
Strategies for Improving the Efficacy of CAR T Cells in Solid Cancers


Abstract
:Simple Summary
Abstract
1. Introduction
2. Co-Stimulatory Domains and Tuning of T Cell Stimulation
3. Strategies for Improved CAR T Cell Persistence
4. Strategies for Countering Tumour Heterogeneity
5. Strategies for Improved Safety
6. Strategies for Overcoming Immune Suppression
7. Perspectives on the Role of CAR T Cell Therapy and the Way Forward
8. Conclusions
Funding
Conflicts of Interest
References
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [Green Version]
- Weber, E.W.; Maus, M.V.; Mackall, C.L. The Emerging Landscape of Immune Cell Therapies. Cell 2020, 181, 46–62. [Google Scholar] [CrossRef]
- Abken, H. Adoptive therapy with CAR redirected T cells: The challenges in targeting solid tumors. Immunotherapy 2015, 7, 535–544. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef] [Green Version]
- Lee, L.; Draper, B.; Chaplin, N.; Philip, B.; Chin, M.; Galas-Filipowicz, D.; Onuoha, S.; Thomas, S.; Baldan, V.; Bughda, R.; et al. An APRIL-based chimeric antigen receptor for dual targeting of BCMA and TACI in multiple myeloma. Blood 2018, 131, 746–758. [Google Scholar] [CrossRef]
- Sadelain, M.; Brentjens, R.; Riviere, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [Green Version]
- Philipson, B.I.; O’Connor, R.S.; May, M.J.; June, C.H.; Albelda, S.M.; Milone, M.C. 4-1BB costimulation promotes CAR T cell survival through noncanonical NF-kappaB signaling. Sci. Signal. 2020, 13. [Google Scholar] [CrossRef]
- Sun, C.; Shou, P.; Du, H.; Hirabayashi, K.; Chen, Y.; Herring, L.E.; Ahn, S.; Xu, Y.; Suzuki, K.; Li, G.; et al. THEMIS-SHP1 Recruitment by 4-1BB Tunes LCK-Mediated Priming of Chimeric Antigen Receptor-Redirected T Cells. Cancer Cell 2020, 37, 216–225.e6. [Google Scholar] [CrossRef]
- Zhao, Z.; Condomines, M.; van der Stegen, S.J.C.; Perna, F.; Kloss, C.C.; Gunset, G.; Plotkin, J.; Sadelain, M. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 2015, 28, 415–428. [Google Scholar] [CrossRef] [Green Version]
- Almasbak, H.; Walseng, E.; Kristian, A.; Myhre, M.R.; Suso, E.M.; Munthe, L.A.; Andersen, J.T.; Wang, M.Y.; Kvalheim, G.; Gaudernack, G.; et al. Inclusion of an IgG1-Fc spacer abrogates efficacy of CD19 CAR T cells in a xenograft mouse model. Gene Ther. 2015, 22, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Kintz, H.; Nylen, E.; Barber, A. Inclusion of Dap10 or 4-1BB costimulation domains in the chPD1 receptor enhances anti-tumor efficacy of T cells in murine models of lymphoma and melanoma. Cell. Immunol. 2020, 351, 104069. [Google Scholar] [CrossRef] [PubMed]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D., Jr.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Feucht, J.; Sun, J.; Eyquem, J.; Ho, Y.J.; Zhao, Z.; Leibold, J.; Dobrin, A.; Cabriolu, A.; Hamieh, M.; Sadelain, M. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med. 2019, 25, 82–88. [Google Scholar] [CrossRef]
