
Correlation between DNA Methylation and Cell Proliferation Identifies New Candidate Predictive Markers in Meningioma

 , , , , and
, , , , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Population and Clinical Data
2.2. Histopathology
2.3. Immunohistochemistry
2.4. DNA Methylation Analysis
3. Results
3.1. Clinicopathological Data
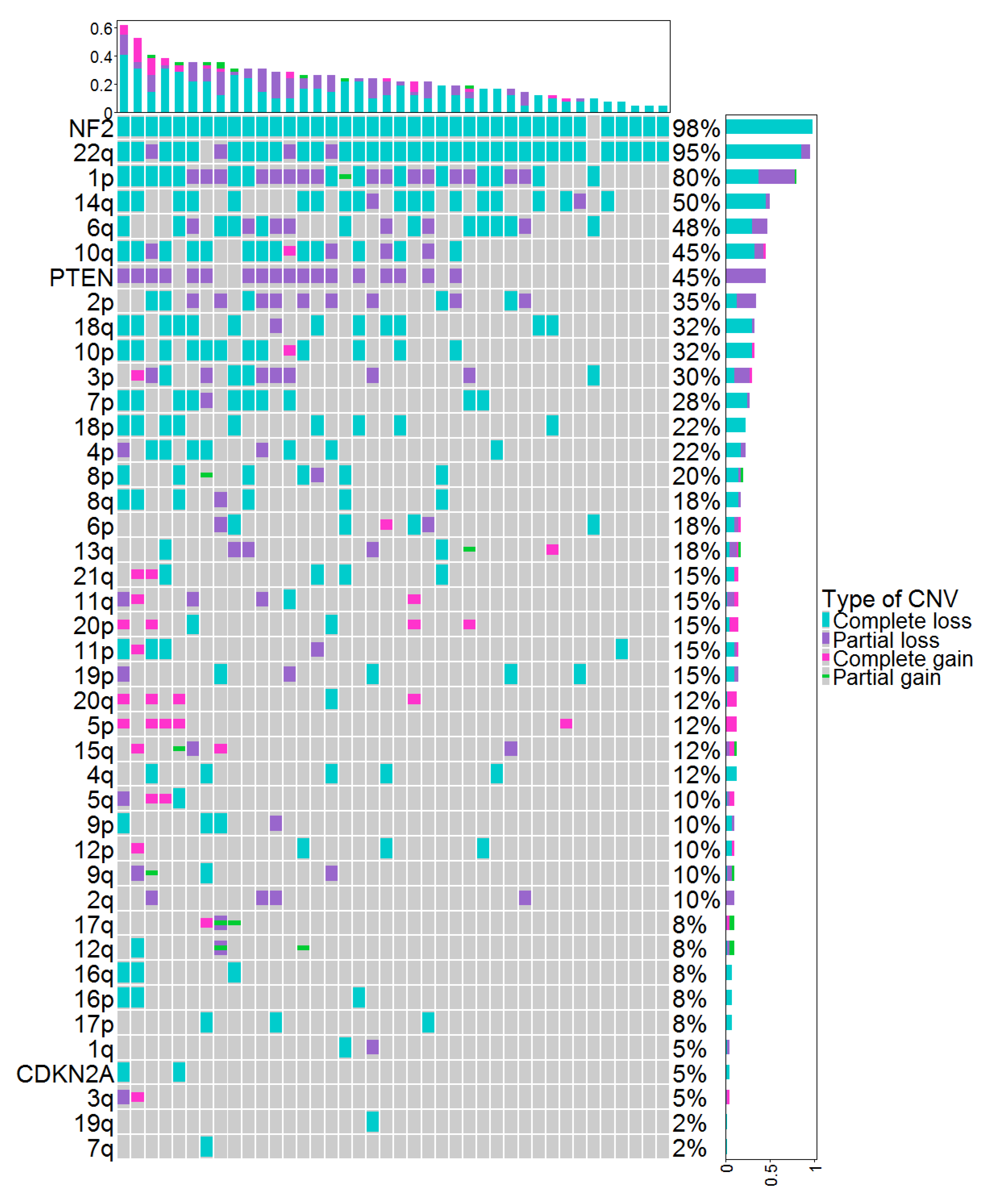
3.2. Molecular Data Based on Molecular Neuropathology Classifiers and Copy-Number Variations
3.3. DNA Methylation and WHO Grade
3.4. Correlation between DNAm and Mitotic Index
3.5. DNAm and Ki-67 Labeling Index
3.6. DNAm and MCM6 Labeling Index
3.7. DNAm Proliferative Signature in Meningiomas
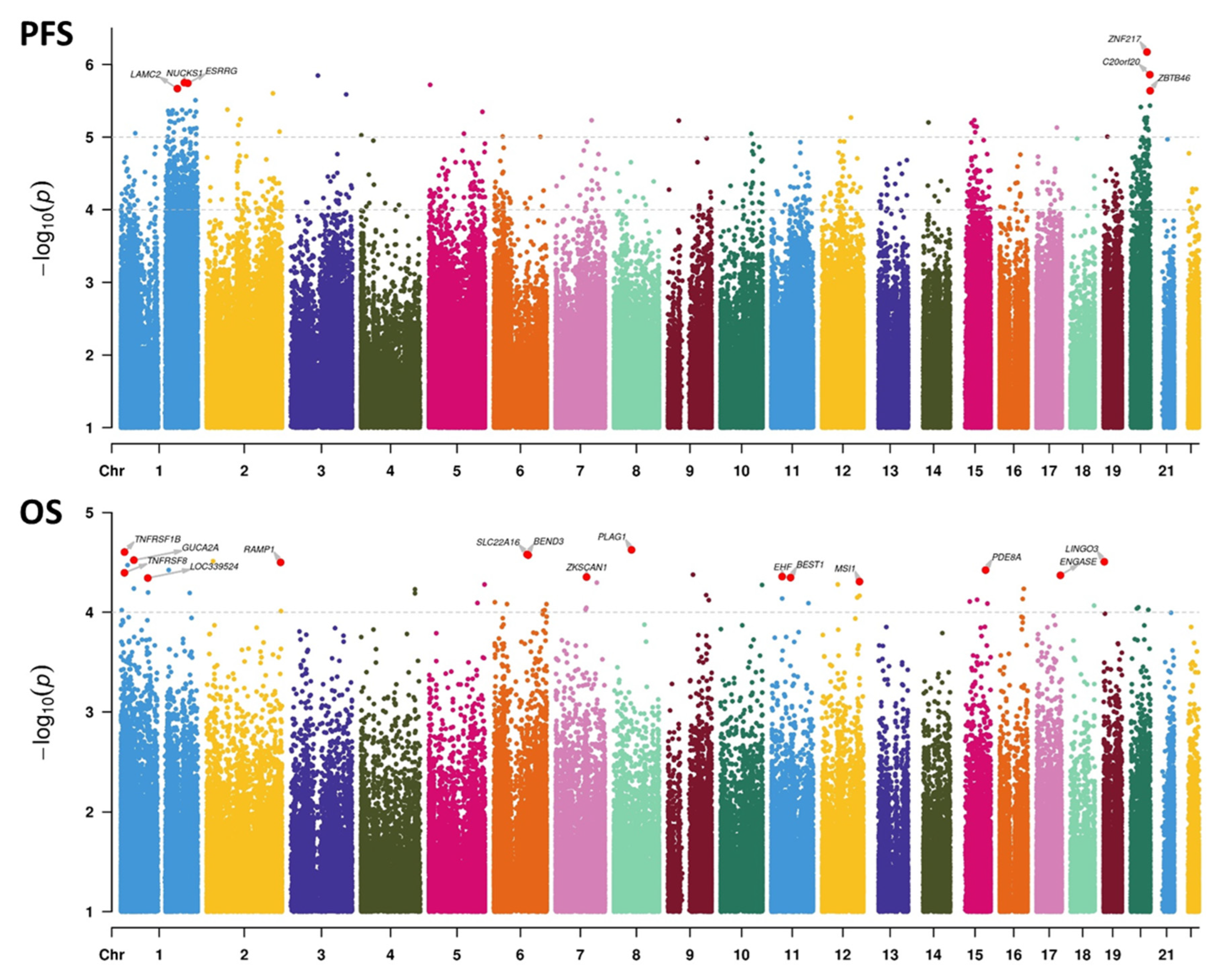
3.8. Associations between DNAm and Survival
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baldi, I.; Engelhardt, J.; Bonnet, C.; Bauchet, L.; Berteaud, E.; Grüber, A.; Loiseau, H. Epidemiology of Meningiomas. Neurochirurgie 2018, 64, 5–14. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer. WHO Classification of Tumours Editorial Board. Central Nervous System Tumours, 5th ed.; WHO Classification of Tumours Series; WHO: Geneva, Switzerland, 2021; Volume 6. [Google Scholar]
- Wang, N.; Osswald, M. Meningiomas: Overview and New Directions in Therapy. Semin. Neurol. 2018, 38, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Aizer, A.A.; Bi, W.L.; Kandola, M.S.; Lee, E.Q.; Nayak, L.; Rinne, M.L.; Norden, A.D.; Beroukhim, R.; Reardon, D.A.; Wen, P.Y.; et al. Extent of Resection and Overall Survival for Patients with Atypical and Malignant Meningioma: Extent of Resection and Recurrence in Meningioma. Cancer 2015, 121, 4376–4381. [Google Scholar] [CrossRef] [PubMed]
- van Alkemade, H.; de Leau, M.; Dieleman, E.M.T.; Kardaun, J.W.P.F.; van Os, R.; Vandertop, W.P.; van Furth, W.R.; Stalpers, L.J.A. Impaired Survival and Long-Term Neurological Problems in Benign Meningioma. Neuro-Oncology 2012, 14, 658–666. [Google Scholar] [CrossRef]
- Pathology Concordance Levels for Meningioma Classification and Grading in NRG Oncology RTOG Trial 0539. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4799683/ (accessed on 20 February 2022).
- Goldbrunner, R.; Minniti, G.; Preusser, M.; Jenkinson, M.D.; Sallabanda, K.; Houdart, E.; von Deimling, A.; Stavrinou, P.; Lefranc, F.; Lund-Johansen, M.; et al. EANO Guidelines for the Diagnosis and Treatment of Meningiomas. Lancet Oncol. 2016, 17, e383–e391. [Google Scholar] [CrossRef]
- Institut National du Cancer. Conduite a Tenir Devant des Patients Atteints de Méningiomes de Grade II et III/Synthèse, septembre 2020. e-cancer.fr. 2020. Available online: https://www.e-cancer.fr/Expertises-et-publications/Catalogue-des-publications/Conduites-a-tenir-devant-des-patients-atteints-d-un-meningiome-de-grade-II-et-III-Synthese (accessed on 1 November 2022).
- Rouleau, G.A.; Merel, P.; Lutchman, M.; Sanson, M.; Zucman, J.; Marineau, C.; Hoang-Xuan, K.; Demczuk, S.; Desmaze, C.; Plougastel, B. Alteration in a New Gene Encoding a Putative Membrane-Organizing Protein Causes Neuro-Fibromatosis Type 2. Nature 1993, 363, 515–521. [Google Scholar] [CrossRef]
- Bi, W.L.; Zhang, M.; Wu, W.W.; Mei, Y.; Dunn, I.F. Meningioma Genomics: Diagnostic, Prognostic, and Therapeutic Applications. Front. Surg. 2016, 3, 40. [Google Scholar] [CrossRef]
- Bi, W.L.; Mei, Y.; Agarwalla, P.K.; Beroukhim, R.; Dunn, I.F. Genomic and Epigenomic Landscape in Meningioma. Neurosurg. Clin. N. Am. 2016, 27, 167–179. [Google Scholar] [CrossRef]
- Proctor, D.T.; Ramachandran, S.; Lama, S.; Sutherland, G.R. Towards Molecular Classification of Meningioma: Evolving Treatment and Diagnostic Paradigms. World Neurosurg. 2018, 119, 366–373. [Google Scholar] [CrossRef]
- Galani, V.; Lampri, E.; Varouktsi, A.; Alexiou, G.; Mitselou, A.; Kyritsis, A.P. Genetic and Epigenetic Alterations in Meningiomas. Clin. Neurol. Neurosurg. 2017, 158, 119–125. [Google Scholar] [CrossRef]
