
Next Steps for Immunotherapy in Glioblastoma

 , ,
, ,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Established Immunotherapeutic Approaches: Enhancing T-Cell Activities
2.1. Vaccine Therapies
2.1.1. Peptide Vaccines
2.1.2. Autologous Vaccines
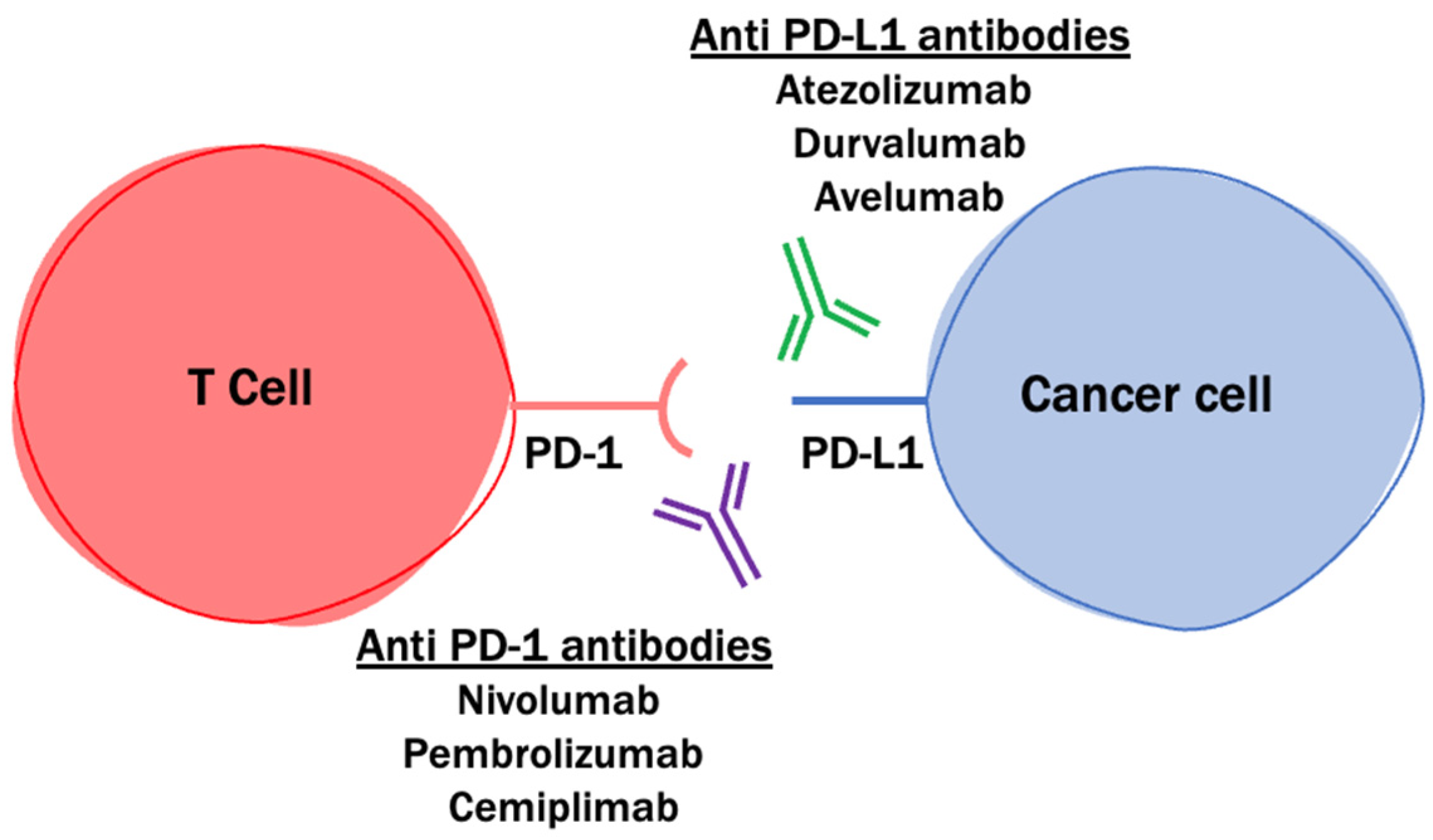
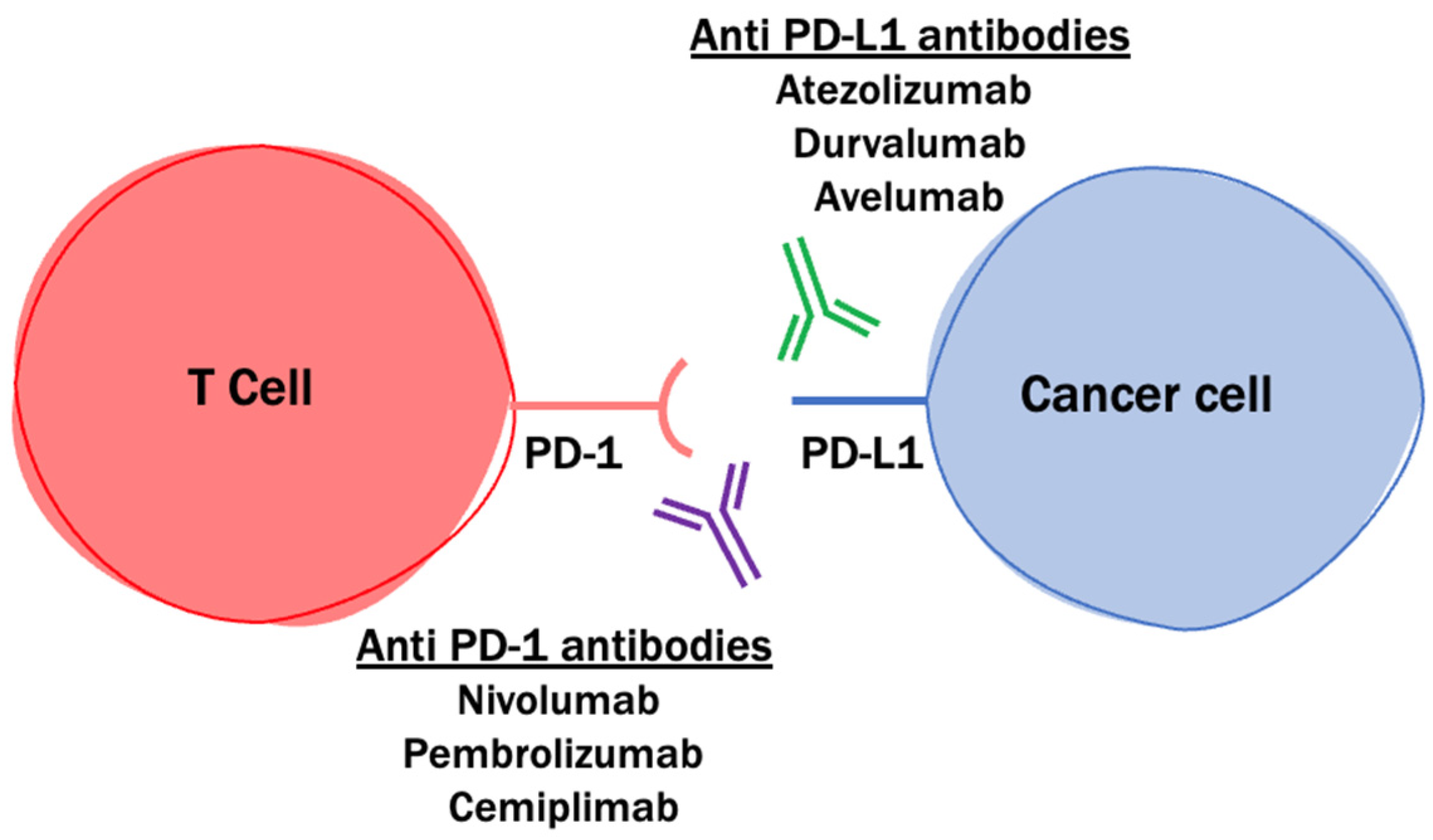
2.2. PD-1 and PD-L1 Blockade
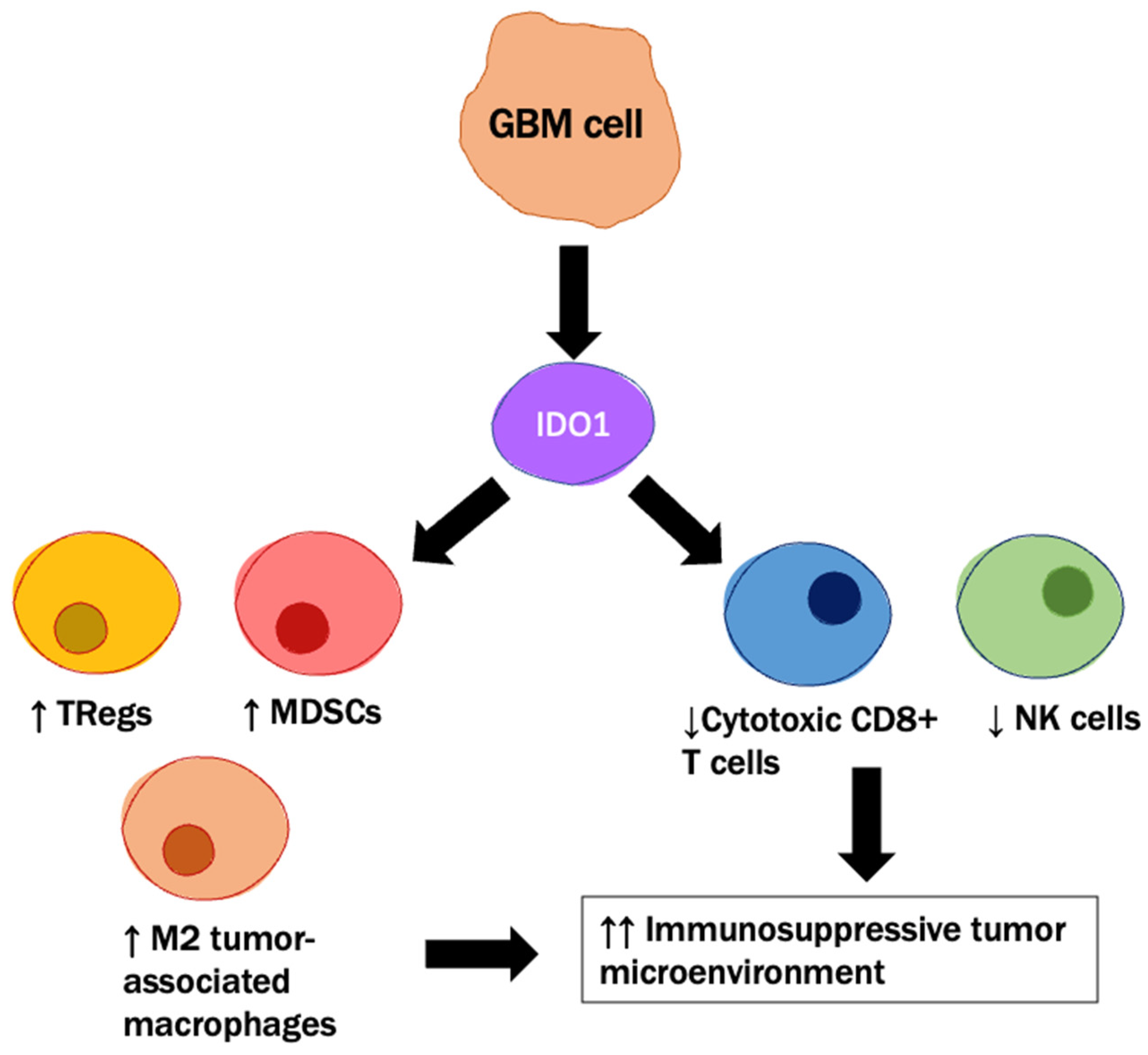
2.3. Targeting Regulatory T Cells and the Associated Immune Suppression Axis
2.4. T-Cell Activators
3. Clinical Factors That Prevent Immunotherapeutic Efficacy in GBM
4. Predictive Biomarkers of Immunotherapeutic Efficacy
4.1. PD-L1 Expression
4.2. Tumor Mutational Burden
4.3. PTEN Mutations
4.4. MAPK Pathway Aberrancies
4.5. Replication Stress Response

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Predicted Response to Checkpoint Blockade | Target | Authors | Primary Results | PMID |
|---|---|---|---|---|
| No effect | PD-L1 expression | Hodges et al. | There is no association between tumor mutational load and PD-1/PD-L1 expression (p = 0.7699, p = 0.8237) | 28371827 [123] |
| No effect/negative response | Tumor mutational burden | McGrail et al. | Gliomas with high TMB had a low ORR (15.3%, 95% CI 92–23.4). ICB-treated glioma patients had worse OS vs. those treated with other modalities (p = 0.23 × 10−5) | 33736924 [125] |
| Touat et al. | Patients with hypermutated gliomas treated with PD-1 blockade had similar PFS and OS vs. those with non-hypermutated gliomas (1.38 vs. 1.87 mo, 8.7 vs. 9.96 mo). Patients with hypermutated gliomas had shorter mOS with PD-1 blockade vs. other treatments (8.07 vs. 16.10 mo, p = 0.02) | 32322066 [127] | ||
| Negative response | PTEN mutations | Zhao et al. | PTEN mutations occur more frequently in non-responders to PD-1 blockade vs. responders (p = 0.0063, OR = 8.5) | 30742119 [126] |
| Improved response | MAPK pathway mutations | Zhao et al. | MAPK pathway alterations (PTPN11, BRAF) are enriched in responders to PD-1 inhibitors (Fisher p = 0.019, OR = 12.8) | 30742119 [126] |
| Arrieta et al. | ERK1/2 activation in recurrent GBM predicts OS after PD1 blockade (HR = 0.18, 95% CI 0.06–0.56) | 35121903 [120] | ||
| Replication stress response defects (RSRD) | McGrail et al. | RSR defects lead to immunostimulatory cytosolic ssDNA and improved ICB response (p = 0.00019 in breast cancer model, p < 0.1 × 10−6 in GBM cohort) | 34705519 [139] |
5. Making Things Hot: Promoting T-Cell Infiltration into GBM
5.1. Oncolytic Viral Therapies
5.2. STING Agonism
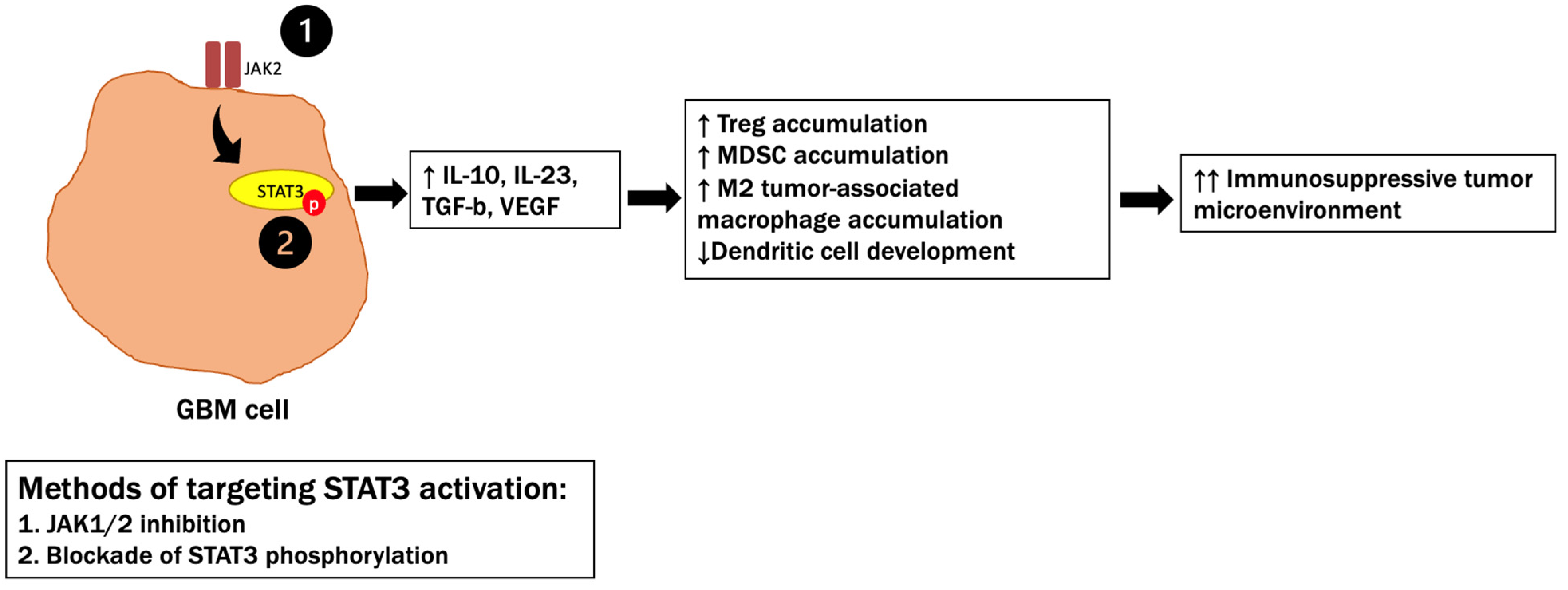
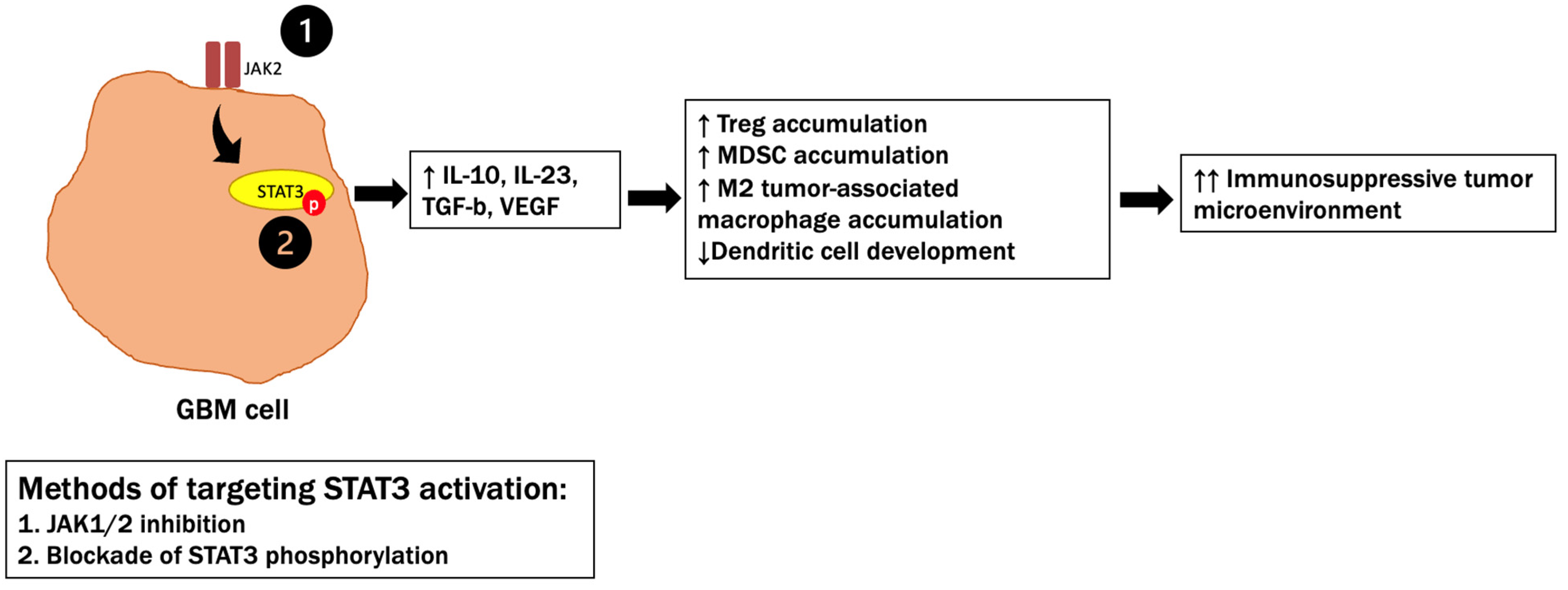
5.3. STAT3 Signaling
5.4. Cytokine Therapies
