
AKT1 Is Required for a Complete Palbociclib-Induced Senescence Phenotype in BRAF-V600E-Driven Human Melanoma

 , , and
, , and
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. CRISPR
2.2. Sequencing and TIDE Analysis
2.3. Cell Culture
2.4. cGAS Agonist Transfection
2.5. CDNA Synthesis and RT-qPCR
2.6. Immunoblot
2.7. Beta-Galactosidase Staining
2.8. 2′3′-cGAMP ELISA
2.9. Cell Cycle Analysis
2.10. Proliferation Assay
2.11. Statistical Analysis
3. Results
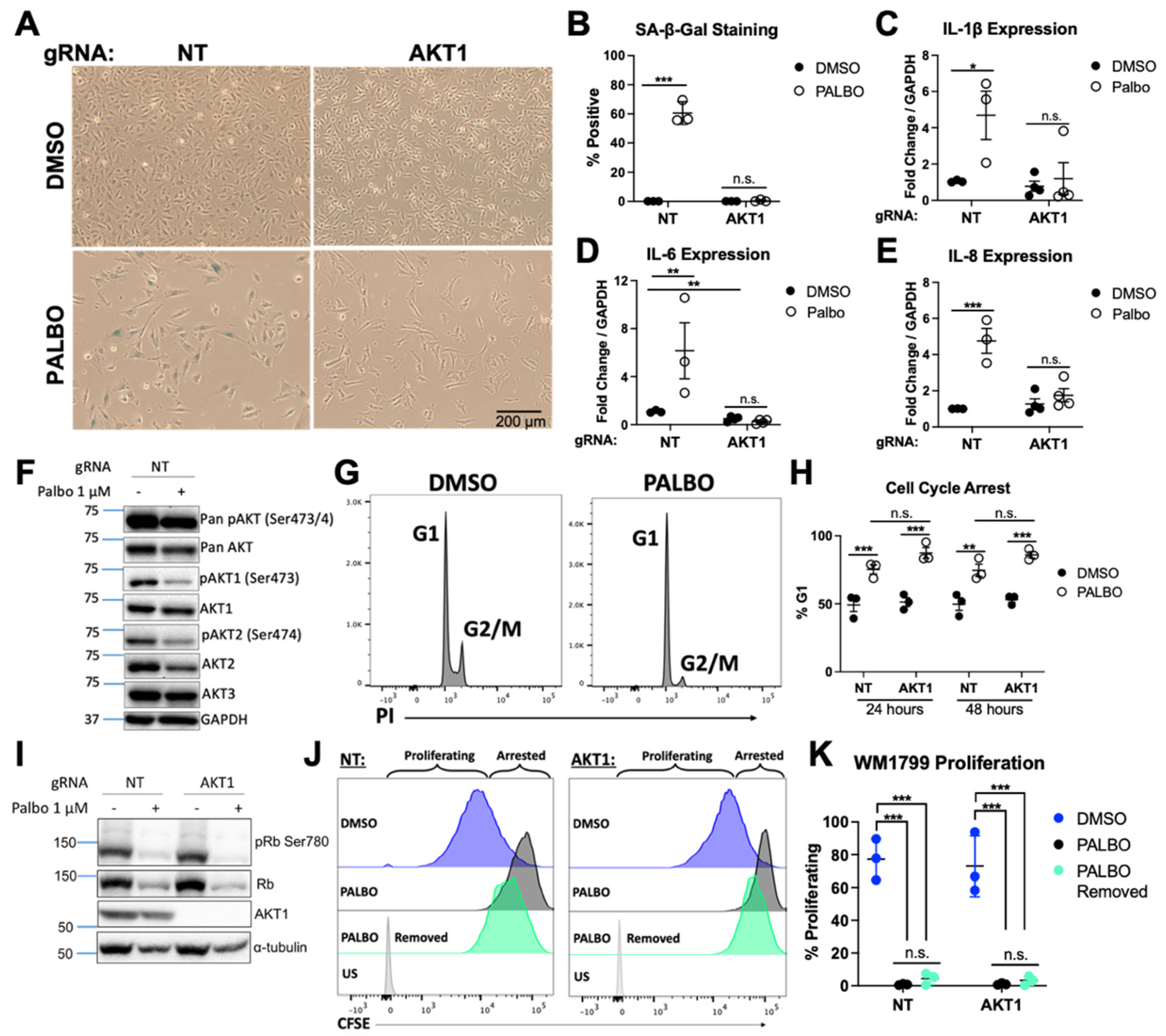
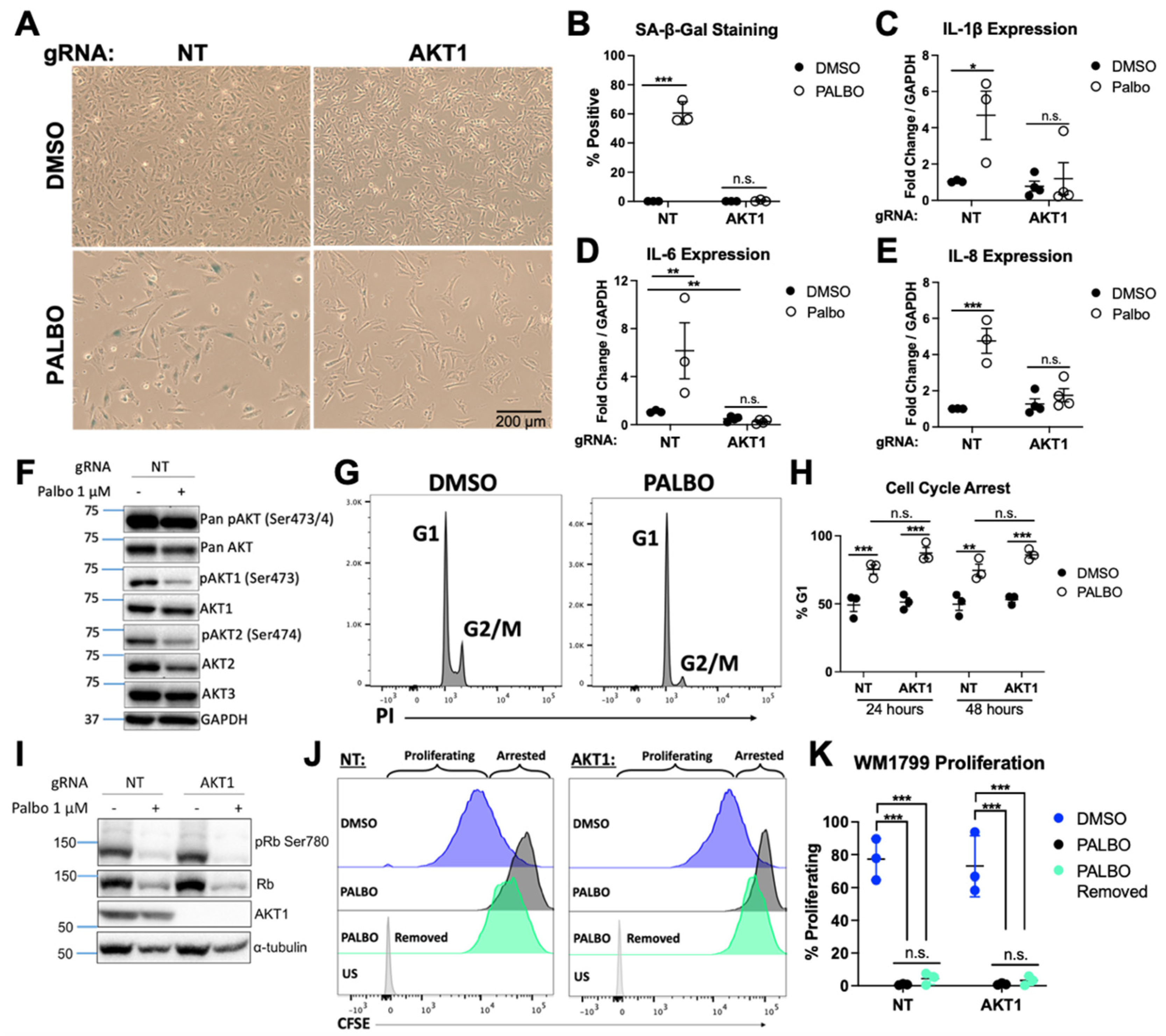
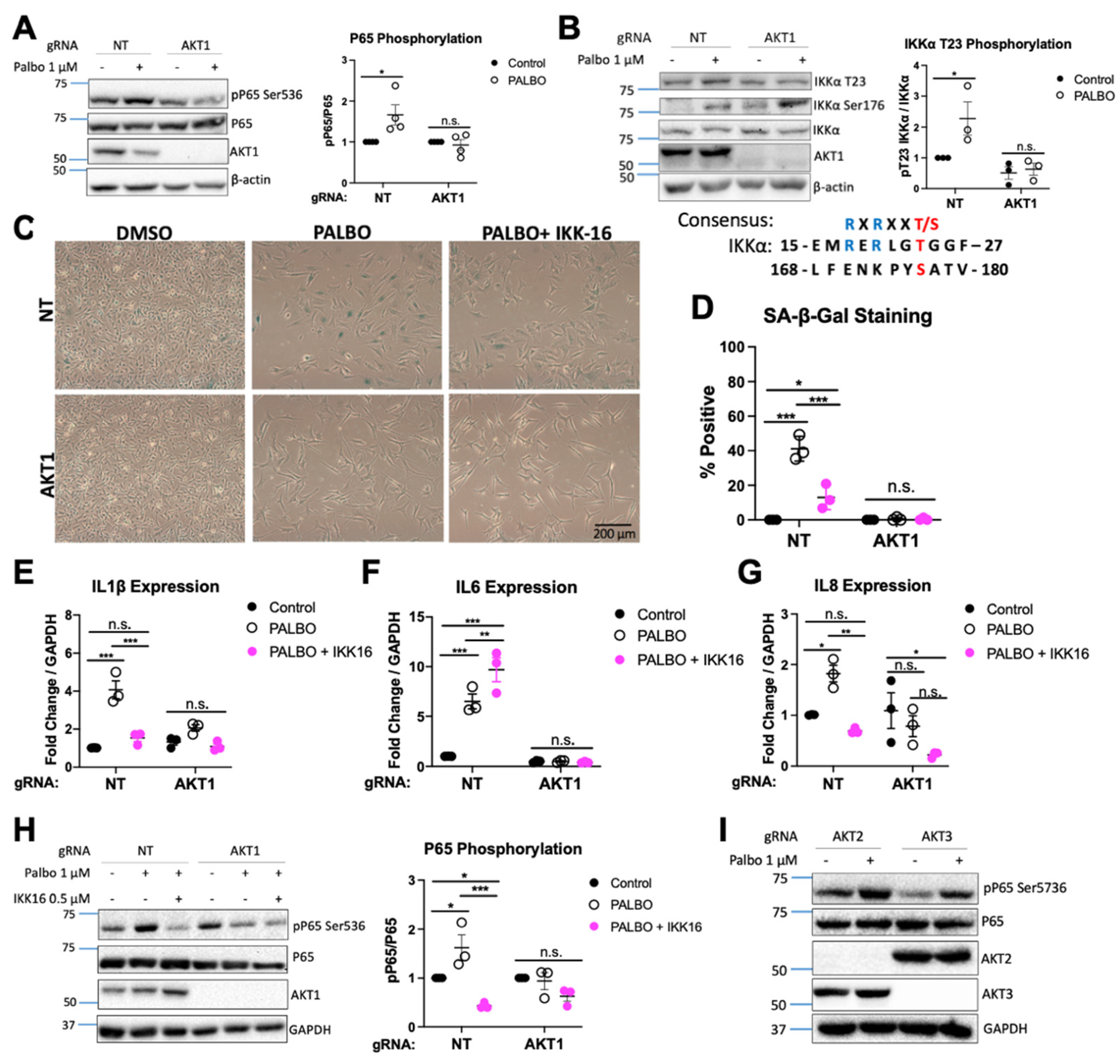
3.1. AKT1 Is Required for Aspects of Palbociclib-Induced Senescence in WM1799 Cells
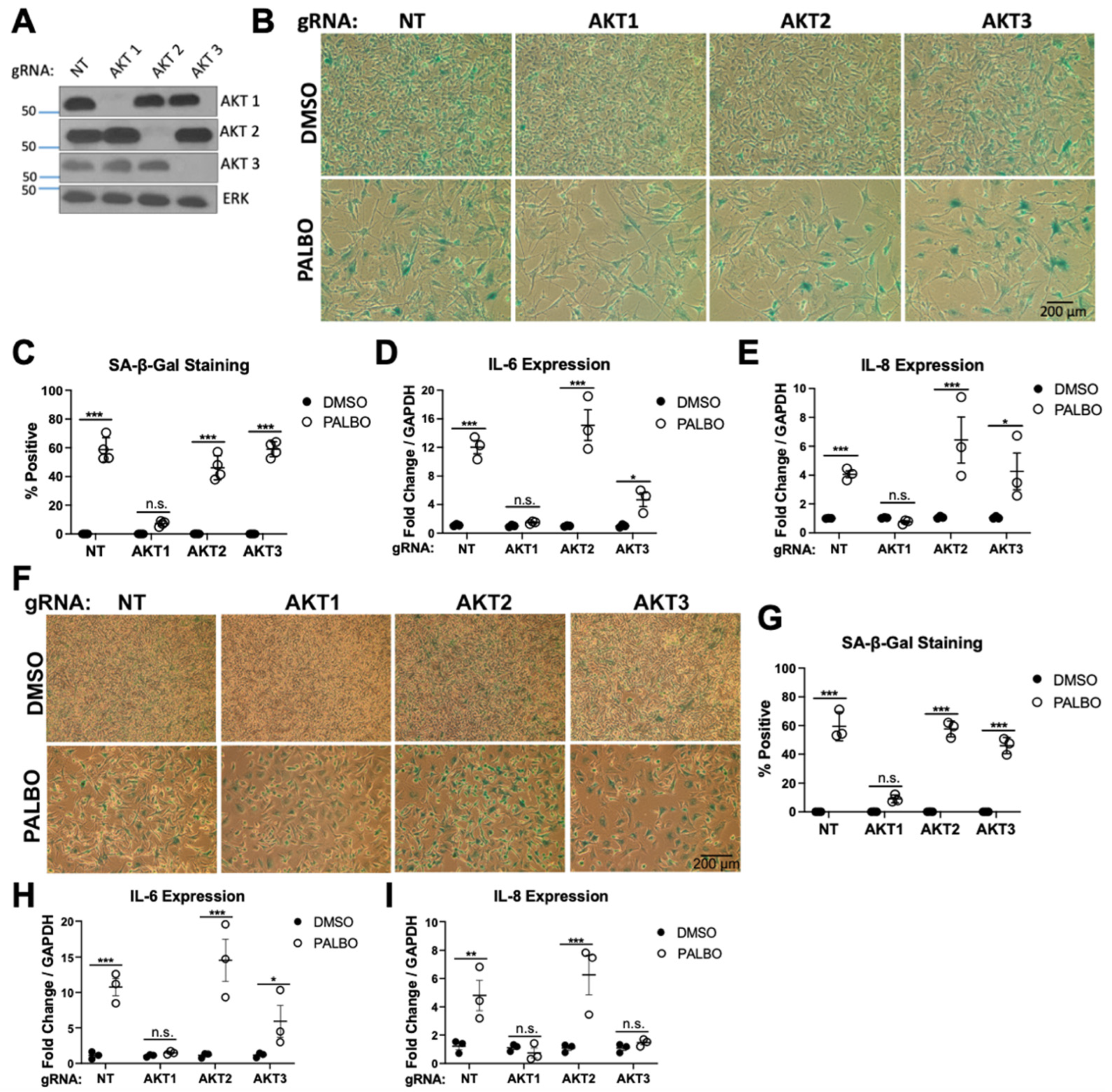
3.2. Clonal AKT1 Knockout Cells Exhibit Impaired Senescence and SASP Induction
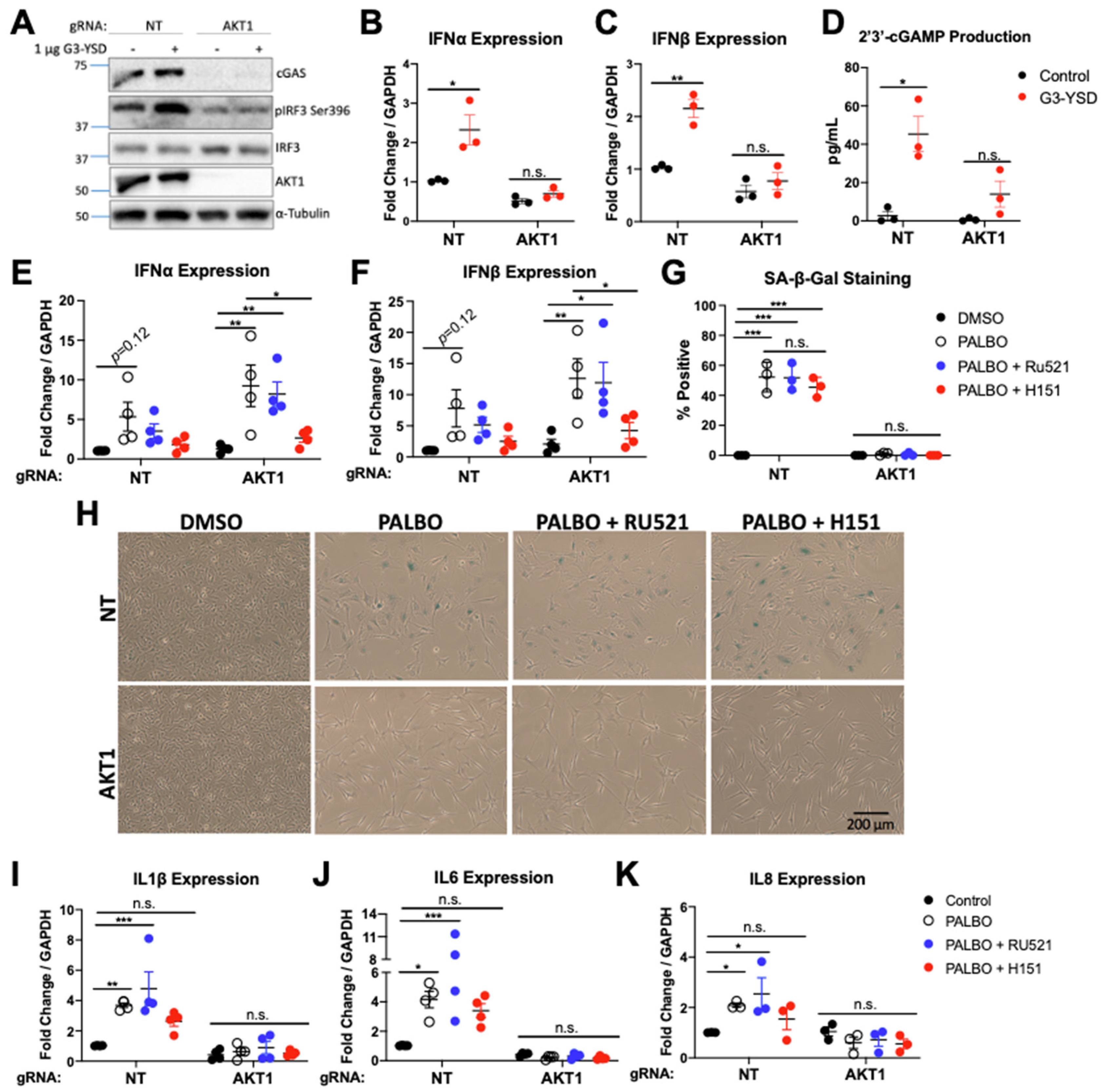
3.3. cGAS-STING Is Impaired in AKT1 Knockout Cells
3.4. cGAS/STING Inhibition Does Not Affect Senescence or SASP Secretion
3.5. NF-κB Function Regulates SASP Factors in an AKT1-Dependent Manner
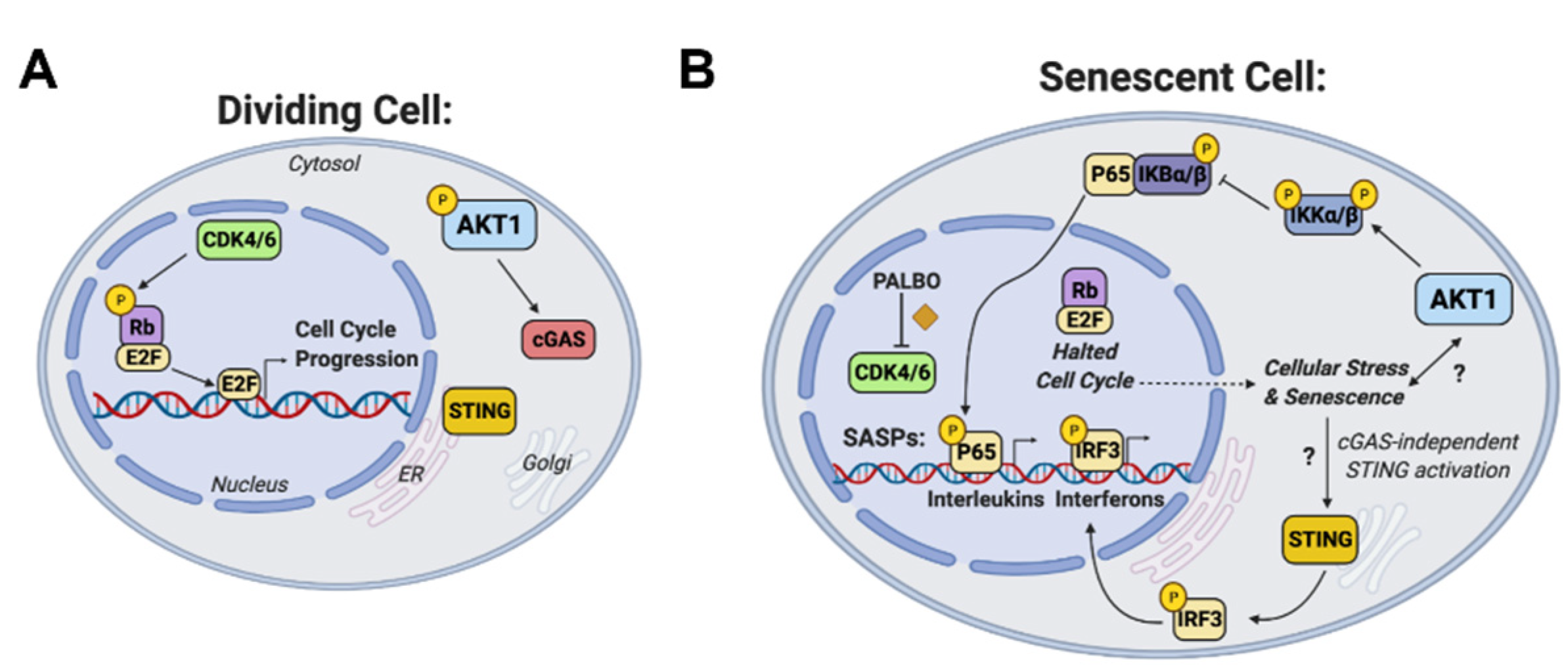
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hayflick, L.; Moorhead, P.S. The Serial Cultivation of Human Diploid Cell Strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Pérez-Mancera, P.A.; Young, A.R.J.; Narita, M. Inside and out: The Activities of Senescence in Cancer. Nat. Rev. Cancer 2014, 14, 547–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Saleh, T.; Tyutynuk-Massey, L.; Cudjoe, E.K.; Idowu, M.O.; Landry, J.W.; Gewirtz, D.A. Non-Cell Autonomous Effects of the Senescence-Associated Secretory Phenotype in Cancer Therapy. Front. Oncol. 2018, 8, 164. [Google Scholar] [CrossRef] [Green Version]