- Yakubov, E.; Ghoochani, A.; Buslei, R.; Buchfelder, M.; Eyüpoglu, I.Y.; Savaskan, N. Hidden Association of Cowden Syndrome, PTEN Mutation and Meningioma Frequency. Oncoscience 2016, 3, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Alexiou, G.A.; Voulgaris, S. The Role of the PTEN Gene in Malignant Gliomas. Neurol. Neurochir. Pol. 2010, 44, 80–86. [Google Scholar] [CrossRef]
- Bi, W.L.; Greenwald, N.F.; Abedalthagafi, M.; Wala, J.; Gibson, W.J.; Agarwalla, P.K.; Horowitz, P.; Schumacher, S.E.; Esaulova, E.; Mei, Y.; et al. Genomic Landscape of High-Grade Meningiomas. NPJ Genomic Med. 2017, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- Brastianos, P.K.; Horowitz, P.M.; Santagata, S.; Jones, R.T.; McKenna, A.; Getz, G.; Ligon, K.L.; Palescandolo, E.; Van Hummelen, P.; Ducar, M.D.; et al. Genomic Sequencing of Meningiomas Identifies Oncogenic SMO and AKT1 Mutations. Nat. Genet. 2013, 45, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Lekanne Deprez, R.H.; Riegman, P.H.; van Drunen, E.; Warringa, U.L.; Groen, N.A.; Stefanko, S.Z.; Koper, J.W.; Avezaat, C.J.; Mulder, P.G.; Zwarthoff, E.C. Cytogenetic, Molecular Genetic and Pathological Analyses in 126 Meningiomas. J. Neuropathol. Exp. Neurol. 1995, 54, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Cordova, C.; Kurz, S.C. Advances in Molecular Classification and Therapeutic Opportunities in Meningiomas. Curr. Oncol. Rep. 2020, 22, 84. [Google Scholar] [CrossRef] [PubMed]
- Suvà, M.L.; Louis, D.N. Next-Generation Molecular Genetics of Brain Tumours. Curr. Opin. Neurol. 2013, 26, 681–687. [Google Scholar] [CrossRef]
- Katz, L.M.; Hielscher, T.; Liechty, B.; Silverman, J.; Zagzag, D.; Sen, R.; Wu, P.; Golfinos, J.G.; Reuss, D.; Neidert, M.C.; et al. Loss of Histone H3K27me3 Identifies a Subset of Meningiomas with Increased Risk of Recurrence. Acta Neuropathol. (Berl.) 2018, 135, 955–963. [Google Scholar] [CrossRef]
- Paramasivam, N.; Hübschmann, D.; Toprak, U.H.; Ishaque, N.; Neidert, M.; Schrimpf, D.; Stichel, D.; Reuss, D.; Sievers, P.; Reinhardt, A.; et al. Mutational Patterns and Regulatory Networks in Epigenetic Subgroups of Meningioma. Acta Neuropathol. (Berl.) 2019, 138, 295–308. [Google Scholar] [CrossRef]
- Sahm, F.; Schrimpf, D.; Stichel, D.; Jones, D.T.W.; Hielscher, T.; Schefzyk, S.; Okonechnikov, K.; Koelsche, C.; Reuss, D.E.; Capper, D.; et al. DNA Methylation-Based Classification and Grading System for Meningioma: A Multicentre, Retrospective Analysis. Lancet Oncol. 2017, 18, 682–694. [Google Scholar] [CrossRef]
- Nassiri, F.; Liu, J.; Patil, V.; Mamatjan, Y.; Wang, J.Z.; Hugh-White, R.; Macklin, A.M.; Khan, S.; Singh, O.; Karimi, S.; et al. A Clinically Applicable Integrative Molecular Classification of Meningiomas. Nature 2021, 597, 119–125. [Google Scholar] [CrossRef]
- Gauchotte, G.; Vigouroux, C.; Rech, F.; Battaglia-Hsu, S.-F.; Soudant, M.; Pinelli, C.; Civit, T.; Taillandier, L.; Vignaud, J.-M.; Bressenot, A. Expression of Minichromosome Maintenance MCM6 Protein in Meningiomas Is Strongly Correlated with Histologic Grade and Clinical Outcome. Am. J. Surg. Pathol. 2012, 36, 283–291. [Google Scholar] [CrossRef]
- Zgheib, R.; Battaglia-Hsu, S.-F.; Hergalant, S.; Quéré, M.; Alberto, J.-M.; Chéry, C.; Rouyer, P.; Gauchotte, G.; Guéant, J.-L.; Namour, F. Folate Can Promote the Methionine-Dependent Reprogramming of Glioblastoma Cells towards Pluripotency. Cell Death Dis. 2019, 10, 596. [Google Scholar] [CrossRef]
- Fortin, J.-P.; Labbe, A.; Lemire, M.; Zanke, B.W.; Hudson, T.J.; Fertig, E.J.; Greenwood, C.M.; Hansen, K.D. Functional Normalization of 450k Methylation Array Data Improves Replication in Large Cancer Studies. Genome Biol. 2014, 15, 503. [Google Scholar] [CrossRef]
- Phipson, B.; Maksimovic, J.; Oshlack, A. MissMethyl: An R Package for Analyzing Data from Illumina’s HumanMethylation450 Platform. Bioinforma. Oxf. Engl. 2016, 32, 286–288. [Google Scholar] [CrossRef]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA Methylation-Based Classification of Central Nervous System Tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef]
- Daoud, E.V.; Zhu, K.; Mickey, B.; Mohamed, H.; Wen, M.; Delorenzo, M.; Tran, I.; Serrano, J.; Hatanpaa, K.J.; Raisanen, J.M.; et al. Epigenetic and Genomic Profiling of Chordoid Meningioma: Implications for Clinical Management. Acta Neuropathol. Commun. 2022, 10, 56. [Google Scholar] [CrossRef]