- Ghorashian, S.; Kramer, A.M.; Onuoha, S.; Wright, G.; Bartram, J.; Richardson, R.; Albon, S.J.; Casanovas-Company, J.; Castro, F.; Popova, B.; et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat. Med. 2019, 25, 1408–1414. [Google Scholar] [CrossRef]
- Maiti, S.N.; Huls, H.; Singh, H.; Dawson, M.; Figliola, M.; Olivares, S.; Rao, P.; Zhao, Y.J.; Multani, A.; Yang, G.; et al. Sleeping beauty system to redirect T-cell specificity for human applications. J. Immunother. 2013, 36, 112–123. [Google Scholar] [CrossRef] [Green Version]
- Monjezi, R.; Miskey, C.; Gogishvili, T.; Schleef, M.; Schmeer, M.; Einsele, H.; Ivics, Z.; Hudecek, M. Enhanced CAR T-cell engineering using non-viral Sleeping Beauty transposition from minicircle vectors. Leukemia 2017, 31, 186–294. [Google Scholar] [CrossRef]
- Singh, H.; Figliola, M.J.; Dawson, M.J.; Huls, H.; Olivares, S.; Switzer, K.; Mi, T.; Maiti, S.; Kebriaei, P.; Lee, D.A.; et al. Reprogramming CD19-specific T cells with IL-21 signaling can improve adoptive immunotherapy of B-lineage malignancies. Cancer Res. 2011, 71, 3516–3527. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, J.M.; Schubert, M.L.; Wang, L.; Huckelhoven, A.; Sellner, L.; Stock, S.; Schmitt, A.; Kleist, C.; Gern, U.; Loskog, A.; et al. Differences in Expansion Potential of Naive Chimeric Antigen Receptor T Cells from Healthy Donors and Untreated Chronic Lymphocytic Leukemia Patients. Front. Immunol. 2017, 8, 1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Naranjo, A.; Brown, C.E.; Bautista, C.; Wong, C.W.; Chang, W.C.; Aguilar, B.; Ostberg, J.R.; Riddell, S.R.; Forman, S.J.; et al. Phenotypic and functional attributes of lentivirus-modified CD19-specific human CD8+ central memory T cells manufactured at clinical scale. J. Immunother. 2012, 35, 689–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, E.W.; Parker, K.R.; Sotillo, E.; Lynn, R.C.; Anbunathan, H.; Lattin, J.; Good, Z.; Belk, J.A.; Daniel, B.; Klysz, D.; et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science 2021, 372, eaba1786. [Google Scholar] [CrossRef]
