinski, P. Targeting cell-cycle machinery in cancer. Cancer Cell 2021, 39, 759–778. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [PubMed]
- Karran, P. DNA double strand break repair in mammalian cells. Curr. Opin. Genet. Dev. 2000, 10, 144–150. [Google Scholar] [CrossRef]
- Chen, C.C.; Feng, W.; Lim, P.X.; Kass, E.M.; Jasin, M. Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer. Annu. Rev. Cancer Biol. 2018, 2, 313–336. [Google Scholar] [CrossRef]
- van Gent, D.C.; Hoeijmakers, J.H.; Kanaar, R. Chromosomal stability and the DNA double-stranded break connection. Nat. Rev. Genet. 2001, 2, 196–206. [Google Scholar] [CrossRef]
- Khanna, K.K.; Jackson, S.P. DNA double-strand breaks: Signaling, repair and the cancer connection. Nat. Genet. 2001, 27, 247–254. [Google Scholar] [CrossRef]
- Caburet, S.; Heddar, A.; Dardillac, E.; Creux, H.; Lambert, M.; Messiaen, S.; Tourpin, S.; Livera, G.; Lopez, B.S.; Misrahi, M. Homozygous hypomorphic BRCA2 variant in primary ovarian insufficiency without cancer or Fanconi anaemia trait. J. Med. Genet. 2020, 58, 125–134. [Google Scholar] [CrossRef]
- Sun, P.; Li, Y.; Chao, X.; Li, J.; Luo, R.; Li, M.; He, J. Clinical characteristics and prognostic implications of BRCA-associated tumors in males: A pan-tumor survey. BMC Cancer 2020, 20, 994. [Google Scholar] [CrossRef]
- Lee, H. Cycling with BRCA2 from DNA repair to mitosis. Exp. Cell Res. 2014, 329, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Moynahan, M.E.; Pierce, A.J.; Jasin, M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol. Cell 2001, 7, 263–272. [Google Scholar] [CrossRef]
- Tutt, A.; Bertwistle, D.; Valentine, J.; Gabriel, A.; Swift, S.; Ross, G.; Griffin, C.; Thacker, J.; Ashworth, A. Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. EMBO J. 2001, 20, 4704–4716. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Doty, T.; Gibson, B.; Heyer, W.D. Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat. Struct. Mol. Biol. 2010, 17, 1260–1262. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, T.R.; Friebel, T.M.; Friedman, E.; Hamann, U.; Huo, D.; Kwong, A.; Olah, E.; Olopade, O.I.; Solano, A.R.; Teo, S.H.; et al. Mutational spectrum in a worldwide study of 29,700 families with BRCA1 or BRCA2 mutations. Hum. Mutat. 2018, 39, 593–620. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Chen, J.; Plug, A.; Xiao, Y.; Weaver, D.; Feunteun, J.; Ashley, T.; Livingston, D.M. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell 1997, 88, 265–275. [Google Scholar] [CrossRef]
- Zhong, Q.; Chen, C.F.; Li, S.; Chen, Y.; Wang, C.C.; Xiao, J.; Chen, P.L.; Sharp, Z.D.; Lee, W.H. Association of BRCA1 with the hRad50-hMre11-p95 complex and the DNA damage response. Science 1999, 285, 747–750. [Google Scholar] [CrossRef]
- Clark, S.L.; Rodriguez, A.M.; Snyder, R.R.; Hankins, G.D.; Boehning, D. Structure-Function Of The Tumor Suppressor BRCA1. Comput. Struct. Biotechnol. J. 2012, 1, e201204005. [Google Scholar] [CrossRef]
- Christou, C.M.; Kyriacou, K. BRCA1 and Its Network of Interacting Partners. Biology 2013, 2, 40–63. [Google Scholar] [CrossRef]
- Bunting, S.F.; Callen, E.; Wong, N.; Chen, H.T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef]