- Vernot, J.P. Senescence-Associated Pro-Inflammatory Cytokines and Tumor Cell Plasticity. Front. Mol. Biosci. 2020, 7, 63. [Google Scholar] [CrossRef]
- Hao, X.; Zhao, B.; Zhou, W.; Liu, H.; Fukumoto, T.; Gabrilovich, D.; Zhang, R. Sensitization of Ovarian Tumor to Immune Checkpoint Blockade by Boosting Senescence-Associated Secretory Phenotype. iScience 2021, 24, 102016. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the P53 Tumor Suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.-Y.; Campisi, J. Senescent Fibroblasts Promote Epithelial Cell Growth and Tumorigenesis: A Link between Cancer and Aging. Proc. Natl. Acad. Sci. USA 2001, 98, 12072–12077. [Google Scholar] [CrossRef] [Green Version]
- Gadsden, N.J.; Fulcher, C.D.; Li, D.; Shrivastava, N.; Thomas, C.; Segall, J.E.; Prystowsky, M.B.; Schlecht, N.F.; Gavathiotis, E.; Ow, T.J. Palbociclib Renders Human Papilloma Virus–Negative Head and Neck Squamous Cell Carcinoma Vulnerable to the Senolytic Agent Navitoclax. Mol. Cancer Res. 2021, 19, 862–873. [Google Scholar] [CrossRef]
- Kolodkin-Gal, D.; Roitman, L.; Ovadya, Y.; Azazmeh, N.; Assouline, B.; Schlesinger, Y.; Kalifa, R.; Horwitz, S.; Khalatnik, Y.; Hochner-Ger, A.; et al. Senolytic Elimination of Cox2-Expressing Senescent Cells Inhibits the Growth of Premalignant Pancreatic Lesions. Gut 2021, 71, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, V.J.; Saleh, T.; Gewirtz, D.A. Senolytics for Cancer Therapy: Is All That Glitters Really Gold? Cancers 2021, 13, 723. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, Y.; Pi, H.; Sheng, Y. Current Therapeutic Progress of CDK4/6 Inhibitors in Breast Cancer. Cancer Manag. Res. 2020, 12, 3477–3487. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non–Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016, 6, 740–753. [Google Scholar] [CrossRef] [Green Version]