- Wang, Y.; Tong, C.; Dai, H.; Wu, Z.; Han, X.; Guo, Y.; Chen, D.; Wei, J.; Ti, D.; Liu, Z.; et al. Low-dose decitabine priming endows CAR T cells with enhanced and persistent antitumour potential via epigenetic reprogramming. Nat. Commun. 2021, 12, 409. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Long, L.; Zheng, W.; Dhungana, Y.; Lim, S.A.; Guy, C.; Wang, Y.; Wang, Y.D.; Qian, C.; Xu, B.; et al. Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature 2019, 576, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, K.; Takeuchi, O.; Standley, D.M.; Kumagai, Y.; Kawagoe, T.; Miyake, T.; Satoh, T.; Kato, H.; Tsujimura, T.; Nakamura, H.; et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature 2009, 458, 1185–1190. [Google Scholar] [CrossRef]
- Xu, J.; Peng, W.; Sun, Y.; Wang, X.; Xu, Y.; Li, X.; Gao, G.; Rao, Z. Structural study of MCPIP1 N-terminal conserved domain reveals a PIN-like RNase. Nucleic Acids Res. 2012, 40, 6957–6965. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Ning, H.; Gu, L.; Peng, H.; Wang, Q.; Hou, R.; Fu, M.; Hoft, D.F.; Liu, J. MCPIP1 Selectively Destabilizes Transcripts Associated with an Antiapoptotic Gene Expression Program in Breast Cancer Cells That Can Elicit Complete Tumor Regression. Cancer Res. 2016, 76, 1429–1440. [Google Scholar] [CrossRef] [Green Version]
- Kurachi, M.; Barnitz, R.A.; Yosef, N.; Odorizzi, P.M.; DiIorio, M.A.; Lemieux, M.E.; Yates, K.; Godec, J.; Klatt, M.G.; Regev, A.; et al. The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat. Immunol. 2014, 15, 373–383. [Google Scholar] [CrossRef]
- Quigley, M.; Pereyra, F.; Nilsson, B.; Porichis, F.; Fonseca, C.; Eichbaum, Q.; Julg, B.; Jesneck, J.L.; Brosnahan, K.; Imam, S.; et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat. Med. 2010, 16, 1147–1151. [Google Scholar] [CrossRef]
- Kuroda, S.; Yamazaki, M.; Abe, M.; Sakimura, K.; Takayanagi, H.; Iwai, Y. Basic leucine zipper transcription factor, ATF-like (BATF) regulates epigenetically and energetically effector CD8 T-cell differentiation via Sirt1 expression. Proc. Natl. Acad. Sci. USA 2011, 108, 14885–14889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishton, R.J.; Sukumar, M.; Restifo, N.P. Metabolic Regulation of T Cell Longevity and Function in Tumor Immunotherapy. Cell Metab. 2017, 26, 94–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quaglino, E.; Conti, L.; Cavallo, F. Breast cancer stem cell antigens as targets for immunotherapy. Semin. Immunol. 2020, 47, 101386. [Google Scholar] [CrossRef]
- Guo, Y.; Feng, K.; Wang, Y.; Han, W. Targeting cancer stem cells by using chimeric antigen receptor-modified T cells: A potential and curable approach for cancer treatment. Protein Cell 2018, 9, 516–526. [Google Scholar] [CrossRef] [Green Version]
- Kyte, J.A.; Fane, A.; Pule, M.; Gaudernack, G. Transient redirection of T cells for adoptive cell therapy with telomerase-specific T helper cell receptors isolated from long term survivors after cancer vaccination. Oncoimmunology 2019, 8, e1565236. [Google Scholar] [CrossRef]