5.5. Disrupting the Blood–Brain Barrier to Enhance Immunotherapy
6. Harnessing the Innate Immune System for GBM Immunotherapy
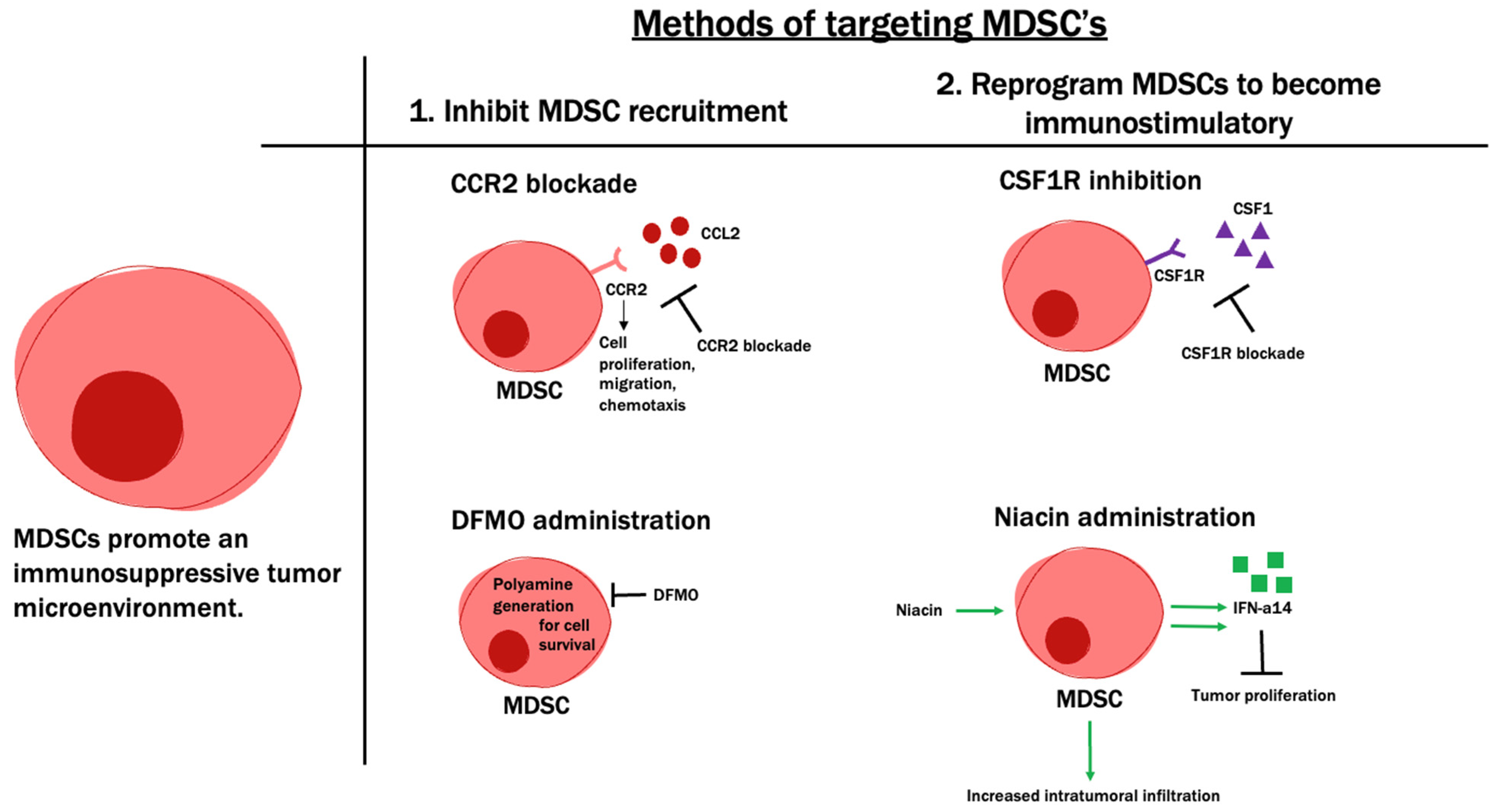
6.1. Tumor-Infiltrating Myeloid Cells
6.2. NK Cells
6.3. Gamma Delta T Cells
6.4. The Role of B Cells
7. Finding the Balance
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lukas, R.V.; Wainwright, D.; Ladomersky, E.; Sachdev, S.; Sonabend, A.M.; Stupp, R. Newly Diagnosed Glioblastoma: A Review on Clinical Management. Oncology 2019, 33, 91. [Google Scholar] [PubMed]
- Lukas, R.V.; Mrugala, M.M. Pivotal therapeutic trials for infiltrating gliomas and how they affect clinical practice. Neuro-Oncol. Pr. 2016, 4, 209–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Lim, M.; Weller, M.; Idbaih, A.; Steinbach, J.; Finocchiaro, G.; Raval, R.R.; Ansstas, G.; Baehring, J.; Taylor, J.W.; Honnorat, J.; et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro-Oncology 2022. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; Brandes, A.; Carpentier, A.F.; Idbaih, A.; Reardon, D.; Cloughesy, T.; Sumrall, A.; Baehring, J.; Bent, M.V.D.; Bähr, O.; et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: An international randomized phase III trial. Neuro-Oncology 2022. [Google Scholar] [CrossRef]
- Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; O’Rourke, D.M.; Tran, D.D.; Fink, K.L.; Nabors, L.B.; Li, G.; Bota, D.A.; Lukas, R.V.; et al. Rindopepimut with bevacizumab for patients with relapsed EGFRvIII-expressing glioblastoma (ReACT): Results of a double-blind randomized phase II trial. Clin. Cancer Res. 2020, 26, 1586–1594. [Google Scholar] [CrossRef]
- Ayasoufi, K.; Pfaller, C.K.; Evgin, L.; Khadka, R.H.; Tritz, Z.P.; Goddery, E.N.; Fain, C.; Yokanovich, L.T.; Himes, B.T.; Jin, F.; et al. Brain cancer induces systemic immunosuppression through release of non-steroid soluble mediators. Brain 2020, 143, 3629–3652. [Google Scholar] [CrossRef]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; DeChant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef]
- Bloch, O.; Crane, C.A.; Kaur, R.; Safaee, M.; Rutkowski, M.J.; Parsa, A.T. Gliomas Promote Immunosuppression through Induction of B7-H1 Expression in Tumor-Associated Macrophages. Clin. Cancer Res. 2013, 19, 3165–3175. [Google Scholar] [CrossRef] [Green Version]
- Ullén, H.; Blom, U.; Blomgren, H.; von Holst, H. Blood lymphocyte subsets in patients with primary intracranial tumours. Correlation to histological tumour type and anatomical site. Acta Neurochir. 1986, 81, 100–105. [Google Scholar] [CrossRef]
- Bodmer, S.; Strommer, K.; Frei, K.; Siepl, C.; De Tribolet, N.; Heid, I.; Fontana, A. Immunosuppression and transforming growth factor-beta in glioblastoma. Preferential production of transforming growth factor-beta 2. J. Immunol. 1989, 143, 3222–3229. [Google Scholar] [PubMed]
