
Harnessing Liquid Biopsies to Guide Immune Checkpoint Inhibitor Therapy

 , ,
, ,  , , ,
, , ,  and
and
Abstract
:Simple Summary
Abstract
1. Background
2. Circulating Tumor DNA (ctDNA)
2.1. ctDNA Levels as an IO Biomarker
2.2. ctDNA Mutations as IO Biomarker
3. Tumor Mutational Burden
4. Circulating Tumor Cells
4.1. CTC Enumeration and IO
4.2. CTC Characterisation and IO
4.3. Crosstalk between CTC and Immune Cells
5. Peripheral Blood Cell-Based Biomarkers
5.1. T-cell Receptor Repertoire
5.2. PD-1 and CD8 Double-Positive T Lymphocytes
5.3. Natural Killer Cells
5.4. Myeloid Derived Suppressor Cells
5.5. Absolute Lymphocyte Counts and Neutrophil to Lymphocyte Ratio (NLR)
6. Other Biomarkers
6.1. Soluble PD1 and PD-L1
6.2. Extracellular Vesicles/Exosomes
7. Challenges in Liquid Biopsy-Based Biomarker Development
7.1. Intrinsic Limitations of Liquid Biopsies as Biomarkers
7.2. Challenges with Markers Based on Immune Cells
Future Aspects
Author Contributions
Funding
Conflicts of Interest
References
- Institute, C.R. 2020 PD-1/PD-L1 Analysis. Available online: https://www.cancerresearch.org/scientists/immuno-oncology-landscape/pd-1-pd-l1-landscape#landscape (accessed on 24 August 2021).
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1480–1492. [Google Scholar] [CrossRef]
- Brozos-Vázquez, E.M.; Díaz-Peña, R.; García-González, J.; León-Mateos, L.; Mondelo-Macía, P.; Peña-Chilet, M.; López-López, R. Immunotherapy in nonsmall-cell lung cancer: Current status and future prospects for liquid biopsy. Cancer Immunol. Immunother. 2020, 70, 1177–1188. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Rini, B.I.; McDermott, D.F.; Aren Frontera, O.; Hammers, H.J.; Carducci, M.A.; Salman, P.; Escudier, B.; Beuselinck, B.; Amin, A.; et al. Nivolumab plus ipilimumab versus sunitinib in first-line treatment for advanced renal cell carcinoma: Extended follow-up of efficacy and safety results from a randomised, controlled, phase 3 trial. Lancet Oncol. 2019, 20, 1370–1385. [Google Scholar] [CrossRef]
- Lee, D.H. Update of early phase clinical trials in cancer immunotherapy. BMB Rep. 2021, 54, 70–88. [Google Scholar] [CrossRef] [PubMed]
- Del Re, M.; Cucchiara, F.; Rofi, E.; Fontanelli, L.; Petrini, I.; Gri, N.; Pasquini, G.; Rizzo, M.; Gabelloni, M.; Belluomini, L.; et al. A multiparametric approach to improve the prediction of response to immunotherapy in patients with metastatic NSCLC. Cancer Immunol. Immunother. 2020, 70, 1667–1678. [Google Scholar] [CrossRef]
- Twomey, J.D.; Zhang, B. Cancer Immunotherapy Update: FDA-Approved Checkpoint Inhibitors and Companion Diagnostics. AAPS J. 2021, 23, 39. [Google Scholar] [CrossRef]
- Tannir, N.M.; Signoretti, S.; Choueiri, T.K.; McDermott, D.F.; Motzer, R.J.; Flaifel, A.; Pignon, J.-C.; Ficial, M.; Frontera, O.A.; George, S.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab versus Sunitinib in First-line Treatment of Patients with Advanced Sarcomatoid Renal Cell Carcinoma. Clin. Cancer Res. 2020, 27, 78–86. [Google Scholar] [CrossRef]
- Nakamura, Y. Biomarkers for Immune Checkpoint Inhibitor-Mediated Tumor Response and Adverse Events. Front. Med. 2019, 6, 119. [Google Scholar] [CrossRef] [Green Version]
- Fenizia, F.; Pasquale, R.; Roma, C.; Bergantino, F.; Iannaccone, A.; Normanno, N. Measuring tumor mutation burden in non-small cell lung cancer: Tissue versus liquid biopsy. Transl. Lung Cancer Res. 2018, 7, 668–677. [Google Scholar] [CrossRef]
- Klempner, S.J.; Fabrizio, D.; Bane, S.; Reinhart, M.; Peoples, T.; Ali, S.M.; Sokol, E.S.; Frampton, G.; Schrock, A.B.; Anhorn, R.; et al. Tumor Mutational Burden as a Predictive Biomarker for Response to Immune Checkpoint Inhibitors: A Review of Current Evidence. Oncologist 2019, 25, e147–e159. [Google Scholar] [CrossRef] [Green Version]
- Wild, P.J. Stellenwert der Testung der Tumormutationslast. Der Pathol. 2019, 40, 366–368. [Google Scholar] [CrossRef] [Green Version]
- Marcus, L.; Fashoyin-Aje, L.A.; Donoghue, M.; Yuan, M.; Rodriguez, L.; Gallagher, P.S.; Philip, R.; Ghosh, S.; Theoret, M.R.; Beaver, J.A.; et al. FDA Approval Summary: Pembrolizumab for the Treatment of Tumor Mutational Burden–High Solid Tumors. Clin. Cancer Res. 2021, 27, 4685–4689. [Google Scholar] [CrossRef]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H., Jr.; et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.-H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Jardim, D.L.; Goodman, A.; Gagliato, D.D.M.; Kurzrock, R. The Challenges of Tumor Mutational Burden as an Immunotherapy Biomarker. Cancer Cell 2020, 39, 154–173. [Google Scholar] [CrossRef]
- Vuijk, F.; van de Water, C.; Vliet, S.L.-V.; van der Valk, M.; Simmer, F.; van de Velde, C.; Vahrmeijer, A.; Nagtegaal, I.; Hilling, D. Intra-Tumoral Genomic Heterogeneity in Rectal Cancer: Mutational Status Is Dependent on Preoperative Biopsy Depth and Location. Cancers 2021, 13, 2271. [Google Scholar] [CrossRef]
- Yamashita, K.; Iwatsuki, M.; Ajani, J.A.; Baba, H. Programmed death ligand-1 expression in gastrointestinal cancer: Clinical significance and future challenges. Ann. Gastroenterol. Surg. 2020, 4, 369–378. [Google Scholar] [CrossRef]
- Keller, L.; Pantel, K. Unravelling tumour heterogeneity by single-cell profiling of circulating tumour cells. Nat. Cancer 2019, 19, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Zhang, C.; Pang, G.; Wang, P. Emerging Blood-Based Biomarkers for Predicting Response to Checkpoint Immunotherapy in Non-Small-Cell Lung Cancer. Front. Immunol. 2020, 11, 603157. [Google Scholar] [CrossRef]
