
KRAS and RAS-MAPK Pathway Deregulation in Mature B Cell Lymphoproliferative Disorders

 , ,
, ,
Abstract
Simple Summary
Abstract
1. Introduction
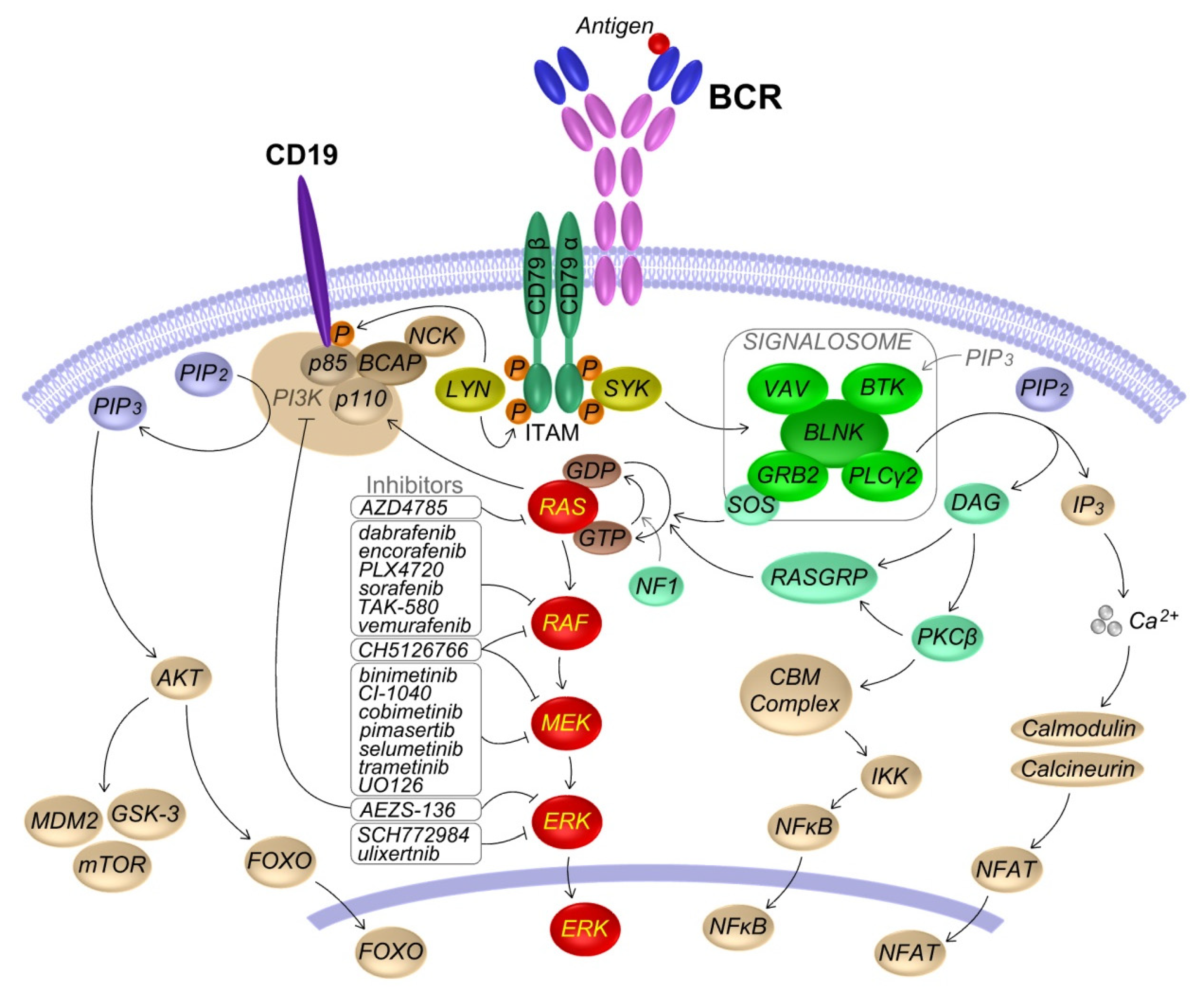
2. KRAS and RAS-MAPK Pathway in B Lymphocytes
2.1. RAS-MAPK Signaling
2.2. Role of the BCR Induced RAS-MAPK Pathway Activation in B Cells
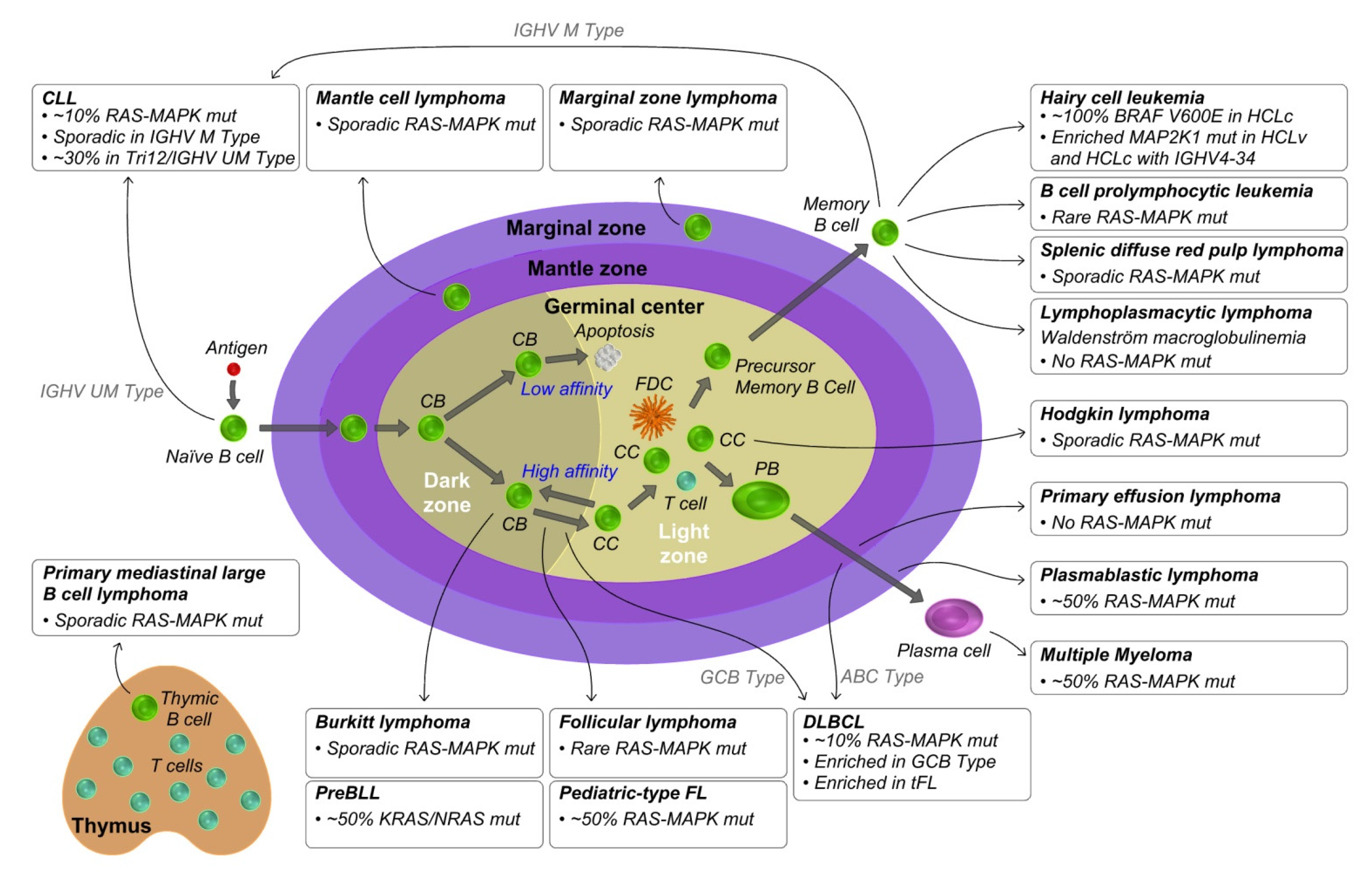
3. KRAS and RAS-MAPK Pathway Aberrations in Mature B Cell Lymphoproliferative Disorders
3.1. Mantle Cell Lymphoma
3.2. Diffuse Large B Cell Lymphoma
3.3. Follicular Lymphoma
3.4. Burkitt Lymphoma
3.5. Hodgkin Lymphoma
3.6. Primary Mediastinal Large B Cell Lymphoma
3.7. Chronic Lymphocytic Leukemia
3.8. Hairy Cell Leukemia
3.9. Other Post Germinal Center Lymphomas
3.10. Multiple Myeloma
3.11. Other Plasma Cell Related Diseases
4. RAS-MAPK Pathway Inhibitors
4.1. RAF Inhibitors
4.1.1. Clinical Setting
4.1.2. Preclinical Studies
4.2. MEK Inhibitors
4.2.1. Clinical Setting
4.2.2. Preclinical Studies
5. Oncogenic RAS/RAF Mouse Model
5.1. KRAS
5.2. NRAS
5.3. BRAF
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Uprety, D.; Adjei, A.A. KRAS: From undruggable to a druggable Cancer Target. Cancer Treat. Rev. 2020, 89. [Google Scholar] [CrossRef] [PubMed]
- Neri, A.; Murphy, J.P.; Cro, L.; Ferrero, D.; Tarella, C.; Baldini, L.; Dalla-Favera, R. Ras oncogene mutation in multiple myeloma. J. Exp. Med. 1989, 170, 1715–1725. [Google Scholar] [CrossRef] [PubMed]
- Degirmenci, U.; Yap, J.; Sim, Y.R.M.; Qin, S.; Hu, J. Drug resistance in targeted cancer therapies with RAF inhibitors. Cancer Drug Resist. 2021, 4, 665–683. [Google Scholar] [CrossRef]
- Cuesta, C.; Arevalo-Alameda, C.; Castellano, E. The Importance of Being PI3K in the RAS Signaling Network. Genes 2021, 12, 1094. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Baba, Y. B Cell Receptor Signaling. Adv. Exp. Med. Biol. 2020, 1254, 23–36. [Google Scholar] [CrossRef]
- McLaurin, J.D.; Weiner, O.D. Multiple sources of signal amplification within the B-cell Ras/MAPK pathway. Mol. Biol. Cell 2019, 30, 1610–1620. [Google Scholar] [CrossRef]
- Shaffer, A.L., 3rd; Young, R.M.; Staudt, L.M. Pathogenesis of human B cell lymphomas. Annu. Rev. Immunol. 2012, 30, 565–610. [Google Scholar] [CrossRef]
- Basso, K.; Dalla-Favera, R. Germinal centres and B cell lymphomagenesis. Nat. Rev. Immunol. 2015, 15, 172–184. [Google Scholar] [CrossRef]
- Nadeu, F.; Martin-Garcia, D.; Clot, G.; Diaz-Navarro, A.; Duran-Ferrer, M.; Navarro, A.; Vilarrasa-Blasi, R.; Kulis, M.; Royo, R.; Gutierrez-Abril, J.; et al. Genomic and epigenomic insights into the origin, pathogenesis, and clinical behavior of mantle cell lymphoma subtypes. Blood 2020, 136, 1419–1432. [Google Scholar] [CrossRef]
- Wu, C.; de Miranda, N.F.; Chen, L.; Wasik, A.M.; Mansouri, L.; Jurczak, W.; Galazka, K.; Dlugosz-Danecka, M.; Machaczka, M.; Zhang, H.; et al. Genetic heterogeneity in primary and relapsed mantle cell lymphomas: Impact of recurrent CARD11 mutations. Oncotarget 2016, 7, 38180–38190. [Google Scholar] [CrossRef]
- Jain, P.; Kanagal-Shamanna, R.; Zhang, S.; Ahmed, M.; Ghorab, A.; Zhang, L.; Ok, C.Y.; Li, S.; Hagemeister, F.; Zeng, D.; et al. Long-term outcomes and mutation profiling of patients with mantle cell lymphoma (MCL) who discontinued ibrutinib. Br. J. Haematol. 2018, 183, 578–587. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018, 24, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.G.; Stojanov, P.; Lawrence, M.S.; Auclair, D.; Chapuy, B.; Sougnez, C.; Cruz-Gordillo, P.; Knoechel, B.; Asmann, Y.W.; Slager, S.L.; et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 3879–3884. [Google Scholar] [CrossRef]
- Reddy, A.; Zhang, J.; Davis, N.S.; Moffitt, A.B.; Love, C.L.; Waldrop, A.; Leppa, S.; Pasanen, A.; Meriranta, L.; Karjalainen-Lindsberg, M.L.; et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 2017, 171, 481–494.e415. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef] [PubMed]
- Okosun, J.; Bodor, C.; Wang, J.; Araf, S.; Yang, C.Y.; Pan, C.; Boller, S.; Cittaro, D.; Bozek, M.; Iqbal, S.; et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat. Genet. 2014, 46, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Louissaint, A., Jr.; Schafernak, K.T.; Geyer, J.T.; Kovach, A.E.; Ghandi, M.; Gratzinger, D.; Roth, C.G.; Paxton, C.N.; Kim, S.; Namgyal, C.; et al. Pediatric-type nodal follicular lymphoma: A Biologically distinct lymphoma with frequent MAPK pathway mutations. Blood 2016, 128, 1093–1100. [Google Scholar] [CrossRef]
- Love, C.; Sun, Z.; Jima, D.; Li, G.; Zhang, J.; Miles, R.; Richards, K.L.; Dunphy, C.H.; Choi, W.W.; Srivastava, G.; et al. The genetic landscape of mutations in Burkitt lymphoma. Nat. Genet. 2012, 44, 1321–1325. [Google Scholar] [CrossRef]
- Grande, B.M.; Gerhard, D.S.; Jiang, A.; Griner, N.B.; Abramson, J.S.; Alexander, T.B.; Allen, H.; Ayers, L.W.; Bethony, J.M.; Bhatia, K.; et al. Genome-wide discovery of somatic coding and noncoding mutations in pediatric endemic and sporadic Burkitt lymphoma. Blood 2019, 133, 1313–1324. [Google Scholar] [CrossRef]
- Schmitz, R.; Young, R.M.; Ceribelli, M.; Jhavar, S.; Xiao, W.; Zhang, M.; Wright, G.; Shaffer, A.L.; Hodson, D.J.; Buras, E.; et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 2012, 490, 116–120. [Google Scholar] [CrossRef]
- Richter, J.; Schlesner, M.; Hoffmann, S.; Kreuz, M.; Leich, E.; Burkhardt, B.; Rosolowski, M.; Ammerpohl, O.; Wagener, R.; Bernhart, S.H.; et al. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat. Genet. 2012, 44, 1316–1320. [Google Scholar] [CrossRef] [PubMed]
- Tiacci, E.; Ladewig, E.; Schiavoni, G.; Penson, A.; Fortini, E.; Pettirossi, V.; Wang, Y.; Rosseto, A.; Venanzi, A.; Vlasevska, S.; et al. Pervasive mutations of JAK-STAT pathway genes in classical Hodgkin lymphoma. Blood 2018, 131, 2454–2465. [Google Scholar] [CrossRef]
- Spina, V.; Bruscaggin, A.; Cuccaro, A.; Martini, M.; Di Trani, M.; Forestieri, G.; Manzoni, M.; Condoluci, A.; Arribas, A.; Terzi-Di-Bergamo, L.; et al. Circulating tumor DNA reveals genetics, clonal evolution, and residual disease in classical Hodgkin lymphoma. Blood 2018, 131, 2413–2425. [Google Scholar] [CrossRef] [PubMed]
- Greil, R.; Pleyer, L.; Jansko, B.; Feierabend, C.; Rettenbacher, L.; Stiefel, O.; Rass, C.; Morre, P.; Neureiter, D.; Greil-Ressler, S. Sequential immunotherapy in a patient with primary refractory Hodgkin lymphoma and novel mutations. Oncotarget 2018, 9, 20928–20940. [Google Scholar] [CrossRef] [PubMed]
- Landau, D.A.; Tausch, E.; Taylor-Weiner, A.N.; Stewart, C.; Reiter, J.G.; Bahlo, J.; Kluth, S.; Bozic, I.; Lawrence, M.; Bottcher, S.; et al. Mutations driving CLL and their evolution in progression and relapse. Nature 2015, 526, 525–530. [Google Scholar] [CrossRef]
- Puente, X.S.; Bea, S.; Valdes-Mas, R.; Villamor, N.; Gutierrez-Abril, J.; Martin-Subero, J.I.; Munar, M.; Rubio-Perez, C.; Jares, P.; Aymerich, M.; et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015, 526, 519–524. [Google Scholar] [CrossRef]
- Herling, C.D.; Klaumunzer, M.; Rocha, C.K.; Altmuller, J.; Thiele, H.; Bahlo, J.; Kluth, S.; Crispatzu, G.; Herling, M.; Schiller, J.; et al. Complex karyotypes and KRAS and POT1 mutations impact outcome in CLL after chlorambucil-based chemotherapy or chemoimmunotherapy. Blood 2016, 128, 395–404. [Google Scholar] [CrossRef]
- Takahashi, K.; Hu, B.; Wang, F.; Yan, Y.; Kim, E.; Vitale, C.; Patel, K.P.; Strati, P.; Gumbs, C.; Little, L.; et al. Clinical implications of cancer gene mutations in patients with chronic lymphocytic leukemia treated with lenalidomide. Blood 2018, 131, 1820–1832. [Google Scholar] [CrossRef]
- Gimenez, N.; Martinez-Trillos, A.; Montraveta, A.; Lopez-Guerra, M.; Rosich, L.; Nadeu, F.; Valero, J.G.; Aymerich, M.; Magnano, L.; Rozman, M.; et al. Mutations in the RAS-BRAF-MAPK-ERK pathway define a specific subgroup of patients with adverse clinical features and provide new therapeutic options in chronic lymphocytic leukemia. Haematologica 2019, 104, 576–586. [Google Scholar] [CrossRef]
- Vendramini, E.; Bomben, R.; Pozzo, F.; Benedetti, D.; Bittolo, T.; Rossi, F.M.; Dal Bo, M.; Rabe, K.G.; Pozzato, G.; Zaja, F.; et al. KRAS, NRAS, and BRAF mutations are highly enriched in trisomy 12 chronic lymphocytic leukemia and are associated with shorter treatment-free survival. Leukemia 2019, 33, 2111–2115. [Google Scholar] [CrossRef]
- Nadeu, F.; Clot, G.; Delgado, J.; Martin-Garcia, D.; Baumann, T.; Salaverria, I.; Bea, S.; Pinyol, M.; Jares, P.; Navarro, A.; et al. Clinical impact of the subclonal architecture and mutational complexity in chronic lymphocytic leukemia. Leukemia 2018, 32, 645–653. [Google Scholar] [CrossRef]