- Esteller, M. Epigenetic Gene Silencing in Cancer: The DNA Hypermethylome. Hum. Mol. Genet. 2007, 16, R50–R59. [Google Scholar] [CrossRef]
- Mack, S.C.; Hubert, C.G.; Miller, T.E.; Taylor, M.D.; Rich, J.N. An Epigenetic Gateway to Brain Tumor Cell Identity. Nat. Neurosci. 2016, 19, 10–19. [Google Scholar] [CrossRef]
- Rechache, N.S.; Wang, Y.; Stevenson, H.S.; Killian, J.K.; Edelman, D.C.; Merino, M.; Zhang, L.; Nilubol, N.; Stratakis, C.A.; Meltzer, P.S.; et al. DNA Methylation Profiling Identifies Global Methylation Differences and Markers of Adrenocortical Tumors. J. Clin. Endocrinol. Metab. 2012, 97, E1004–E1013. [Google Scholar] [CrossRef]
- Song, M.-A.; Tiirikainen, M.; Kwee, S.; Okimoto, G.; Yu, H.; Wong, L.L. Elucidating the Landscape of Aberrant DNA Methylation in Hepatocellular Carcinoma. PLoS ONE 2013, 8, e55761. [Google Scholar] [CrossRef]
- Rodriguez, J.; Frigola, J.; Vendrell, E.; Risques, R.-A.; Fraga, M.F.; Morales, C.; Moreno, V.; Esteller, M.; Capellà, G.; Ribas, M.; et al. Chromosomal Instability Correlates with Genome-Wide DNA Demethylation in Human Primary Colorectal Cancers. Cancer Res. 2006, 66, 8462–9468. [Google Scholar] [CrossRef]
- Nomura, M.; Saito, K.; Aihara, K.; Nagae, G.; Yamamoto, S.; Tatsuno, K.; Ueda, H.; Fukuda, S.; Umeda, T.; Tanaka, S.; et al. DNA Demethylation Is Associated with Malignant Progression of Lower-Grade Gliomas. Sci. Rep. 2019, 9, 1903. [Google Scholar] [CrossRef]
- Lietz, C.E.; Newman, E.T.; Kelly, A.D.; Xiang, D.H.; Zhang, Z.; Luscko, C.A.; Lozano-Calderon, S.A.; Ebb, D.H.; Raskin, K.A.; Cote, G.M.; et al. Genome-Wide DNA Methylation Patterns Reveal Clinically Relevant Predictive and Prognostic Subtypes in Human Osteosarcoma. Commun. Biol. 2022, 5, 213. [Google Scholar] [CrossRef]
- Cancino, G.I.; Yiu, A.P.; Fatt, M.P.; Dugani, C.B.; Flores, E.R.; Frankland, P.W.; Josselyn, S.A.; Miller, F.D.; Kaplan, D.R. P63 Regulates Adult Neural Precursor and Newly Born Neuron Survival to Control Hippocampal-Dependent Behavior. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 12569–12585. [Google Scholar] [CrossRef]
- Dugani, C.B.; Paquin, A.; Fujitani, M.; Kaplan, D.R.; Miller, F.D. P63 Antagonizes P53 to Promote the Survival of Embryonic Neural Precursor Cells. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 6710–6721. [Google Scholar] [CrossRef]
- Feng, X.-D.; Song, Q.; Li, C.-W.; Chen, J.; Tang, H.-M.; Peng, Z.-H.; Wang, X.-C. Structural Maintenance of Chromosomes 4 Is a Predictor of Survival and a Novel Therapeutic Target in Colorectal Cancer. Asian Pac. J. Cancer Prev. APJCP 2014, 15, 9459–9465. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, F.; Deng, L.; Yuan, X.; Tao, Q.; Wang, T.; Li, D.; Fan, Y.; Peng, Q.; Tang, D. HIF-1-MiR-219-SMC4 Regulatory Pathway Promoting Proliferation and Migration of HCC under Hypoxic Condition. BioMed Res. Int. 2019, 2019, 8983704. [Google Scholar] [CrossRef]
- Yan, Y.; Liu, C.; Zhang, J.; Li, W.; Yin, X.; Dong, L.; Pang, S.; Li, X. SMC4 Knockdown Inhibits Malignant Biological Behaviors of Endometrial Cancer Cells by Regulation of FoxO1 Activity. Arch. Biochem. Biophys. 2021, 712, 109026. [Google Scholar] [CrossRef]
- Ma, R.-M.; Yang, F.; Huang, D.-P.; Zheng, M.; Wang, Y.-L. The Prognostic Value of the Expression of SMC4 MRNA in Breast Cancer. Dis. Markers 2019, 2019, 2183057. [Google Scholar] [CrossRef]
- Zhou, B.; Chen, H.; Wei, D.; Kuang, Y.; Zhao, X.; Li, G.; Xie, J.; Chen, P. A Novel MiR-219-SMC4-JAK2/Stat3 Regulatory Pathway in Human Hepatocellular Carcinoma. J. Exp. Clin. Cancer Res. CR 2014, 33, 55. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Zhou, J.; Zhong, D.; Zhou, Y.; Zhang, W.; Wu, W.; Zhao, Z.; Wang, W.; Xu, W.; He, L.; et al. Overexpression of SMC4 Activates TGFβ/Smad Signaling and Promotes Aggressive Phenotype in Glioma Cells. Oncogenesis 2017, 6, e301. [Google Scholar] [CrossRef] [PubMed]