- Castro-Giner, F.; Gkountela, S.; Donato, C.; Alborelli, I.; Quagliata, L.; Ng, C.K.Y.; Piscuoglio, S.; Aceto, N. Cancer Diagnosis Using a Liquid Biopsy: Challenges and Expectations. Diagnostics 2018, 8, 31. [Google Scholar] [CrossRef] [Green Version]
- Fattore, L.; Ruggiero, C.F.; Liguoro, D.; Castaldo, V.; Catizone, A.; Ciliberto, G.; Mancini, R. The Promise of Liquid Biopsy to Predict Response to Immunotherapy in Metastatic Melanoma. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- Möller, M.; Turzer, S.; Schütte, W.; Seliger, B.; Riemann, D. Blood Immune Cell Biomarkers in Patient With Lung Cancer Undergoing Treatment With Checkpoint Blockade. J. Immunother. 2019, 43, 57–66. [Google Scholar] [CrossRef]
- Liu, Y.; Zugazagoitia, J.; Ahmed, F.S.; Henick, B.S.; Gettinger, S.N.; Herbst, R.S.; Schalper, K.A.; Rimm, D.L. Immune Cell PD-L1 Colocalizes with Macrophages and Is Associated with Outcome in PD-1 Pathway Blockade Therapy. Clin. Cancer Res. 2019, 26, 970–977. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Wang, C.; Wang, Y.; Dai, L. Soluble PD-L1 as a predictive biomarker in lung cancer: A systematic review and meta-analysis. Future Oncol. 2021. [Google Scholar] [CrossRef]
- Wei, H.; Wu, F.; Mao, Y.; Zhang, Y.; Leng, G.; Wang, J.; Zhang, W.; Wang, T. Measurement of soluble PD-1 and soluble PD-L1 as well as PD-L1 and PD-1 from perioperative patients with gastric carcinoma. Jpn. J. Clin. Oncol. 2022. [Google Scholar] [CrossRef]
- Peters, S.; Cho, B.C.; Reinmuth, N. Tumor mutational burden (TMB) as a biomarker of survival in metastatic non-small cell lung cancer (mNSCLC): Blood and tissue TMB analysis from MYSTIC, a Phase III study of first-line durvalumab ± tremelimumab vs. chemotherapy. Cancer Res. 2019, 79. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kang, Y.; Kim, J.S.; Sung, H.H.; Jeon, H.G.; Jeong, B.C.; Seo, S.I.; Jeon, S.S.; Lee, H.M.; Park, D.; et al. Potential of circulating tumor DNA as a predictor of therapeutic responses to immune checkpoint blockades in metastatic renal cell carcinoma. Sci. Rep. 2021, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ricciuti, B.; Jones, G.; Severgnini, M.; Alessi, J.V.; Recondo, G.; Lawrence, M.; Forshew, T.; Lydon, C.; Nishino, M.; Cheng, M.; et al. Early plasma circulating tumor DNA (ctDNA) changes predict response to first-line pembrolizumab-based therapy in non-small cell lung cancer (NSCLC). J. Immunother. Cancer 2021, 9, e001504. [Google Scholar] [CrossRef] [PubMed]
- Forschner, A.; Battke, F.; Hadaschik, D.; Schulze, M.; Weißgraeber, S.; Han, C.-T.; Kopp, M.; Frick, M.; Klumpp, B.; Tietze, N.; et al. Tumor mutation burden and circulating tumor DNA in combined CTLA-4 and PD-1 antibody therapy in metastatic melanoma—Results of a prospective biomarker study. J. Immunother. Cancer 2019, 7, 180. [Google Scholar] [CrossRef] [PubMed]
- Akinleye, A.; Rasool, Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J. Hematol. Oncol. 2019, 12, 92. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.T.; Cristescu, R.; Bass, A.J.; Kim, K.-M.; Odegaard, J.I.; Kim, K.; Liu, X.Q.; Sher, X.; Jung, H.; Lee, M.; et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat. Med. 2018, 24, 1449–1458. [Google Scholar] [CrossRef]
- Call, S.G.; Huang, H.J.; Rodon, J.; Karp, D.D.; Tsimberidou, A.M.; Naing, A.; Janku, F. Abstract PO058: Targeted next-generation sequencing (NGS) of 105 cancer-related genes in circulating tumor DNA (ctDNA) from patients with advanced cancers treated with immune checkpoint inhibitors (iCPI). Cancer Immunol. Res. 2021, 9, PO058. [Google Scholar] [CrossRef]
- Sharabi, A.; Kim, S.S.; Kato, S.; Sanders, P.D.; Patel, S.P.; Sanghvi, P.; Weihe, E.; Kurzrock, R. Exceptional Response to Nivolumab and Stereotactic Body Radiation Therapy (SBRT) in Neuroendocrine Cervical Carcinoma with High Tumor Mutational Burden: Management Considerations from the Center For Personalized Cancer Therapy at UC San Diego Moores Cancer Center. Oncologist 2017, 22, 631–637. [Google Scholar] [CrossRef] [Green Version]
- Pavan, A.; Bragadin, A.B.; Calvetti, L.; Ferro, A.; Zulato, E.; Attili, I.; Nardo, G.; Maso, A.D.; Frega, S.; Menin, A.G.; et al. Role of next generation sequencing-based liquid biopsy in advanced non-small cell lung cancer patients treated with immune checkpoint inhibitors: Impact of STK11, KRAS and TP53 mutations and co-mutations on outcome. Transl. Lung Cancer Res. 2021, 10, 202–220. [Google Scholar] [CrossRef]
- Guibert, N.; Delaunay, M.; Lusque, A.; Boubekeur, N.; Rouquette, I.; Clermont, E.; Mourlanette, J.; Gouin, S.; Dormoy, I.; Favre, G.; et al. PD-L1 expression in circulating tumor cells of advanced non-small cell lung cancer patients treated with nivolumab. Lung Cancer 2018, 120, 108–112. [Google Scholar] [CrossRef]
- Dall’Olio, F.G.; Gelsomino, F.; Conci, N.; Marcolin, L.; De Giglio, A.; Grilli, G.; Sperandi, F.; Fontana, F.; Terracciano, M.; Fragomeno, B.; et al. PD-L1 Expression in Circulating Tumor Cells as a Promising Prognostic Biomarker in Advanced Non–small-cell Lung Cancer Treated with Immune Checkpoint Inhibitors. Clin. Lung Cancer 2021, 22, 423–431. [Google Scholar] [CrossRef]
- Yue, C.; Jiang, Y.; Li, P.; Wang, Y.; Xue, J.; Li, N.; Li, D.; Wang, R.; Dang, Y.; Hu, Z.; et al. Dynamic change of PD-L1 expression on circulating tumor cells in advanced solid tumor patients undergoing PD-1 blockade therapy. OncoImmunology 2018, 7, e1438111. [Google Scholar] [CrossRef]
