
Characterization of Glioblastoma Cells Response to Regorafenib

 ,
,  , , , ,
, , , ,  , , , ,
, , , , 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Viability Assay
2.3. IC50 Calculation
2.4. Cell-Cycle Analysis
2.5. Annexin V Staining
2.6. Clonogenesis Assay
2.7. Reverse Transcription and Real Time PCR Analyses
2.8. Western Blot Analysis
2.9. Tumor Spheroid Three-Dimensional Invasion Assay
2.10. Fluorescent Staining Protocol
2.11. X-ray Microtomography (microCT)
2.12. Statistical Analysis
3. Results
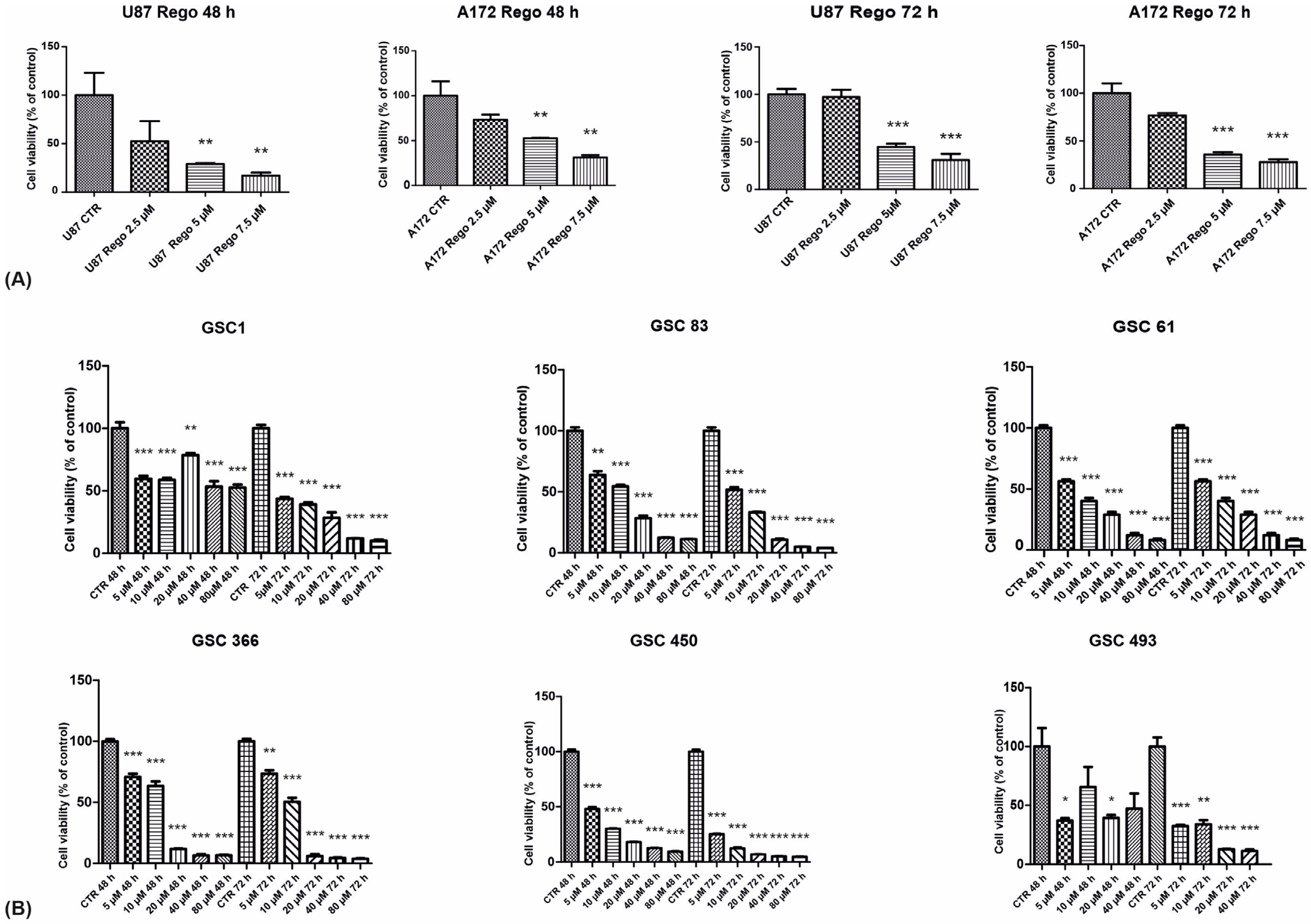
3.1. Cell Viability, Cell-Cycle Regulation, and Neurosphere-Forming Ability upon Regorafenib Exposure
3.2. Evaluation of Apoptosis in Regorafenib-Treated Cells
3.3. Transcriptional Regulation of Genes of Epithelial to Mesenchymal Transition (EMT) in Regorafenib-Treated Cells
3.4. Characterization of U87, A172, GSC#1, and GSC#83 3D Spheroids to Regorafenib
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.; Dubrovska, A.; Linge, A.; Baumann, M. Cancer stem cells: Radioresistance, prediction of radiotherapy outcome and specific targets for combined treatments. Adv. Drug Deliv. Rev. 2017, 109, 63–73. [Google Scholar] [CrossRef]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef]
- Reardon, D.A.; Turner, S.; Peters, K.B.; Desjardins, A.; Gururangan, S.; Sampson, J.H.; McLendon, R.E.; Herndon, J.E.; Jones, L.W.; Kirkpatrick, J.P.; et al. A review of VEGF/VEGFR-targeted therapeutics for recurrent glioblastoma. J. Natl. Compr. Canc Netw. 2011, 9, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Zeiner, P.S.; Kinzig, M.; Divé, I.; Maurer, G.D.; Filipski, K.; Harter, P.N.; Senft, C.; Bähr, O.; Hattingen, E.; Steinbach, J.P.; et al. Regorafenib CSF Penetration, Efficacy, and MRI Patterns in Recurrent Malignant Glioma Patients. J. Clin. Med. 2019, 8, 2031. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, T.T.; Gerstner, E.R.; Ye, X.; Desideri, S.; Duda, D.G.; Peereboom, D.; Lesser, G.J.; Chowdhary, S.; Wen, P.Y.; Grossman, S.; et al. Feasibility, phase I, and phase II studies of tandutinib, an oral platelet-derived growth factor receptor-β tyrosine kinase inhibitor, in patients with recurrent glioblastoma. Neuro Oncol. 2017, 19, 567–575. [Google Scholar] [CrossRef]
- Kalpathy-Cramer, J.; Chandra, V.; Da, X.; Ou, Y.; Emblem, K.E.; Muzikansky, A.; Cai, X.; Douw, L.; Evans, J.G.; Dietrich, J.; et al. Phase II study of tivozanib, an oral VEGFR inhibitor, in patients with recurrent glioblastoma. J. Neurooncol. 2017, 131, 603–610. [Google Scholar] [CrossRef]