- Dix, A.R.; Brooks, W.H.; Roszman, T.L.; Morford, L. Immune defects observed in patients with primary malignant brain tumors. J. Neuroimmunol. 1999, 100, 216–232. [Google Scholar] [CrossRef]
- Land, C.A.; Musich, P.R.; Haydar, D.; Krenciute, G.; Xie, Q. Chimeric antigen receptor T-cell therapy in glioblastoma: Charging the T cells to fight. J. Transl. Med. 2020, 18, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sayegh, E.T.; Oh, T.; Fakurnejad, S.; Bloch, O.; Parsa, A.T. Vaccine therapies for patients with glioblastoma. J. Neuro-Oncol. 2014, 119, 531–546. [Google Scholar] [CrossRef] [PubMed]
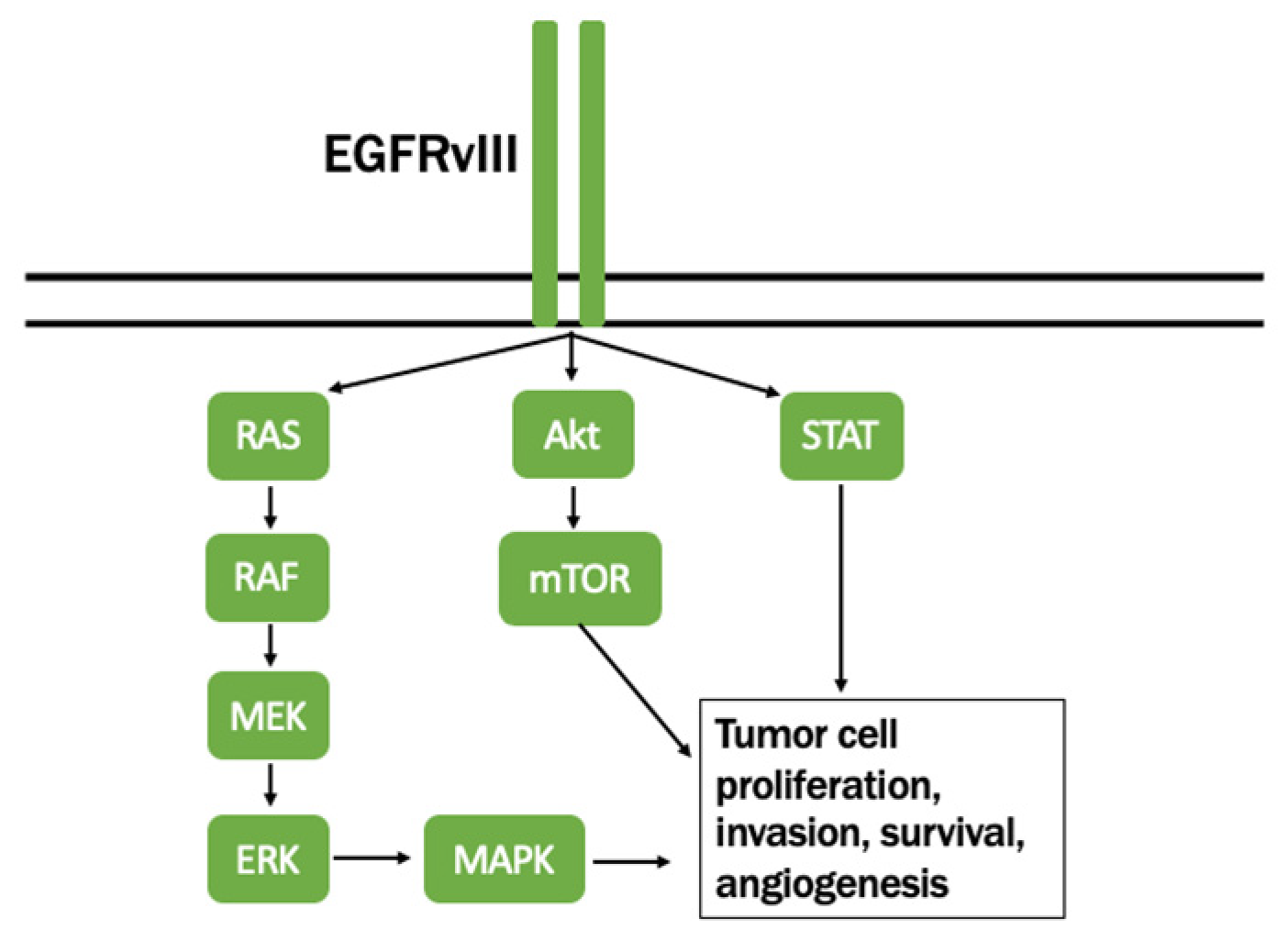
- Batra, S.K.; Castelino-Prabhu, S.; Wikstrand, C.J.; Zhu, X.; Humphrey, P.A.; Friedman, H.S.; Bigner, D.D. Epidermal growth factor ligand-independent, unregulated, cell- transforming potential of a naturally occurring human mutant EGFRvIII gene. Cell Growth Differ. 1995, 6, 1251–1259. [Google Scholar]
- Lal, A.; Glazer, C.; Martinson, H.; Friedman, H.S.; Archer, G.; Sampson, J.H.; Riggins, G.J. Mutant epidermal growth factor receptor up-regulates molecular effectors of tumor invasion. Cancer Res. 2002, 62, 3335–3339. [Google Scholar]
- Nagane, M.; Coufal, F.; Lin, H.; Bogler, O.; Cavenee, W.K.; Huang, H.J. A common mutant epidermal growth factor receptor confers enhanced tumorigenicity on human glioblastoma cells by increasing proliferation and reducing apoptosis. Cancer Res. 1996, 56, 5079–5086. [Google Scholar]
- Montgomery, R.B.; Guzman, J.; O’Rourke, D.M.; Stahl, W.L. Expression of onco- genic epidermal growth factor receptor family kinases induces paclitaxel resistance and alters beta-tubulin isotype expression. J. Biol. Chem. 2000, 275, 17358–17363. [Google Scholar] [CrossRef] [Green Version]
- Lammering, G.; Valerie, K.; Lin, P.S.; Hewit, T.H.; Schmidt-Ullrich, R.K. Radiation- induced activation of a common variant of EGFR confers enhanced radio- resistance. Radiother. Oncol. 2004, 72, 267–273. [Google Scholar] [CrossRef]
- Huang, H.-J.S.; Nagane, M.; Klingbeil, C.K.; Lin, H.; Nishikawa, R.; Ji, X.-D.; Huang, C.-M.; Gill, G.N.; Wiley, H.S.; Cavenee, W.K. The Enhanced Tumorigenic Activity of a Mutant Epidermal Growth Factor Receptor Common in Human Cancers Is Mediated by Threshold Levels of Constitutive Tyrosine Phosphorylation and Unattenuated Signaling. J. Biol. Chem. 1997, 272, 2927–2935. [Google Scholar] [CrossRef] [Green Version]