- Dickson, M.A.; Schwartz, G.K.; Keohan, M.L.; D’Angelo, S.P.; Gounder, M.M.; Chi, P.; Antonescu, C.R.; Landa, J.; Qin, L.-X.; Crago, A.M.; et al. Phase 2 Trial of the CDK4 Inhibitor Palbociclib (PD0332991) at 125 Mg Dose in Well-Differentiated or Dedifferentiated Liposarcoma. JAMA Oncol. 2016, 2, 937–940. [Google Scholar] [CrossRef] [Green Version]
- Silvestri, M.; Cristaudo, A.; Morrone, A.; Messina, C.; Bennardo, L.; Nisticò, S.P.; Mariano, M.; Cameli, N. Emerging Skin Toxicities in Patients with Breast Cancer Treated with New Cyclin-Dependent Kinase 4/6 Inhibitors: A Systematic Review. Drug Saf. 2021, 44, 725–732. [Google Scholar] [CrossRef]
- Bueno, A.M.; Aramillo, J.B.; Mosquera, J.J.G.; Capitan, A.G.; Codony-Servat, J.; Aguilar, A.; Román, S.G.; de las Casas, C.M.; Viteri, S.; Molina, M.A. Palbociclib-Induced Senescence Upregulates the Expression of IL-8 and May Enhance the Response to Inmunotherapy. Ann. Oncol. 2019, 30, iii15–iii16. [Google Scholar] [CrossRef]
- Kovatcheva, M.; Liu, D.D.; Dickson, M.A.; Klein, M.E.; O’Connor, R.; Wilder, F.O.; Socci, N.D.; Tap, W.D.; Schwartz, G.K.; Singer, S.; et al. MDM2 Turnover and Expression of ATRX Determine the Choice between Quiescence and Senescence in Response to CDK4 Inhibition. Oncotarget 2015, 6, 8226–8243. [Google Scholar] [CrossRef] [Green Version]
- Puyol, M.; Martín, A.; Dubus, P.; Mulero, F.; Pizcueta, P.; Khan, G.; Guerra, C.; Santamaría, D.; Barbacid, M. A Synthetic Lethal Interaction between K-Ras Oncogenes and Cdk4 Unveils a Therapeutic Strategy for Non-Small Cell Lung Carcinoma. Cancer Cell 2010, 18, 63–73. [Google Scholar] [CrossRef]