- Tiacci, E.; Trifonov, V.; Schiavoni, G.; Holmes, A.; Kern, W.; Martelli, M.P.; Pucciarini, A.; Bigerna, B.; Pacini, R.; Wells, V.A.; et al. BRAF mutations in hairy-cell leukemia. N. Engl. J. Med. 2011, 364, 2305–2315. [Google Scholar] [CrossRef]
- Waterfall, J.J.; Arons, E.; Walker, R.L.; Pineda, M.; Roth, L.; Killian, J.K.; Abaan, O.D.; Davis, S.R.; Kreitman, R.J.; Meltzer, P.S. High prevalence of MAP2K1 mutations in variant and IGHV4-34-expressing hairy-cell leukemias. Nat. Genet. 2014, 46, 8–10. [Google Scholar] [CrossRef]
- Durham, B.H.; Getta, B.; Dietrich, S.; Taylor, J.; Won, H.; Bogenberger, J.M.; Scott, S.; Kim, E.; Chung, Y.R.; Chung, S.S.; et al. Genomic analysis of hairy cell leukemia identifies novel recurrent genetic alterations. Blood 2017, 130, 1644–1648. [Google Scholar] [CrossRef]
- Tschernitz, S.; Flossbach, L.; Bonengel, M.; Roth, S.; Rosenwald, A.; Geissinger, E. Alternative BRAF mutations in BRAF V600E-negative hairy cell leukaemias. Br. J. Haematol. 2014, 165, 529–533. [Google Scholar] [CrossRef]
- Parry, M.; Rose-Zerilli, M.J.; Ljungstrom, V.; Gibson, J.; Wang, J.; Walewska, R.; Parker, H.; Parker, A.; Davis, Z.; Gardiner, A.; et al. Genetics and Prognostication in Splenic Marginal Zone Lymphoma: Revelations from Deep Sequencing. Clin. Cancer Res. 2015, 21, 4174–4183. [Google Scholar] [CrossRef]
- Rossi, D.; Trifonov, V.; Fangazio, M.; Bruscaggin, A.; Rasi, S.; Spina, V.; Monti, S.; Vaisitti, T.; Arruga, F.; Fama, R.; et al. The coding genome of splenic marginal zone lymphoma: Activation of NOTCH2 and other pathways regulating marginal zone development. J. Exp. Med. 2012, 209, 1537–1551. [Google Scholar] [CrossRef]
- Jallades, L.; Baseggio, L.; Sujobert, P.; Huet, S.; Chabane, K.; Callet-Bauchu, E.; Verney, A.; Hayette, S.; Desvignes, J.P.; Salgado, D.; et al. Exome sequencing identifies recurrent BCOR alterations and the absence of KLF2, TNFAIP3 and MYD88 mutations in splenic diffuse red pulp small B-cell lymphoma. Haematologica 2017, 102, 1758–1766. [Google Scholar] [CrossRef]
- Jaramillo Oquendo, C.; Parker, H.; Oscier, D.; Ennis, S.; Gibson, J.; Strefford, J.C. Systematic Review of Somatic Mutations in Splenic Marginal Zone Lymphoma. Sci. Rep. 2019, 9, 10444. [Google Scholar] [CrossRef]
- Pillonel, V.; Juskevicius, D.; Ng, C.K.Y.; Bodmer, A.; Zettl, A.; Jucker, D.; Dirnhofer, S.; Tzankov, A. High-throughput sequencing of nodal marginal zone lymphomas identifies recurrent BRAF mutations. Leukemia 2018, 32, 2412–2426. [Google Scholar] [CrossRef]
- Hyeon, J.; Lee, B.; Shin, S.H.; Yoo, H.Y.; Kim, S.J.; Kim, W.S.; Park, W.Y.; Ko, Y.H. Targeted deep sequencing of gastric marginal zone lymphoma identified alterations of TRAF3 and TNFAIP3 that were mutually exclusive for MALT1 rearrangement. Mod. Pathol. 2018, 31, 1418–1428. [Google Scholar] [CrossRef] [PubMed]
- Cascione, L.; Rinaldi, A.; Bruscaggin, A.; Tarantelli, C.; Arribas, A.J.; Kwee, I.; Pecciarini, L.; Mensah, A.A.; Spina, V.; Chung, E.Y.L.; et al. Novel insights into the genetics and epigenetics of MALT lymphoma unveiled by next generation sequencing analyses. Haematologica 2019, 104, e558–e561. [Google Scholar] [CrossRef] [PubMed]
- Martinez, D.; Navarro, A.; Martinez-Trillos, A.; Molina-Urra, R.; Gonzalez-Farre, B.; Salaverria, I.; Nadeu, F.; Enjuanes, A.; Clot, G.; Costa, D.; et al. NOTCH1, TP53, and MAP2K1 Mutations in Splenic Diffuse Red Pulp Small B-cell Lymphoma Are Associated With Progressive Disease. Am. J. Surg. Pathol. 2016, 40, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Langabeer, S.E.; Quinn, F.; O’Brien, D.; McElligott, A.M.; Kelly, J.; Browne, P.V.; Vandenberghe, E. Incidence of the BRAF V600E mutation in chronic lymphocytic leukaemia and prolymphocytic leukaemia. Leuk. Res. 2012, 36, 483–484. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef]
- Walker, B.A.; Boyle, E.M.; Wardell, C.P.; Murison, A.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Johnson, D.C.; Kaiser, M.F.; Melchor, L.; et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients With Newly Diagnosed Myeloma. J. Clin. Oncol. 2015, 33, 3911–3920. [Google Scholar] [CrossRef]
- Xu, J.; Pfarr, N.; Endris, V.; Mai, E.K.; Md Hanafiah, N.H.; Lehners, N.; Penzel, R.; Weichert, W.; Ho, A.D.; Schirmacher, P.; et al. Molecular signaling in multiple myeloma: Association of RAS/RAF mutations and MEK/ERK pathway activation. Oncogenesis 2017, 6, e337. [Google Scholar] [CrossRef]
- Liu, Z.; Filip, I.; Gomez, K.; Engelbrecht, D.; Meer, S.; Lalloo, P.N.; Patel, P.; Perner, Y.; Zhao, J.; Wang, J.; et al. Genomic characterization of HIV-associated plasmablastic lymphoma identifies pervasive mutations in the JAK-STAT pathway. Blood Cancer Discov. 2020, 1, 112–125. [Google Scholar] [CrossRef]
- Ramis-Zaldivar, J.E.; Gonzalez-Farre, B.; Nicolae, A.; Pack, S.; Clot, G.; Nadeu, F.; Mottok, A.; Horn, H.; Song, J.Y.; Fu, K.; et al. MAP-kinase and JAK-STAT pathways dysregulation in plasmablastic lymphoma. Haematologica 2021, 10, 2682. [Google Scholar] [CrossRef]
- Garcia-Reyero, J.; Martinez Magunacelaya, N.; Gonzalez de Villambrosia, S.; Loghavi, S.; Gomez Mediavilla, A.; Tonda, R.; Beltran, S.; Gut, M.; Perena Gonzalez, A.; d’Amore, E.; et al. Genetic lesions in MYC and STAT3 drive oncogenic transcription factor overexpression in plasmablastic lymphoma. Haematologica 2021, 106, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Frontzek, F.; Staiger, A.M.; Zapukhlyak, M.; Xu, W.; Bonzheim, I.; Borgmann, V.; Sander, P.; Baptista, M.J.; Heming, J.N.; Berning, P.; et al. Molecular and functional profiling identifies therapeutically targetable vulnerabilities in plasmablastic lymphoma. Nat. Commun. 2021, 12, 5183. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Bea, S.; Jares, P.; Campo, E. Molecular Pathogenesis of Mantle Cell Lymphoma. Hematol. Oncol. Clin. N. Am. 2020, 34, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Jares, P.; Colomer, D.; Campo, E. Molecular pathogenesis of mantle cell lymphoma. J. Clin. Investig. 2012, 122, 3416–3423. [Google Scholar] [CrossRef] [PubMed]
- Navarro, A.; Clot, G.; Royo, C.; Jares, P.; Hadzidimitriou, A.; Agathangelidis, A.; Bikos, V.; Darzentas, N.; Papadaki, T.; Salaverria, I.; et al. Molecular subsets of mantle cell lymphoma defined by the IGHV mutational status and SOX11 expression have distinct biologic and clinical features. Cancer Res. 2012, 72, 5307–5316. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Dalla-Favera, R. Genetics of diffuse large B-cell lymphoma. Blood 2018, 131, 2307–2319. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef]
- Rosenwald, A.; Wright, G.; Chan, W.C.; Connors, J.M.; Campo, E.; Fisher, R.I.; Gascoyne, R.D.; Muller-Hermelink, H.K.; Smeland, E.B.; Giltnane, J.M.; et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N. Engl. J. Med. 2002, 346, 1937–1947. [Google Scholar] [CrossRef]
- Davis, R.E.; Ngo, V.N.; Lenz, G.; Tolar, P.; Young, R.M.; Romesser, P.B.; Kohlhammer, H.; Lamy, L.; Zhao, H.; Yang, Y.; et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 2010, 463, 88–92. [Google Scholar] [CrossRef]
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.H.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470, 115–119. [Google Scholar] [CrossRef]
- Mandelbaum, J.; Bhagat, G.; Tang, H.; Mo, T.; Brahmachary, M.; Shen, Q.; Chadburn, A.; Rajewsky, K.; Tarakhovsky, A.; Pasqualucci, L.; et al. BLIMP1 is a tumor suppressor gene frequently disrupted in activated B cell-like diffuse large B cell lymphoma. Cancer Cell 2010, 18, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, M.; Grau, M.; Lenze, D.; Wenzel, S.S.; Wolf, A.; Wollert-Wulf, B.; Dietze, K.; Nogai, H.; Storek, B.; Madle, H.; et al. PTEN loss defines a PI3K/AKT pathway-dependent germinal center subtype of diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 12420–12425. [Google Scholar] [CrossRef] [PubMed]
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.J.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Muppidi, J.R.; Schmitz, R.; Green, J.A.; Xiao, W.; Larsen, A.B.; Braun, S.E.; An, J.; Xu, Y.; Rosenwald, A.; Ott, G.; et al. Loss of signalling via Galpha13 in germinal centre B-cell-derived lymphoma. Nature 2014, 516, 254–258. [Google Scholar] [CrossRef]
- Iqbal, J.; Sanger, W.G.; Horsman, D.E.; Rosenwald, A.; Pickering, D.L.; Dave, B.; Dave, S.; Xiao, L.; Cao, K.; Zhu, Q.; et al. BCL2 translocation defines a unique tumor subset within the germinal center B-cell-like diffuse large B-cell lymphoma. Am. J. Pathol. 2004, 165, 159–166. [Google Scholar] [CrossRef]
- Intlekofer, A.M.; Joffe, E.; Batlevi, C.L.; Hilden, P.; He, J.; Seshan, V.E.; Zelenetz, A.D.; Palomba, M.L.; Moskowitz, C.H.; Portlock, C.; et al. Integrated DNA/RNA targeted genomic profiling of diffuse large B-cell lymphoma using a clinical assay. Blood Cancer J. 2018, 8, 60. [Google Scholar] [CrossRef]