- Tada, H.; Takahashi, H.; Kawabata-Iwakawa, R.; Nagata, Y.; Uchida, M.; Shino, M.; Ida, S.; Mito, I.; Matsuyama, T.; Chikamatsu, K. Molecular phenotypes of circulating tumor cells and efficacy of nivolumab treatment in patients with head and neck squamous cell carcinoma. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef]
- Lin, S.Y.; Chang, S.-C.; Lam, S.; Ramos, R.I.; Tran, K.; Ohe, S.; Salomon, M.P.; Bhagat, A.A.S.; Lim, C.T.; Fischer, T.D.; et al. Prospective Molecular Profiling of Circulating Tumor Cells from Patients with Melanoma Receiving Combinatorial Immunotherapy. Clin. Chem. 2019, 66, 169–177. [Google Scholar] [CrossRef]
- Lin, M.; Liang, S.-Z.; Shi, J.; Niu, L.-Z.; Chen, J.-B.; Zhang, M.-J.; Xu, K.-C. Circulating tumor cell as a biomarker for evaluating allogenic NK cell immunotherapy on stage IV non-small cell lung cancer. Immunol. Lett. 2017, 191, 10–15. [Google Scholar] [CrossRef]
- Qin, Z.; Chen, J.; Zeng, J.; Niu, L.; Xie, S.; Wang, X.; Liang, Y.; Wu, Z.; Zhang, M. Effect of NK cell immunotherapy on immune function in patients with hepatic carcinoma: A preliminary clinical study. Cancer Biol. Ther. 2017, 18, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Boudadi, K.; Suzman, D.L.; Anagnostou, V.; Fu, W.; Luber, B.; Wang, H.; Niknafs, N.; White, J.R.; Silberstein, J.L.; Sullivan, R.; et al. Ipilimumab plus nivolumab and DNA-repair defects in AR-V7-expressing metastatic prostate cancer. Oncotarget 2018, 9, 28561–28571. [Google Scholar] [CrossRef] [Green Version]
- Adams, S.; Loi, S.; Toppmeyer, D.; Cescon, D.W.; De Laurentiis, M.; Nanda, R.; Winer, E.P.; Mukai, H.; Tamura, K.; Armstrong, A.; et al. Pembrolizumab monotherapy for previously untreated, PD-L1-positive, metastatic triple-negative breast cancer: Cohort B of the phase II KEYNOTE-086 study. Ann. Oncol. 2019, 30, 405–411. [Google Scholar] [CrossRef] [Green Version]
- Janning, M.; Kobus, F.; Babayan, A.; Wikman, H.; Velthaus, J.-L.; Bergmann, S.; Schatz, S.; Falk, M.; Berger, L.-A.; Böttcher, L.-M.; et al. Determination of PD-L1 Expression in Circulating Tumor Cells of NSCLC Patients and Correlation with Response to PD-1/PD-L1 Inhibitors. Cancers 2019, 11, 835. [Google Scholar] [CrossRef] [Green Version]
- Postow, M.A.; Manuel, M.; Wong, P.; Yuan, J.; Dong, Z.; Liu, C.; Perez, S.; Tanneau, I.; Noel, M.; Courtier, A.; et al. Peripheral T cell receptor diversity is associated with clinical outcomes following ipilimumab treatment in metastatic melanoma. J. Immunother. Cancer 2015, 3, 23. [Google Scholar] [CrossRef] [Green Version]
- Hogan, S.A.; Courtier, A.; Cheng, P.F.; Jaberg-Bentele, N.F.; Goldinger, S.M.; Manuel, M.; Perez, S.; Plantier, N.; Mouret, J.-F.; Nguyen-Kim, T.D.L.; et al. Peripheral Blood TCR Repertoire Profiling May Facilitate Patient Stratification for Immunotherapy against Melanoma. Cancer Immunol. Res. 2018, 7, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.R.; Park, S.-M.; Seo, S.-U.; Jung, I.; Yoon, H.I.; Gabrilovich, D.I.; Cho, B.C.; Seong, S.-Y.; Ha, S.-J.; Youn, J.-I. The Ratio of Peripheral Regulatory T Cells to Lox-1+Polymorphonuclear Myeloid-derived Suppressor Cells Predicts the Early Response to Anti–PD-1 Therapy in Patients with Non–Small Cell Lung Cancer. Am. J. Respir. Crit. Care Med. 2019, 199, 243–246. [Google Scholar] [CrossRef]
- Sordo-Bahamonde, C.; Lorenzo-Herrero, S.; González-Rodríguez, A.; Payer, Á.; González-García, E.; López-Soto, A.; Gonzalez, S. LAG-3 Blockade with Relatlimab (BMS-986016) Restores Anti-Leukemic Responses in Chronic Lymphocytic Leukemia. Cancers 2021, 13, 2112. [Google Scholar] [CrossRef]
- Chu, F.; Westin, J.R.; Zhang, M.; Feng, L.; Baladandayuthapani, V.; Wang, Z.; Allen, R.; Wallace, M.; Vence, L.; Radvanyi, L.; et al. Immunological Effects and Predictive Gene Signatures in Patients with Relapsed Follicular Lymphoma Treated with CT-011, a Humanized Anti-PD-1 Monoclonal Antibody. Blood 2012, 120, 162. [Google Scholar] [CrossRef]
- Gobbini, E.; Swalduz, A.; Levra, M.G.; Ortiz-Cuaran, S.; Toffart, A.-C.; Pérol, M.; Moro-Sibilot, D.; Saintigny, P. Implementing ctDNA Analysis in the Clinic: Challenges and Opportunities in Non-Small Cell Lung Cancer. Cancers 2020, 12, 3112. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.H.; Bender, S.; Krahn, T.; Schlange, T. ctDNA and CTCs in Liquid Biopsy—Current Status and Where We Need to Progress. Comput. Struct. Biotechnol. J. 2018, 16, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Assaf, Z.J.; Davarpanah, N.; Banchereau, R.; Szabados, B.E.; Yuen, K.C.; Grivas, P.; Hussain, M.; Oudard, S.; Gschwend, J.E.; et al. ctDNA guiding adjuvant immunotherapy in urothelial carcinoma. Nature 2021, 595, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Raja, R.; Kuziora, M.; Brohawn, P.Z.; Higgs, B.; Gupta, A.; Dennis, P.A.; Ranade, K. Early Reduction in ctDNA Predicts Survival in Patients with Lung and Bladder Cancer Treated with Durvalumab. Clin. Cancer Res. 2018, 24, 6212–6222. [Google Scholar] [CrossRef] [Green Version]
- Clouthier, D.L.; Lien, S.C.; Yang, S.Y.C.; Nguyen, L.T.; Manem, V.S.K.; Gray, D.; Ryczko, M.; Razak, A.R.A.; Lewin, J.; Lheureux, S.; et al. An interim report on the investigator-initiated phase 2 study of pembrolizumab immunological response evaluation (INSPIRE). J. Immunother. Cancer 2019, 7, 72. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Hui, Y.; Zhang, Y.; Zhang, M.; Ji, B.; Tian, G.; Guo, Y.; Tang, M.; Li, L.; Guo, B.; et al. Application of Circulating Tumor DNA as a Biomarker for Non-Small Cell Lung Cancer. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- Lee, J.H.; Long, G.V.; Boyd, S.; Lo, S.; Menzies, A.M.; Tembe, V.; Guminski, A.; Jakrot, V.; Scolyer, R.A.; Mann, G.J.; et al. Circulating tumour DNA predicts response to anti-PD1 antibodies in metastatic melanoma. Ann. Oncol. 2017, 28, 1130–1136. [Google Scholar] [CrossRef]