- Lombardi, G.; De Salvo, G.L.; Brandes, A.A.; Eoli, M.; Rudà, R.; Faedi, M.; Lolli, I.; Pace, A.; Daniele, B.; Pasqualetti, F.; et al. Regorafenib compared with lomustine in patients with relapsed glioblastoma (REGOMA): A multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 2019, 20, 110–119. [Google Scholar] [CrossRef]
- Lombardi, G.; Caccese, M.; Padovan, M.; Cerretti, G.; Pintacuda, G.; Manara, R.; Di Sarra, F.; Zagonel, V. Regorafenib in Recurrent Glioblastoma Patients: A Large and Monocentric Real-Life Study. Cancers 2021, 13, 4731. [Google Scholar] [CrossRef]
- Lombardi, G.; Del Bianco, P.; Brandes, A.A.; Eoli, M.; Rudà, R.; Ibrahim, T.; Lolli, I.; Rizzato, S.; Daniele, B.; Pace, A.; et al. Patient-reported outcomes in a phase II randomised study of regorafenib compared with lomustine in patients with relapsed glioblastoma (the REGOMA trial). Eur. J. Cancer 2021, 155, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Santangelo, A.; Rossato, M.; Lombardi, G.; Benfatto, S.; Lavezzari, D.; De Salvo, G.L.; Indraccolo, S.; Dechecchi, M.C.; Prandini, P.; Gambari, R.; et al. A molecular signature associated with prolonged survival in glioblastoma patients treated with regorafenib. Neuro Oncol. 2021, 23, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Indraccolo, S.; De Salvo, G.L.; Verza, M.; Caccese, M.; Esposito, G.; Piga, I.; Del Bianco, P.; Pizzi, M.; Gardiman, M.P.; Eoli, M.; et al. Phosphorylated Acetyl-CoA Carboxylase Is Associated with Clinical Benefit with Regorafenib in Relapsed Glioblastoma: REGOMA Trial Biomarker Analysis. Clin. Cancer Res. 2020, 26, 4478–4484. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Zhang, L.; Chen, H.; Lei, Y.; Zhang, T.; Wang, Y.; Jin, P.; Lan, J.; Zhou, L.; Huang, Z.; et al. Regorafenib induces lethal autophagy arrest by stabilizing PSAT1 in glioblastoma. Autophagy 2020, 16, 106–122. [Google Scholar] [CrossRef] [PubMed]
- Detti, B.; Scoccianti, S.; Lucidi, S.; Maragna, V.; Teriaca, M.A.; Ganovelli, M.; Desideri, I.; Lorenzetti, V.; Scoccimarro, E.; Greto, D.; et al. Regorafenib in glioblastoma recurrence: A case report. Cancer Treat Res. Commun. 2021, 26, 100263. [Google Scholar] [CrossRef]
- Marziali, G.; Signore, M.; Buccarelli, M.; Grande, S.; Palma, A.; Biffoni, M.; Rosi, A.; D’Alessandris, Q.G.; Martini, M.; Larocca, L.M.; et al. Metabolic/Proteomic Signature Defines Two Glioblastoma Subtypes With Different Clinical Outcome. Sci. Rep. 2016, 6, 21557. [Google Scholar] [CrossRef]