- Vijayaraghavan, S.; Karakas, C.; Doostan, I.; Chen, X.; Bui, T.; Yi, M.; Raghavendra, A.S.; Zhao, Y.; Bashour, S.I.; Ibrahim, N.K.; et al. CDK4/6 and Autophagy Inhibitors Synergistically Induce Senescence in Rb Positive Cytoplasmic Cyclin E Negative Cancers. Nat. Commun. 2017, 8, 15916. [Google Scholar] [CrossRef]
- Schettini, F.; De Santo, I.; Rea, C.G.; De Placido, P.; Formisano, L.; Giuliano, M.; Arpino, G.; De Laurentiis, M.; Puglisi, F.; De Placido, S.; et al. CDK 4/6 Inhibitors as Single Agent in Advanced Solid Tumors. Front. Oncol. 2018, 8, 608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 Inhibition Triggers Anti-Tumour Immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Charles, A.; Bourne, C.; Aretz, Z.E.; Mun, S.S.; Korontsvit, T.; Dao, T.; Klatt, M.G.; Scheinberg, D.A. Low-Dose CDK4/6 Inhibitors Induce Presentation of Pathway Specific MHC Ligands as Targets for Cancer Immunotherapy. bioRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Fisher, D.E.; Garbe, C.; Gershenwald, J.E.; Grob, J.-J.; Halpern, A.; Herlyn, M.; Marchetti, M.A.; McArthur, G.; Ribas, A.; et al. Melanoma. Nat. Rev. Dis. Primers 2015, 1, 15003. [Google Scholar] [CrossRef]
- Rothermel, L.D.; Sarnaik, A.A.; Khushalani, N.I.; Sondak, V.K. Current Immunotherapy Practices in Melanoma. Surg. Oncol. Clin. 2019, 28, 403–418. [Google Scholar] [CrossRef] [PubMed]
- Kollmann, K.; Briand, C.; Bellutti, F.; Schicher, N.; Blunder, S.; Zojer, M.; Hoeller, C. The Interplay of CDK4 and CDK6 in Melanoma. Oncotarget 2019, 10, 1346–1359. [Google Scholar] [CrossRef]
- Mao, L.; Cao, Y.; Sheng, X.; Bai, X.; Chi, Z.; Cui, C.; Wang, X.; Tang, B.; Lian, B.; Yan, X.; et al. Palbociclib (P) in Advanced Acral Lentiginous Melanoma (ALM) with CDK4 Pathway Gene Aberrations. JCO 2019, 37, 9528. [Google Scholar] [CrossRef]