- Nedergaard, T.; Guldberg, P.; Ralfkiaer, E.; Zeuthen, J. A one-step DGGE scanning method for detection of mutations in the K-, N-, and H-ras oncogenes: Mutations at codons 12, 13 and 61 are rare in B-cell non-Hodgkin’s lymphoma. Int. J. Cancer 1997, 71, 364–369. [Google Scholar] [CrossRef]
- Rahrmann, E.P.; Wolf, N.K.; Otto, G.M.; Heltemes-Harris, L.; Ramsey, L.B.; Shu, J.; LaRue, R.S.; Linden, M.A.; Rathe, S.K.; Starr, T.K.; et al. Sleeping Beauty Screen Identifies RREB1 and Other Genetic Drivers in Human B-cell Lymphoma. Mol. Cancer Res. 2019, 17, 567–582. [Google Scholar] [CrossRef]
- Knief, J.; Gebauer, N.; Bernard, V.; Schemme, J.; Reddemann, K.; Gebauer, J.; Rades, D.; Brabant, G.; Lehnert, H.; Feller, A.C.; et al. Oncogenic mutations and chromosomal aberrations in primary extranodal diffuse large B-cell lymphomas of the thyroid--a study of 21 cases. J. Clin. Endocrinol. Metab. 2015, 100, 754–762. [Google Scholar] [CrossRef]
- Aggarwal, N.; Swerdlow, S.H.; Kelly, L.M.; Ogilvie, J.B.; Nikiforova, M.N.; Sathanoori, M.; Nikiforov, Y.E. Thyroid carcinoma-associated genetic mutations also occur in thyroid lymphomas. Mod. Pathol. 2012, 25, 1203–1211. [Google Scholar] [CrossRef]
- Milpied, P.; Gandhi, A.K.; Cartron, G.; Pasqualucci, L.; Tarte, K.; Nadel, B.; Roulland, S. Follicular lymphoma dynamics. Adv. Immunol. 2021, 150, 43–103. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Roulland, S.; Gloghini, A.; Younes, A.; von Keudell, G.; Lopez-Guillermo, A.; Fitzgibbon, J. Follicular lymphoma. Nat. Rev. Dis. Primers 2019, 5, 83. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Jurinovic, V.; Kridel, R.; Hoster, E.; Staiger, A.M.; Szczepanowski, M.; Pott, C.; Kopp, N.; Murakami, M.; Horn, H.; et al. Integration of gene mutations in risk prognostication for patients receiving first-line immunochemotherapy for follicular lymphoma: A retrospective analysis of a prospective clinical trial and validation in a population-based registry. Lancet Oncol. 2015, 16, 1111–1122. [Google Scholar] [CrossRef]
- Green, M.R.; Kihira, S.; Liu, C.L.; Nair, R.V.; Salari, R.; Gentles, A.J.; Irish, J.; Stehr, H.; Vicente-Duenas, C.; Romero-Camarero, I.; et al. Mutations in early follicular lymphoma progenitors are associated with suppressed antigen presentation. Proc. Natl. Acad. Sci. USA 2015, 112, E1116–E1125. [Google Scholar] [CrossRef] [PubMed]
- Egan, C.; Lack, J.; Skarshaug, S.; Pham, T.A.; Abdullaev, Z.; Xi, L.; Pack, S.; Pittaluga, S.; Jaffe, E.S.; Raffeld, M. The mutational landscape of histiocytic sarcoma associated with lymphoid malignancy. Mod. Pathol. 2021, 34, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Pericart, S.; Waysse, C.; Siegfried, A.; Struski, S.; Delabesse, E.; Laurent, C.; Evrard, S. Subsequent development of histiocytic sarcoma and follicular lymphoma: Cytogenetics and next-generation sequencing analyses provide evidence for transdifferentiation of early common lymphoid precursor-a case report and review of literature. Virchows Arch. 2020, 476, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Elenitoba-Johnson, K.S.J.; Jenson, S.D.; Abbott, R.T.; Palais, R.A.; Bohling, S.D.; Lin, Z.; Tripp, S.; Shami, P.J.; Wang, L.Y.; Coupland, R.W.; et al. Involvement of multiple signaling pathways in follicular lymphoma transformation: p38-mitogen-activated protein kinase as a target for therapy. Proc. Natl. Acad. Sci. USA 2003, 100, 7259–7264. [Google Scholar] [CrossRef]
- Schmidt, J.; Ramis-Zaldivar, J.E.; Nadeu, F.; Gonzalez-Farre, B.; Navarro, A.; Egan, C.; Montes-Mojarro, I.A.; Marafioti, T.; Cabecadas, J.; van der Walt, J.; et al. Mutations of MAP2K1 are frequent in pediatric-type follicular lymphoma and result in ERK pathway activation. Blood 2017, 130, 323–327. [Google Scholar] [CrossRef]
- Dozzo, M.; Carobolante, F.; Donisi, P.M.; Scattolin, A.; Maino, E.; Sancetta, R.; Viero, P.; Bassan, R. Burkitt lymphoma in adolescents and young adults: Management challenges. Adolesc. Health Med. 2017, 8, 11–29. [Google Scholar] [CrossRef]
- Molyneux, E.M.; Rochford, R.; Griffin, B.; Newton, R.; Jackson, G.; Menon, G.; Harrison, C.J.; Israels, T.; Bailey, S. Burkitt’s lymphoma. Lancet 2012, 379, 1234–1244. [Google Scholar] [CrossRef]
- Zhang, J.; Meng, L.; Jiang, W.; Zhang, H.; Zhou, A.; Zeng, N. Identification of clinical molecular targets for childhood Burkitt lymphoma. Transl. Oncol. 2020, 13, 100855. [Google Scholar] [CrossRef]
- Wagener, R.; Lopez, C.; Kleinheinz, K.; Bausinger, J.; Aukema, S.M.; Nagel, I.; Toprak, U.H.; Seufert, J.; Altmuller, J.; Thiele, H.; et al. IG-MYC (+) neoplasms with precursor B-cell phenotype are molecularly distinct from Burkitt lymphomas. Blood 2018, 132, 2280–2285. [Google Scholar] [CrossRef]
- Mathas, S.; Hartmann, S.; Kuppers, R. Hodgkin lymphoma: Pathology and Biology. Semin. Hematol. 2016, 53, 139–147. [Google Scholar] [CrossRef]
- Kuppers, R. The biology of Hodgkin’s lymphoma. Nat. Rev. Cancer 2009, 9, 15–27. [Google Scholar] [CrossRef]
- Reichel, J.; Chadburn, A.; Rubinstein, P.G.; Giulino-Roth, L.; Tam, W.; Liu, Y.; Gaiolla, R.; Eng, K.; Brody, J.; Inghirami, G.; et al. Flow sorting and exome sequencing reveal the oncogenome of primary Hodgkin and Reed-Sternberg cells. Blood 2015, 125, 1061–1072. [Google Scholar] [CrossRef]
- Wienand, K.; Chapuy, B.; Stewart, C.; Dunford, A.J.; Wu, D.; Kim, J.; Kamburov, A.; Wood, T.R.; Cader, F.Z.; Ducar, M.D.; et al. Genomic analyses of flow-sorted Hodgkin Reed-Sternberg cells reveal complementary mechanisms of immune evasion. Blood Adv. 2019, 3, 4065–4080. [Google Scholar] [CrossRef]
- Venanzi, A.; Marra, A.; Schiavoni, G.; Milner, S.G.; Limongello, R.; Santi, A.; Pettirossi, V.; Ultimo, S.; Tasselli, L.; Pucciarini, A.; et al. Dissecting Clonal Hematopoiesis in Tissues of Classical Hodgkin Lymphoma Patients. Blood Cancer Discov. 2021, 2, 216–225. [Google Scholar] [CrossRef]
- Ahmed, Z.; Afridi, S.S.; Shahid, Z.; Zamani, Z.; Rehman, S.; Aiman, W.; Khan, M.; Mir, M.A.; Awan, F.T.; Anwer, F.; et al. Primary Mediastinal B-Cell Lymphoma: A 2021 Update on Genetics, Diagnosis, and Novel Therapeutics. Clin. Lymphoma Myeloma Leuk. 2021, 21, e865–e875. [Google Scholar] [CrossRef]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Wienand, K.; Kamburov, A.; Griffin, G.K.; Chen, P.H.; Lako, A.; Redd, R.A.; et al. Genomic analyses of PMBL reveal new drivers and mechanisms of sensitivity to PD-1 blockade. Blood 2019, 134, 2369–2382. [Google Scholar] [CrossRef]
- Mottok, A.; Hung, S.S.; Chavez, E.A.; Woolcock, B.; Telenius, A.; Chong, L.C.; Meissner, B.; Nakamura, H.; Rushton, C.; Vigano, E.; et al. Integrative genomic analysis identifies key pathogenic mechanisms in primary mediastinal large B-cell lymphoma. Blood 2019, 134, 802–813. [Google Scholar] [CrossRef]
- Fabbri, G.; Dalla-Favera, R. The molecular pathogenesis of chronic lymphocytic leukaemia. Nat. Rev. Cancer 2016, 16, 145–162. [Google Scholar] [CrossRef]
- Tadmor, T.; Levy, I. Richter Transformation in Chronic Lymphocytic Leukemia: Update in the Era of Novel Agents. Cancers 2021, 13, 5141. [Google Scholar] [CrossRef]
- Guieze, R.; Wu, C.J. Genomic and epigenomic heterogeneity in chronic lymphocytic leukemia. Blood 2015, 126, 445–453. [Google Scholar] [CrossRef]
- Dal Bo, M.; Bulian, P.; Bomben, R.; Zucchetto, A.; Rossi, F.M.; Pozzo, F.; Tissino, E.; Benedetti, D.; Bittolo, T.; Nanni, P.; et al. CD49d prevails over the novel recurrent mutations as independent prognosticator of overall survival in chronic lymphocytic leukemia. Leukemia 2016, 30, 2011–2018. [Google Scholar] [CrossRef]
- Bulian, P.; Shanafelt, T.D.; Fegan, C.; Zucchetto, A.; Cro, L.; Nuckel, H.; Baldini, L.; Kurtova, A.V.; Ferrajoli, A.; Burger, J.A.; et al. CD49d is the strongest flow cytometry-based predictor of overall survival in chronic lymphocytic leukemia. J. Clin. Oncol. 2014, 32, 897–904. [Google Scholar] [CrossRef]
- Leeksma, A.C.; Taylor, J.; Wu, B.; Gardner, J.R.; He, J.; Nahas, M.; Gonen, M.; Alemayehu, W.G.; Te Raa, D.; Walther, T.; et al. Clonal diversity predicts adverse outcome in chronic lymphocytic leukemia. Leukemia 2019, 33, 390–402. [Google Scholar] [CrossRef]
- Sellar, R.S.; Fend, F.; Akarca, A.U.; Agostinelli, C.; Shende, V.; Quintanilla-Martinez, L.; Stein, H.; Pileri, S.A.; Linch, D.; Marafioti, T. BRAF(V600E) mutations are found in Richter syndrome and may allow targeted therapy in a subset of patients. Br. J. Haematol. 2015, 170, 282–285. [Google Scholar] [CrossRef]
- Roos-Weil, D.; Nguyen-Khac, F.; Chevret, S.; Touzeau, C.; Roux, C.; Lejeune, J.; Cosson, A.; Mathis, S.; Feugier, P.; Lepretre, S.; et al. Mutational and cytogenetic analyses of 188 CLL patients with trisomy 12: A retrospective study from the French Innovative Leukemia Organization (FILO) working group. Genes Chromosomes Cancer 2018, 57, 533–540. [Google Scholar] [CrossRef]