- Buccarelli, M.; D’Alessandris, Q.G.; Matarrese, P.; Mollinari, C.; Signore, M.; Cappannini, A.; Martini, M.; D’Aliberti, P.; De Luca, G.; Pedini, F.; et al. Elesclomol-induced increase of mitochondrial reactive oxygen species impairs glioblastoma stem-like cell survival and tumor growth. J. Exp. Clin. Cancer Res. 2021, 40, 228. [Google Scholar] [CrossRef] [PubMed]
- Kalejta, R.F.; Brideau, A.D.; Banfield, B.W.; Beavis, A.J. An integral membrane green fluorescent protein marker, Us9-GFP, is quantitatively retained in cells during propidium iodide-based cell cycle analysis by flow cytometry. Exp. Cell Res. 1999, 248, 322–328. [Google Scholar] [CrossRef][Green Version]
- Lucà, R.; Assenza, M.R.; Maiullari, F.; Pieroni, L.; Maiullari, S.; Federici, G.; Marini, F.; Rizzi, R.; Urbani, A.; Soddu, S.; et al. Inhibition of the mTOR pathway and reprogramming of protein synthesis by MDM4 reduce ovarian cancer metastatic properties. Cell Death Dis. 2021, 12, 558. [Google Scholar] [CrossRef]
- Karimi, H.; Leszczyński, B.; Kołodziej, T.; Kubicz, E.; Przybyło, M.; Stępień, E. X-ray microtomography as a new approach for imaging and analysis of tumor spheroids. Micron 2020, 137, 102917. [Google Scholar] [CrossRef]
- Barbagallo, F.; Rotilio, V.; Assenza, M.R.; Aguanno, S.; Orsini, T.; Putti, S.; Isidori, A.M.; Lenzi, A.; Naro, F.; De Angelis, L.; et al. PDE2A Is Indispensable for Mouse Liver Development and Hematopoiesis. Int. J. Mol. Sci. 2020, 21, 2902. [Google Scholar] [CrossRef] [PubMed]
- Pontén, J.; Macintyre, E.H. Long term culture of normal and neoplastic human glia. Acta Pathol. Microbiol. Scand. 1968, 74, 465–486. [Google Scholar] [CrossRef] [PubMed]
- Giard, D.J.; Aaronson, S.A.; Todaro, G.J.; Arnstein, P.; Kersey, J.H.; Dosik, H.; Parks, W.P. In vitro cultivation of human tumors: Establishment of cell lines derived from a series of solid tumors. J. Natl. Cancer Inst. 1973, 51, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J. Cancer stem cells and chemoresistance: The smartest survives the raid. Pharmacol. Ther. 2016, 160, 145–158. [Google Scholar] [CrossRef]
- Beier, D.; Schulz, J.B.; Beier, C.P. Chemoresistance of glioblastoma cancer stem cells--much more complex than expected. Mol. Cancer 2011, 10, 128. [Google Scholar] [CrossRef]
- Alves, A.L.V.; Gomes, I.N.F.; Carloni, A.C.; Rosa, M.N.; da Silva, L.S.; Evangelista, A.F.; Reis, R.M.; Silva, V.A.O. Role of glioblastoma stem cells in cancer therapeutic resistance: A perspective on antineoplastic agents from natural sources and chemical derivatives. Stem Cell Res. Ther. 2021, 12, 206. [Google Scholar] [CrossRef]