- Guo, L.; Qi, J.; Wang, H.; Jiang, X.; Liu, Y. Getting under the Skin: The Role of CDK4/6 in Melanomas. Eur. J. Med. Chem. 2020, 204, 112531. [Google Scholar] [CrossRef]
- Gonzalez, E.; McGraw, T.E. The Akt Kinases: Isoform Specificity in Metabolism and Cancer. Cell Cycle 2009, 8, 2502–2508. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [Green Version]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, D.L.; Martinka, M.; Li, G. Prognostic Significance of Activated Akt Expression in Melanoma: A Clinicopathologic Study of 292 Cases. JCO 2005, 23, 1473–1482. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Robinson, J.P.; Arave, R.A.; Burnett, W.J.; Kircher, D.A.; Chen, G.; Davies, M.A.; Grossmann, A.H.; VanBrocklin, M.W.; McMahon, M.; et al. AKT1 Activation Promotes the Development of Melanoma Metastases. Cell Rep. 2015, 13, 898–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Dai, M.; Lu, A.; Yu, E.; Merlino, G. PHLPP1 Mediates Melanoma Metastasis Suppression through Repressing AKT2 Activation. Oncogene 2018, 37, 2225. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.Y.; Jee, H.J.; Um, J.-H.; Kim, Y.M.; Bae, S.S.; Yun, J. Cooperation between P21 and Akt Is Required for P53-Dependent Cellular Senescence. Aging Cell 2017, 16, 1094–1103. [Google Scholar] [CrossRef]
- Astle, M.V.; Hannan, K.M.; Ng, P.Y.; Lee, R.S.; George, A.J.; Hsu, A.K.; Haupt, Y.; Hannan, R.D.; Pearson, R.B. AKT Induces Senescence in Human Cells via MTORC1 and P53 in the Absence of DNA Damage: Implications for Targeting MTOR during Malignancy. Oncogene 2011, 31, 1949–1962. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.G.; Deshpande, A.; Enos, M.; Mao, D.; Hinds, E.A.; Hu, G.; Chang, R.; Guo, Z.; Dose, M.; Mao, C.; et al. A Requirement for CDK6 in Thymocyte Development and Tumorigenesis. Cancer Res. 2009, 69, 810–818. [Google Scholar] [CrossRef] [Green Version]