- Leisegang, M.; Wilde, S.; Spranger, S.; Milosevic, S.; Frankenberger, B.; Uckert, W.; Schendel, D.J. MHC-restricted fratricide of human lymphocytes expressing survivin-specific transgenic T cell receptors. J. Clin. Investig. 2010, 120, 3869–3877. [Google Scholar] [CrossRef]
- Kyte, J.A.; Gaudernack, G.; Faane, A.; Lislerud, K.; Inderberg, E.M.; Brunsvig, P.; Aamdal, S.; Kvalheim, G.; Walchli, S.; Pule, M. T-helper cell receptors from long-term survivors after telomerase cancer vaccination for use in adoptive cell therapy. Oncoimmunology 2016, 5, e1249090. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Fu, X.; Lopez, J.I.; Rowan, A.; Au, L.; Fendler, A.; Hazell, S.; Xu, H.; Horswell, S.; Shepherd, S.T.C.; et al. Selection of metastasis competent subclones in the tumour interior. Nat. Ecol. Evol. 2021, 5, 1033–1045. [Google Scholar] [CrossRef]
- Dentro, S.C.; Leshchiner, I.; Haase, K.; Tarabichi, M.; Wintersinger, J.; Deshwar, A.G.; Yu, K.; Rubanova, Y.; Macintyre, G.; Demeulemeester, J.; et al. Characterizing genetic intra-tumor heterogeneity across 2,658 human cancer genomes. Cell 2021, 184, 2239–2254.e39. [Google Scholar] [CrossRef]
- Bailey, C.; Black, J.R.M.; Reading, J.L.; Litchfield, K.; Turajlic, S.; McGranahan, N.; Jamal-Hanjani, M.; Swanton, C. Tracking Cancer Evolution through the Disease Course. Cancer Discov. 2021, 11, 916–932. [Google Scholar] [CrossRef]
- Kloss, C.C.; Condomines, M.; Cartellieri, M.; Bachmann, M.; Sadelain, M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol. 2013, 31, 71–75. [Google Scholar] [CrossRef]
- Roybal, K.T.; Rupp, L.J.; Morsut, L.; Walker, W.J.; McNally, K.A.; Park, J.S.; Lim, W.A. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 2016, 164, 770–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019, 37, 1049–1058. [Google Scholar] [CrossRef]
- Lai, J.; Mardiana, S.; House, I.G.; Sek, K.; Henderson, M.A.; Giuffrida, L.; Chen, A.X.Y.; Todd, K.L.; Petley, E.V.; Chan, J.D.; et al. Adoptive cellular therapy with T cells expressing the dendritic cell growth factor Flt3L drives epitope spreading and antitumor immunity. Nat. Immunol. 2020, 21, 914–926. [Google Scholar] [CrossRef]
- Knutson, K.L.; Disis, M.L. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol. Immunother. 2005, 54, 721–728. [Google Scholar] [CrossRef]
- Slaney, C.Y.; von Scheidt, B.; Davenport, A.J.; Beavis, P.A.; Westwood, J.A.; Mardiana, S.; Tscharke, D.C.; Ellis, S.; Prince, H.M.; Trapani, J.A.; et al. Dual-specific Chimeric Antigen Receptor T Cells and an Indirect Vaccine Eradicate a Variety of Large Solid Tumors in an Immunocompetent, Self-antigen Setting. Clin. Cancer Res. 2017, 23, 2478–2490. [Google Scholar] [CrossRef] [Green Version]