- Del Giudice, I.; Rossi, D.; Chiaretti, S.; Marinelli, M.; Tavolaro, S.; Gabrielli, S.; Laurenti, L.; Marasca, R.; Rasi, S.; Fangazio, M.; et al. NOTCH1 mutations in +12 chronic lymphocytic leukemia (CLL) confer an unfavorable prognosis, induce a distinctive transcriptional profiling and refine the intermediate prognosis of +12 CLL. Haematologica 2012, 97, 437–441. [Google Scholar] [CrossRef]
- Herling, C.D.; Abedpour, N.; Weiss, J.; Schmitt, A.; Jachimowicz, R.D.; Merkel, O.; Cartolano, M.; Oberbeck, S.; Mayer, P.; Berg, V.; et al. Clonal dynamics towards the development of venetoclax resistance in chronic lymphocytic leukemia. Nat. Commun. 2018, 9, 727. [Google Scholar] [CrossRef]
- Pandzic, T.; Larsson, J.; He, L.; Kundu, S.; Ban, K.; Akhtar-Ali, M.; Hellstrom, A.R.; Schuh, A.; Clifford, R.; Blakemore, S.J.; et al. Transposon Mutagenesis Reveals Fludarabine Resistance Mechanisms in Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2016, 22, 6217–6227. [Google Scholar] [CrossRef]
- Murali, I.; Kasar, S.; Naeem, A.; Tyekucheva, S.; Khalsa, J.K.; Thrash, E.M.; Itchaki, G.; Livitz, D.; Leshchiner, I.; Dong, S.; et al. Activation of the MAPK pathway mediates resistance to PI3K inhibitors in chronic lymphocytic leukemia. Blood 2021, 138, 44–56. [Google Scholar] [CrossRef]
- Tiacci, E.; Pettirossi, V.; Schiavoni, G.; Falini, B. Genomics of Hairy Cell Leukemia. J. Clin. Oncol. 2017, 35, 1002–1010. [Google Scholar] [CrossRef]
- Grever, M.R.; Blachly, J.S.; Andritsos, L.A. Hairy cell leukemia: Update on molecular profiling and therapeutic advances. Blood Rev. 2014, 28, 197–203. [Google Scholar] [CrossRef]
- Tiacci, E.; Schiavoni, G.; Martelli, M.P.; Boveri, E.; Pacini, R.; Tabarrini, A.; Zibellini, S.; Santi, A.; Pettirossi, V.; Fortini, E.; et al. Constant activation of the RAF-MEK-ERK pathway as a diagnostic and therapeutic target in hairy cell leukemia. Haematologica 2013, 98, 635–639. [Google Scholar] [CrossRef]
- Kamiguti, A.S.; Harris, R.J.; Slupsky, J.R.; Baker, P.K.; Cawley, J.C.; Zuzel, M. Regulation of hairy-cell survival through constitutive activation of mitogen-activated protein kinase pathways. Oncogene 2003, 22, 2272–2284. [Google Scholar] [CrossRef]
- Pettirossi, V.; Santi, A.; Imperi, E.; Russo, G.; Pucciarini, A.; Bigerna, B.; Schiavoni, G.; Fortini, E.; Spanhol-Rosseto, A.; Sportoletti, P.; et al. BRAF inhibitors reverse the unique molecular signature and phenotype of hairy cell leukemia and exert potent antileukemic activity. Blood 2015, 125, 1207–1216. [Google Scholar] [CrossRef]
- Xi, L.; Arons, E.; Navarro, W.; Calvo, K.R.; Stetler-Stevenson, M.; Raffeld, M.; Kreitman, R.J. Both variant and IGHV4-34-expressing hairy cell leukemia lack the BRAF V600E mutation. Blood 2012, 119, 3330–3332. [Google Scholar] [CrossRef]
- Weston-Bell, N.J.; Tapper, W.; Gibson, J.; Bryant, D.; Moreno, Y.; John, M.; Ennis, S.; Kluin-Nelemans, H.C.; Collins, A.R.; Sahota, S.S. Exome Sequencing in Classic Hairy Cell Leukaemia Reveals Widespread Variation in Acquired Somatic Mutations between Individual Tumours Apart from the Signature BRAF V(600)E Lesion. PLoS ONE 2016, 11, e0149162. [Google Scholar] [CrossRef]
- Dietrich, S.; Hullein, J.; Lee, S.C.; Hutter, B.; Gonzalez, D.; Jayne, S.; Dyer, M.J.; Oles, M.; Else, M.; Liu, X.; et al. Recurrent CDKN1B (p27) mutations in hairy cell leukemia. Blood 2015, 126, 1005–1008. [Google Scholar] [CrossRef]
- Spina, V.; Rossi, D. Molecular pathogenesis of splenic and nodal marginal zone lymphoma. Best Pract. Res. Clin. Haematol. 2017, 30, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Spina, V.; Khiabanian, H.; Messina, M.; Monti, S.; Cascione, L.; Bruscaggin, A.; Spaccarotella, E.; Holmes, A.B.; Arcaini, L.; Lucioni, M.; et al. The genetics of nodal marginal zone lymphoma. Blood 2016, 128, 1362–1373. [Google Scholar] [CrossRef] [PubMed]
- Du, M.Q. MALT lymphoma: Genetic abnormalities, Immunol.ogical stimulation and molecular mechanism. Best Pract. Res. Clin. Haematol. 2017, 30, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Cross, M.; Dearden, C. B and T cell prolymphocytic leukaemia. Best Pract. Res. Clin. Haematol. 2019, 32, 217–228. [Google Scholar] [CrossRef]
- Collignon, A.; Wanquet, A.; Maitre, E.; Cornet, E.; Troussard, X.; Aurran-Schleinitz, T. Prolymphocytic Leukemia: New Insights in Diagnosis and in Treatment. Curr. Oncol. Rep. 2017, 19, 29. [Google Scholar] [CrossRef]
- Chapiro, E.; Pramil, E.; Diop, M.; Roos-Weil, D.; Dillard, C.; Gabillaud, C.; Maloum, K.; Settegrana, C.; Baseggio, L.; Lesesve, J.F.; et al. Genetic characterization of B-cell prolymphocytic leukemia: A prognostic model involving MYC and TP53. Blood 2019, 134, 1821–1831. [Google Scholar] [CrossRef]
- Treon, S.P.; Xu, L.; Guerrera, M.L.; Jimenez, C.; Hunter, Z.R.; Liu, X.; Demos, M.; Gustine, J.; Chan, G.; Munshi, M.; et al. Genomic Landscape of Waldenstrom Macroglobulinemia and Its Impact on Treatment Strategies. J. Clin. Oncol. 2020, 38, 1198–1208. [Google Scholar] [CrossRef]
- Hunter, Z.R.; Yang, G.; Xu, L.; Liu, X.; Castillo, J.J.; Treon, S.P. Genomics, Signaling, and Treatment of Waldenstrom Macroglobulinemia. J. Clin. Oncol. 2017, 35, 994–1001. [Google Scholar] [CrossRef]
- Garcia-Sanz, R.; Jimenez, C.; Puig, N.; Paiva, B.; Gutierrez, N.C.; Rodriguez-Otero, P.; Almeida, J.; San Miguel, J.; Orfao, A.; Gonzalez, M.; et al. Origin of Waldenstrom’s macroglobulinaemia. Best Pract. Res. Clin. Haematol. 2016, 29, 136–147. [Google Scholar] [CrossRef]
- Treon, S.P.; Xu, L.; Yang, G.; Zhou, Y.; Liu, X.; Cao, Y.; Sheehy, P.; Manning, R.J.; Patterson, C.J.; Tripsas, C.; et al. MYD88 L265P somatic mutation in Waldenstrom’s macroglobulinemia. N. Engl. J. Med. 2012, 367, 826–833. [Google Scholar] [CrossRef]
- Hunter, Z.R.; Xu, L.; Tsakmaklis, N.; Demos, M.G.; Kofides, A.; Jimenez, C.; Chan, G.G.; Chen, J.; Liu, X.; Munshi, M.; et al. Insights into the genomic landscape of MYD88 wild-type Waldenstrom macroglobulinemia. Blood Adv. 2018, 2, 2937–2946. [Google Scholar] [CrossRef] [PubMed]
- Roos-Weil, D.; Decaudin, C.; Armand, M.; Della-Valle, V.; Diop, M.K.; Ghamlouch, H.; Ropars, V.; Herate, C.; Lara, D.; Durot, E.; et al. A recurrent Activating Missense Mutation in Waldenstrom Macroglobulinemia Affects the DNA Binding of the ETS Transcription Factor SPI1 and Enhances Proliferation. Cancer Discov. 2019, 9, 796–811. [Google Scholar] [CrossRef] [PubMed]
- Awada, H.; Thapa, B.; Awada, H.; Dong, J.; Gurnari, C.; Hari, P.; Dhakal, B. A Comprehensive Review of the Genomics of Multiple Myeloma: Evolutionary Trajectories, Gene Expression Profiling, and Emerging Therapeutics. Cells 2021, 10, 1961. [Google Scholar] [CrossRef]
- Hoang, P.H.; Dobbins, S.E.; Cornish, A.J.; Chubb, D.; Law, P.J.; Kaiser, M.; Houlston, R.S. Whole-genome sequencing of multiple myeloma reveals oncogenic pathways are targeted somatically through multiple mechanisms. Leukemia 2018, 32, 2459–2470. [Google Scholar] [CrossRef]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.E.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 2018, 132, 587–597. [Google Scholar] [CrossRef]
- Boyle, E.M.; Ashby, C.; Tytarenko, R.G.; Deshpande, S.; Wang, H.; Wang, Y.; Rosenthal, A.; Sawyer, J.; Tian, E.; Flynt, E.; et al. BRAF and DIS3 Mutations Associate with Adverse Outcome in a Long-term Follow-up of Patients with Multiple Myeloma. Clin. Cancer Res. 2020, 26, 2422–2432. [Google Scholar] [CrossRef]
- Morgan, G.J.; He, J.; Tytarenko, R.; Patel, P.; Stephens, O.W.; Zhong, S.; Deshpande, S.; Bauer, M.; Weinhold, N.; Schinke, C.; et al. Kinase domain activation through gene rearrangement in multiple myeloma. Leukemia 2018, 32, 2435–2444. [Google Scholar] [CrossRef]
- Podar, K.; Chauhan, D.; Anderson, K.C. Bone marrow microenvironment and the identification of new targets for myeloma therapy. Leukemia 2009, 23, 10–24. [Google Scholar] [CrossRef]
- Boyle, E.M.; Deshpande, S.; Tytarenko, R.; Ashby, C.; Wang, Y.; Bauer, M.A.; Johnson, S.K.; Wardell, C.P.; Thanendrarajan, S.; Zangari, M.; et al. The molecular make up of smoldering myeloma highlights the evolutionary pathways leading to multiple myeloma. Nat. Commun. 2021, 12, 293. [Google Scholar] [CrossRef]
- Kortum, K.M.; Mai, E.K.; Hanafiah, N.H.; Shi, C.X.; Zhu, Y.X.; Bruins, L.; Barrio, S.; Jedlowski, P.; Merz, M.; Xu, J.; et al. Targeted sequencing of refractory myeloma reveals a high incidence of mutations in CRBN and Ras pathway genes. Blood 2016, 128, 1226–1233. [Google Scholar] [CrossRef]