- Zhang, J.; Yan, Z.; Wang, Y.; Wang, Y.; Guo, X.; Jing, J.; Dong, X.; Dong, S.; Liu, X.; Yu, X.; et al. Cancer-associated 53BP1 mutations induce DNA damage repair defects. Cancer Lett. 2021, 501, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Panier, S.; Boulton, S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014, 15, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2008, 7, 2902–2906. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Sandhu, S.K.; Carden, C.P.; de Bono, J.S. Poly(ADP-ribose) polymerase (PARP) inhibitors: Exploiting a synthetic lethal strategy in the clinic. CA Cancer J. Clin. 2011, 61, 31–49. [Google Scholar] [CrossRef] [PubMed]
- Furgason, J.M.; Bahassi, E.M. Targeting DNA repair mechanisms in cancer. Pharmacol. Ther. 2013, 137, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; O’Carrigan, B.; Jackson, S.P.; Yap, T.A. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov. 2017, 7, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef]
- Deeks, E.D. Olaparib: First global approval. Drugs 2015, 75, 231–240. [Google Scholar] [CrossRef]
- Scott, L.J. Niraparib: First Global Approval. Drugs 2017, 77, 1029–1034. [Google Scholar] [CrossRef]
- Syed, Y.Y. Rucaparib: First Global Approval. Drugs 2017, 77, 585–592. [Google Scholar] [CrossRef]
- Hoy, S.M. Talazoparib: First Global Approval. Drugs 2018, 78, 1939–1946. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Balasubramaniam, S.; Zhang, H.; Berman, T.; Narayan, P.; Suzman, D.; Bloomquist, E.; Tang, S.; Gong, Y.; Sridhara, R.; et al. FDA Approval Summary: Olaparib Monotherapy or in Combination with Bevacizumab for the Maintenance Treatment of Patients with Advanced Ovarian Cancer. Oncologist 2021, 26, e164–e172. [Google Scholar] [CrossRef] [PubMed]
- Kwok, M.; Davies, N.; Agathanggelou, A.; Smith, E.; Oldreive, C.; Petermann, E.; Stewart, G.; Brown, J.; Lau, A.; Pratt, G.; et al. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood 2016, 127, 582–595. [Google Scholar] [CrossRef]
- Menolfi, D.; Zha, S. ATM, ATR and DNA-PKcs kinases-the lessons from the mouse models: Inhibition not equal deletion. Cell Biosci. 2020, 10, 8. [Google Scholar] [CrossRef]
- Topatana, W.; Juengpanich, S.; Li, S.; Cao, J.; Hu, J.; Lee, J.; Suliyanto, K.; Ma, D.; Zhang, B.; Chen, M.; et al. Advances in synthetic lethality for cancer therapy: Cellular mechanism and clinical translation. J. Hematol. Oncol. 2020, 13, 118. [Google Scholar] [CrossRef]
- David, C.J.; Manley, J.L. Alternative pre-mRNA splicing regulation in cancer: Pathways and programs unhinged. Genes Dev. 2010, 24, 2343–2364. [Google Scholar] [CrossRef] [PubMed]
- Dvinge, H.; Kim, E.; Abdel-Wahab, O.; Bradley, R.K. RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer 2016, 16, 413–430. [Google Scholar] [CrossRef]
- Climente-Gonzalez, H.; Porta-Pardo, E.; Godzik, A.; Eyras, E. The Functional Impact of Alternative Splicing in Cancer. Cell Rep. 2017, 20, 2215–2226. [Google Scholar] [CrossRef]
- Rahman, M.A.; Krainer, A.R.; Abdel-Wahab, O. SnapShot: Splicing Alterations in Cancer. Cell 2020, 180, 208.e201. [Google Scholar] [CrossRef]