- You, A.; Rao, G.; Wang, J.; Li, J.; Zhang, Y.; Gu, J.; Ge, X.; Zhang, K.; Gao, X.; Wu, X.; et al. MiR-433-3p Restrains the Proliferation, Migration and Invasion of Glioma Cells via Targeting SMC4. Brain Res. 2021, 1767, 147563. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, Z. The Clinical Significance and Transcription Regulation of a DNA Damage Repair Gene, SMC4, in Low-Grade Glioma via Integrated Bioinformatic Analysis. Front. Oncol. 2021, 11, 761693. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.Y.; Hu, Y.; Siegel, E.; Stanley, L.; Zhou, Y.-H. PAX6 Increases Glioma Cell Susceptibility to Detachment and Oxidative Stress. J. Neurooncol. 2007, 84, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Pavlakis, E.; Tonchev, A.B.; Kaprelyan, A.; Enchev, Y.; Stoykova, A. Interaction between Transcription Factors PAX6/PAX6-5a and Specific Members of MiR-183-96-182 Cluster, May Contribute to Glioma Progression in Glioblastoma Cell Lines. Oncol. Rep. 2017, 37, 1579–1592. [Google Scholar] [CrossRef]
- Zhou, Y.-H.; Wu, X.; Tan, F.; Shi, Y.-X.; Glass, T.; Liu, T.J.; Wathen, K.; Hess, K.R.; Gumin, J.; Lang, F.; et al. PAX6 Suppresses Growth of Human Glioblastoma Cells. J. Neurooncol. 2005, 71, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.V.; Maniatis, T. Clustered Protocadherins. Dev. Camb. Engl. 2013, 140, 3297–3302. [Google Scholar] [CrossRef]
- Tasic, B.; Nabholz, C.E.; Baldwin, K.K.; Kim, Y.; Rueckert, E.H.; Ribich, S.A.; Cramer, P.; Wu, Q.; Axel, R.; Maniatis, T. Promoter Choice Determines Splice Site Selection in Protocadherin Alpha and Gamma Pre-MRNA Splicing. Mol. Cell 2002, 10, 21–33. [Google Scholar] [CrossRef]
- Vega-Benedetti, A.F.; Loi, E.; Moi, L.; Blois, S.; Fadda, A.; Antonelli, M.; Arcella, A.; Badiali, M.; Giangaspero, F.; Morra, I.; et al. Clustered Protocadherins Methylation Alterations in Cancer. Clin. Epigenet. 2019, 11, 100. [Google Scholar] [CrossRef]
- Jun, P.; Hong, C.; Lal, A.; Wong, J.M.; McDermott, M.W.; Bollen, A.W.; Plass, C.; Held, W.A.; Smiraglia, D.J.; Costello, J.F. Epigenetic Silencing of the Kinase Tumor Suppressor WNK2 Is Tumor-Type and Tumor-Grade Specific. Neuro-Oncology 2009, 11, 414–422. [Google Scholar] [CrossRef]
- He, S.; Pham, M.H.; Pease, M.; Zada, G.; Giannotta, S.L.; Wang, K.; Mack, W.J. A Review of Epigenetic and Gene Expression Alterations Associated with Intracranial Meningiomas. Neurosurg. Focus 2013, 35, E5. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Wang, X.; Liu, J.; He, Y.; Liang, Z.; Xia, Z.; Cai, Y.; Zhou, L.; Zhu, H.; Liang, S. Downregulation of ARHGDIA Contributes to Human Glioma Progression through Activation of Rho GTPase Signaling Pathway. Tumour Biol. 2016, 37, 15783–15793. [Google Scholar] [CrossRef]
- Wang, H.; Wang, B.; Liao, Q.; An, H.; Li, W.; Jin, X.; Cui, S.; Zhao, L. Overexpression of RhoGDI, a Novel Predictor of Distant Metastasis, Promotes Cell Proliferation and Migration in Hepatocellular Carcinoma. FEBS Lett. 2014, 588, 503–508. [Google Scholar] [CrossRef]
- Yamashita, T.; Okamura, T.; Nagano, K.; Imai, S.; Abe, Y.; Nabeshi, H.; Yoshikawa, T.; Yoshioka, Y.; Kamada, H.; Tsutsumi, Y.; et al. Rho GDP-Dissociation Inhibitor Alpha Is Associated with Cancer Metastasis in Colon and Prostate Cancer. Pharm.-Int. J. Pharm. Sci. 2012, 67, 253–255. [Google Scholar]
- Bozza, W.P.; Zhang, Y.; Hallett, K.; Rosado, L.A.R.; Zhang, B. RhoGDI Deficiency Induces Constitutive Activation of Rho GTPases and COX-2 Pathways in Association with Breast Cancer Progression. Oncotarget 2015, 6, 32723–32736. [Google Scholar] [CrossRef][Green Version]
- Harding, M.A.; Theodorescu, D. RhoGDI Signaling Provides Targets for Cancer Therapy. Eur. J. Cancer Oxf. Engl. 1990 2010, 46, 1252–1259. [Google Scholar] [CrossRef]
- Shirkavand, A.; Boroujeni, Z.N.; Aleyasin, S.A. Examination of Methylation Changes of VIM, CXCR4, DOK7, and SPDEF Genes in Peripheral Blood DNA in Breast Cancer Patients. Indian J. Cancer 2018, 55, 366–371. [Google Scholar] [CrossRef]
- Heyn, H.; Carmona, F.J.; Gomez, A.; Ferreira, H.J.; Bell, J.T.; Sayols, S.; Ward, K.; Stefansson, O.A.; Moran, S.; Sandoval, J.; et al. DNA Methylation Profiling in Breast Cancer Discordant Identical Twins Identifies DOK7 as Novel Epigenetic Biomarker. Carcinogenesis 2013, 34, 102–108. [Google Scholar] [CrossRef]