- Heimberger, A.B.; Crotty, L.; Archer, G.; Hess, K.R.; Wikstrand, C.J.; Friedman, A.H.; Friedman, H.S.; Bigner, D.D.; Sampson, J.H. Epidermal growth factor receptor VIII peptide vaccination is efficacious against established intracerebral tumors. Clin. Cancer Res. 2003, 9, 4247–4254. [Google Scholar] [PubMed]
- Heimberger, A.B.; Hlatky, R.; Suki, D.; Yang, D.; Weinberg, J.; Gilbert, M.; Sawaya, R.; Aldape, K. Prognostic Effect of Epidermal Growth Factor Receptor and EGFRvIII in Glioblastoma Multiforme Patients. Clin. Cancer Res. 2005, 11, 1462–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonabend, A.M.; Dana, K.; Lesniak, M.S. Targeting epidermal growth factor receptor variant III: A novel strategy for the therapy of malignant glioma. Expert Rev. Anticancer Ther. 2007, 7, S45–S50. [Google Scholar] [CrossRef]
- Sampson, J.H.; Archer, G.E.; Mitchell, D.A.; Heimberger, A.B.; Herndon, J.E.; Lally-Goss, D.; McGehee-Norman, S.; Paolino, A.; Reardon, D.A.; Friedman, A.H.; et al. An epidermal growth factor receptor variant III–targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol. Cancer Ther. 2009, 8, 2773–2779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampson, J.H.; Heimberger, A.B.; Archer, G.E.; Aldape, K.D.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Ii, J.E.H.; McLendon, R.E.; Mitchell, D.A.; et al. Immunologic Escape After Prolonged Progression-Free Survival With Epidermal Growth Factor Receptor Variant III Peptide Vaccination in Patients With Newly Diagnosed Glioblastoma. J. Clin. Oncol. 2010, 28, 4722–4729. [Google Scholar] [CrossRef]
- Schuster, J.; Lai, R.K.; Recht, L.D.; Reardon, D.A.; Paleologos, N.A.; Groves, M.D.; Mrugala, M.M.; Jensen, R.; Baehring, J.M.; Sloan, A.; et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: The ACT III study. Neuro-Oncology 2015, 17, 854–861. [Google Scholar] [CrossRef] [Green Version]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Sampson, J.H.; Aldape, K.D.; Archer, G.E.; Coan, A.; Desjardins, A.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Herndon, J.E.; McLendon, R.E.; et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro-Oncology 2011, 13, 324–333. [Google Scholar] [CrossRef] [Green Version]
- van den Bent, M.J.; Gao, Y.; Kerkhof, M.; Kros, J.M.; Gorlia, T.; Van Zwieten, K.; Prince, J.; Van Duinen, S.; Sillevis Smitt, P.A.; Taphoorn, M.; et al. Changes in the EGFR amplification and EGFRvIII expression between paired primary and recurrent glioblastomas. Neuro-Oncology 2015, 17, 935–941. [Google Scholar] [CrossRef] [Green Version]