- Jena, N.; Sheng, J.; Hu, J.K.; Li, W.; Zhou, W.; Lee, G.; Tsichlis, N.; Pathak, A.; Brown, N.; Deshpande, A.; et al. CDK6-Mediated Repression of CD25 Is Required for Induction and Maintenance of Notch1- Induced T Cell Acute Lymphoblastic Leukemia. Leukemia 2016, 30, 1033–1043. [Google Scholar] [CrossRef] [Green Version]
- Cretella, D.; Ravelli, A.; Fumarola, C.; La Monica, S.; Digiacomo, G.; Cavazzoni, A.; Alfieri, R.; Biondi, A.; Generali, D.; Bonelli, M.; et al. The Anti-Tumor Efficacy of CDK4/6 Inhibition Is Enhanced by the Combination with PI3K/AKT/MTOR Inhibitors through Impairment of Glucose Metabolism in TNBC Cells. J. Exp. Clin. Cancer Res. 2018, 37, 72. [Google Scholar] [CrossRef]
- Bonelli, M.A.; Digiacomo, G.; Fumarola, C.; Alfieri, R.; Quaini, F.; Falco, A.; Madeddu, D.; La Monica, S.; Cretella, D.; Ravelli, A.; et al. Combined Inhibition of CDK4/6 and PI3K/AKT/MTOR Pathways Induces a Synergistic Anti-Tumor Effect in Malignant Pleural Mesothelioma Cells. Neoplasia 2017, 19, 637–648. [Google Scholar] [CrossRef]
- Seo, G.J.; Yang, A.; Tan, B.; Kim, S.; Liang, Q.; Choi, Y.; Yuan, W.; Feng, P.; Park, H.-S.; Jung, J.U. Akt Kinase-Mediated Checkpoint of CGAS DNA Sensing Pathway. Cell Rep. 2015, 13, 440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Zhang, Q.; Zhang, F.; Meng, F.; Liu, S.; Zhou, R.; Wu, Q.; Li, X.; Shen, L.; Huang, J.; et al. HER2 Recruits AKT1 to Disrupt STING Signalling and Suppress Antiviral Defence and Antitumour Immunity. Nat. Cell Biol. 2019, 21, 1027–1040. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. CGAS Is Essential for Cellular Senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loo, T.M.; Miyata, K.; Tanaka, Y.; Takahashi, A. Cellular Senescence and Senescence-associated Secretory Phenotype via the CGAS-STING Signaling Pathway in Cancer. Cancer Sci. 2020, 111, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Chen, P.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; Ye, L.; He, Y.; et al. CGAS-STING, an Important Pathway in Cancer Immunotherapy. J. Hematol. Oncol. 2020, 13, 81. [Google Scholar] [CrossRef]
- Saeed, A.F.; Ruan, X.; Guan, H.; Su, J.; Ouyang, S. Regulation of CGAS-Mediated Immune Responses and Immunotherapy. Adv. Sci. 2020, 7, 1902599. [Google Scholar] [CrossRef]
- Brinkman, E.K.; Chen, T.; Amendola, M.; van Steensel, B. Easy Quantitative Assessment of Genome Editing by Sequence Trace Decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef] [PubMed]
- McRee, S.; Pietruska, J.; Bayer, A.L.; Tsichlis, P.; Hinds, P.W. AKT2 Loss Impairs BRAF Mutant Melanoma Metastasis. 2022; In Preparation. [Google Scholar]
- Glück, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.-W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate Immune Sensing of Cytosolic Chromatin Fragments through CGAS Promotes Senescence. Nat. Cell Biol. 2017, 19, 1061. [Google Scholar] [CrossRef]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the Senescence-Associated Secretory Phenotype by NF-ΚB Promotes Senescence and Enhances Chemosensitivity. Genes Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef] [Green Version]
- Rovillain, E.; Mansfield, L.; Caetano, C.; Alvarez-Fernandez, M.; Caballero, O.L.; Medema, R.H.; Hummerich, H.; Jat, P.S. Activation of Nuclear Factor-Kappa B Signalling Promotes Cellular Senescence. Oncogene 2011, 30, 2356–2366. [Google Scholar] [CrossRef] [Green Version]