- van Nieuwenhuijzen, N.; Spaan, I.; Raymakers, R.; Peperzak, V. From MGUS to Multiple Myeloma, a Paradigm for Clonal Evolution of Premalignant Cells. Cancer Res. 2018, 78, 2449–2456. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, G.; Lichter, D.I.; Di Bacco, A.; Blakemore, S.J.; Berger, A.; Koenig, E.; Bernard, H.; Trepicchio, W.; Li, B.; Neuwirth, R.; et al. Mutation of NRAS but not KRAS significantly reduces myeloma sensitivity to single-agent bortezomib therapy. Blood 2014, 123, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Leeman-Neill, R.J.; Soderquist, C.R.; Montanari, F.; Raciti, P.; Park, D.; Radeski, D.; Mansukhani, M.M.; Murty, V.V.; Hsiao, S.; Alobeid, B.; et al. Phenogenomic heterogeneity of post-transplant plasmablastic lymphomas. Haematologica, 2020; online ahead of print. [Google Scholar] [CrossRef]
- Shimada, K.; Hayakawa, F.; Kiyoi, H. Biology and management of primary effusion lymphoma. Blood 2018, 132, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Gaidano, G.; Capello, D.; Cilia, A.M.; Gloghini, A.; Perin, T.; Quattrone, S.; Migliazza, A.; Lo Coco, F.; Saglio, G.; Ascoli, V.; et al. Genetic characterization of HHV-8/KSHV-positive primary effusion lymphoma reveals frequent mutations of BCL6: Implications for disease pathogenesis and histogenesis. Genes Chromosomes Cancer 1999, 24, 16–23. [Google Scholar] [CrossRef]
- Roy, D.; Sin, S.H.; Damania, B.; Dittmer, D.P. Tumor suppressor genes FHIT and WWOX are deleted in primary effusion lymphoma (PEL) cell lines. Blood 2011, 118, e32–e39. [Google Scholar] [CrossRef]
- Wagener, R.; Alexandrov, L.B.; Montesinos-Rongen, M.; Schlesner, M.; Haake, A.; Drexler, H.G.; Richter, J.; Bignell, G.R.; McDermott, U.; Siebert, R. Analysis of mutational signatures in exomes from B-cell lymphoma cell lines suggest APOBEC3 family members to be involved in the pathogenesis of primary effusion lymphoma. Leukemia 2015, 29, 1612–1615. [Google Scholar] [CrossRef]
- Calvani, J.; Gerard, L.; Fadlallah, J.; Poullot, E.; Galicier, L.; Robe, C.; Garzaro, M.; Bertinchamp, R.; Boutboul, D.; Cuccuini, W.; et al. A Comprehensive Clinicopathologic and Molecular Study of 19 Primary Effusion Lymphomas in HIV-infected Patients. Am. J. Surg. Pathol. 2021. [Google Scholar] [CrossRef]
- Tiacci, E.; Park, J.H.; De Carolis, L.; Chung, S.S.; Broccoli, A.; Scott, S.; Zaja, F.; Devlin, S.; Pulsoni, A.; Chung, Y.R.; et al. Targeting Mutant BRAF in Relapsed or Refractory Hairy-Cell Leukemia. N. Engl. J. Med. 2015, 373, 1733–1747. [Google Scholar] [CrossRef]
- Dietrich, S.; Glimm, H.; Andrulis, M.; von Kalle, C.; Ho, A.D.; Zenz, T. BRAF inhibition in refractory hairy-cell leukemia. N. Engl. J. Med. 2012, 366, 2038–2040. [Google Scholar] [CrossRef]
- Hyman, D.M.; Puzanov, I.; Subbiah, V.; Faris, J.E.; Chau, I.; Blay, J.Y.; Wolf, J.; Raje, N.S.; Diamond, E.L.; Hollebecque, A.; et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N. Engl. J. Med. 2015, 373, 726–736. [Google Scholar] [CrossRef]
- Raje, N.; Chau, I.; Hyman, D.M.; Ribrag, V.; Blay, J.Y.; Tabernero, J.; Elez, E.; Wolf, J.; Yee, A.J.; Kaiser, M.; et al. Vemurafenib in Patients with Relapsed Refractory Multiple Myeloma Harboring BRAF (V600) Mutations: A Cohort of the Histology-Independent VE-BASKET Study. JCO Precis. Oncol. 2018, 2, PO.18.00070. [Google Scholar] [CrossRef] [PubMed]
- Andrulis, M.; Lehners, N.; Capper, D.; Penzel, R.; Heining, C.; Huellein, J.; Zenz, T.; von Deimling, A.; Schirmacher, P.; Ho, A.D.; et al. Targeting the BRAF V600E mutation in multiple myeloma. Cancer Discov. 2013, 3, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.S.; Lehners, N.; Xu, J.; Ho, A.D.; Schirmacher, P.; Goldschmidt, H.; Andrulis, M. Spatially divergent clonal evolution in multiple myeloma: Overcoming resistance to BRAF inhibition. Blood 2016, 127, 2155–2157. [Google Scholar] [CrossRef] [PubMed]
- Sharman, J.P.; Chmielecki, J.; Morosini, D.; Palmer, G.A.; Ross, J.S.; Stephens, P.J.; Stafl, J.; Miller, V.A.; Ali, S.M. Vemurafenib response in 2 patients with posttransplant refractory BRAF V600E-mutated multiple myeloma. Clin. Lymphoma Myeloma Leuk. 2014, 14, e161–e163. [Google Scholar] [CrossRef]
- Bohn, O.L.; Hsu, K.; Hyman, D.M.; Pignataro, D.S.; Giralt, S.; Teruya-Feldstein, J. BRAF V600E mutation and clonal evolution in a patient with relapsed refractory myeloma with plasmablastic differentiation. Clin. Lymphoma Myeloma Leuk. 2014, 14, e65–e68. [Google Scholar] [CrossRef]
- Caeser, R.; Collord, G.; Yao, W.Q.; Chen, Z.; Vassiliou, G.S.; Beer, P.A.; Du, M.Q.; Scott, M.A.; Follows, G.A.; Hodson, D.J. Targeting MEK in vemurafenib-resistant hairy cell leukemia. Leukemia 2019, 33, 541–545. [Google Scholar] [CrossRef]
- Mey, U.J.M.; Renner, C.; von Moos, R. Vemurafenib in combination with cobimetinib in relapsed and refractory extramedullary multiple myeloma harboring the BRAF V600E mutation. Hematol. Oncol. 2017, 35, 890–893. [Google Scholar] [CrossRef]
- Otieno, S.B.; Nasir, S.; Weir, A.; Johnson, R. Rapid Response in a Patient with Relapsed/Refractory Multiple Myeloma Treated with BRAF/MEK Inhibitors. Case Rep. Hematol. 2020, 2020, 8821415. [Google Scholar] [CrossRef]
- Tiacci, E.; De Carolis, L.; Simonetti, E.; Capponi, M.; Ambrosetti, A.; Lucia, E.; Antolino, A.; Pulsoni, A.; Ferrari, S.; Zinzani, P.L.; et al. Vemurafenib plus Rituximab in Refractory or Relapsed Hairy-Cell Leukemia. N. Engl. J. Med. 2021, 384, 1810–1823. [Google Scholar] [CrossRef]
- Tiacci, E.; De Carolis, L.; Simonetti, E.; Merluzzi, M.; Bennati, A.; Perriello, V.M.; Pucciarini, A.; Santi, A.; Venanzi, A.; Pettirossi, V.; et al. Safety and efficacy of the BRAF inhibitor dabrafenib in relapsed or refractory hairy cell leukemia: A pilot phase-2 clinical trial. Leukemia 2021, 35, 3314–3318. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Moreau, P.; Hutchings, M.; Gazzah, A.; Blay, J.-Y.; Wainberg, Z.A.; Stein, A.; Dietrich, S.; de Jonge, M.J.A.; Willenbacher, W.; et al. Treatment with Combination of Dabrafenib and Trametinib in Patients with Recurrent/Refractory BRAF V600E-Mutated Hairy Cell Leukemia (HCL). Blood 2018, 132 (Suppl. 1), 391. [Google Scholar] [CrossRef]
- Suzuki, R.; Kitamura, Y.; Nakamura, Y.; Akashi, H.; Ogawa, Y.; Kawada, H.; Ando, K. Anti-tumor activities of the new oral pan-RAF inhibitor, TAK-580, used as monotherapy or in combination with novel agents in multiple myeloma. Oncotarget 2020, 11, 3984–3997. [Google Scholar] [CrossRef] [PubMed]
- Srkalovic, G.; Hussein, M.A.; Hoering, A.; Zonder, J.A.; Popplewell, L.L.; Trivedi, H.; Mazzoni, S.; Sexton, R.; Orlowski, R.Z.; Barlogie, B. A phase II trial of BAY 43-9006 (sorafenib) (NSC-724772) in patients with relapsing and resistant multiple myeloma: SWOG S0434. Cancer Med. 2014, 3, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Yordanova, A.; Hose, D.; Neben, K.; Witzens-Harig, M.; Gutgemann, I.; Raab, M.S.; Moehler, T.; Goldschmidt, H.; Schmidt-Wolf, I.G. Sorafenib in patients with refractory or recurrent multiple myeloma. Hematol. Oncol. 2013, 31, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Messmer, D.; Fecteau, J.F.; O’Hayre, M.; Bharati, I.S.; Handel, T.M.; Kipps, T.J. Chronic lymphocytic leukemia cells receive RAF-dependent survival signals in response to CXCL12 that are sensitive to inhibition by sorafenib. Blood 2011, 117, 882–889. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huber, S.; Oelsner, M.; Decker, T.; zum Buschenfelde, C.M.; Wagner, M.; Lutzny, G.; Kuhnt, T.; Schmidt, B.; Oostendorp, R.A.; Peschel, C.; et al. Sorafenib induces cell death in chronic lymphocytic leukemia by translational downregulation of Mcl-1. Leukemia 2011, 25, 838–847. [Google Scholar] [CrossRef][Green Version]
- Fecteau, J.F.; Bharati, I.S.; O’Hayre, M.; Handel, T.M.; Kipps, T.J.; Messmer, D. Sorafenib-induced apoptosis of chronic lymphocytic leukemia cells is associated with downregulation of RAF and myeloid cell leukemia sequence 1 (Mcl-1). Mol. Med. 2012, 18, 19–28. [Google Scholar] [CrossRef]
- Lopez-Guerra, M.; Xargay-Torrent, S.; Perez-Galan, P.; Saborit-Villarroya, I.; Rosich, L.; Villamor, N.; Aymerich, M.; Roue, G.; Campo, E.; Montserrat, E.; et al. Sorafenib targets BCR kinases and blocks migratory and microenvironmental survival signals in CLL cells. Leukemia 2012, 26, 1429–1432. [Google Scholar] [CrossRef][Green Version]
- Greenwald, D.R.; Li, H.; Luger, S.M.; Go, R.S.; King, D.; Patel, T.; Gascoyne, R.D.; Kolesar, J.; Kahl, B.S.; Horning, S. A phase II study of sorafenib (BAY 43-9006) in recurrent diffuse large B cell lymphoma: An eastern cooperative oncology group study (E1404). J. Hematol. Oncol. 2013, 6, 46. [Google Scholar] [CrossRef]
- Guidetti, A.; Carlo-Stella, C.; Locatelli, S.L.; Malorni, W.; Pierdominici, M.; Barbati, C.; Mortarini, R.; Devizzi, L.; Matteucci, P.; Marchiano, A.; et al. Phase II study of sorafenib in patients with relapsed or refractory lymphoma. Br. J. Haematol. 2012, 158, 108–119. [Google Scholar] [CrossRef]