- Zhang, Q.; Luo, J.; Wu, S.; Si, H.; Gao, C.; Xu, W.; Abdullah, S.E.; Higgs, B.W.; Dennis, P.A.; van der Heijden, M.S.; et al. Prognostic and Predictive Impact of Circulating Tumor DNA in Patients with Advanced Cancers Treated with Immune Checkpoint Blockade. Cancer Discov. 2020, 10, 1842–1853. [Google Scholar] [CrossRef]
- Indini, A.; Rijavec, E.; Grossi, F. Circulating Biomarkers of Response and Toxicity of Immunotherapy in Advanced Non-Small Cell Lung Cancer (NSCLC): A Comprehensive Review. Cancers 2021, 13, 1794. [Google Scholar] [CrossRef]
- Powles, T.; O’Donnell, P.H.; Massard, C.; Arkenau, H.-T.; Friedlander, T.W.; Hoimes, C.; Lee, J.L.; Ong, M.; Sridhar, S.S.; Vogelzang, N.J.; et al. Efficacy and Safety of Durvalumab in Locally Advanced or Metastatic Urothelial Carcinoma. JAMA Oncol. 2017, 3, e172411. [Google Scholar] [CrossRef]
- Garassino, M.C.; Cho, B.-C.; Kim, J.-H.; Mazières, J.; Vansteenkiste, J.; Lena, H.; Jaime, J.C.; Gray, J.E.; Powderly, J.; Chouaid, C.; et al. Final overall survival and safety update for durvalumab in third- or later-line advanced NSCLC: The phase II ATLANTIC study. Lung Cancer 2020, 147, 137–142. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Cho, B.C.; Reinmuth, N.; Lee, K.H.; Luft, A.; Ahn, M.-J.; Van Den Heuvel, M.M.; Cobo, M.; Vicente, D.; Smolin, A.; et al. Durvalumab With or Without Tremelimumab vs. Standard Chemotherapy in First-line Treatment of Metastatic Non-Small Cell Lung Cancer: The MYSTIC Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 661–674. [Google Scholar] [CrossRef] [Green Version]
- Emancipator, K. Keytruda and PD-L1: A Real-World Example of Co-development of a Drug with a Predictive Biomarker. AAPS J. 2020, 23, 1–11. [Google Scholar] [CrossRef]
- Letter, T.M. In brief: Pembrolizumab (Keytruda) for cancers with biomarkers. 1523-2859 (Electronic) 0025-732X (Linking). USA, 1 January 2018; e8. [Google Scholar]
- Wakelee, H.A.; Altorki, N.K.; Zhou, C.; Csőszi, T.; Vynnychenko, I.O.; Goloborodko, O.; Luft, A.; Akopov, A.; Martinez-Marti, A.; Kenmotsu, H.; et al. IMpower010: Primary results of a phase III global study of atezolizumab versus best supportive care after adjuvant chemotherapy in resected stage IB-IIIA non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2021, 39, 8500. [Google Scholar] [CrossRef]
- Felip, E.; Altorki, N.; Zhou, C.; Csőszi, T.; Vynnychenko, I.; Goloborodko, O.; Luft, A.; Akopov, A.; Martinez-Marti, A.; Kenmotsu, H.; et al. Adjuvant atezolizumab after adjuvant chemotherapy in resected stage IB–IIIA non-small-cell lung cancer (IMpower010): A randomised, multicentre, open-label, phase 3 trial. Lancet 2021, 398, 1344–1357. [Google Scholar] [CrossRef]
- Thompson, J.C.; Carpenter, E.L.; Silva, B.A.; Rosenstein, J.; Chien, A.L.; Quinn, K.; Espenschied, C.R.; Mak, A.; Kiedrowski, L.A.; Lefterova, M.; et al. Serial Monitoring of Circulating Tumor DNA by Next-Generation Gene Sequencing as a Biomarker of Response and Survival in Patients With Advanced NSCLC Receiving Pembrolizumab-Based Therapy. JCO Precis. Oncol. 2021, 5, 510–524. [Google Scholar] [CrossRef]
- Guibert, N.; Jones, G.; Beeler, J.F.; Plagnol, V.; Morris, C.; Mourlanette, J.; Delaunay, M.; Keller, L.; Rouquette, I.; Favre, G.; et al. Targeted sequencing of plasma cell-free DNA to predict response to PD1 inhibitors in advanced non-small cell lung cancer. Lung Cancer 2019, 137, 1–6. [Google Scholar] [CrossRef]
- Basher, F.; Saravia, D.; Fanfan, D.; Cotta, J.A.; Lopes, G. Impact of STK11 and KRAS co-mutations on outcomes with immunotherapy in non-small cell lung cancer. J. Clin. Oncol. 2020, 38, e15135. [Google Scholar] [CrossRef]
- Zhu, H.; Yu, Y.; Zheng, Y.; Xu, B.; Zheng, S.; Zeng, F.; Xie, W.; Huang, L.; Li, F.; Lin, W.; et al. KEAP1/NFE2L2 as a prognostic biomarker on immunotherapy and correlation with immune infiltrates in non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2020, 38, e21551. [Google Scholar] [CrossRef]
- Sun, D.; Tian, L.; Zhu, Y.; Wo, Y.; Liu, Q.; Liu, S.; Li, H.; Hou, H. Subunits of ARID1 serve as novel biomarkers for the sensitivity to immune checkpoint inhibitors and prognosis of advanced non-small cell lung cancer. Mol. Med. 2020, 26, 1–9. [Google Scholar] [CrossRef]
- Reece, M.; Saluja, H.; Hollington, P.; Karapetis, C.; Vatandoust, S.; Young, G.; Symonds, E.L. The Use of Circulating Tumor DNA to Monitor and Predict Response to Treatment in Colorectal Cancer. Front. Genet. 2019, 10, 1118. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; Wu, D.-H.; Ma, S.-C.; Wang, J.; Tang, X.-R.; Kang, S.; Fu, Q.J.; Cao, C.-H.; Luo, H.-S.; Chen, Y.-H.; et al. Development and validation of a genomic mutation signature to predict response to PD-1 inhibitors in non-squamous NSCLC: A multicohort stud. J. Immunother. Cancer 2020, 8, e000381. [Google Scholar] [CrossRef]
- Fabrizio, D. Setting the pace for blood tumor mutational burden. Found. Med. 2018. [Google Scholar]
- Snyder, A.; Morrissey, M.P.; Hellmann, M.D. Use of Circulating Tumor DNA for Cancer Immunotherapy. Clin. Cancer Res. 2019, 25, 6909–6915. [Google Scholar] [CrossRef] [Green Version]