- Zhao, H.; Chen, G.; Ye, L.; Yu, H.; Li, S.; Jiang, W.G. DOK7V1 Influences the Malignant Phenotype of Lung Cancer Cells through PI3K/AKT/MTOR and FAK/Paxillin Signaling Pathways. Int. J. Oncol. 2019, 54, 381–389. [Google Scholar] [CrossRef]
- Chen, G.; Yu, H.; Satherley, L.; Zabkiewicz, C.; Resaul, J.; Zhao, H.; Mu, H.; Zhi, X.; He, J.; Ye, L.; et al. The Downstream of Tyrosine Kinase 7 Is Reduced in Lung Cancer and Is Associated with Poor Survival of Patients with Lung Cancer. Oncol. Rep. 2017, 37, 2695–2701. [Google Scholar] [CrossRef] [PubMed]
- Hua, C.-D.; Bian, E.-B.; Chen, E.-F.; Yang, Z.-H.; Tang, F.; Wang, H.-L.; Zhao, B. Repression of Dok7 Expression Mediated by DNMT1 Promotes Glioma Cells Proliferation. Biomed. Pharmacother. Biomedecine Pharmacother. 2018, 106, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.-H.; Choi, H.; Oshima, M.; Cheong, J.-H.; Kim, S.; Lee, J.H.; Park, Y.S.; Choi, H.-S.; Kweon, M.-N.; Pack, C.-G.; et al. Estrogen-Related Receptor Gamma Functions as a Tumor Suppressor in Gastric Cancer. Nat. Commun. 2018, 9, 1920. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Hu, Y.; Zhou, C.; Yuan, J.; Xu, J.; Hao, W.; Deng, H.; Ye, D. ESRRG Promoter Hypermethylation as a Diagnostic and Prognostic Biomarker in Laryngeal Squamous Cell Carcinoma. J. Clin. Lab. Anal. 2019, 33, e22899. [Google Scholar] [CrossRef]
- Acampora, D.; Mazan, S.; Avantaggiato, V.; Barone, P.; Tuorto, F.; Lallemand, Y.; Brûlet, P.; Simeone, A. Epilepsy and Brain Abnormalities in Mice Lacking the Otx1 Gene. Nat. Genet. 1996, 14, 218–222. [Google Scholar] [CrossRef]
- Huang, B.; Li, X.; Tu, X.; Zhao, W.; Zhu, D.; Feng, Y.; Si, X.; Chen, J.-G. OTX1 Regulates Cell Cycle Progression of Neural Progenitors in the Developing Cerebral Cortex. J. Biol. Chem. 2018, 293, 2137–2148. [Google Scholar] [CrossRef]
- Jiang, L.; Zuo, Z.; Lin, J.; Yang, C. Orthodenticle Homeobox OTX1 Is a Potential Prognostic Biomarker for Bladder Cancer. Bioengineered 2021, 12, 6559–6571. [Google Scholar] [CrossRef]
- Tu, X.-P.; Li, H.; Chen, L.-S.; Luo, X.-N.; Lu, Z.-M.; Zhang, S.-Y.; Chen, S.-H. OTX1 Exerts an Oncogenic Role and Is Negatively Regulated by MiR129-5p in Laryngeal Squamous Cell Carcinoma. BMC Cancer 2020, 20, 794. [Google Scholar] [CrossRef]
- Jiang, Y.; Prabakaran, I.; Wan, F.; Mitra, N.; Furstenau, D.K.; Hung, R.K.; Cao, S.; Zhang, P.J.; Fraker, D.L.; Guvakova, M.A. Vav2 Protein Overexpression Marks and May Predict the Aggressive Subtype of Ductal Carcinoma in Situ. Biomark. Res. 2014, 2, 22. [Google Scholar] [CrossRef]
- Citterio, C.; Menacho-Márquez, M.; García-Escudero, R.; Larive, R.M.; Barreiro, O.; Sánchez-Madrid, F.; Paramio, J.M.; Bustelo, X.R. The Rho Exchange Factors Vav2 and Vav3 Control a Lung Metastasis-Specific Transcriptional Program in Breast Cancer Cells. Sci. Signal. 2012, 5, ra71. [Google Scholar] [CrossRef]
- Tan, B.-B.; Li, Y.; Fan, L.-Q.; Zhao, Q.; Liu, Q.-W.; Liu, Y.; Wang, D.; Jia, N. Upregulated Vav2 in Gastric Cancer Tissues Promotes Tumor Invasion and Metastasis. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2017, 39, 1010428317698392. [Google Scholar] [CrossRef]
- Lorenzo-Martín, L.F.; Fernández-Parejo, N.; Menacho-Márquez, M.; Rodríguez-Fdez, S.; Robles-Valero, J.; Zumalave, S.; Fabbiano, S.; Pascual, G.; García-Pedrero, J.M.; Abad, A.; et al. VAV2 Signaling Promotes Regenerative Proliferation in Both Cutaneous and Head and Neck Squamous Cell Carcinoma. Nat. Commun. 2020, 11, 4788. [Google Scholar] [CrossRef]
- Liu, W.; Miao, C.; Zhang, S.; Liu, Y.; Niu, X.; Xi, Y.; Guo, W.; Chu, J.; Lin, A.; Liu, H.; et al. VAV2 Is Required for DNA Repair and Implicated in Cancer Radiotherapy Resistance. Signal Transduct. Target. Ther. 2021, 6, 322. [Google Scholar] [CrossRef]
- Jones, D.T.W.; Jäger, N.; Kool, M.; Zichner, T.; Hutter, B.; Sultan, M.; Cho, Y.-J.; Pugh, T.J.; Hovestadt, V.; Stütz, A.M.; et al. Dissecting the Genomic Complexity Underlying Medulloblastoma. Nature 2012, 488, 100–105. [Google Scholar] [CrossRef]