- Grande, S.; Palma, A.; Ricci-Vitiani, L.; Luciani, A.M.; Buccarelli, M.; Biffoni, M.; Molinari, A.; Calcabrini, A.; D’Amore, E.; Guidoni, L.; et al. Metabolic Heterogeneity Evidenced by MRS among Patient-Derived Glioblastoma Multiforme Stem-Like Cells Accounts for Cell Clustering and Different Responses to Drugs. Stem Cells Int. 2018, 2018, 3292704. [Google Scholar] [CrossRef]
- Marziali, G.; Buccarelli, M.; Giuliani, A.; Ilari, R.; Grande, S.; Palma, A.; D’Alessandris, Q.G.; Martini, M.; Biffoni, M.; Pallini, R.; et al. A three-microRNA signature identifies two subtypes of glioblastoma patients with different clinical outcomes. Mol. Oncol. 2017, 11, 1115–1129. [Google Scholar] [CrossRef][Green Version]
- Guidoni, L.; Ricci-Vitiani, L.; Rosi, A.; Palma, A.; Grande, S.; Luciani, A.M.; Pelacchi, F.; di Martino, S.; Colosimo, C.; Biffoni, M.; et al. 1H NMR detects different metabolic profiles in glioblastoma stem-like cells. NMR Biomed. 2014, 27, 129–145. [Google Scholar] [CrossRef]
- McCord, A.M.; Jamal, M.; Williams, E.S.; Camphausen, K.; Tofilon, P.J. CD133+ glioblastoma stem-like cells are radiosensitive with a defective DNA damage response compared with established cell lines. Clin. Cancer Res. 2009, 15, 5145–5153. [Google Scholar] [CrossRef]
- Deshors, P.; Arnauduc, F.; Boëlle, B.; Cohen-Jonathan Moyal, E.; Courtade-Saïdi, M.; Evrard, S.M. Impact of Regorafenib on Endothelial Transdifferentiation of Glioblastoma Stem-like Cells. Cancers 2022, 14, 1551. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dowbenko, D.; Lasky, L.A. AKT/PKB phosphorylation of p21Cip/WAF1 enhances protein stability of p21Cip/WAF1 and promotes cell survival. J. Biol. Chem. 2002, 277, 11352–11361. [Google Scholar] [CrossRef] [PubMed]
- Rössig, L.; Jadidi, A.S.; Urbich, C.; Badorff, C.; Zeiher, A.M.; Dimmeler, S. Akt-dependent phosphorylation of p21(Cip1) regulates PCNA binding and proliferation of endothelial cells. Mol. Cell Biol. 2001, 21, 5644–5657. [Google Scholar] [CrossRef]
- Zhou, B.P.; Liao, Y.; Xia, W.; Spohn, B.; Lee, M.H.; Hung, M.C. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat. Cell Biol. 2001, 3, 245–252. [Google Scholar] [CrossRef] [PubMed]
- de Groot, J.F.; Fuller, G.; Kumar, A.J.; Piao, Y.; Eterovic, K.; Ji, Y.; Conrad, C.A. Tumor invasion after treatment of glioblastoma with bevacizumab: Radiographic and pathologic correlation in humans and mice. Neuro Oncol. 2010, 12, 233–242. [Google Scholar] [CrossRef]