- Nobusawa, S.; Watanabe, T.; Kleihues, P.; Ohgaki, H. IDH1 Mutations as Molecular Signature and Predictive Factor of Secondary Glioblastomas. Clin. Cancer Res. 2009, 15, 6002–6007. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Liu, Y.; Cai, S.J.; Qian, M.; Ding, J.; Larion, M.; Gilbert, M.R.; Yang, C. IDH mutation in glioma: Molecular mechanisms and potential therapeutic targets. Br. J. Cancer 2020, 122, 1580–1589. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Schilling, D.; Bunse, L.; Wick, A.; Bunse, T.; Riehl, D.; Karapanagiotou-Schenkel, I.; Harting, I.; Sahm, F.; Schmitt, A.; et al. A mutation-specific peptide vaccine targeting IDH1R132H in patients with newly diagnosed malignant astrocytomas: A first-in-man multicenter phase I clinical trial of the German Neurooncology Working Group (NOA-16). J. Clin. Oncol. 2018, 36, 2001. [Google Scholar] [CrossRef]
- Kijima, N.; Hosen, N.; Kagawa, N.; Hashimoto, N.; Kinoshita, M.; Oji, Y.; Sugiyama, H.; Yoshimine, T. Wilms’ tumor 1 is involved in tumorigenicity of glioblastoma by regulating cell proliferation and apoptosis. Anticancer Res. 2014, 34, 61–67. [Google Scholar]
- Izumoto, S.; Tsuboi, A.; Oka, Y.; Suzuki, T.; Hashiba, T.; Kagawa, N.; Hashimoto, N.; Maruno, M.; Elisseeva, O.A.; Shirakata, T.; et al. Phase II clinical trial of Wilms tumor 1 peptide vaccination for patients with recurrent glioblastoma multiforme. J. Neurosurg. 2008, 108, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Oji, Y.; Hashimoto, N.; Tsuboi, A.; Murakami, Y.; Iwai, M.; Kagawa, N.; Chiba, Y.; Izumoto, S.; Elisseeva, O.; Ichinohasama, R.; et al. Association of WT1 IgG antibody against WT1 peptide with prolonged survival in glioblastoma multiforme patients vaccinated with WT1 peptide. Int. J. Cancer 2016, 139, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Takashima, S.; Oka, Y.; Fujiki, F.; Morimoto, S.; Nakajima, H.; Nakae, Y.; Nakata, J.; Nishida, S.; Hosen, N.; Tatsumi, N.; et al. Syndecan-4 as a biomarker to predict clinical outcome for glioblastoma multiforme treated with WT1 peptide vaccine. Futur. Sci. OA 2016, 2, FSO96. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.; Piccioni, D.E.; Liu, H.; Lukas, R.V.; Kesari, S.; Aregawi, D.; Hong, D.S.; Yamaguchi, K.; Whicher, K.; Zhang, Y.; et al. A phase I study of the WT2725 dosing emulsion in patients with advanced malignancies. Sci. Rep. 2021, 11, 22355. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, Y.; Udaka, K.; Yawata, T.; Nakai, E.; Fukuda, H.; Fukui, N.; Ueba, T. Imt-03 Clinical Trial For Newly Diagnosed Malignant Glioma With Wt1-W10 Vaccination. Neuro-Oncology Adv. 2019, 1, ii17. [Google Scholar] [CrossRef]
- Cobbs, C.S.; Harkins, L.; Samanta, M.; Gillespie, G.Y.; Bharara, S.; King, P.H.; Nabors, L.B.; Cobbs, C.G.; Britt, W.J. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res. 2002, 62, 3347–3350. [Google Scholar]