- Nakahara, Y.; Shiraishi, T.; Okamoto, H.; Mineta, T.; Oishi, T.; Sasaki, K.; Tabuchi, K. Detrended Fluctuation Analysis of Genome-Wide Copy Number Profiles of Glioblastomas Using Array-Based Comparative Genomic Hybridization. Neuro-Oncol. 2004, 6, 281–289. [Google Scholar] [CrossRef]
- McGarry, T.J.; Kirschner, M.W. Geminin, an Inhibitor of DNA Replication, Is Degraded during Mitosis. Cell 1998, 93, 1043–1053. [Google Scholar] [CrossRef]
- Miotto, B.; Struhl, K. HBO1 Histone Acetylase Activity Is Essential for DNA Replication Licensing and Inhibited by Geminin. Mol. Cell 2010, 37, 57–66. [Google Scholar] [CrossRef]
- Sugimoto, N.; Tatsumi, Y.; Tsurumi, T.; Matsukage, A.; Kiyono, T.; Nishitani, H.; Fujita, M. Cdt1 Phosphorylation by Cyclin A-Dependent Kinases Negatively Regulates Its Function without Affecting Geminin Binding. J. Biol. Chem. 2004, 279, 19691–19697. [Google Scholar] [CrossRef]
- Caillat, C.; Pefani, D.-E.; Gillespie, P.J.; Taraviras, S.; Blow, J.J.; Lygerou, Z.; Perrakis, A. The Geminin and Idas Coiled Coils Preferentially Form a Heterodimer That Inhibits Geminin Function in DNA Replication Licensing. J. Biol. Chem. 2013, 288, 31624–31634. [Google Scholar] [CrossRef]
- Zhou, B.; Liu, C.; Xu, Z.; Zhu, G. Structural Basis for Homeodomain Recognition by the Cell-Cycle Regulator Geminin. Proc. Natl. Acad. Sci. USA 2012, 109, 8931–8936. [Google Scholar] [CrossRef]
- Hao, Y.; Li, G. Role of EFNA1 in Tumorigenesis and Prospects for Cancer Therapy. Biomed. Pharmacother. 2020, 130, 110567. [Google Scholar] [CrossRef]
- Pötschke, R.; Haase, J.; Glaß, M.; Simmermacher, S.; Misiak, C.; Penalva, L.O.F.; Kühnöl, C.D.; Hüttelmaier, S. MSI1 Promotes the Expression of the GBM Stem Cell Marker CD44 by Impairing MiRNA-Dependent Degradation. Cancers 2020, 12, 3654. [Google Scholar] [CrossRef]
- Vo, D.T.; Abdelmohsen, K.; Martindale, J.L.; Qiao, M.; Tominaga, K.; Burton, T.L.; Gelfond, J.A.L.; Brenner, A.J.; Patel, V.; Trageser, D.; et al. The Oncogenic RNA-Binding Protein Musashi1 Is Regulated by HuR via MRNA Translation and Stability in Glioblastoma Cells. Mol. Cancer Res. MCR 2012, 10, 143–155. [Google Scholar] [CrossRef]
- Gauchotte, G.; Hergalant, S.; Vigouroux, C.; Casse, J.-M.; Houlgatte, R.; Kaoma, T.; Helle, D.; Brochin, L.; Rech, F.; Peyre, M.; et al. Cytoplasmic Overexpression of RNA-Binding Protein HuR Is a Marker of Poor Prognosis in Meningioma, and HuR Knockdown Decreases Meningioma Cell Growth and Resistance to Hypoxia. J. Pathol. 2017, 242, 421–434. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WHO Grade (2021 Classification) | n = 48 |
|---|---|
| Grade 1 | 10 (21%) |
| Grade 2 | 33 (69%) |
| Grade 3 | 5 (10%) |
| Age | 57 (48; 67) |
| Sex | |
| Female | 32 (67%) |
| Male | 16 (33%) |
| Localization | |
| Skull base | 12 (25%) |
| Convexity | 33 (69%) |
| Ventricular | 2 (4%) |
| Spinal | 1 (2%) |
| Complete resection | |
| Yes | 26 (54%) |
| No | 16 (33%) |
| Unknown | 6 (13%) |
| Adjuvant chemotherapy | 1 (2%) |
| Adjuvant radiotherapy | 22 (46%) |
| Progression | 19 (40%) |
| Median progression-free survival (months) | 39 (16; 55) |
| Death | 9 (19%) |
| Median overall survival (months) | 52 (31; 95) |
| Ki67 (%) | 21 (9; 38) |
| MCM6 (%) | 51 (29; 73) |
| Mitoses/1.6 mm2 | 2 (1; 6) |
| WHO Grade | Grade 1 (n = 10) | Grade 2 (n = 33) | Grade 3 (n = 5) | Total (n = 48) |
|---|---|---|---|---|
| Methylation class | ||||
| Benign | 8 (80%) | 5 (15%) | 0 (0%) | 13 (27%) |
| Intermediate | 2 (20%) | 12 (36%) | 0 (0%) | 14 (29%) |
| Malignant | 0 (0%) | 2 (6%) | 2 (40%) | 4 (8%) |
| No match (calibrated score < 0.9) | 0 (0%) | 14 (42%) | 3 (60%) | 17 (35%) |
| Methylation sub-class | ||||
| 1 | 2 (20%) | 1 (3%) | 0 (0%) | 3 (6%) |
| 2 | 4 (40%) | 3 (9%) | 0 (0%) | 7 (15%) |
| 3 | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| 4 | 1 (10%) | 9 (27%) | 0 (0%) | 10 (21%) |
| 5 | 0 (0%) | 2 (6%) | 0 (0%) | 2 (4%) |
| 6 | 0 (0%) | 2 (6%) | 2 (40%) | 4 (8%) |
| No match (calibrated score < 0.9) | 3 (30%) | 16 (48%) | 3 (60%) | 22 (46%) |
| CDKN2A/B homozygous loss | 0 (0%) | 0 (0%) | 2 (40%) | 2 (4%) |
| PTEN loss | 0 (0%) | 14 (42%) | 4 (100%) | 18 (38%) |
| NF2 loss | 5 (50%) | 30 (91%) | 4 (100%) | 39 (81%) |