- Bai, D.; Ueno, L.; Vogt, P.K. Akt-Mediated Regulation of NFκB and the Essentialness of NFκB for the Oncogenicity of PI3K and Akt. Int. J. Cancer 2009, 125, 2863–2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, A.; Lee, E.K.; Diehl, J.A. Induction of Therapeutic Senescence in Vemurafenib-Resistant Melanoma by Extended Inhibition of CDK4/6. Cancer Res. 2016, 76, 2990–3002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, T.; Barber, G.N. Cytosolic-DNA-Mediated, STING-Dependent Proinflammatory Gene Induction Necessitates Canonical NF-ΚB Activation through TBK1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bayer, A.L.; Pietruska, J.; Farrell, J.; McRee, S.; Alcaide, P.; Hinds, P.W. AKT1 Is Required for a Complete Palbociclib-Induced Senescence Phenotype in BRAF-V600E-Driven Human Melanoma. Cancers 2022, 14, 572. https://doi.org/10.3390/cancers14030572
Bayer AL, Pietruska J, Farrell J, McRee S, Alcaide P, Hinds PW. AKT1 Is Required for a Complete Palbociclib-Induced Senescence Phenotype in BRAF-V600E-Driven Human Melanoma. Cancers. 2022; 14(3):572. https://doi.org/10.3390/cancers14030572
Chicago/Turabian StyleBayer, Abraham L., Jodie Pietruska, Jaymes Farrell, Siobhan McRee, Pilar Alcaide, and Philip W. Hinds. 2022. "AKT1 Is Required for a Complete Palbociclib-Induced Senescence Phenotype in BRAF-V600E-Driven Human Melanoma" Cancers 14, no. 3: 572. https://doi.org/10.3390/cancers14030572
APA StyleBayer, A. L., Pietruska, J., Farrell, J., McRee, S., Alcaide, P., & Hinds, P. W. (2022). AKT1 Is Required for a Complete Palbociclib-Induced Senescence Phenotype in BRAF-V600E-Driven Human Melanoma. Cancers, 14(3), 572. https://doi.org/10.3390/cancers14030572