- Rahman, M.; Dastmalchi, F.; Karachi, A.; Mitchell, D. The role of CMV in glioblastoma and implications for immunotherapeutic strategies. OncoImmunology 2018, 8, e1514921. [Google Scholar] [CrossRef]
- Lucas, K.G.; Bao, L.; Bruggeman, R.; Dunham, K.; Specht, C. The detection of CMV pp65 and IE1 in glioblastoma multiforme. J. Neuro-Oncol. 2010, 103, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Batich, K.A.; Reap, E.A.; Archer, G.E.; Sanchez-Perez, L.; Nair, S.K.; Schmittling, R.J.; Norberg, P.; Xie, W.; Herndon, J.E., II; Healy, P.; et al. Long-term Survival in Glioblastoma with Cytomegalovirus pp65-Targeted Vaccination. Clin. Cancer Res. 2017, 23, 1898–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weathers, S.-P.S.; Penas-Prado, M.; Banerjee, P.P.; Bdiwi, M.; Shaim, H.; Alsuliman, A.; Shanley, M.; Long, J.; De Groot, J.F.; O’Brien, B.J.; et al. A phase I/II clinical trial of autologous CMV-specific T cells in glioblastoma (GBM) patients to reveal a lack of immune effector function. J. Clin. Oncol. 2020, 38, 2515. [Google Scholar] [CrossRef]
- Smith, C.; Lineburg, K.E.; Martins, J.P.; Ambalathingal, G.R.; Neller, M.A.; Morrison, B.; Matthews, K.K.; Rehan, S.; Crooks, P.; Panikkar, A.; et al. Autologous CMV-specific T cells are a safe adjuvant immunotherapy for primary glioblastoma multiforme. J. Clin. Investig. 2020, 130, 6041–6053. [Google Scholar] [CrossRef] [PubMed]
- Batich, K.A.; Mitchell, D.A.; Healy, P.; Herndon, J.E., 2nd; Sampson, J.H. Once, Twice, Three Times a Finding: Reproducibility of Dendritic Cell Vaccine Trials Targeting Cytomegalovirus in Glioblastoma. Clin. Cancer Res. 2020, 26, 5297–5303. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.H.; Batich, K.; Mitchell, D.A.; Herndon, J.E.; Broadwater, G.; Healy, P.; Sanchez-Perez, L.; Nair, S.; Congdon, K.; Norberg, P.; et al. Reproducibility of outcomes in sequential trials using CMV-targeted dendritic cell vaccination for glioblastoma. J. Clin. Oncol. 2022, 40, 2005. [Google Scholar] [CrossRef]
- Mitchell, D.A.; Batich, K.A.; Gunn, M.D.; Huang, M.-N.; Sanchez-Perez, L.; Nair, S.K.; Congdon, K.L.; Reap, E.A.; Archer, G.E.; Desjardins, A.; et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 2015, 519, 366–369. [Google Scholar] [CrossRef]
- Garg, H.; Suri, P.; Gupta, J.C.; Talwar, G.P.; Dubey, S. Survivin: A unique target for tumor therapy. Cancer Cell Int. 2016, 16, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Fenstermaker, R.A.; Ciesielski, M.J.; Qiu, J.; Yang, N.; Frank, C.L.; Lee, K.P.; Mechtler, L.R.; Belal, A.; Ahluwalia, M.S.; Hutson, A.D. Clinical study of a survivin long peptide vaccine (SurVaxM) in patients with recurrent malignant glioma. Cancer Immunol. Immunother. 2016, 65, 1339–1352. [Google Scholar] [CrossRef] [Green Version]