- Parkhurst, M.R.; Yang, J.C.; Langan, R.C.; Dudley, M.E.; Nathan, D.A.; Feldman, S.A.; Davis, J.L.; Morgan, R.A.; Merino, M.J.; Sherry, R.M.; et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. 2011, 19, 620–626. [Google Scholar] [CrossRef]
- Linette, G.P.; Stadtmauer, E.A.; Maus, M.V.; Rapoport, A.P.; Levine, B.L.; Emery, L.; Litzky, L.; Bagg, A.; Carreno, B.M.; Cimino, P.J.; et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 2013, 122, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013, 36, 133–151. [Google Scholar] [CrossRef] [Green Version]
- Lamers, C.H.; Sleijfer, S.; van Steenbergen, S.; van Elzakker, P.; van Krimpen, B.; Groot, C.; Vulto, A.; den Bakker, M.; Oosterwijk, E.; Debets, R.; et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: Clinical evaluation and management of on-target toxicity. Mol. Ther. 2013, 21, 904–912. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Di Stasi, A.; Tey, S.K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef] [Green Version]
- Philip, B.; Kokalaki, E.; Mekkaoui, L.; Thomas, S.; Straathof, K.; Flutter, B.; Marin, V.; Marafioti, T.; Chakraverty, R.; Linch, D.; et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood 2014, 124, 1277–1287. [Google Scholar] [CrossRef]
- Beatty, G.L.; Haas, A.R.; Maus, M.V.; Torigian, D.A.; Soulen, M.C.; Plesa, G.; Chew, A.; Zhao, Y.; Levine, B.L.; Albelda, S.M.; et al. Mesothelin-specific Chimeric Antigen Receptor mRNA-Engineered T cells Induce Anti-Tumor Activity in Solid Malignancies. Cancer Immunol. Res. 2014, 2, 112–120. [Google Scholar] [CrossRef] [Green Version]
- Oei, V.Y.S.; Siernicka, M.; Graczyk-Jarzynka, A.; Hoel, H.J.; Yang, W.; Palacios, D.; Almasbak, H.; Bajor, M.; Clement, D.; Brandt, L.; et al. Intrinsic Functional Potential of NK-Cell Subsets Constrains Retargeting Driven by Chimeric Antigen Receptors. Cancer Immunol. Res. 2018, 6, 467–480. [Google Scholar] [CrossRef] [Green Version]
- Maus, M.V.; June, C.H. Making Better Chimeric Antigen Receptors for Adoptive T-cell Therapy. Clin. Cancer Res. 2016, 22, 1875–1884. [Google Scholar] [CrossRef] [Green Version]
- Hartley, J.; Abken, H. Chimeric antigen receptors designed to overcome transforming growth factor-beta-mediated repression in the adoptive T-cell therapy of solid tumors. Clin. Transl. Immunol. 2019, 8, e1064. [Google Scholar] [CrossRef]
- Newick, K.; O’Brien, S.; Sun, J.; Kapoor, V.; Maceyko, S.; Lo, A.; Pure, E.; Moon, E.; Albelda, S.M. Augmentation of CAR T-cell Trafficking and Antitumor Efficacy by Blocking Protein Kinase A Localization. Cancer Immunol. Res. 2016, 4, 541–551. [Google Scholar] [CrossRef] [Green Version]