| Linked Gene | Chromosome | # CpGs | FDR | Mean. Diff. | Max. Diff. | |
|---|---|---|---|---|---|---|
| Hypermethylated in high grades | CYP26B1 | chr2 | 3 | 5.21 × 10−15 | +29% | +42% |
| REC8 | chr14 | 13 | 1.09 × 10−38 | +29% | +37% | |
| C2CD4D | chr1 | 5 | 6.10 × 10−13 | +28% | +35% | |
| KIFC2 | chr8 | 4 | 2.45 × 10−14 | +28% | +35% | |
| CALCB | chr11 | 5 | 4.77 × 10−14 | +28% | +38% | |
| HEPACAM | chr11 | 4 | 1.02 × 10−16 | +28% | +38% | |
| DCDC2C | chr2 | 5 | 7.55 × 10−14 | +27% | +41% | |
| PAX6 | chr11 | 15 | 7.63 × 10−23 | +27% | +42% | |
| SPEG | chr2 | 6 | 8.06 × 10−22 | +27% | +35% | |
| LTBP4 | chr19 | 5 | 2.84 × 10−11 | +26% | +32% | |
| WNK2 | chr9 | 8 | 1.11 × 10−35 | +25% | +43% | |
| PITX1 | chr5 | 6 | 5.17 × 10−14 | +25% | +34% | |
| KLB | chr4 | 4 | 2.85 × 10−11 | +25% | +33% | |
| B4GALNT1 | chr12 | 5 | 1.41 × 10−14 | +24% | +36% | |
| IRX1 | chr5 | 8 | 2.14 × 10−15 | +24% | +37% | |
| Hypomethylated in high grades | SMC4/miR16 | chr3 | 4 | 2.30 × 10−16 | −41% | −46% |
| ARHGAP23 | chr17 | 3 | 6.68 × 10−24 | −36% | −44% | |
| PATJ | chr1 | 3 | 6.07 × 10−12 | −31% | −33% | |
| CACNA1H | chr16 | 3 | 3.94 × 10−14 | −28% | −35% | |
| THSD4 | chr15 | 3 | 4.52 × 10−12 | −25% | −27% | |
| DNAJB6 | chr7 | 7 | 2.61 × 10−13 | −23% | −36% | |
| TP63 | chr3 | 9 | 2.38 × 10−19 | −23% | −42% | |
| LINC01589 | chr22 | 4 | 1.30 × 10−16 | −23% | −32% | |
| DHX30 | chr3 | 3 | 1.14 × 10−13 | −20% | −28% | |
| RBM47 | chr4 | 11 | 6.62 × 10−19 | −19% | −28% |
| Reference | Ontology Term | N | DE (%) | P.DE | FDR |
|---|---|---|---|---|---|
| GO:0009653 | anatomical structure morphogenesis | 2629 | 77.7% | 1.44 × 10−27 | 3.27 × 10−23 |
| GO:0007399 | nervous system development | 2264 | 79.1% | 6.68 × 10−27 | 7.60 × 10−23 |
| GO:0048856 | anatomical structure development | 5697 | 73.0% | 3.36 × 10−26 | 2.55 × 10−22 |
| GO:0032502 | developmental process | 6073 | 72.6% | 1.65 × 10−25 | 9.36 × 10−22 |
| GO:0016043 | cellular component organization | 6081 | 73.2% | 5.20 × 10−25 | 2.37 × 10−21 |
| GO:0007275 | multicellular organism development | 5227 | 73.1% | 2.03 × 10−24 | 7.71 × 10−21 |
| GO:0071840 | cellular component organization or biogenesis | 6261 | 72.9% | 3.91 × 10−24 | 1.11 × 10−20 |
| GO:0048518 | positive regulation of biological process | 5830 | 72.6% | 2.35 × 10−23 | 5.94 × 10−20 |
| GO:0048522 | positive regulation of cellular process | 5143 | 73.1% | 5.74 × 10−23 | 1.31 × 10−19 |
| GO:0048731 | system development | 4689 | 73.2% | 3.42 × 10−22 | 7.08 × 10−19 |
| GO:0048468 | cell development | 2096 | 77.8% | 2.10 × 10−20 | 3.97 × 10−17 |
| GO:0048869 | cellular developmental process | 4222 | 73.0% | 1.74 × 10−19 | 3.05 × 10−16 |
| GO:0022008 | neurogenesis | 1563 | 79.5% | 3.76 × 10−19 | 6.11 × 10−16 |
| GO:0009893 | positive regulation of metabolic process | 3450 | 73.9% | 4.98 × 10−19 | 7.56 × 10−16 |
| GO:0048699 | generation of neurons | 1465 | 79.7% | 1.95 × 10−18 | 2.77 × 10−15 |
| GO:0010604 | positive regulation of macromolecular metabolic process | 3193 | 74.1% | 3.98 × 10−18 | 5.33 × 10−15 |
| GO:0030182 | neuron differentiation | 1310 | 80.4% | 5.16 × 10−18 | 6.52 × 10−15 |
| GO:0030154 | cell differentiation | 4040 | 72.7% | 1.28 × 10−17 | 1.53 × 10−14 |
| GO:0031325 | positive regulation of cellular metabolic process | 3166 | 74.0% | 1.79 × 10−17 | 2.04 × 10−14 |
| GO:0010646 | regulation of cell communication | 3381 | 73.9% | 2.44 × 10−17 | 2.64 × 10−14 |
| CpG | Gene (# Hits) | Chr. | Rho | p-Value | DNAm Median Level (Beta-Value) | DNAm Diff. Range (%) | ||
|---|---|---|---|---|---|---|---|---|
| Mitotic index | negative | cg21942721 | ASB4 (1) | chr7 | −0.71 | 1.27 × 10−8 | High (0.84) | 60 (26) |
| cg18568061 | PTRF (1) | chr17 | −0.69 | 4.94 × 10−8 | Medium (0.37) | 63 (21) | ||
| cg01764105 | SMC4/miR16 (7) | chr3 | −0.69 | 7.46 × 10−8 | Low (0.08) | 76 (21) | ||
| cg17605814 | CD82 (2) | chr11 | −0.68 | 1.22 × 10−7 | Medium (0.57) | 75 (25) | ||
| cg22624818 | SDPR (2) | chr2 | −0.68 | 1.34 × 10−7 | High (0.80) | 66 (17) | ||
| cg16166651 | DEPDC1 (3) | chr1 | −0.67 | 1.53 × 10−7 | Low (0.16) | 62 (12) | ||