- Ahluwalia, M.; Peereboom, D.; Rauf, Y.; Schilero, C.; Ciolfi, M.; Ciesielski, M.; Fenstermaker, R. Phase II study of pembrolizumab plus SurVaxM for glioblastoma at first recurrence. Neuro-Oncology 2020, 22, ii34–ii35. [Google Scholar] [CrossRef]
- Ciesielski, M.J.; Ahluwalia, M.S.; Reardon, D.A.; Abad, A.P.; Curry, W.T.; Wong, E.T.; Peereboom, D.M.; Figel, S.A.; Hutson, A.; Groman, A.; et al. Final data from the phase 2a single-arm trial of SurVaxM for newly diagnosed glioblastoma. J. Clin. Oncol. 2022, 40, 2037. [Google Scholar] [CrossRef]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A., Jr.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Koh, J.; Kim, S.-I.; Won, J.K.; Park, C.-K.; Choi, S.H.; Park, S.-H. The frequency and prognostic effect of TERT promoter mutation in diffuse gliomas. Acta Neuropathol. Commun. 2017, 5, 1–11. [Google Scholar] [CrossRef]
- Arakawa, Y.; Yoshiko, O.; Yoshitaka, N. Efficacy finding cohort of a cancer peptide vaccine, TAS0313, in treating recurrent glioblastoma. J. Clin. Oncol. 2021, 39, 2038. [Google Scholar] [CrossRef]
- Wen, P.Y.; Reardon, D.A.; Armstrong, T.S.; Phuphanich, S.; Aiken, R.D.; Landolfi, J.C.; Curry, W.T.; Zhu, J.J.; Glantz, M.; Peereboom, D.M.; et al. A Randomized Double-Blind Placebo-Controlled Phase II Trial of Dendritic Cell Vaccine ICT-107 in Newly Diagnosed Patients with GlioblastomaICT-107 Vaccine for Newly Diagnosed Glioblastoma. Clin. Cancer Res. 2019, 25, 5799–5807. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Idbaih, A.; Tabatabai, G.; Vieito, M.; Stradella, A.; Ghiringhelli, F.; Burger, M.C.; Mildenberger, I.; Herrlinger, U.; Renovanz, M.; et al. EO2401, a novel microbiome-derived therapeutic vaccine for patients with recurrent glioblastoma: ROSALIE study. J. Clin. Oncol. 2022, 40, 2034. [Google Scholar] [CrossRef]
- Schumacher, N.T.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.-R.; Hildebrand, W.H.; Mardis, E.R.; et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef] [Green Version]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.-P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2018, 565, 234–239. [Google Scholar] [CrossRef]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First results on survival from a large phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.L.; Omofoye, O.A.; Rudnick, J.D.; Kim, S.; Tighiouart, M.; Phuphanich, S.; Wang, H.; Mazer, M.; Ganaway, T.; Chu, R.M.; et al. A Phase I Study of Autologous Dendritic Cell Vaccine Pulsed with Allogeneic Stem-like Cell Line Lysate in Patients with Newly Diagnosed or Recurrent GlioblastomaA Study of Dendritic Cell Vaccine in Patients with Glioblastoma. Clin. Cancer Res. 2022, 28, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, M.; Seiji, T.; Yoshinobu, T. Heat shock protein derivatives for delivery of antigens to antigen presenting cells. Int. J. Pharm. 2008, 354, 23–27. [Google Scholar] [CrossRef]