- Prosser, M.E.; Brown, C.E.; Shami, A.F.; Forman, S.J.; Jensen, M.C. Tumor PD-L1 co-stimulates primary human CD8(+) cytotoxic T cells modified to express a PD1:CD28 chimeric receptor. Mol. Immunol. 2012, 51, 263–272. [Google Scholar] [CrossRef]
- Liu, X.; Ranganathan, R.; Jiang, S.; Fang, C.; Sun, J.; Kim, S.; Newick, K.; Lo, A.; June, C.H.; Zhao, Y.; et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res. 2016, 76, 1578–1590. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, P.; Wang, T.; Fang, Y.; Ding, Y.; Qian, Q. Chimeric antigen receptor T cells engineered to secrete CD40 agonist antibodies enhance antitumor efficacy. J. Transl. Med. 2021, 19, 82. [Google Scholar] [CrossRef]
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011, 71, 5697–5706. [Google Scholar] [CrossRef] [Green Version]
- Yeku, O.O.; Purdon, T.J.; Koneru, M.; Spriggs, D.; Brentjens, R.J. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci. Rep. 2017, 7, 10541. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Ren, J.; Luo, Y.; Keith, B.; Young, R.M.; Scholler, J.; Zhao, Y.; June, C.H. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep. 2017, 20, 3025–3033. [Google Scholar] [CrossRef] [Green Version]
- Chinnasamy, D.; Yu, Z.; Kerkar, S.P.; Zhang, L.; Morgan, R.A.; Restifo, N.P.; Rosenberg, S.A. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin. Cancer Res. 2012, 18, 1672–1683. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Morgan, R.A.; Beane, J.D.; Zheng, Z.; Dudley, M.E.; Kassim, S.H.; Nahvi, A.V.; Ngo, L.T.; Sherry, R.M.; Phan, G.Q.; et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin. Cancer Res. 2015, 21, 2278–2288. [Google Scholar] [CrossRef] [Green Version]
- Martin, C.J.; Datta, A.; Littlefield, C.; Kalra, A.; Chapron, C.; Wawersik, S.; Dagbay, K.B.; Brueckner, C.T.; Nikiforov, A.; Danehy, F.T., Jr.; et al. Selective inhibition of TGFbeta1 activation overcomes primary resistance to checkpoint blockade therapy by altering tumor immune landscape. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Principe, D.R.; Park, A.; Dorman, M.J.; Kumar, S.; Viswakarma, N.; Rubin, J.; Torres, C.; McKinney, R.; Munshi, H.G.; Grippo, P.J.; et al. TGFbeta Blockade Augments PD-1 Inhibition to Promote T-Cell-Mediated Regression of Pancreatic Cancer. Mol. Cancer Ther. 2019, 18, 613–620. [Google Scholar] [CrossRef] [Green Version]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Lacuesta, K.; Buza, E.; Hauser, H.; Granville, L.; Pule, M.; Corboy, G.; Finegold, M.; Weiss, H.; Chen, S.Y.; Brenner, M.K.; et al. Assessing the safety of cytotoxic T lymphocytes transduced with a dominant negative transforming growth factor-beta receptor. J. Immunother. 