- Nguyen, T.K.; Jordan, N.; Friedberg, J.; Fisher, R.I.; Dent, P.; Grant, S. Inhibition of MEK/ERK1/2 sensitizes lymphoma cells to sorafenib-induced apoptosis. Leuk. Res. 2010, 34, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, K.; Wurster, K.D.; Lamprecht, B.; Kochert, K.; Engert, A.; Dorken, B.; Janz, M.; Mathas, S. BAY 43-9006/Sorafenib blocks CSF1R activity and induces apoptosis in various classical Hodgkin lymphoma cell lines. Br. J. Haematol. 2011, 155, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Holz, M.S.; Janning, A.; Renne, C.; Gattenlohner, S.; Spieker, T.; Brauninger, A. Induction of endoplasmic reticulum stress by sorafenib and activation of NF-kappaB by lestaurtinib as a novel resistance mechanism in Hodgkin lymphoma cell lines. Mol. Cancer 2013, 12, 173–183. [Google Scholar] [CrossRef]
- Xargay-Torrent, S.; Lopez-Guerra, M.; Montraveta, A.; Saborit-Villarroya, I.; Rosich, L.; Navarro, A.; Perez-Galan, P.; Roue, G.; Campo, E.; Colomer, D. Sorafenib inhibits cell migration and stroma-mediated bortezomib resistance by interfering B-cell receptor signaling and protein translation in mantle cell lymphoma. Clin. Cancer Res. 2013, 19, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, V.; Timm, M.; Haug, J.L.; Kimlinger, T.K.; Halling, T.; Wellik, L.E.; Witzig, T.E.; Rajkumar, S.V.; Adjei, A.A.; Kumar, S. Sorafenib, a multikinase inhibitor, is effective in vitro against non-Hodgkin lymphoma and synergizes with the mTOR inhibitor rapamycin. Am. J. Hematol. 2012, 87, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, S.L.; Cleris, L.; Stirparo, G.G.; Tartari, S.; Saba, E.; Pierdominici, M.; Malorni, W.; Carbone, A.; Anichini, A.; Carlo-Stella, C. BIM upregulation and ROS-dependent necroptosis mediate the antitumor effects of the HDACi Givinostat and Sorafenib in Hodgkin lymphoma cell line xenografts. Leukemia 2014, 28, 1861–1871. [Google Scholar] [CrossRef]
- Locatelli, S.L.; Giacomini, A.; Guidetti, A.; Cleris, L.; Mortarini, R.; Anichini, A.; Gianni, A.M.; Carlo-Stella, C. Perifosine and sorafenib combination induces mitochondrial cell death and antitumor effects in NOD/SCID mice with Hodgkin lymphoma cell line xenografts. Leukemia 2013, 27, 1677–1687. [Google Scholar] [CrossRef]
- Guidetti, A.; Carlo-Stella, C.; Locatelli, S.L.; Malorni, W.; Mortarini, R.; Viviani, S.; Russo, D.; Marchiano, A.; Sorasio, R.; Dodero, A.; et al. Phase II study of perifosine and sorafenib dual-targeted therapy in patients with relapsed or refractory lymphoproliferative diseases. Clin. Cancer Res. 2014, 20, 5641–5651. [Google Scholar] [CrossRef]
- Sacco, A.; Federico, C.; Todoerti, K.; Ziccheddu, B.; Palermo, V.; Giacomini, A.; Ravelli, C.; Maccarinelli, F.; Bianchi, G.; Belotti, A.; et al. Specific targeting of the KRAS mutational landscape in myeloma as a tool to unveil the elicited anti-tumor activity. Blood 2021, 138, 1705–1720. [Google Scholar] [CrossRef]
- Guo, C.; Chenard-Poirier, M.; Roda, D.; de Miguel, M.; Harris, S.J.; Candilejo, I.M.; Sriskandarajah, P.; Xu, W.; Scaranti, M.; Constantinidou, A.; et al. Intermittent schedules of the oral RAF-MEK inhibitor CH5126766/VS-6766 in patients with RAS/RAF-mutant solid tumours and multiple myeloma: A single-centre, open-label, phase 1 dose-escalation and basket dose-expansion study. Lancet Oncol. 2020, 21, 1478–1488. [Google Scholar] [CrossRef]
- Andritsos, L.A.; Grieselhuber, N.R.; Anghelina, M.; Rogers, K.A.; Roychowdhury, S.; Reeser, J.W.; Timmers, C.D.; Freud, A.G.; Blachly, J.S.; Lucas, D.M.; et al. Trametinib for the treatment of IGHV4-34, MAP2K1-mutant variant hairy cell leukemia. Leuk. Lymphoma 2018, 59, 1008–1011. [Google Scholar] [CrossRef] [PubMed]
- Heuck, C.J.; Jethava, Y.; Khan, R.; van Rhee, F.; Zangari, M.; Chavan, S.; Robbins, K.; Miller, S.E.; Matin, A.; Mohan, M.; et al. Inhibiting MEK in MAPK pathway-activated myeloma. Leukemia 2016, 30, 976–980. [Google Scholar] [CrossRef] [PubMed]
- Shirazi, F.; Jones, R.J.; Singh, R.K.; Zou, J.; Kuiatse, I.; Berkova, Z.; Wang, H.; Lee, H.C.; Hong, S.; Dick, L.; et al. Activating KRAS, NRAS, and BRAF mutants enhance proteasome capacity and reduce endoplasmic reticulum stress in multiple myeloma. Proc. Natl. Acad. Sci. USA 2020, 117, 20004–20014. [Google Scholar] [CrossRef] [PubMed]
- Kozaki, R.; Vogler, M.; Walter, H.S.; Jayne, S.; Dinsdale, D.; Siebert, R.; Dyer, M.J.S.; Yoshizawa, T. Responses to the Selective Bruton’s Tyrosine Kinase (BTK) Inhibitor Tirabrutinib (ONO/GS-4059) in Diffuse Large B-cell Lymphoma Cell Lines. Cancers 2018, 10, 127. [Google Scholar] [CrossRef] [PubMed]
- Schjesvold, F.; Ribrag, V.; Rodriguez-Otero, P.; Robak, P.J.; Hansson, M.; Hajek, R.; Amor, A.A.; Martinez-López, J.; Onishi, M.; Gallo, J.D.; et al. Safety and Preliminary Efficacy Results from a Phase Ib/II Study of Cobimetinib As a Single Agent and in Combination with Venetoclax with or without Atezolizumab in Patients with Relapsed/Refractory Multiple Myeloma. Blood 2020, 136 (Suppl. 1), 45–46. [Google Scholar] [CrossRef]
- Crassini, K.; Shen, Y.; Stevenson, W.S.; Christopherson, R.; Ward, C.; Mulligan, S.P.; Best, O.G. MEK1/2 inhibition by binimetinib is effective as a single agent and potentiates the actions of Venetoclax and ABT-737 under conditions that mimic the chronic lymphocytic leukaemia (CLL) tumour microenvironment. Br. J. Haematol. 2018, 182, 360–372. [Google Scholar] [CrossRef]
- Shen, Y.; Crassini, K.; Sandhu, S.; Fatima, N.; Christopherson, R.I.; Mulligan, S.P.; Best, O.G. Dual inhibition of MEK1/2 and AKT by binimetinib and MK2206 induces apoptosis of chronic lymphocytic leukemia cells under conditions that mimic the tumor microenvironment. Leuk. Lymphoma 2019, 60, 1632–1643. [Google Scholar] [CrossRef]
- Holkova, B.; Zingone, A.; Kmieciak, M.; Bose, P.; Badros, A.Z.; Voorhees, P.M.; Baz, R.; Korde, N.; Lin, H.Y.; Chen, J.Q.; et al. A Phase II Trial of AZD6244 (Selumetinib, ARRY-142886), an Oral MEK1/2 Inhibitor, in Relapsed/Refractory Multiple Myeloma. Clin. Cancer Res. 2016, 22, 1067–1075. [Google Scholar] [CrossRef]
- Galanina, N.; Smith, S.M.; Liao, C.; Petrich, A.; Libao, B.; Gartenhaus, R.; Westin, J.R.; Cohen, K.S.; Knost, J.A.; Stadler, W.M.; et al. University of Chicago phase II consortium trial of selumetinib (MEKi) demonstrates low tolerability and efficacy in relapsed DLBCL. Br. J. Haematol. 2018, 181, 264–267. [Google Scholar] [CrossRef]
- Bhalla, S.; Evens, A.M.; Dai, B.; Prachand, S.; Gordon, L.I.; Gartenhaus, R.B. The novel anti-MEK small molecule AZD6244 induces BIM-dependent and AKT-independent apoptosis in diffuse large B-cell lymphoma. Blood 2011, 118, 1052–1061. [Google Scholar] [CrossRef]
- Ramakrishnan, V.G.; Miller, K.C.; Macon, E.P.; Kimlinger, T.K.; Haug, J.; Kumar, S.; Gonsalves, W.I.; Rajkumar, S.V.; Kumar, S.K. Histone deacetylase inhibition in combination with MEK or BCL-2 inhibition in multiple myeloma. Haematologica 2019, 104, 2061–2074. [Google Scholar] [CrossRef] [PubMed]
- Gaudio, E.; Tarantelli, C.; Kwee, I.; Barassi, C.; Bernasconi, E.; Rinaldi, A.; Ponzoni, M.; Cascione, L.; Targa, A.; Stathis, A.; et al. Combination of the MEK inhibitor pimasertib with BTK or PI3K-delta inhibitors is active in preclinical models of aggressive lymphomas. Ann. Oncol. 2016, 27, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Fiumara, P.; Li, Y.V.; Georgakis, G.; Snell, V.; Younes, M.; Vauthey, J.N.; Carbone, A.; Younes, A. MEK/ERK pathway is aberrantly active in Hodgkin disease: A signaling pathway shared by CD30, CD40, and RANK that regulates cell proliferation and survival. Blood 2003, 102, 1019–1027. [Google Scholar] [CrossRef]
- Locatelli, S.L.; Careddu, G.; Stirparo, G.G.; Castagna, L.; Santoro, A.; Carlo-Stella, C. Dual PI3K/ERK inhibition induces necroptotic cell death of Hodgkin Lymphoma cells through IER3 downregulation. Sci. Rep. 2016, 6, 35745. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Targeting oncogenic Raf protein-serine/threonine kinases in human cancers. Pharm. Res. 2018, 135, 239–258. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Chauhan, D.; Richardson, P.; Mitsiades, C.; Mitsiades, N.; Hayashi, T.; Munshi, N.; Dang, L.; Castro, A.; Palombella, V.; et al. NF-kappa B as a therapeutic target in multiple myeloma. J. Biol. Chem. 2002, 277, 16639–16647. [Google Scholar] [CrossRef]
- Dietrich, S.; Pircher, A.; Endris, V.; Peyrade, F.; Wendtner, C.M.; Follows, G.A.; Hullein, J.; Jethwa, A.; Ellert, E.; Walther, T.; et al. BRAF inhibition in hairy cell leukemia with low-dose vemurafenib. Blood 2016, 127, 2847–2855. [Google Scholar] [CrossRef]
- Dietrich, S.; Oles, M.; Lu, J.; Sellner, L.; Anders, S.; Velten, B.; Wu, B.; Hullein, J.; da Silva Liberio, M.; Walther, T.; et al. Drug-perturbation-based stratification of blood cancer. J. Clin. Investig. 2018, 128, 427–445. [Google Scholar] [CrossRef]