- Legare, S.; Cavallone, L.; Mamo, A.; Chabot, C.; Sirois, I.; Magliocco, A.; Klimowicz, A.; Tonin, P.N.; Buchanan, M.; Keilty, D.; et al. The Estrogen Receptor Cofactor SPEN Functions as a Tumor Suppressor and Candidate Biomarker of Drug Responsiveness in Hormone-Dependent Breast Cancers. Cancer Res. 2015, 75, 4351–4363. [Google Scholar] [CrossRef]
- Ariyoshi, M.; Schwabe, J.W. A conserved structural motif reveals the essential transcriptional repression function of Spen proteins and their role in developmental signaling. Genes Dev. 2003, 17, 1909–1920. [Google Scholar] [CrossRef] [PubMed]
- Hiriart, E.; Gruffat, H.; Buisson, M.; Mikaelian, I.; Keppler, S.; Meresse, P.; Mercher, T.; Bernard, O.A.; Sergeant, A.; Manet, E. Interaction of the Epstein-Barr virus mRNA export factor EB2 with human Spen proteins SHARP, OTT1, and a novel member of the family, OTT3, links Spen proteins with splicing regulation and mRNA export. J. Biol. Chem. 2005, 280, 36935–36945. [Google Scholar] [CrossRef]
- Bartkowiak, B.; Greenleaf, A.L. Expression, purification, and identification of associated proteins of the full-length hCDK12/CyclinK complex. J. Biol. Chem. 2015, 290, 1786–1795. [Google Scholar] [CrossRef] [PubMed]
- Ekumi, K.M.; Paculova, H.; Lenasi, T.; Pospichalova, V.; Bosken, C.A.; Rybarikova, J.; Bryja, V.; Geyer, M.; Blazek, D.; Barboric, M. Ovarian carcinoma CDK12 mutations misregulate expression of DNA repair genes via deficient formation and function of the Cdk12/CycK complex. Nucleic Acids Res. 2015, 43, 2575–2589. [Google Scholar] [CrossRef]
- Blazek, D.; Kohoutek, J.; Bartholomeeusen, K.; Johansen, E.; Hulinkova, P.; Luo, Z.; Cimermancic, P.; Ule, J.; Peterlin, B.M. The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev. 2011, 25, 2158–2172. [Google Scholar] [CrossRef]
- Krajewska, M.; Dries, R.; Grassetti, A.V.; Dust, S.; Gao, Y.; Huang, H.; Sharma, B.; Day, D.S.; Kwiatkowski, N.; Pomaville, M.; et al. CDK12 loss in cancer cells affects DNA damage response genes through premature cleavage and polyadenylation. Nat. Commun. 2019, 10, 1757. [Google Scholar] [CrossRef] [PubMed]
- Tien, J.F.; Mazloomian, A.; Cheng, S.G.; Hughes, C.S.; Chow, C.C.T.; Canapi, L.T.; Oloumi, A.; Trigo-Gonzalez, G.; Bashashati, A.; Xu, J.; et al. CDK12 regulates alternative last exon mRNA splicing and promotes breast cancer cell invasion. Nucleic Acids Res. 2017, 45, 6698–6716. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas, N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Schneeweiss, A.; Park-Simon, T.-W.; Albanell, J.; Lassen, U.; Cortés, J.; Dieras, V.; May, M.; Schindler, C.; Marmé, F.; Cejalvo, J.M.; et al. Phase Ib study evaluating safety and clinical activity of the anti-HER3 antibody lumretuzumab combined with the anti-HER2 antibody pertuzumab and paclitaxel in HER3-positive, HER2-low metastatic breast cancer. Investig. New Drugs 2018, 36, 848–859. [Google Scholar] [CrossRef]
- Duncan, R.; Bazar, L.; Michelotti, G.; Tomonaga, T.; Krutzsch, H.; Avigan, M.; Levens, D. A sequence-specific, single-strand binding protein activates the far upstream element of c-myc and defines a new DNA-binding motif. Genes Dev. 1994, 8, 465–480. [Google Scholar] [CrossRef]