2006, 29, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-beta Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayan, V.; Barber-Rotenberg, J.; Fraietta, J.; Hwang, W.-T.; Lacey, S.F.; Plesa, G.; Carpenter, E.L.; Maude, S.L.; Lal, P.; Vapiwala, N.; et al. A phase I clinical trial of PSMA-directed/TGFβ-insensitive CAR-T cells in metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2021, 39, 125. [Google Scholar] [CrossRef]
- Chang, Z.L.; Lorenzini, M.H.; Chen, X.; Tran, U.; Bangayan, N.J.; Chen, Y.Y. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat. Chem. Biol. 2018, 14, 317–324. [Google Scholar] [CrossRef]
- Sukumaran, S.; Watanabe, N.; Bajgain, P.; Raja, K.; Mohammed, S.; Fisher, W.E.; Brenner, M.K.; Leen, A.M.; Vera, J.F. Enhancing the potency and specificity of engineered T cells for cancer treatment. Cancer Discov. 2018, 8, 972–987. [Google Scholar] [CrossRef] [Green Version]
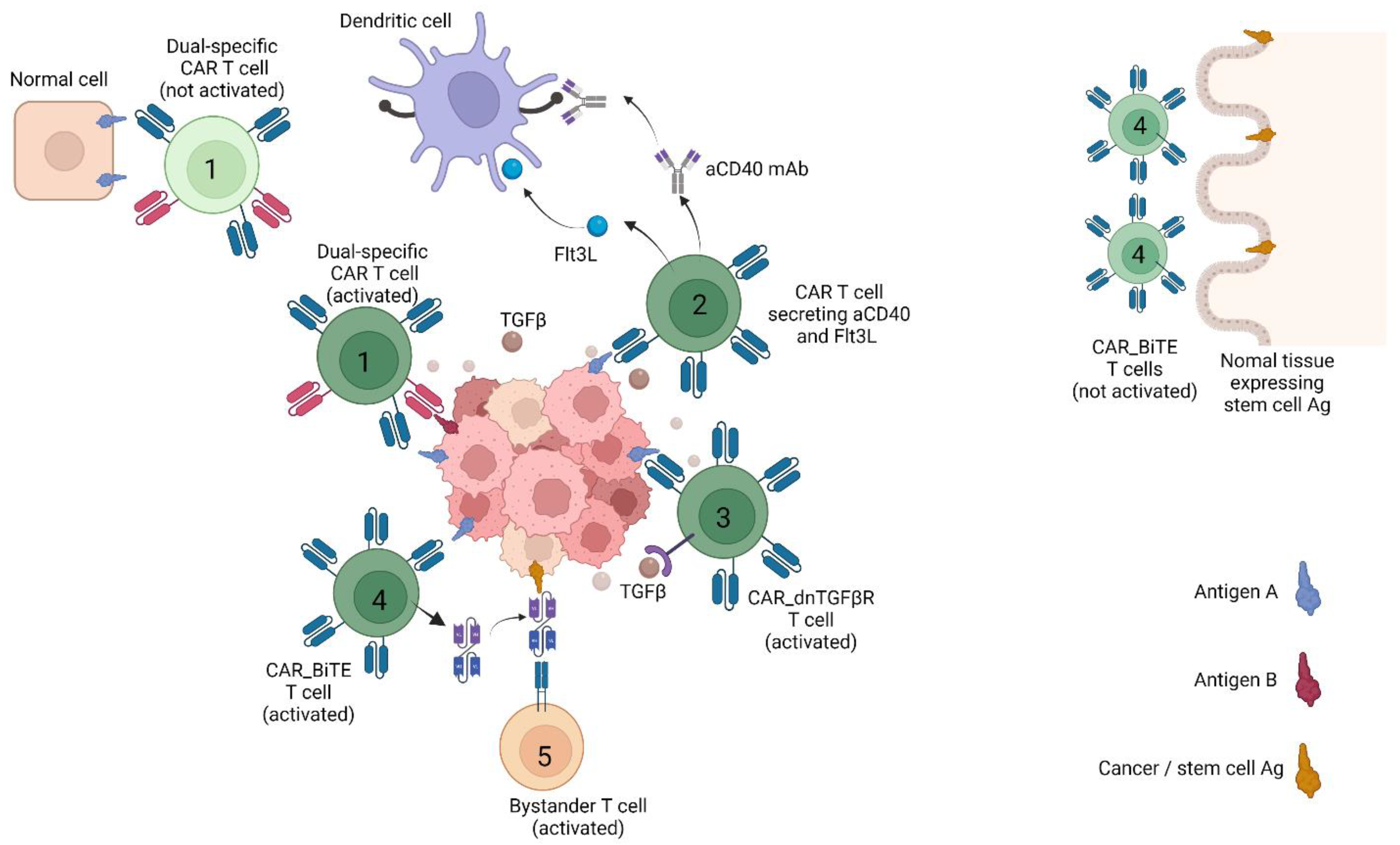

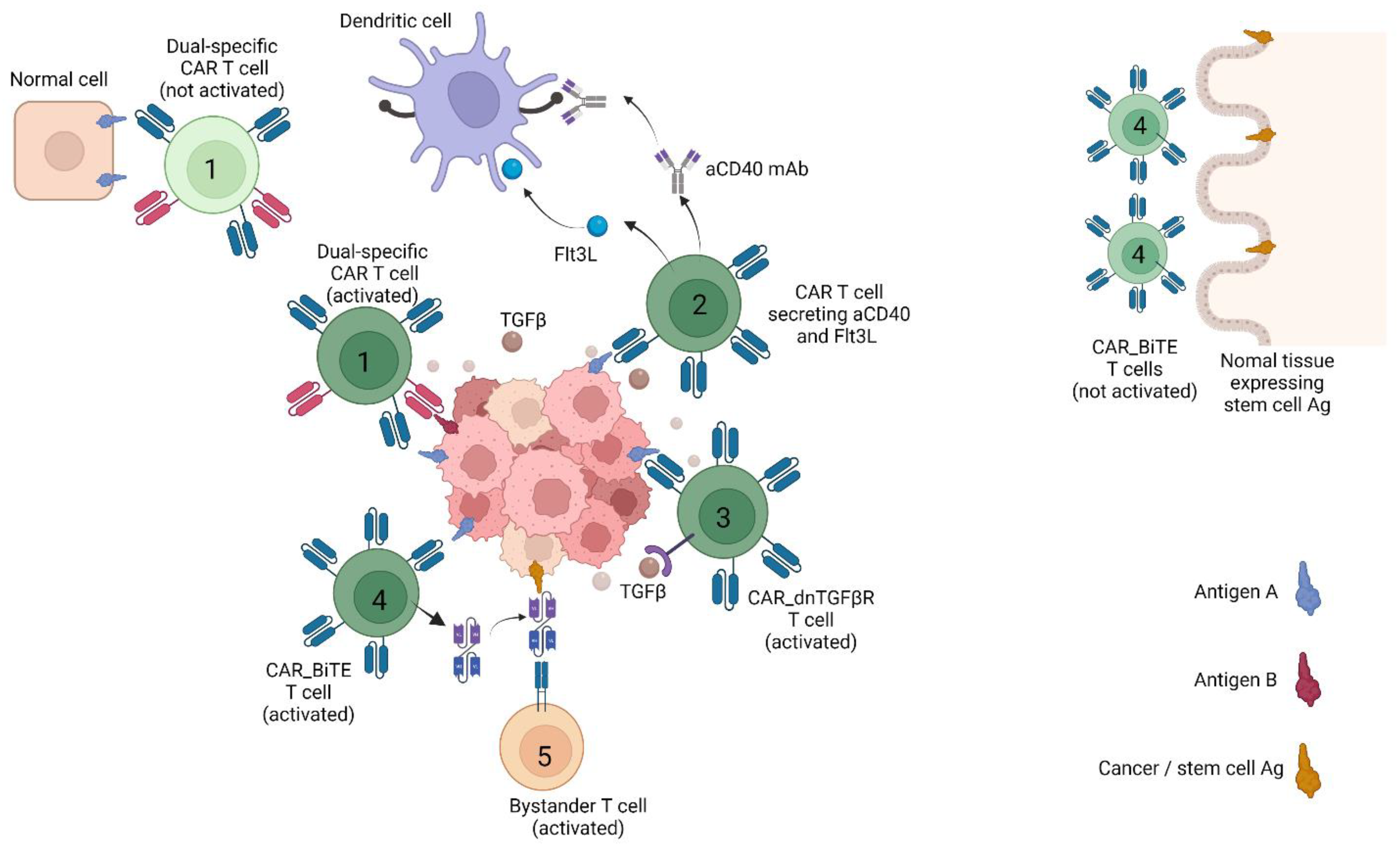{kind=link}
| Antigen Target | Clinical Trial Identifiers |
|---|---|
| AXL | NCT03198052, NCT03393936, NCT05128786 |
| B7-H3 | NCT03198052, NCT04385173, NCT04185038, NCT04077866, NCT04483778, NCT04483778, NCT04432649, NCT04670068, NCT04077866 |
| CAIX | NCT04969354 |
| CD147 | NCT03993743, NCT04045847 |
| CD171 | NCT02311621, NCT02311621, NCT02311621 |
| CD20 | NCT03893019 |
| CD44v6 | NCT04430595, NCT04427449 |
| CD70 | NCT02830724, NCT04438083 |
| CEA | NCT03818165, NCT04348643, NCT03682744, NCT02850536, NCT04513431, NCT04037241, NCT01373047, NCT01212887, NCT02349724 |
| CLDN18.2 | NCT04404595, NCT04467853, NCT03874897, NCT04977193, NCT04966143, NCT03159819 |
| CLDN6 | NCT04503278 |
| c-met | NCT01837602, NCT03060356 |
| DLL3 | NCT03392064 |
| DR5 | NCT03638206, NCT03941626 |
| EGFRvIII | NCT03638206, NCT03941626, NCT02209376, NCT03726515, NCT03283631, NCT02664363, NCT01454596 |
| EpCAM | NCT03563326, NCT03013712, NCT02915445, NCT04151186 |
| ErbB | NCT01818323 |
| FRα | NCT03585764, NCT03185468 |
| GD2 | NCT03356795, NCT04196413, NCT04539366, NCT02761915, NCT03373097, NCT02765243, NCT04099797, NCT03635632, NCT04430595, NCT03721068, NCT02992210, NCT01953900, NCT01822652, NCT05070156 |
| gp100 (MHC-I) | NCT03649529 |
| GFRα4 | NCT04877613 |
| GPC3 | NCT03198052, NCT04506983, NCT03198546, NCT03198546, NCT04121273, NCT04377932, NCT02905188, NCT02932956, NCT03980288, NCT03884751, NCT05003895 |
| HER2 | NCT03198052, NCT03500991, NCT03696030, NCT04430595, NCT02442297, NCT04511871, NCT00902044, NCT01109095, NCT01935843 |