- Matsumura-Kimoto, Y.; Tsukamoto, T.; Shimura, Y.; Chinen, Y.; Tanba, K.; Kuwahara-Ota, S.; Fujibayashi, Y.; Nishiyama, D.; Isa, R.; Yamaguchi, J.; et al. Serine-227 in the N-terminal kinase domain of RSK2 is a potential therapeutic target for mantle cell lymphoma. Cancer Med. 2020, 9, 5185–5199. [Google Scholar] [CrossRef]
- Tuveson, D.A.; Shaw, A.T.; Willis, N.A.; Silver, D.P.; Jackson, E.L.; Chang, S.; Mercer, K.L.; Grochow, R.; Hock, H.; Crowley, D.; et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell 2004, 5, 375–387. [Google Scholar] [CrossRef]
- Kindler, T.; Cornejo, M.G.; Scholl, C.; Liu, J.; Leeman, D.S.; Haydu, J.E.; Frohling, S.; Lee, B.H.; Gilliland, D.G. K-RasG12D-induced T-cell lymphoblastic lymphoma/leukemias harbor Notch1 mutations and are sensitive to gamma-secretase inhibitors. Blood 2008, 112, 3373–3382. [Google Scholar] [CrossRef] [PubMed]
- Mullins, C.D.; Su, M.Y.; Hucthagowder, V.; Chu, L.; Lu, L.; Kulkarni, S.; Novack, D.; Vij, R.; Tomasson, M.H. Germinal center B-cells resist transformation by Kras independently of tumor suppressor Arf. PLoS ONE 2013, 8, e67941. [Google Scholar] [CrossRef] [PubMed]
- Poulin, E.J.; Bera, A.K.; Lu, J.; Lin, Y.J.; Strasser, S.D.; Paulo, J.A.; Huang, T.Q.; Morales, C.; Yan, W.; Cook, J.; et al. Tissue-Specific Oncogenic Activity of KRAS(A146T). Cancer Discov. 2019, 9, 738–755. [Google Scholar] [CrossRef]
- Hernandez-Porras, I.; Fabbiano, S.; Schuhmacher, A.J.; Aicher, A.; Canamero, M.; Camara, J.A.; Cusso, L.; Desco, M.; Heeschen, C.; Mulero, F.; et al. K-RasV14I recapitulates Noonan syndrome in mice. Proc. Natl. Acad. Sci. USA 2014, 111, 16395–16400. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, Y.; Li, Z.; Wang, Z.; Tan, L.X.; Ryu, M.J.; Meline, B.; Du, J.; Young, K.H.; Ranheim, E.; et al. Endogenous oncogenic Nras mutation initiates hematopoietic malignancies in a dose- and cell type-dependent manner. Blood 2011, 118, 368–379. [Google Scholar] [CrossRef]
- Wang, J.; Liu, Y.; Li, Z.; Du, J.; Ryu, M.J.; Taylor, P.R.; Fleming, M.D.; Young, K.H.; Pitot, H.; Zhang, J. Endogenous oncogenic Nras mutation promotes aberrant GM-CSF signaling in granulocytic/monocytic precursors in a murine model of chronic myelomonocytic leukemia. Blood 2010, 116, 5991–6002. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Rajagopalan, A.; Flietner, E.D.; Yun, G.; Chesi, M.; Furumo, Q.; Burns, R.T.; Papadas, A.; Ranheim, E.A.; Pagenkopf, A.C.; et al. Expression of NrasQ61R and MYC transgene in germinal center B cells induces a highly malignant multiple myeloma in mice. Blood 2021, 137, 61–74. [Google Scholar] [CrossRef]
- Mercer, K.; Giblett, S.; Green, S.; Lloyd, D.; DaRocha Dias, S.; Plumb, M.; Marais, R.; Pritchard, C. Expression of endogenous oncogenic V600EB-raf induces proliferation and developmental defects in mice and transformation of primary fibroblasts. Cancer Res. 2005, 65, 11493–11500. [Google Scholar] [CrossRef]
- Chung, S.S.; Kim, E.; Park, J.H.; Chung, Y.R.; Lito, P.; Teruya-Feldstein, J.; Hu, W.; Beguelin, W.; Monette, S.; Duy, C.; et al. Hematopoietic stem cell origin of BRAFV600E mutations in hairy cell leukemia. Sci. Transl. Med. 2014, 6, 238ra71. [Google Scholar] [CrossRef]
- Tsai, Y.T.; Lakshmanan, A.; Lehman, A.; Harrington, B.K.; Lucas, F.M.; Tran, M.; Sass, E.J.; Long, M.; Flechtner, A.D.; Jaynes, F.; et al. BRAF(V600E) accelerates disease progression and enhances immune suppression in a mouse model of B-cell leukemia. Blood Adv. 2017, 1, 2147–2160. [Google Scholar] [CrossRef]
- Kong, G.; Du, J.; Liu, Y.; Meline, B.; Chang, Y.I.; Ranheim, E.A.; Wang, J.; Zhang, J. Notch1 gene mutations target KRAS G12D-expressing CD8+ cells and contribute to their leukemogenic transformation. J. Biol. Chem. 2013, 288, 18219–18227. [Google Scholar] [CrossRef] [PubMed]
- Kong, G.; You, X.; Wen, Z.; Chang, Y.I.; Qian, S.; Ranheim, E.A.; Letson, C.; Zhang, X.; Zhou, Y.; Liu, Y.; et al. Downregulating Notch counteracts Kras(G12D)-induced ERK activation and oxidative phosphorylation in myeloproliferative neoplasm. Leukemia 2019, 33, 671–685. [Google Scholar] [CrossRef] [PubMed]
- Karreth, F.A.; Reschke, M.; Ruocco, A.; Ng, C.; Chapuy, B.; Leopold, V.; Sjoberg, M.; Keane, T.M.; Verma, A.; Ala, U.; et al. The BRAF pseudogene functions as a competitive endogenous RNA and induces lymphoma in vivo. Cell 2015, 161, 319–332. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene (RefSeq Accession Number) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Disease | KRAS | NRAS | HRAS | BRAF | RAF1 | MAP2K1 | MAP2K2 | MAPK3 | MAPK1 | NF1 | References |
| (NM_033360) | (NM_002524) | (NM_001130442) | (NM_004333) | (NM_002880) | (NM_002755) | (NM_030662) | (NM_002746) | (NM_138957) | (NM_001042492) | ||
| MCL | G12D, L19F | F156L | L485W, L485_V487del, N581I | V162L | G661fs | [9,10,11] | |||||
| DLBCL | L6P, G12A/S/R/D, G13D, V14I, L19F, Q22K, L23R, E31K, T58I, Q61H, A83V, M111V, K117N, D119G/N, A146T, V160M, G174S (NM_004985), P178T, P178Vfs*2, C180X | G13D, I24N, Q61R, N116S, E153K | R41W, S145L, R153C/H (NM_176795), H166Y, P166S (NM_176795), R169Q, K170X | D22N, V120I, H223P, R260C, G327_splice, Q344L, R347Q, D352E, R360Q, N378S, R389H, R424Q, T440P, G466R, S467T, F468S, G469A, K483E, L485F, L525R, N581S/H, D594G/A/N, F595L, L597R/Q, T599R, V600E, K601E/N, R671Q, R719C | H105D, L149F, R333C, T353I, R391S, L476F, E478K | A26V, Q46L, R47Q, F53L/S/V/C/Y, K57T/E, D67N, A106T, C121S, N122D, Y130H/C, G176S, A283V, T292I, R291K, R305Q | Q60P, D71N, V202M | E367G, E203K | Y131H, I240V, E322A, A327S | S15N, Q28H, G57S, R156H, C245_E247del, R262H, Q282R, G312E, R681Q, R711H, E924X, R1066_splice, G1166_splice, I1186L, A1202S, R1276X, N1338Y, G1403S, R1462Q, V1674I, I1755V, A1858T, L1892X, H1962Pfs, I2026T, I2057V, I2127V, S2152T, T2204I, T2222A, I2267R, N2432S, V2511L, V2657_splice, Y2702F, P2717L, I2739T, Y2742S, T2773A, G2793E | [12,13,14,15] |
| FL | F53Y a, Q56P a, K57E/R a, C121S a | N297D a, D321G a | M1539Wfs*35 | [16,17] | |||||||
| BL | E501A, T508I, D594G, L597R/Q | E478K | S248L | [18,19,20,21] | |||||||
| HL | L19F | V114L | D454G, S335Y D380_splice | L30F, L342X | C324S, R765C | [22,23,24] | |||||
| CLL | V7G, G12D/C/V/S/R, G13D/C, V14I, A18V, L19F, Q22K/E, T50I, T58I, Q61H/L/R, K117N/R, A146T/V/P | G12C/D, G13D/R/V, S17T, Q61K/R/H/L, A146T | G466E, F468S, G469A/V/E/R, K483E, L485F, N486_P491del, E501K, G534R, N581S/I, D594G/N, F595L, L597R/Q, V600E, K601N/E, D638E | R391W | F53L/V/C, K57N, C121S, P124L, G128V/D, R201H, E203K | Q60P, Y134C | D291G | Y489C, T676fs, R1276Q, K1444N, R2594P | [25,26,27,28,29,30,31] | ||
| HCL | G12D, K117N | G12C | V600E, D449E, F468C, S602T | F53L, Q56P, K57E/T/N, I103N, C121S, L42_K57del | [32,33,34,35] | ||||||
| SMZL | E174K | V157I, V600E | R282X, S605F | K57E, F53L | R371Q | L1208F | [36,37,38,39] | ||||
| NMZL | N58I, L597Q, V600E | [40] | |||||||||
| EMZL-MALT | Y141C | Q709_splice | F2830L, I941V, W267X | [41,42] | |||||||
| SDRPL | G12V | V187A | G469A, K601Q | P261S | K57E/N, V60E, I103N, C121S | [38,43] | |||||
| B-PLL | V600E | [44] | |||||||||
| MM | G12A/V/D/C/R/S, G13D/V, V14I, A18D/V, L19F, Q22K/E, I24N, N26K, I36M, A59G, G60R, Q61H/L/R/K/P, E63K, Y64D/N, I84M, K117N/R, D119H, A146T/V, K147E | G12D/A/V/S/C/R, G13D/R/C/V, I46M, Q61H/L/R/K/P, Y64D/N, F82L, A91V, A146T, K147N | T241M, D287_splice, D380Y, G464V, G466E/V/A, S467L, G469V/A/R/E, K483Q, N486_T491 > K, I554T, N581S/I, E586K, D594G/N/H/E/A, G596V, L597Q, V600E, K601E | R47Q | T25K, Y49H, F51V, S168P, L1631fs*1, G1649E, S1938_splice, E2729fs*10 | [45,46,47,48] | |||||
| PBL | V7G, G12R/V/D, G13D/C, A59G, Q61H/R, E63K, K117N, D119N, A146T/V | G12D/R/V, G13D/C/R/V, A59D, Q61H/K/R, K117R, A146T | R123P, Q61K | G464E, G466V/R G469V/A/R, V471F, T599TT, V600E, K601N, M689V | S257L, V512E | K57N, C121S, P124R/L, T378N | M310Cfs*2 | G16V | A2321D, S620I, A928S, R2637Q | [49,50,51,52] | |
| Compound | Combination Therapy | Target Mechanism | Disease | Molecular Inclusion Criteria a | Clinical Trial | Clinical Inclusion Criteria | Other Settings | References |
|---|---|---|---|---|---|---|---|---|
| vemurafenib | none | BRAF V600E inh | HCL | BRAF V600E | phase II: EudraCT 2011-005487-13 and NCT01711632 | r/r | case report | [139,140] |
| MM | BRAF V600E/K | phase II: NCT01524978 | r/r | case report | [141,142,143,144,145,146] | |||
| cobimetinib | BRAF V600E inh + MEK inh | HCL | BRAF V600E | case report | [147] | |||
| MM | BRAF V600 | phase II: NCT03297606 | r/r | case report | [148,149] | |||
| other B- NHL | BRAF V600 | phase II: NCT03297606 | r/r | NA | ||||
| rituximab | BRAF V600E inh + anti-CD20 mab | HCL | BRAF V600E | phase II: EudraCT-2014-003046-27 | r/r | [150] | ||
| obinutuzumab | BRAF V600E inh + anti-CD20 mab | HCL | BRAF V600E | phase II: NCT03410875 | untreated | NA | ||