- Gašperšič, J.; Paska, A.V. Potential of modern circulating cell-free DNA diagnostic tools for detection of specific tumour cells in clinical practice. Biochem. Medica 2020, 30, 409–421. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Nathanson, T.; Rizvi, H.; Creelan, B.C.; Sanchez-Vega, F.; Ahuja, A.; Ni, A.; Novik, J.B.; Mangarin, L.M.; Abu-Akeel, M.; et al. Genomic Features of Response to Combination Immunotherapy in Patients with Advanced Non-Small-Cell Lung Cancer. Cancer Cell 2018, 33, 843–852.e4. [Google Scholar] [CrossRef] [Green Version]
- Socinski, M.A.; Paul, S.M.; Yun, C.; Hu, S.; Shen, V.; Velcheti, V. CT194/8-Exploratory Subgroup Analysis of Atezolizumab (Atezo) Clinical Characteristics in Patients (pts) with Low Circulating Tumor DNA (ctDNA) in B-F1RST—A Phase II Trial Evaluating Blood-based Tumor Mutational Burden (bTMB) in NSCLC; AACR: Philadelphia, PA, USA, 2019. [Google Scholar]
- Zou, W.; Yaung, S.J.; Fuhlbrück, F.; Ballinger, M.; Peters, E.; Palma, J.F.; Shames, D.S.; Gandara, D.; Jiang, Y.; Patil, N.S. ctDNA Predicts Overall Survival in Patients With NSCLC Treated With PD-L1 Blockade or With Chemotherapy. JCO Precis. Oncol. 2021, 827–838. [Google Scholar] [CrossRef]
- Nabet, B.Y.; Esfahani, M.S.; Moding, E.J.; Hamilton, E.G.; Chabon, J.J.; Rizvi, H.; Steen, C.B.; Chaudhuri, A.A.; Liu, C.L.; Hui, A.B.; et al. Noninvasive Early Identification of Therapeutic Benefit from Immune Checkpoint Inhibition. Cell 2020, 183, 363–376.e13. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Duan, J.; Cai, S.; Han, M.; Dong, H.; Zhao, J.; Zhu, B.; Wang, S.; Zhuo, M.; Sun, J.; et al. Assessment of Blood Tumor Mutational Burden as a Potential Biomarker for Immunotherapy in Patients With Non–Small Cell Lung Cancer With Use of a Next-Generation Sequencing Cancer Gene Panel. JAMA Oncol. 2019, 5, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Albiges, L.; Tannir, N.M.; Burotto, M.; McDermott, D.; Plimack, E.R.; Barthélémy, P.; Porta, C.; Powles, T.; Donskov, F.; George, S.; et al. Nivolumab plus ipilimumab versus sunitinib for first-line treatment of advanced renal cell carcinoma: Extended 4-year follow-up of the phase III CheckMate 214 trial. ESMO Open 2020, 5, e001079. [Google Scholar] [CrossRef]
- Friedlaender, A.; Nouspikel, T.; Christinat, Y.; Ho, L.; McKee, T.; Addeo, A. Tissue-Plasma TMB Comparison and Plasma TMB Monitoring in Patients With Metastatic Non-small Cell Lung Cancer Receiving Immune Checkpoint Inhibitors. Front. Oncol. 2020, 10, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Dai, J.; Wang, D.; Lu, H.; Dai, H.; Ye, H.; Gu, J.; Chen, S.; Huang, B. Assessment of tumor mutation burden calculation from gene panel sequencing data. OncoTargets Ther. 2019, 12, 3401–3409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenzinger, A.; Endris, V.; Budczies, J.; Merkelbach-Bruse, S.; Kazdal, D.; Dietmaier, W.; Pfarr, N.; Siebolts, U.; Hummel, M.; Herold, S.; et al. Harmonization and Standardization of Panel-Based Tumor Mutational Burden Measurement: Real-World Results and Recommendations of the Quality in Pathology Study. J. Thorac. Oncol. 2020, 15, 1177–1189. [Google Scholar] [CrossRef]
- Wu, X.; Zhu, L.; Ma, P.C. Next-Generation Novel Noninvasive Cancer Molecular Diagnostics Platforms Beyond Tissues. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 964–977. [Google Scholar] [CrossRef] [Green Version]
- Cabel, L.; Proudhon, C.; Mariani, P.; Tzanis, D.; Beinse, G.; Bieche, I.; Pierga, J.-Y.; Bidard, F.-C. Circulating tumor cells and circulating tumor DNA: What surgical oncologists need to know? Eur. J. Surg. Oncol. (EJSO) 2017, 43, 949–962. [Google Scholar] [CrossRef]
- Cabel, L.; Proudhon, C.; Gortais, H.; Loirat, D.; Coussy, F.; Pierga, J.-Y.; Bidard, F.-C. Circulating tumor cells: Clinical validity and utility. Int. J. Clin. Oncol. 2017, 22, 421–430. [Google Scholar] [CrossRef]
- Ding, L.; Gosh, A.; Lee, D.J.; Emri, G.; Huss, W.J.; Bogner, P.N.; Paragh, G. Prognostic biomarkers of cutaneous melanoma. Photodermatol. Photoimmunol. Photomed. 2022. [Google Scholar] [CrossRef]
- Hoshimoto, S.; Faries, M.B.; Morton, D.L.; Shingai, T.; Kuo, C.; Wang, H.J.; Elashoff, R.; Mozzillo, N.; Kelley, M.C.; Thompson, J.F.; et al. Assessment of prognostic circulating tumor cells in a phase III trial of adjuvant immunotherapy after complete resection of stage IV melanoma. Ann. Surg. 2012, 255, 357–362. [Google Scholar] [CrossRef] [Green Version]
- Ramsköld, D.; Luo, S.; Wang, Y.-C.; Li, R.; Deng, Q.; Faridani, O.R.; Daniels, G.A.; Khrebtukova, I.; Loring, J.F.; Laurent, L.C.; et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol. 2012, 30, 777–782. [Google Scholar] [CrossRef] [Green Version]
- Lian, S.; Xie, R.; Ye, Y.; Lu, Y.; Cheng, Y.; Xie, X.; Li, S.; Jia, L. Dual blockage of both PD-L1 and CD47 enhances immunotherapy against circulating tumor cells. Sci. Rep. 2019, 9, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Khair, D.; Bax, H.J.; Mele, S.; Crescioli, S.; Pellizzari, G.; Khiabany, A.; Nakamura, M.; Harris, R.J.; French, E.; Hoffmann, R.M.; et al. Combining Immune Checkpoint Inhibitors: Established and Emerging Targets and Strategies to Improve Outcomes in Melanoma. Front. Immunol. 2019, 10, 453. [Google Scholar] [CrossRef] [Green Version]
- Papadaki, M.A.; Koutsopoulos, A.V.; Tsoulfas, P.G.; Lagoudaki, E.; Aggouraki, D.; Monastirioti, A.; Koutoulaki, C.; Apostolopoulou, C.A.; Merodoulaki, A.C.; Papadaki, C.; et al. Clinical Relevance of Immune Checkpoints on Circulating Tumor Cells in Breast Cancer. Cancers 2020, 12, 376. [Google Scholar] [CrossRef] [Green Version]