- Gomez-Manzano, C.; Holash, J.; Fueyo, J.; Xu, J.; Conrad, C.A.; Aldape, K.D.; de Groot, J.F.; Bekele, B.N.; Yung, W.K. VEGF Trap induces antiglioma effect at different stages of disease. Neuro Oncol. 2008, 10, 940–945. [Google Scholar] [CrossRef] [PubMed]
- Pàez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Viñals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar] [CrossRef]
- Falchetti, M.L.; D’Alessandris, Q.G.; Pacioni, S.; Buccarelli, M.; Morgante, L.; Giannetti, S.; Lulli, V.; Martini, M.; Larocca, L.M.; Vakana, E.; et al. Glioblastoma endothelium drives bevacizumab-induced infiltrative growth via modulation of PLXDC1. Int. J. Cancer 2019, 144, 1331–1344. [Google Scholar] [CrossRef]
- Zhang, N.; Zhang, S.; Wu, W.; Lu, W.; Jiang, M.; Zheng, N.; Huang, J.; Wang, L.; Liu, H.; Zheng, M.; et al. Regorafenib inhibits migration, invasion, and vasculogenic mimicry of hepatocellular carcinoma via targeting ID1-mediated EMT. Mol. Carcinog. 2021, 60, 151–163. [Google Scholar] [CrossRef]
- Costa, E.C.; Moreira, A.F.; de Melo-Diogo, D.; Gaspar, V.M.; Carvalho, M.P.; Correia, I.J. 3D tumor spheroids: An overview on the tools and techniques used for their analysis. Biotechnol. Adv. 2016, 34, 1427–1441. [Google Scholar] [CrossRef]
- Orcheston-Findlay, L.; Bax, S.; Utama, R.; Engel, M.; Govender, D.; O’Neill, G. Advanced Spheroid, Tumouroid and 3D Bioprinted In-Vitro Models of Adult and Paediatric Glioblastoma. Int. J. Mol. Sci. 2021, 22, 2962. [Google Scholar] [CrossRef] [PubMed]
- Kessel, S.; Cribbes, S.; Déry, O.; Kuksin, D.; Sincoff, E.; Qiu, J.; Chan, L.L. High-Throughput 3D Tumor Spheroid Screening Method for Cancer Drug Discovery Using Celigo Image Cytometry. SLAS Technol. 2017, 22, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; Reichardt, P.; Kang, Y.K.; Blay, J.Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; von Mehren, M.; Joensuu, H.; et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Grothey, A.; Van Cutsem, E.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouché, O.; Mineur, L.; Barone, C.; et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, G.; Spimpolo, A.; Berti, S.; Campi, C.; Anglani, M.G.; Simeone, R.; Evangelista, L.; Causin, F.; Zorzi, G.; Gorgoni, G.; et al. PET/MR in recurrent glioblastoma patients treated with regorafenib: [18F]FET and DWI-ADC for response assessment and survival prediction. Br. J. Radiol. 2021, 95, 20211018. [Google Scholar] [CrossRef]
- Chiesa, S.; Mangraviti, A.; Martini, M.; Cenci, T.; Mazzarella, C.; Gaudino, S.; Bracci, S.; Martino, A.; Della Pepa, G.M.; Offi, M.; et al. Clinical and NGS predictors of response to regorafenib in recurrent glioblastoma. Sci. Rep. 2022, 12, 16265. [Google Scholar] [CrossRef]