| IL-13Rα2 | NCT04510051, NCT02208362, NCT04661384 |
| KLK2 kallikrein 2 | NCT05022849 |
| LeY | NCT03851146, NCT03198052 |
| LFA1 | NCT04420754 |
| MMP2 | NCT04214392 |
| Mesothelin | NCT03198052, NCT03638206, NCT03356795, NCT03941626, NCT04503980, NCT04489862, NCT03747965, NCT03814447, NCT03916679, NCT03638193, NCT03799913, NCT03545815, NCT03497819, NCT03323944, NCT02414269, NCT03054298, NCT02792114, NCT01897415, NCT04981691, NCT03615313, NCT03054298, NCT02414269, NCT02792114 |
| MUC1 | NCT03198052, NCT03356795, NCT03633773, NCT03706326, NCT03525782, NCT04020575 |
| MUC16 | NCT03907527 |
| MUC16ecto | NCT02498912 |
| NECTIN4/FAP | NCT03932565 |
| NKG2D | NCT03692429, NCT05131763 |
| NKG2DL | NCT04270461, NCT04107142 |
| PSCA | NCT03198052, NCT03873805, NCT02744287, NCT02744287 |
| PSMA | NCT03356795, NCT04053062, NCT04227275, NCT03089203, NCT03185468, NCT04429451 |
| ROR1 | NCT02706392 |
| ROR2 | NCT03960060, NCT03393936 |
| TM4SF1 | NCT04151186 |
| TnMUC1 | NCT04025216 |
| Target Antigen | Clinical Trial Identifiers | Novel Feature |
|---|---|---|
| EGFR | NCT04153799 | CAR T cells modified to express C-X-C Chemokine receptor type 5 |
| NCT03618381 | CAR T cells directed at EGFR and CD19, based on the hypothesis that CD19+ B cells will promote CAR T persistence | |
| NCT03542799 | CAR T with NFAT transcription factors inducing expression of IL—12 | |
| GPC3 | NCT04377932 | CAR T cells modified to secrete interleukin-15; safety/killing switch |
| MUC16 | NCT03907527 | CAR T cells expressing membrane-bound IL-15; safety/killing switch |
| MUC16ecto | NCT02498912 | Intravenous and intraperitoneal infusion of CAR T cells modified to secrete IL-12 |
| NECTIN4/FAP | NCT03932565 | Intratumoural injection of Nectin4/FAP-targeted fourth-generation CAR T cells expressing IL7 and CCL19, or IL12 |
| PSMA | NCT03089203 | CAR T cells expressing dominant negative TGFβ receptor |
| NCT04227275 | CAR T cells expressing dominant negative TGFβ receptor |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kyte, J.A. Strategies for Improving the Efficacy of CAR T Cells in Solid Cancers. Cancers 2022, 14, 571. https://doi.org/10.3390/cancers14030571
Kyte JA. Strategies for Improving the Efficacy of CAR T Cells in Solid Cancers. Cancers. 2022; 14(3):571. https://doi.org/10.3390/cancers14030571
Chicago/Turabian StyleKyte, Jon Amund. 2022. "Strategies for Improving the Efficacy of CAR T Cells in Solid Cancers" Cancers 14, no. 3: 571. https://doi.org/10.3390/cancers14030571
APA StyleKyte, J. A. (2022). Strategies for Improving the Efficacy of CAR T Cells in Solid Cancers. Cancers, 14(3), 571. https://doi.org/10.3390/cancers14030571