- Seidel, J.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Relecom, A.; Merhi, M.; Inchakalody, V.; Uddin, S.; Rinchai, D.; Bedognetti, D.; Dermime, S. Emerging dynamics pathways of response and resistance to PD-1 and CTLA-4 blockade: Tackling uncertainty by confronting complexity. J. Exp. Clin. Cancer Res. 2021, 40, 1–19. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Qi, Q.; Liu, Y.; Cheng, Y.; Glanville, J.; Zhang, D.; Lee, J.-Y.; Olshen, R.A.; Weyand, C.M.; Boyd, S.D.; Goronzy, J.J. Diversity and clonal selection in the human T-cell repertoire. Proc. Natl. Acad. Sci. USA 2014, 111, 13139–13144. [Google Scholar] [CrossRef] [Green Version]
- Robert, L.; Tsoi, J.; Wang, X.; Emerson, R.; Homet, B.; Chodon, T.; Mok, S.; Huang, R.R.; Cochran, A.J.; Comin-Anduix, B.; et al. CTLA4 Blockade Broadens the Peripheral T-Cell Receptor Repertoire. Clin. Cancer Res. 2014, 20, 2424–2432. [Google Scholar] [CrossRef] [Green Version]
- Britanova, O.V.; Putintseva, E.V.; Shugay, M.; Merzlyak, E.M.; Turchaninova, M.A.; Staroverov, D.B.; Bolotin, D.A.; Lukyanov, S.; Bogdanova, E.A.; Mamedov, I.Z.; et al. Age-related decrease in TCR repertoire diversity measured with deep and normalized sequence profiling. J. Immunol. 2014, 192, 2689–2698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishna, C.; Chowell, D.; Gönen, M.; Elhanati, Y.; Chan, T.A. Genetic and environmental determinants of human TCR repertoire diversity. Immun. Ageing 2020, 17, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Duan, J.; Bai, H.; Wang, Y.; Wan, R.; Wang, X.; Chen, S.; Tian, Y.; Wang, D.; Fei, K.; et al. TCR Repertoire Diversity of Peripheral PD-1+CD8+ T Cells Predicts Clinical Outcomes after Immunotherapy in Patients with Non–Small Cell Lung Cancer. Cancer Immunol. Res. 2019, 8, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Mazzaschi, G.; Facchinetti, F.; Missale, G.; Canetti, D.; Madeddu, D.; Zecca, A.; Veneziani, M.; Gelsomino, F.; Goldoni, M.; Buti, S.; et al. The circulating pool of functionally competent NK and CD8+ cells predicts the outcome of anti-PD1 treatment in advanced NSCLC. Lung Cancer 2019, 127, 153–163. [Google Scholar] [CrossRef]
- Kamphorst, A.O.; Pillai, R.N.; Yang, S.; Nasti, T.H.; Akondy, R.S.; Wieland, A.; Sica, G.L.; Yu, K.; Koenig, L.; Patel, N.T.; et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1–targeted therapy in lung cancer patients. Proc. Natl. Acad. Sci. USA 2017, 114, 4993–4998. [Google Scholar] [CrossRef] [Green Version]
- Ben-Shmuel, A.; Biber, G.; Barda-Saad, M. Unleashing Natural Killer Cells in the Tumor Microenvironment—The Next Generation of Immunotherapy? Front. Immunol. 2020, 11, 275. [Google Scholar] [CrossRef] [Green Version]
- Niu, C.; Li, M.; Zhu, S.; Chen, Y.; Zhou, L.; Xu, D.; Xu, J.; Li, Z.; Li, W.; Cui, J. PD-1-positive Natural Killer Cells have a weaker antitumor function than that of PD-1-negative Natural Killer Cells in Lung Cancer. Int. J. Med Sci. 2020, 17, 1964–1973. [Google Scholar] [CrossRef]
- Quatrini, L.; Mariotti, F.; Munari, E.; Tumino, N.; Vacca, P.; Moretta, L. The Immune Checkpoint PD-1 in Natural Killer Cells: Expression, Function and Targeting in Tumour Immunotherapy. Cancers 2020, 12, 3285. [Google Scholar] [CrossRef]
- Cho, Y.-H.; Choi, M.G.; Kim, D.H.; Choi, Y.J.; Kim, S.Y.; Sung, K.J.; Lee, J.C.; Kim, S.-Y.; Rho, J.K.; Choi, C.-M. Natural Killer Cells as a Potential Biomarker for Predicting Immunotherapy Efficacy in Patients with Non-Small Cell Lung Cancer. Target. Oncol. 2020, 15, 241–247. [Google Scholar] [CrossRef]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, Y.; Safi, S.; Blattner, C.; Rathinasamy, A.; Umansky, L.; Juenger, S.; Warth, A.; Eichhorn, M.; Muley, T.; Herth, F.J.F.; et al. Circulating and Tumor Myeloid-derived Suppressor Cells in Resectable Non–Small Cell Lung Cancer. Am. J. Respir. Crit. Care Med. 2018, 198, 777–787. [Google Scholar] [CrossRef]
- Zhang, H.; Ye, Y.-L.; Li, M.-X.; Ye, S.-B.; Huang, W.-R.; Cai, T.-T.; He, J.; Peng, J.-Y.; Duan, T.-H.; Cui, J.; et al. CXCL2/MIF-CXCR2 signaling promotes the recruitment of myeloid-derived suppressor cells and is correlated with prognosis in bladder cancer. Oncogene 2016, 36, 2095–2104. [Google Scholar] [CrossRef]
- Passaro, A.; Mancuso, P.; Gandini, S.; Spitaleri, G.; Labanca, V.; Guerini-Rocco, E.; Barberis, M.; Catania, C.; Del Signore, E.; De Marinis, F.; et al. Gr-MDSC-linked asset as a potential immune biomarker in pretreated NSCLC receiving nivolumab as second-line therapy. Clin. Transl. Oncol. 2019, 22, 603–611. [Google Scholar] [CrossRef]
- Criscitiello, C.; Marra, A.; Morganti, S.; Zagami, P.; Viale, G.; Esposito, A.; Curigliano, G. Pretreatment Blood Parameters Predict Efficacy from Immunotherapy Agents in Early Phase Clinical Trials. Oncol. 2020, 25, e1732–e1742. [Google Scholar] [CrossRef]