- Kreisl, T.N.; Kim, L.; Moore, K.; Duic, P.; Royce, C.; Stroud, I.; Garren, N.; Mackey, M.; Butman, J.A.; Camphausen, K.; et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J. Clin. Oncol. 2009, 27, 740–745. [Google Scholar] [CrossRef]
- Friedman, H.S.; Prados, M.D.; Wen, P.Y.; Mikkelsen, T.; Schiff, D.; Abrey, L.E.; Yung, W.K.; Paleologos, N.; Nicholas, M.K.; Jensen, R.; et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J. Clin. Oncol. 2009, 27, 4733–4740. [Google Scholar] [CrossRef]
- Chamberlain, M.C. Radiographic patterns of relapse in glioblastoma. J. Neurooncol. 2011, 101, 319–323. [Google Scholar] [CrossRef]
- Lucio-Eterovic, A.K.; Piao, Y.; de Groot, J.F. Mediators of glioblastoma resistance and invasion during antivascular endothelial growth factor therapy. Clin. Cancer Res. 2009, 15, 4589–4599. [Google Scholar] [CrossRef] [PubMed]
- Fischer, I.; Cunliffe, C.H.; Bollo, R.J.; Raza, S.; Monoky, D.; Chiriboga, L.; Parker, E.C.; Golfinos, J.G.; Kelly, P.J.; Knopp, E.A.; et al. High-grade glioma before and after treatment with radiation and Avastin: Initial observations. Neuro Oncol. 2008, 10, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.D.; Aghi, M.K. Mechanisms of evasion to antiangiogenic therapy in glioblastoma. Clin. Neurosurg. 2010, 57, 123–128. [Google Scholar]
- Nowosielski, M.; Wiestler, B.; Goebel, G.; Hutterer, M.; Schlemmer, H.P.; Stockhammer, G.; Wick, W.; Bendszus, M.; Radbruch, A. Progression types after antiangiogenic therapy are related to outcome in recurrent glioblastoma. Neurology 2014, 82, 1684–1692. [Google Scholar] [CrossRef]
- Brandsma, D.; van den Bent, M.J. Pseudoprogression and pseudoresponse in the treatment of gliomas. Curr. Opin. Neurol. 2009, 22, 633–638. [Google Scholar] [CrossRef]
- Nowosielski, M.; Wen, P.Y. Imaging Criteria in Neuro-oncology. Semin. Neurol. 2018, 38, 24–31. [Google Scholar] [CrossRef]
- Gatto, L.; Franceschi, E.; Tosoni, A.; Di Nunno, V.; Maggio, I.; Tonon, C.; Lodi, R.; Agati, R.; Bartolini, S.; Brandes, A.A. Distinct MRI pattern of “pseudoresponse” in recurrent glioblastoma multiforme treated with regorafenib: Case report and literature review. Clin. Case Rep. 2021, 9, e04604. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID Gene | Primer Forward | Primer Reverse |
|---|---|---|
| TBP | TGCCCGAAACGCCGAATATAATC | TGGTTCGTGGCTCTCTTATCCTC |
| VEGFA | CCTTGCTGCTCTACCTCCAC | CAACTTCGTGATGATTCTGC |