| cg06003566 | METTL24 (2) | chr6 | −0.67 | 2.19 × 10−7 | High (0.86) | 79 (14) | ||
| positive | cg25588576 | MIR7641-2 (1) | chr14 | 0.66 | 3.52 × 10−7 | High (0.79) | 69 (31) | |
| cg21784383 | ESRRG (6) | chr1 | 0.65 | 5.83 × 10−7 | Low (0.18) | 71 (27) | ||
| cg18361098 | PAX9 (2) | chr14 | 0.62 | 2.80 × 10−6 | Low (0.10) | 80 (43) | ||
| cg10640333 | OTX1 (2) | chr2 | 0.61 | 4.33 × 10−6 | High (0.76) | 90 (20) | ||
| cg13244312 | TTC9 (1) | chr14 | 0.61 | 4.81 × 10−6 | Medium (0.58) | 55 (19) | ||
| Ki-67 LI% | negative | cg01464849 | SMC4/miR16 (9) | chr3 | −0.73 | 3.33 × 10−9 | Low (0.26) | 84 (58) |
| cg18943088 | IQCJ-SCHIP1 (16) | chr3 | −0.71 | 1.78 × 10−8 | Low (0.30) | 83 (42) | ||
| cg22800629 | RAB33B (2) | chr4 | −0.69 | 7.13 × 10−8 | Low (0.09) | 53 (15) | ||
| cg17253087 | HIPK3 (1) | chr11 | −0.68 | 7.97 × 10−8 | Medium (0.55) | 78 (45) | ||
| cg11629830 | IQGAP2 (3) | chr5 | −0.68 | 1.04 × 10−7 | Medium (0.69) | 68 (36) | ||
| cg13944632 | VAV2 (4) | chr9 | −0.66 | 2.97 × 10−7 | Medium (0.37) | 75 (15) | ||
| positive | cg03126579 | ZFR2 (1) | chr19 | 0.71 | 1.27 × 10−8 | Medium (0.65) | 93 (37) | |
| cg10269365 | CCDC140 (10) | chr2 | 0.69 | 6.92 × 10−8 | Medium (0.55) | 84 (37) | ||
| cg08139247 | CLEC14A (5) | chr14 | 0.68 | 9.42 × 10−8 | Medium (0.53) | 83 (48) | ||
| cg21784383 | ESRRG (7) | chr1 | 0.66 | 3.34 × 10−7 | Low (0.18) | 71 (27) | ||
| cg26418900 | NPY (4) | chr7 | 0.66 | 3.34 × 10−7 | Medium (0.40) | 87 (32) | ||
| cg10640333 | OTX1 (4) | chr2 | 0.65 | 5.83 × 10−7 | High (0.76) | 90 (20) | ||
| MCM6 LI% | negative | cg04570316 | GMNN (1) | chr6 | −0.62 | 2.45 × 10−6 | High (0.89) | 71 (11) |
| cg09130952 | CCDC108 (1) | chr2 | −0.62 | 2.52 × 10−6 | High (0.71) | 68 (25) | ||
| cg16959792 | SLC50A1/EFNA1 (1) | chr1 | −0.62 | 2.57 × 10−6 | Low (0.23) | 39 (18) | ||
| cg24310126 | FLJ46361 (1) | chr10 | −0.62 | 2.99 × 10−6 | High (0.85) | 78 (16) | ||
| cg03805253 | CACNA1G (1) | chr17 | −0.62 | 3.05 × 10−6 | Medium (0.68) | 61 (19) | ||
| cg10422455 | MRGPRX2 (1) | chr11 | −0.61 | 3.69 × 10−6 | Medium (0.66) | 82 (33) | ||
| positive | cg03126579 | ZFR2 (1) | chr19 | 0.68 | 8.32 × 10−8 | Medium (0.65) | 93 (37) | |
| cg03552103 | SEPT10/ANKRD57 (1) | chr2 | 0.67 | 1.70 × 10−7 | Medium (0.69) | 50 (13) | ||
| cg15415136 | ZNF540 (2) | chr19 | 0.66 | 3.60 × 10−7 | Low (0.15) | 56 (10) | ||
| cg06488443 | TBR1 (2) | chr2 | 0.66 | 3.93 × 10−7 | Low (0.29) | 61 (22) | ||
| cg05008496 | SSPN (1) | chr12 | 0.64 | 1.20 × 10−6 | Medium (0.56) | 52 (22) | ||
| cg22151446 | PCDHabg clusters (27) | chr5 | 0.63 | 1.61 × 10−6 | Medium (0.31) | 66 (38) | ||
| cg12052661 | CACNA1B (2) | chr9 | 0.63 | 1.72 × 10−6 | Medium (0.39) | 52 (23) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hergalant, S.; Saurel, C.; Divoux, M.; Rech, F.; Pouget, C.; Godfraind, C.; Rouyer, P.; Lacomme, S.; Battaglia-Hsu, S.-F.; Gauchotte, G. Correlation between DNA Methylation and Cell Proliferation Identifies New Candidate Predictive Markers in Meningioma. Cancers 2022, 14, 6227. https://doi.org/10.3390/cancers14246227
Hergalant S, Saurel C, Divoux M, Rech F, Pouget C, Godfraind C, Rouyer P, Lacomme S, Battaglia-Hsu S-F, Gauchotte G. Correlation between DNA Methylation and Cell Proliferation Identifies New Candidate Predictive Markers in Meningioma. Cancers. 2022; 14(24):6227. https://doi.org/10.3390/cancers14246227
Chicago/Turabian StyleHergalant, Sébastien, Chloé Saurel, Marion Divoux, Fabien Rech, Celso Pouget, Catherine Godfraind, Pierre Rouyer, Stéphanie Lacomme, Shyue-Fang Battaglia-Hsu, and Guillaume Gauchotte. 2022. "Correlation between DNA Methylation and Cell Proliferation Identifies New Candidate Predictive Markers in Meningioma" Cancers 14, no. 24: 6227. https://doi.org/10.3390/cancers14246227
APA StyleHergalant, S., Saurel, C., Divoux, M., Rech, F., Pouget, C., Godfraind, C., Rouyer, P., Lacomme, S., Battaglia-Hsu, S.-F., & Gauchotte, G. (2022). Correlation between DNA Methylation and Cell Proliferation Identifies New Candidate Predictive Markers in Meningioma. Cancers, 14(24), 6227. https://doi.org/10.3390/cancers14246227