| dabrafenib | none | BRAF V600E/K inh | HCL | BRAF V600E | phase II: EudraCT-2014-001379-29 | r/r | [151] | |
| CLL | RAS-MAPK | pre-clinical in vitro model | [29] | |||||
| trametinib | BRAF V600E/K inh + MEK inh | HCL | BRAF V600E | phase II: NCT02034110 | r/r | [152] | ||
| MM | KRAS, NRAS, BRAF | phase I: NCT03091257 | r/r | NA | ||||
| encorafenib | binimetinib | BRAF V600E/K/D inh + MEK inh | MM | BRAF V600E/K | phase II: NCT02834364 | r/r | NA | |
| TAK-580 | pan-RAF inh | MM | pre-clinical in vitro model | [153] | ||||
| sorafenib | none | RAF/multikinase inh | MM | phase II: NSC-724772 | r/r | [154,155] | ||
| CLL | phase II: NCT00303966; NCT01510756 | r/r | pre-clinical in vitro model | [156,157,158,159] | ||||
| DLBCL | phase II: eastern cooperative oncology group study (E1404) | r/r | pre-clinical in vitro model | [160,161,162] | ||||
| HL | pre-clinical in vitro model | [163,164] | ||||||
| everolimus | RAF/multikinase inh + mTOR inh | MM /other B-NHL | phase I/II: NCT00474929 | r/r | NA | |||
| bortezomid, R406 | RAF/multikinase inh + Proteasome inh or Syk inh | MCL | pre-clinical in vitro and in vivo model | [165] | ||||
| rapamycin | RAF/multikinase inh + mTOR inh | FL/other B-NHL | pre-clinical in vitro model | [166] | ||||
| Givinostat | RAF/multikinase inh + HDAC inh | HL | pre-clinical in vitro and in vivo model | [167] | ||||
| perifosine | RAF/multikinase inh + AKT inh | HL/CLL | phase II | r/r | pre-clinical in vitro and in vivo model | [168,169] | ||
| AZD4785 | antisense oligonucleotide targeting KRAS | MM | KRAS | pre-clinical in vitro model | [170] | |||
| CH5126766 | MEK-pan-RAF inh | MM | KRAS and BRAF | phase I: NCT02407509 | r/r | [171] | ||
| trametinib | none | MEK inh | HCL | MAP2K1 | case report | [172] | ||
| MM | KRAS, NRAS, BRAF | r/r | retrospective review of trametinib treated patients | [173] | ||||
| bortezomib | MEK inh + Proteasome inh | MM | NRAS | pre-clinical in vivo model | [174] | |||
| tirabrutinib | MEK inh + BTK inh | DLBCL | pre-clinical in vitro model | [175] | ||||
| cobimetinib | venetoclax, atezolizumab | MEK inh + BCL2 inh + anti-PD-L1 mab | MM | phase I/II: NCT03312530 | r/r | [176] | ||
| binimetinib | ABT-737, venetoclax | MEK inh + BH3-mimetic or BCL2 inh | CLL | pre-clinical in vitro model | [177] | |||
| MK2206, idelalisib | MEK inh + AKT inh or PI3K delta inh | CLL | pre-clinical in vitro model | [178] | ||||
| selumetinib | none | MEK inh | MM | phase II: NCT01085214 | r/r | [179] | ||
| DLBCL | phase II: NCT01278615 | r/r | pre-clinical in vitro and in vivo model | [180,181] | ||||
| LBH589, FK228 | MEK inh + HDAC inh | MM | KRAS, NRAS, BRAF | pre-clinical in vitro model | [182] | |||
| pimasertib | ibrutinib, idelalisib | MEK inh + BTK inh or PI3K-delta inh | DLBCL/other B-NHL | pre-clinical in vitro model | [183] | |||
| UO126 | MEK inh | HL | pre-clinical in vitro model | [184] | ||||
| AEZS-136 | PI3K/ERK dual inh | HL | pre-clinical in vitro and in vivo model | [185] | ||||
| ulixertinib | ERK inh | CLL | RAS-MAPK | pre-clinical in vitro model | [29] | |||
| SCH772984 | CI-1040, trametinib, idelalisib | ERK inh or MEK inh and/or PI3K- delta inh | CLL | MAP2K1 | pre-clinical in vitro model | [102] |
| Target Gene | Mouse Model | Genetic Background | Model Type | Phenotype | Reference |
|---|---|---|---|---|---|
| KRAS | CMV-cre; LSL-KrasG12D | C57BL/6 | Conditional KrasG12D expression in all tissues (mosaic pattern) at early embryonic stage | Embryonic lethality | [191] |
| Mx1-Cre; LSL-KrasG12D | C57BL/6 | Conditional KrasG12D expression in HSC | Development of MPD closely resembling CMML/JMML in all mice. Co-occurrence of T-ALL in minor fraction of mice. BM cell transplantation in primary recipient mice lead mostly to acute T-ALL enriched with Notch1 mutations | [192] | |
| Cγ1-Cre; LSL-KrasG12D | C57BL/6 | Conditional KrasG12D expression in post-GC B cells | Development of thymic lymphomas and lung adenomas | [193] | |
| AID-Cre-YFP; LSL-KrasG12D | 129/SvJ x C57BL/6 | Conditional KrasG12D expression in B cells undergoing GC reaction | No hematopoietic phenotype, development of focal epidermal papillomas | [193] | |
| AID-Cre-YFP; LSL-KrasG12D; Arf-/- | 129/SvJ x C57BL/6 | Conditional KrasG12D expression in B cells undergoing GC reaction in the context of tumor-prone Arf-null background (KRAS cooperating mutation). | Impairment of splenic architecture with deficiency of GC, increased polyclonal antibody responses over time. Development of fatal epidermal papillomas and cutaneous sarcomas | [193] | |
| Mx1-Cre; LSL-KrasA146T | C57BL/6 | Conditional KrasA146T expression in HSC | Development of MDS/MPN with expansion of immature myeloid cells in the BM and spleen. Delayed disease onset and death in comparison to Mx1-Cre; KrasG12D mice | [194] | |
| Kras+/V14I or KrasV14I/V14I | 129S2/Sv, C57BL/6J or mixed B6/129 | Constitutive KrasV14I expression | Noonan syndrome phenotype and development of MPD reminiscent of human JMML in KrasV14I/V14I; milder phenotype in the heterozygous model | [195] | |
| NRAS | Mox2-Cre/+; LSL-NrasG12D/+ | C57BL/6 | Conditional NrasG12D expression in epiblasts beginning at E5 | Embryonic lethality | [196] |
| Mox2-Cre/+; LSL-Nras G12Dhypo/+ or LSL-Nras G12Dhypo/G12Dhypo | C57BL/6 | Conditional NrasG12D hypomorphic allele expression (equivalent to 25–40% of single copy Nras wild type allele) in epiblasts beginning at E5 | No hematopoietic phenotype | [196] | |
| Mx1-Cre; LSL-NrasG12D/+ or LSL-NrasG12D/G12D | C57BL/6 | Conditional NrasG12D expression in HSC | In the homozygous model, development of acute MPD with ERK hyperactivation at 12 months. BM cells transplantation in primary recipient mice lead to 100% acute T-ALL enriched with Notch1 mutations; milder phenotype in the heterozygous model: development of histiocytic sarcoma (predominant) or chronic MPD (occasional) resembling CMML at 12 months. BM cells transplantation in primary recipient mice lead to 95% CMML and 8% acute T ALL. | [196,197] | |
| IgG1-Cre; LSL-NrasQ61R/+ | C57BL/6 | Conditional NrasQ61R expression in GC B cells | Development of MM or other lymphoid disease in a fraction of mice | [198] | |
| Vκ*MYC; IgG1-Cre; LSL-NrasQ61R/+ | C57BL/6 | Conditional NrasQ61R expression in GC B cells of Vκ*MYC mice (indolent MM mouse model) | Development of highly malignant MM characterized by high proliferation index, hyperactivation of ERK and AKT signaling, impaired hematopoiesis, extramedullary disease and expression of human MM gene signatures. | [198] | |
| BRAF | CMV-Cre; LSL-BrafV600E | C57BL/6 | Conditional BrafV600E expression in all tissue (mosaic pattern) at early embryonic stage | Embryonic lethality | [199] |
| Vav-cre; LSL-BrafV600E | C57BL/6 | Conditional BrafV600E expression in prenatal hematopoietic cells | In utero hematopoietic transformation and embryonic lethality beyond day 12.5 | [200] | |
| Mx1-cre; LSL-BrafV600E | C57BL/6 | Conditional BrafV600E expression in HSC | Development of HCL-like disorder characterized by extramedullary hematopoiesis, impaired erythroid differentiation, increased clonogenic capacity of B lineage cells and increased circulating soluble CD25. No hairy cells morphologic phenotype | [200] | |
| Cd19-cre; LSL-BrafV600E | C57BL/6 | Conditional BrafV600E expression in B lineage cells | No hematopoietic phenotype. MAPK signaling activation in B lineage cells, minimal elevation of soluble CD25 | [200] | |
| Eµ-TCL1; Cd19-cre; LSL-BrafV600E | Conditional BrafV600E expression in B lineage cells of Eµ-TCL1 mice (CLL mouse model) | Acceleration of CLL onset with decreased spontaneous apoptosis, enhanced immune suppression and shortening of mice survival | [201] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vendramini, E.; Bomben, R.; Pozzo, F.; Bittolo, T.; Tissino, E.; Gattei, V.; Zucchetto, A. KRAS and RAS-MAPK Pathway Deregulation in Mature B Cell Lymphoproliferative Disorders. Cancers 2022, 14, 666. https://doi.org/10.3390/cancers14030666
Vendramini E, Bomben R, Pozzo F, Bittolo T, Tissino E, Gattei V, Zucchetto A. KRAS and RAS-MAPK Pathway Deregulation in Mature B Cell Lymphoproliferative Disorders. Cancers. 2022; 14(3):666. https://doi.org/10.3390/cancers14030666
Chicago/Turabian StyleVendramini, Elena, Riccardo Bomben, Federico Pozzo, Tamara Bittolo, Erika Tissino, Valter Gattei, and Antonella Zucchetto. 2022. "KRAS and RAS-MAPK Pathway Deregulation in Mature B Cell Lymphoproliferative Disorders" Cancers 14, no. 3: 666. https://doi.org/10.3390/cancers14030666
APA StyleVendramini, E., Bomben, R., Pozzo, F., Bittolo, T., Tissino, E., Gattei, V., & Zucchetto, A. (2022). KRAS and RAS-MAPK Pathway Deregulation in Mature B Cell Lymphoproliferative Disorders. Cancers, 14(3), 666. https://doi.org/10.3390/cancers14030666