| Id1 | TGAACGGCTGTTACTCACG | CAACTGAAGGTCCCTGATG |
| Id3 | GAGAGGCACTCAGCTTAGCC | TCCTTTTGTCGTTGGAGATGAC |
| SERPINE1 | GCAAGGCACCTCTGAGAACT | TCACCAAAGACAAGGGCCAG |
| SnaI1 | AAGCCTAACTACAGCGAGCT | GAGTCCCAGATGAGCATTGG |
| SnaI2 | AGCATTTCAACGCCTCCAAA | TGGTTGTGGTATGACAGGCA |
| TEM7 | GTCAAAACCGGCCTATCGGA | GATGCTCCTTCGCCGAGATT |
| TWIST | AGTCTTACGAGGAGCTGCAG | ATCTTGCTCAGCTTGTCCGA |
| GSC# | Age/Sex | Tumor Location | Histology (WHO Grade) | Molecular Profile | PFS */OS (mos) |
|---|---|---|---|---|---|
| 1 | 40/M | Right frontal | Glioblastoma (IV) | MGMT M, IDH wt, EGFRvIII neg, VEGF hyper | 6.5/12.5 |
| 61 | 59/M | Occipital | Glioblastoma (IV) | MGMT UM, IDH wt, EGFRvIII pos, VEGF normal | 3/6 |
| 83 | 52/M | Temporal | Glioblastoma (IV) | MGMT UM, IDH wt, EGFRvIII pos, VEGF hyper | 3/8 |
| 366 | 54/M | Temporal | Glioblastoma (IV) | MGMT UM, IDH wt, EGFRvIII neg, VEGF hyper | 42/60.5 |
| 450 | 76/M | Temporal | Glioblastoma (IV) | MGMT M, IDH wt, EGFRvIII neg, VEGF hyper | 3/6 |
| 493 | 49/F | Parietal | Glioblastoma (IV) | MGMT M, IDH wt, EGFRvIII pos, VEGF hyper | 11/22.5 |
| Cell Line | IC50 (μM) |
|---|---|
| A172 | 2.4 |
| U87 | 6.3 |
| GSC#1 | 4.7 |
| GSC#61 | 6.2 |
| GSC#83 | 5.4 |
| GSC#450 | 3.3 |
| GSC#366 | 6.2 |
| GSC#439 | 3.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mongiardi, M.P.; Buccarelli, M.; Formato, A.; Orecchini, E.; Salbini, M.; Ricci, V.; Orsini, T.; Putti, S.; Chiesa, S.; Ricci-Vitiani, L.; et al. Characterization of Glioblastoma Cells Response to Regorafenib. Cancers 2022, 14, 6193. https://doi.org/10.3390/cancers14246193
Mongiardi MP, Buccarelli M, Formato A, Orecchini E, Salbini M, Ricci V, Orsini T, Putti S, Chiesa S, Ricci-Vitiani L, et al. Characterization of Glioblastoma Cells Response to Regorafenib. Cancers. 2022; 14(24):6193. https://doi.org/10.3390/cancers14246193
Chicago/Turabian StyleMongiardi, Maria Patrizia, Mariachiara Buccarelli, Alessia Formato, Elisa Orecchini, Maria Salbini, Valentina Ricci, Tiziana Orsini, Sabrina Putti, Silvia Chiesa, Lucia Ricci-Vitiani, and et al. 2022. "Characterization of Glioblastoma Cells Response to Regorafenib" Cancers 14, no. 24: 6193. https://doi.org/10.3390/cancers14246193
APA StyleMongiardi, M. P., Buccarelli, M., Formato, A., Orecchini, E., Salbini, M., Ricci, V., Orsini, T., Putti, S., Chiesa, S., Ricci-Vitiani, L., D’Alessandris, Q. G., Pallini, R., Levi, A., & Falchetti, M. L. (2022). Characterization of Glioblastoma Cells Response to Regorafenib. Cancers, 14(24), 6193. https://doi.org/10.3390/cancers14246193