- Hsiao, H.H.; Nath, A.; Lin, C.Y.; Folta-Stogniew, E.J.; Rhoades, E.; Braddock, D.T. Quantitative characterization of the interactions among c-myc transcriptional regulators FUSE, FBP, and FIR. Biochemistry 2010, 49, 4620–4634. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.G.; Singh, R.K.; Mohammad, F.; Bebee, T.W.; Chandler, D.S. The splicing factor FUBP1 is required for the efficient splicing of oncogene MDM2 pre-mRNA. J. Biol. Chem. 2014, 289, 17350–17364. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Abdel-Wahab, O. Therapeutic targeting of splicing in cancer. Nat. Med. 2016, 22, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Urbanski, L.M.; Leclair, N.; Anczukow, O. Alternative-splicing defects in cancer: Splicing regulators and their downstream targets, guiding the way to novel cancer therapeutics. Wiley Interdiscip. Rev. RNA 2018, 9, e1476. [Google Scholar] [CrossRef]
- Zhang, Y.; Qian, J.; Gu, C.; Yang, Y. Alternative splicing and cancer: A systematic review. Signal Transduct. Target. Ther. 2021, 6, 78. [Google Scholar] [CrossRef]
- Elman, J.S.; Ni, T.K.; Mengwasser, K.E.; Jin, D.; Wronski, A.; Elledge, S.J.; Kuperwasser, C. Identification of FUBP1 as a Long Tail Cancer Driver and Widespread Regulator of Tumor Suppressor and Oncogene Alternative Splicing. Cell Rep. 2019, 28, 3435–3449.e3435. [Google Scholar] [CrossRef]
- Sailo, B.L.; Banik, K.; Girisa, S.; Bordoloi, D.; Fan, L.; Halim, C.E.; Wang, H.; Kumar, A.P.; Zheng, D.; Mao, X.; et al. FBXW7 in Cancer: What Has Been Unraveled Thus Far? Cancers 2019, 11, 246. [Google Scholar] [CrossRef]
- Wyatt, D.W.; Feng, W.; Conlin, M.P.; Yousefzadeh, M.J.; Roberts, S.A.; Mieczkowski, P.; Wood, R.D.; Gupta, G.P.; Ramsden, D.A. Essential Roles for Polymerase theta-Mediated End Joining in the Repair of Chromosome Breaks. Mol. Cell 2016, 63, 662–673. [Google Scholar] [CrossRef]
- Feng, W.; Simpson, D.A.; Carvajal-Garcia, J.; Price, B.A.; Kumar, R.J.; Mose, L.E.; Wood, R.D.; Rashid, N.; Purvis, J.E.; Parker, J.S.; et al. Genetic determinants of cellular addiction to DNA polymerase theta. Nat. Commun. 2019, 10, 4286. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Signaling | Drugs |
| ABL1 | Bosutinib, Brigatinib, Dasatinib, Ibrutinib, Imatinib, Niotinib, Pazopanib, Ponatinib, Regorafenib, Sunitinib, Tivozanib, Vandetanib |
| ALK | Alectinib, Brigatinib, Ceritinib, Crizotinib, Entrectinib, Gilteritinib, Lorlatinib, Sunitinib |
| BRAF | Binimetinib, Cobimetinib, Dabrafenib, Dasatinib, Encorafenib, Regorafenib, Sorafenib, Trametinib, Vemurafenib |
| EGFR | Afatinib, Brigatinib, Ceritinib, Cetuximab, Dacomitinib, Erlotinib, Gefitinib, Ibrutinib, Lapatinib, Lorlatinib, Mobocertinib, Necitumumab, Neratinib, Osimertinib, Panitumumab, Vandetanib. |
| ERBB2 | Afatinib, Dacomitinib, Everolimus, Gefitinib, Ibrutinib, Lapatinib, Margetuximab, Metformin, Mobocertinib, Nelfinavir, Neratinib, Pertuzumab, Sirolimus, Temsirolimus, Trastuzumab, Trastuzumab Deruxtecan, Trastuzumab Emtansine, Tucatinib |
| FGFR1 | Brigatinib, Dasatinib, Erdafitinib, Infigratinib, Lenvatinib, Nintedanib, Pazopanib, Ponatinib, Regorafenib, Sorafenib, Sunitinib, Tivozanib, Vandetanib |
| FGFR2 | Brigatinib, Ceritinib, Erdafitinib, Infigratinib, Lenvatinib, Nintedanib, Pazopanib, Regorafenib, Sorafenib, Sunitinib, Vandetanib |