- Cao, D.; Xu, H.; Xu, X.; Guo, T.; Ge, W. A reliable and feasible way to predict the benefits of Nivolumab in patients with non-small cell lung cancer: A pooled analysis of 14 retrospective studies. OncoImmunology 2018, 7, e1507262. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhang, Z.; Hu, Y.; Yan, X.; Song, Q.; Wang, G.; Chen, R.; Jiao, S.; Wang, J. Pretreatment Neutrophil-to-Lymphocyte Ratio (NLR) May Predict the Outcomes of Advanced Non-small-cell Lung Cancer (NSCLC) Patients Treated With Immune Checkpoint Inhibitors (ICIs). Front. Oncol. 2020, 10, 654. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Mahoney, K.M.; Giobbie-Hurder, A.; Zhao, F.; Lee, S.; Liao, X.; Rodig, S.; Li, J.; Wu, X.; Butterfield, L.H.; et al. Soluble PD-L1 as a Biomarker in Malignant Melanoma Treated with Checkpoint Blockade. Cancer Immunol. Res. 2017, 5, 480–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, S.Y.; Kim, S.; Keam, B.; Kim, T.M.; Kim, D.-W.; Heo, D.S. Soluble PD-L1 is a predictive and prognostic biomarker in advanced cancer patients who receive immune checkpoint blockade treatment. Sci. Rep. 2021, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ohkuma, R.; Ieguchi, K.; Watanabe, M.; Takayanagi, D.; Goshima, T.; Onoue, R.; Hamada, K.; Kubota, Y.; Horiike, A.; Ishiguro, T.; et al. Increased Plasma Soluble PD-1 Concentration Correlates with Disease Progression in Patients with Cancer Treated with Anti-PD-1 Antibodies. Biomedicines 2021, 9, 1929. [Google Scholar] [CrossRef] [PubMed]
- Mathew, M.; Zade, M.; Mezghani, N.; Patel, R.; Wang, Y.; Momen-Heravi, F. Extracellular Vesicles as Biomarkers in Cancer Immunotherapy. Cancers 2020, 12, 2825. [Google Scholar] [CrossRef] [PubMed]
- Gehrmann, U.; Näslund, T.I.; Hiltbrunner, S.; Larssen, P.; Gabrielsson, S. Harnessing the exosome-induced immune response for cancer immunotherapy. Semin. Cancer Biol. 2014, 28, 58–67. [Google Scholar] [CrossRef]
- Yu, W.; Hurley, J.; Roberts, D.; Chakrabortty, S.; Enderle, D.; Noerholm, M.; Breakefield, X.; Skog, J. Exosome-based liquid biopsies in cancer: Opportunities and challenges. Ann. Oncol. 2021, 32, 466–477. [Google Scholar] [CrossRef]
- Serratì, S.; Guida, M.; Di Fonte, R.; De Summa, S.; Strippoli, S.; Iacobazzi, R.M.; Quarta, A.; De Risi, I.; Guida, G.; Paradiso, A.; et al. Circulating extracellular vesicles expressing PD1 and PD-L1 predict response and mediate resistance to checkpoint inhibitors immunotherapy in metastatic melanoma. Mol. Cancer 2022, 21, 1–18. [Google Scholar] [CrossRef]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef]
- Dou, X.; Hua, Y.; Chen, Z.; Chao, F.; Li, M. Extracellular vesicles containing PD-L1 contribute to CD8+ T-cell immune suppression and predict poor outcomes in small cell lung cancer. Clin. Exp. Immunol. 2022. [Google Scholar] [CrossRef]
- Theodoraki, M.-N.; Yerneni, S.S.; Hoffmann, T.K.; Gooding, W.E.; Whiteside, T.L. Clinical Significance of PD-L1+ Exosomes in Plasma of Head and Neck Cancer Patients. Clin. Cancer Res. 2018, 24, 896–905. [Google Scholar] [CrossRef] [Green Version]
- Daassi, D.; Mahoney, K.M.; Freeman, G.J. The importance of exosomal PDL1 in tumour immune evasion. Nat. Rev. Immunol. 2020, 20, 209–215. [Google Scholar] [CrossRef]
- Whiteside, T.L. Immune modulation of T-cell and NK (natural killer) cell activities by TEXs (tumour-derived exosomes). Biochem. Soc. Trans. 2013, 41, 245–251. [Google Scholar] [CrossRef]
- Augustus, E.; Zwaenepoel, K.; Siozopoulou, V.; Raskin, J.; Jordaens, S.; Baggerman, G.; Sorber, L.; Roeyen, G.; Peeters, M.; Pauwels, P. Prognostic and Predictive Biomarkers in Non-Small Cell Lung Cancer Patients on Immunotherapy—The Role of Liquid Biopsy in Unraveling the Puzzle. Cancers 2021, 13, 1675. [Google Scholar] [CrossRef]
- Becker, T.M.; Caixeiro, N.J.; Lim, S.H.; Tognela, A.; Kienzle, N.; Scott, K.; Spring, K.J.; De Souza, P. New frontiers in circulating tumor cell analysis: A reference guide for biomolecular profiling toward translational clinical use. Int. J. Cancer 2013, 134, 2523–2533. [Google Scholar] [CrossRef] [Green Version]
- Whale, A.S.; Devonshire, A.S.; Karlin-Neumann, G.A.; Regan, J.; Javier, L.; Cowen, S.; Fernandez-Gonzalez, A.; Jones, G.M.; Redshaw, N.; Beck, J.; et al. International Interlaboratory Digital PCR Study Demonstrating High Reproducibility for the Measurement of a Rare Sequence Variant. Anal. Chem. 2017, 89, 1724–1733. [Google Scholar] [CrossRef]
- Mehnert, J.M.; Monjazeb, A.M.; Beerthuijzen, J.M.; Collyar, D.; Rubinstein, L.; Harris, L.N. The Challenge for Development of Valuable Immuno-oncology Biomarkers. Clin. Cancer Res. 2017, 23, 4970–4979. [Google Scholar] [CrossRef] [Green Version]

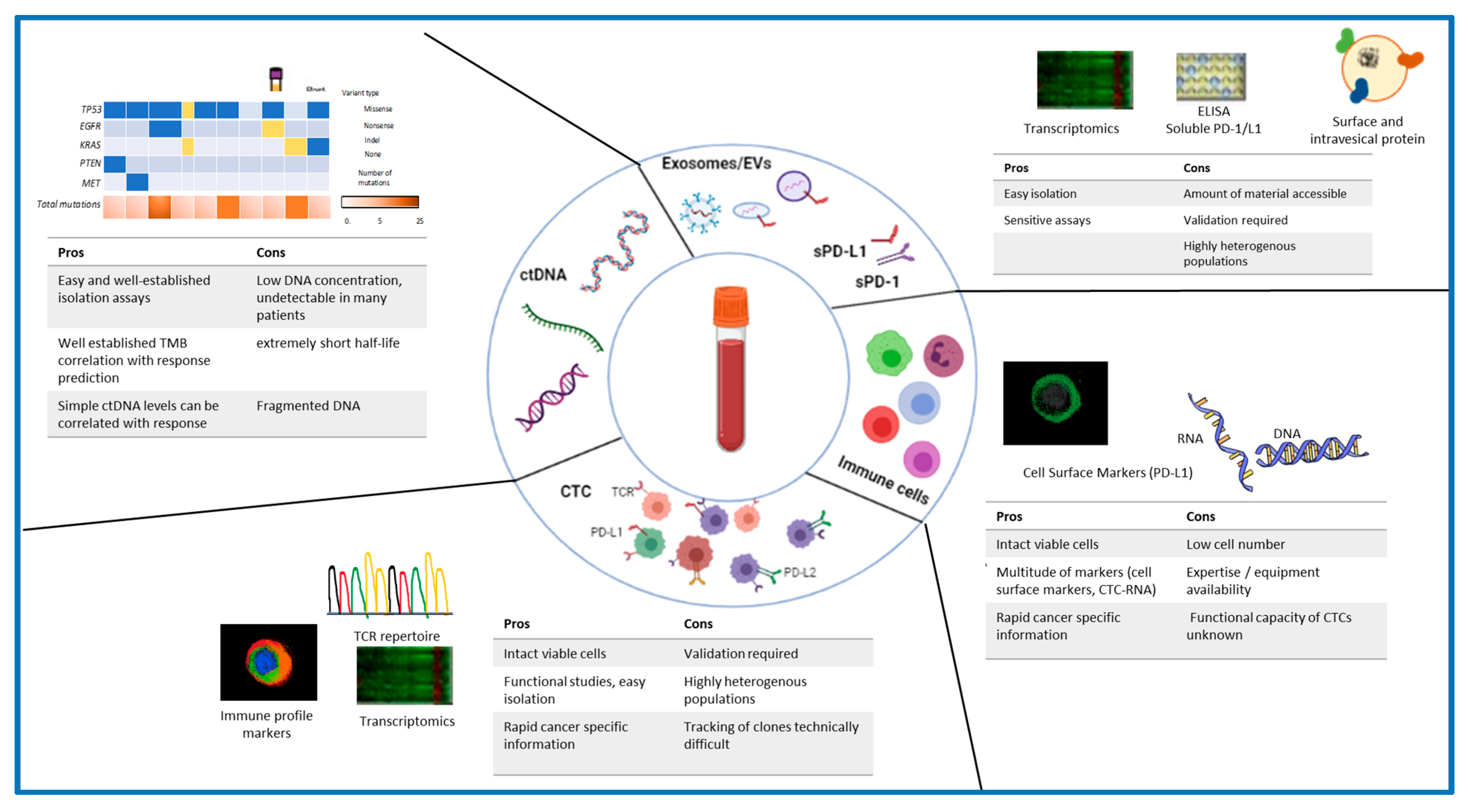

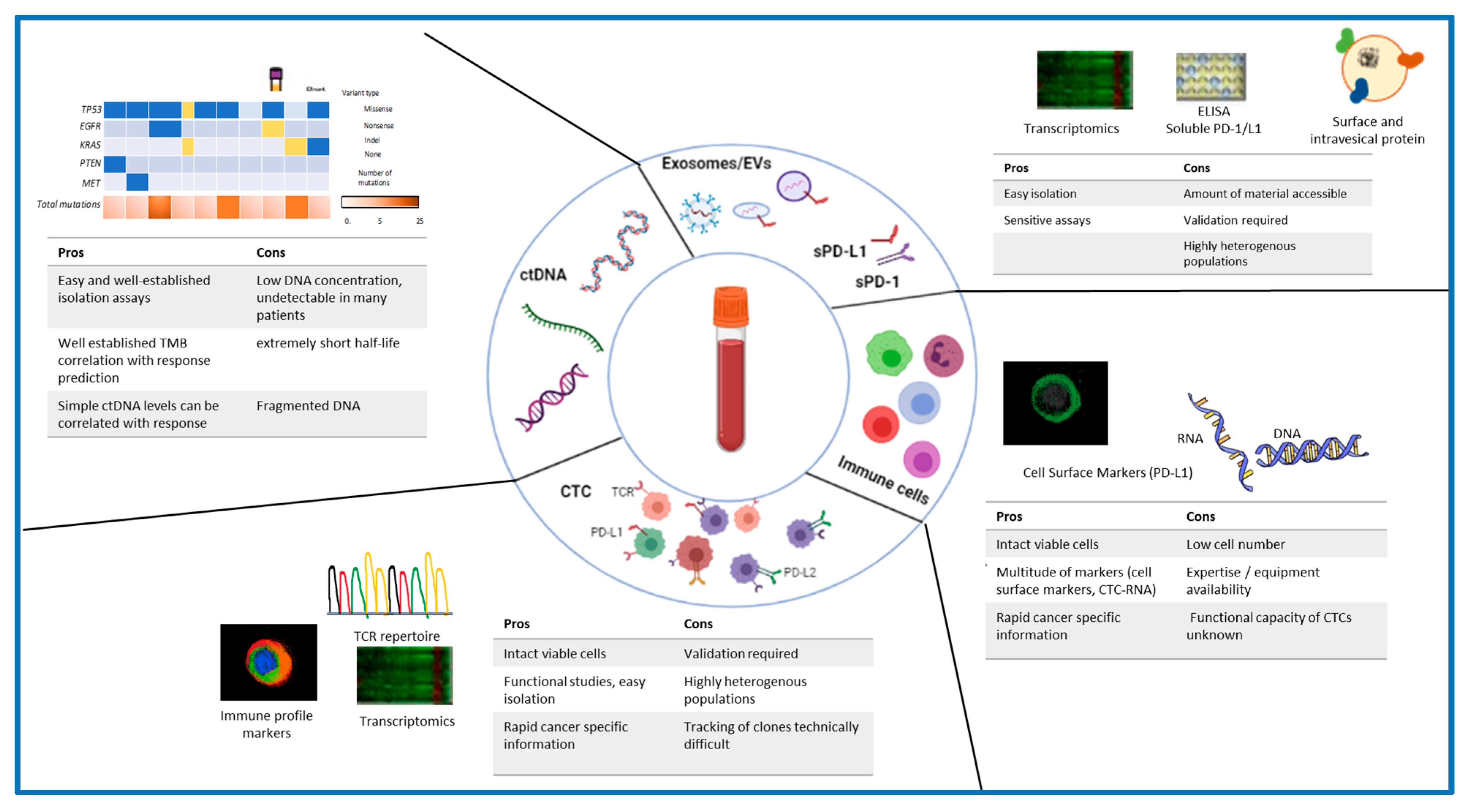
{kind=link}
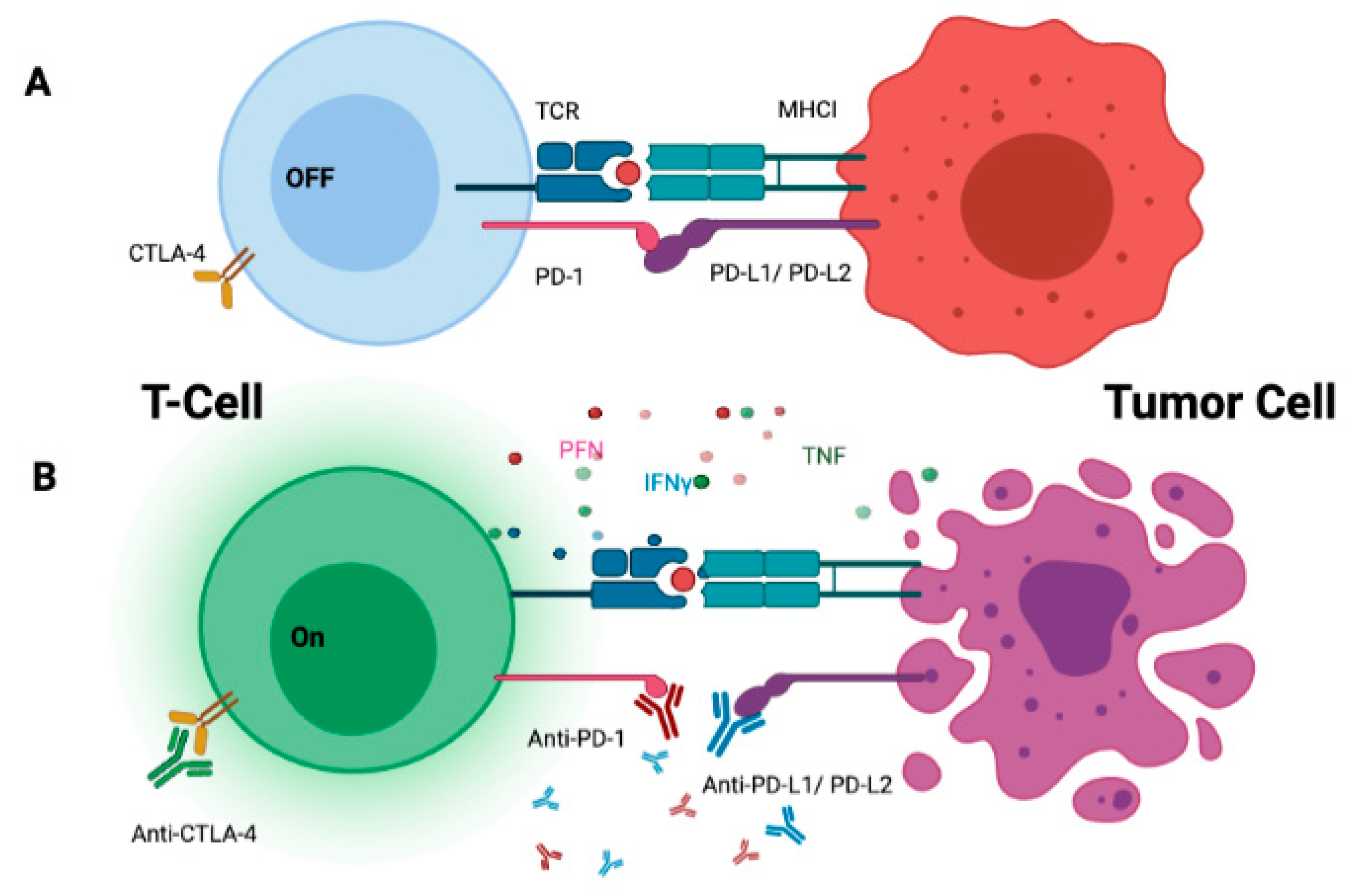
{kind=link}
| Cancer Type | Immunotherapeutic Strategies | Measurement Index | Data Outcomes | Reference |
|---|---|---|---|---|
| ctDNA | ||||
| Metastatic NSCLC | Durvalumab ± tremelimumab | GuardantOMNI ctDNA platform | ctDNA-based TMB (cTMB ≥16 mut/Mb) correlates positively with tissue (t) TMB (≥10 mut/Mb) and is predictive of survival benefit | [28] |
| Advanced renal cell carcinoma | Nivolumab plus ipilimumab | Targeted deep sequencing | Concordance of variants with biopsy-based sequencing tests, increased ctDNA levels post 1 month of treatment showed intrinsic resistance, decreased ctDNA levels showed higher response | [29] |
| NSCLC | Pembrolizumab ± platinum doublet chemotherapy | Tagged-amplicon sequencing of hotspots and coding regions from 36 genes | Rapid decrease in ctDNA post 21 days treatment prior to radiological assessment correlated with higher radiographic responses and long-term clinical outcomes | [30] |
| Melanoma | Ipilimumab (as anti-CTLA-4) and nivolumab (as anti-PD-1) | NGS | Simultaneous reduction of both tissue TMB and cTMB parameters after 3 weeks of starting treatment was able to identify responders vs. non-responders | [31] |
| Metastatic gastric cancer | Pembrolizumab | 73-gene ctDNA NGS assay | Decreased ctDNA post treatment was associated with improved outcomes | [32,33] |
| Advanced solid cancers | ICIs | Targeted NGS panel | Higher quantity of ctDNA was associated with shorter time to failure and shorter OS compared to patients with lower quantity of ctDNA | [34] |
| Neuroendocrine Cervical Carcinoma | Nivolumab and Stereotactic Body Radiation Therapy (SBRT) | Deep sequencing | High cTMB correlated with observed high tTMB, as a consequence of a mismatch repair gene defect. This observation led to a therapeutic “match” with an anti-PD 1 antibody combined with radiation therapy, resulting in a durable response (10 + months). | [35] |
| NSCLC | Pembrolizumab±platinum/pemetrexed | Guardant360® test, and MAGIC (Monitoring Advanced NSCLC through plasma Genotyping | Presence of TP53 mutations in ctDNA, confers poor survival both on ICI and chemotherapy. STK11 mutated patients (n = 9) showed worse OS (only if treated with ICIs). The presence of KRAS/STK11 co-mutation and KRAS/STK11/TP53 co-mutation affected OS only in patients treated with ICIs indicating a predictive role | [36] |
| CTCs | ||||
| NSCLC | Nivolumab | CTCs number, PD-L1+ CTCs | Contradictory result on the correlation of PD-L1 CTC with poor outcome | [37,38] |
| Gastrointestinal tumours | IBI308 (PD-1 monoclonal antibody) | PD-L1 expression | Responders have high expression of PDL1 on CTCs. After treatment: significant reduction of PD-L1+ CTCs and high-positive PD-L1 CTCs | [39] |
| HNC | Nivolumab | CTCs number, PD-L1+ CTCs | Post treatment CTC-positive patients had a shorter PFS, and PD-L1-positive CTCs were significantly associated with worse outcomes | [40] |
| Melanoma | Combinatorial immunotherapy (pembrolizumab, bevacizumab nivolumab/dabrafenib/trametinib) | CTC mRNA and DNA biomarker panels | Incorporation of a DNA biomarker with mRNA profiling increased overall CTC-detection capability. CTC-CTNNB1 was associated with progressive disease/stable disease compared to complete-responder patient status | [41] |
| NSCLC, Hepatic carcinoma | NK cell therapy | CTCs number | The decrease of CTC number is related to NK cell therapy efficacy | [42,43] |
| AR-V7 positive metastatic PCa | Ipilimumab and nivolumab | ARV7 expression, DNA-repair gene mutations and phenotypic heterogeneity | Higher mutations in DNA-repair-related genes in isolated CTCs and their phenotypic heterogeneity were associated with improved clinical outcomes in ARV-7 positive patients Presence of AR-V7+ CTCs was associated with CTCs heterogeneity, which correlated with the likelihood of a favourable response to immune-checkpoint inhibition | [44] |
| Breast cancer | Pembrolizumab | CTC mRNA | Patients with overexpression of PD-L1 have shown a better response to mono-immunotherapy | [45] |
| NSCLC | Pembrolizumab, nivolumab, atezolizumab | PD-L1 expression of CTCs | Upon disease progression, all patients demonstrated an increase in PD-L1+ CTCs, while no change or a decrease in PD-L1+ CTCs was observed in responding patients. An increase of PD-L1+ CTCs had the potential to predict resistance to PD-1/PD-L1 inhibitor | [46] |
| Immune cells | ||||
| Metastatic melanoma | Ipilimumab | T-cell repertoire (sequencing of the CD region 3 of the T-cell receptors) | TCR diversity was evaluated using a polymerase chain reaction assay, which measures TCR combinatorial diversity between V and J genes from genomic DNA. TCR repertoire TCR diversity was associated with clinical benefit but not overall survival | [47] |
| NSCLC | Pembrolizumab | The expression level of PD-L1 repertoire of T-cell were analysed using the PCR-based method) | Patients with low T-cell receptor diversity at baseline responded better to anti-PD-L1 therapy | [48] |
| NSCLC | Nivolumab | Frequency of immune-suppressive cells, including Tregs and MDSCs | The number of polymorphonuclear MDCSCs and TREG in peripheral blood was significantly higher in non-responders than in responders using flow cytometry | [49] |
| Chronic lymphatic leukaemia | Relatlimab | Lymphocyte activation gene 3 (LAG-3 expression level in T cells and NK cells) | The expression level of LAG-3 was analysed by in Silico mRNA analysis and serum level of LAG-3 by ELISA | [50] |
| Relapsed Follicular Lymphoma | Pidilizumab | The number of PB immune cells, the expression of the activating receptor NKG2D on NK cells | Increased number of PB immune cell observed using multiparametric flow cytometry. Whole genome gene expression profiling (GEP) was performed on core needle biopsies | [51] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fatima, S.; Ma, Y.; Safrachi, A.; Haider, S.; Spring, K.J.; Vafaee, F.; Scott, K.F.; Roberts, T.L.; Becker, T.M.; de Souza, P. Harnessing Liquid Biopsies to Guide Immune Checkpoint Inhibitor Therapy. Cancers 2022, 14, 1669. https://doi.org/10.3390/cancers14071669
Fatima S, Ma Y, Safrachi A, Haider S, Spring KJ, Vafaee F, Scott KF, Roberts TL, Becker TM, de Souza P. Harnessing Liquid Biopsies to Guide Immune Checkpoint Inhibitor Therapy. Cancers. 2022; 14(7):1669. https://doi.org/10.3390/cancers14071669
Chicago/Turabian StyleFatima, Shadma, Yafeng Ma, Azadeh Safrachi, Sana Haider, Kevin J. Spring, Fatemeh Vafaee, Kieran F. Scott, Tara L. Roberts, Therese M. Becker, and Paul de Souza. 2022. "Harnessing Liquid Biopsies to Guide Immune Checkpoint Inhibitor Therapy" Cancers 14, no. 7: 1669. https://doi.org/10.3390/cancers14071669
APA StyleFatima, S., Ma, Y., Safrachi, A., Haider, S., Spring, K. J., Vafaee, F., Scott, K. F., Roberts, T. L., Becker, T. M., & de Souza, P. (2022). Harnessing Liquid Biopsies to Guide Immune Checkpoint Inhibitor Therapy. Cancers, 14(7), 1669. https://doi.org/10.3390/cancers14071669