| FLT3 | Brigatinib, Cabozatinib, Ceritinib, Fedratinib, Gilteritinib, Ibrutinib, Midostaurin, Nintedanib, Pexidartinib, Sorafenib, Sunitinib, Vandetanib |
| KIT | Axitinib, Cabozatinib, Dasatinib, Fedratinib, Imatinib, Infigratinib, Lenvatinib, Midostaurin, Nilotinib, Pazopanib, Pexidartinib, Ponatinib, Regorafenib, Sorafenib, Sunitinib, Tivozanib |
| KRAS | Binimetinib, Cobimetinib, Sotorasib, Trametinib |
| MET | Cabozatinib, Capmatinib, Crizotinib, Tepotinib, Tivozanib |
| NRAS | Binimetinib, Cobimetinib, Trametinib |
| NTRK1 | Cabozatinib, Crizotinib, Entrectinib, Larotrectinib, Lorlatinib, Regorafenib, Sorafenib, Sunitinib |
| NTRK2 | Cabozatinib, Entrectinib, Larotrectinib, Lorlatinib, Sorafenib, Sunitinib |
| PDGFRA | Axitinib, Dasatinib, Ibrutinib, Imatinib, Lenvatinib, Midostaurin, Nilotinib, Nintedanib, Olaratumab, Pazopanib, Ponatinib, Regorafenib, Sorafenib, Sunitinib, Tivozanib |
| PTPN11 | Binimetinib, Cobimetinib, Trametinib |
| RET | Alectinib, Brigatinib, Cabozatinib, Ceritinib, Fedratinib, Ibrutinib, Lenvatinib, Pazopanib, Ponatinib, Pralsetinib, Regorafenib, Selpercatinib, Sorafenib, Sunitinib, Vandetanib |
| ROS1 | Brigatinib, Cabozatinib, Ceritinib, Crizotinib, Entrectinib, Lorlatinib |
| Gene Signaling | Drugs |
|---|---|
| IDH1 | Ivositenib |
| PIK3CA | Alpelisib, Copanlisib, Duvelisib, Everolimus, Metformin, Midostaurin, Sirolimus, Temsirolimus |
| PIK3R1 | Alpelisib, Copanlisib, Duvelisib, Everolimus, Idelalisib, Midostaurin, Sirolimus, Temsirolimus, Umbralisib |
| PTEN | Alpelisib, Copanlisib, Duvelisib, Everolimus, Metformin, Midostaurin, Sirolimus, Temsirolimus |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karagiannakos, A.; Adamaki, M.; Tsintarakis, A.; Vojtesek, B.; Fåhraeus, R.; Zoumpourlis, V.; Karakostis, K. Targeting Oncogenic Pathways in the Era of Personalized Oncology: A Systemic Analysis Reveals Highly Mutated Signaling Pathways in Cancer Patients and Potential Therapeutic Targets. Cancers 2022, 14, 664. https://doi.org/10.3390/cancers14030664
Karagiannakos A, Adamaki M, Tsintarakis A, Vojtesek B, Fåhraeus R, Zoumpourlis V, Karakostis K. Targeting Oncogenic Pathways in the Era of Personalized Oncology: A Systemic Analysis Reveals Highly Mutated Signaling Pathways in Cancer Patients and Potential Therapeutic Targets. Cancers. 2022; 14(3):664. https://doi.org/10.3390/cancers14030664
Chicago/Turabian StyleKaragiannakos, Alexandros, Maria Adamaki, Antonis Tsintarakis, Borek Vojtesek, Robin Fåhraeus, Vassilis Zoumpourlis, and Konstantinos Karakostis. 2022. "Targeting Oncogenic Pathways in the Era of Personalized Oncology: A Systemic Analysis Reveals Highly Mutated Signaling Pathways in Cancer Patients and Potential Therapeutic Targets" Cancers 14, no. 3: 664. https://doi.org/10.3390/cancers14030664
APA StyleKaragiannakos, A., Adamaki, M., Tsintarakis, A., Vojtesek, B., Fåhraeus, R., Zoumpourlis, V., & Karakostis, K. (2022). Targeting Oncogenic Pathways in the Era of Personalized Oncology: A Systemic Analysis Reveals Highly Mutated Signaling Pathways in Cancer Patients and Potential Therapeutic Targets. Cancers, 14(3), 664. https://doi.org/10.3390/cancers14030664