- Bloch, O.; Crane, C.A.; Fuks, Y.; Kaur, R.; Aghi, M.K.; Berger, M.S.; Butowski, N.A.; Chang, S.M.; Clarke, J.L.; McDermott, M.W.; et al. Heat-shock protein peptide complex–96 vaccination for recurrent glioblastoma: A phase II, single-arm trial. Neuro-Oncology 2014, 16, 274–279. [Google Scholar] [CrossRef]
- Bloch, O.; Shi, Q.; Anderson, S.K.; Knopp, M.; Raizer, J.; Clarke, J.; Waziri, A.; Colman, H.; Bruce, J.; Olson, J.J.; et al. ATIM-14. Alliance A071101: A phase II randomized trial comparing the efficacy of heat shock protein peptide complex-96 (HSPPC-96) vaccine given with bevacizumab versus bevacizumab alone in the treatment of surgically resectable recurrent glioblastoma. Neuro-Oncology 2017, 19 (Suppl. 6), vi29. [Google Scholar] [CrossRef] [Green Version]
- Galanis, E.; Wu, W.; Cloughesy, T.; Lamborn, K.; Mann, B.; Wen, P.Y.; Reardon, D.A.; Wick, W.; Macdonald, D.; Armstrong, T.S.; et al. Phase 2 trial design in neuro-oncology revisited: A report from the RANO group. Lancet Oncol. 2012, 13, e196–e204. [Google Scholar] [CrossRef]
- Grossman, S.A.; Schreck, K.; Ballman, K.; Alexander, B. Point/counterpoint: Randomized versus single-arm phase II clinical trials for patients with newly diagnosed glioblastoma. Neuro-Oncology 2017, 19, 469–474. [Google Scholar] [CrossRef] [Green Version]
- Sheikh, S.; Radivoyevitch, T.; Barnholtz-Sloan, J.S.; Vogelbaum, M. Long-term trends in glioblastoma survival: Implications for historical control groups in clinical trials. Neuro-Oncol. Pr. 2019. [Google Scholar] [CrossRef]
- Liu, Y.; Carlsson, R.; Ambjørn, M.; Hasan, M.; Badn, W.; Darabi, A.; Siesjö, P.; Issazadeh-Navikas, S. PD-L1 Expression by Neurons Nearby Tumors Indicates Better Prognosis in Glioblastoma Patients. J. Neurosci. 2013, 33, 14231–14245. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immuno-therapy. Nat. Rev. Cancer. 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binder, D.C.; Davis, A.A.; Wainwright, D.A. Immunotherapy for cancer in the central nervous system: Current and future directions. OncoImmunology 2015, 5, e1082027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: The CheckMate 143 phase 3 randomized trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Duerinck, J.; Schwarze, J.K.; Awada, G.; Tijtgat, J.; Vaeyens, F.; Bertels, C.; Geens, W.; Klein, S.; Seynaeve, L.; Cras, L.; et al. Intracerebral administration of CTLA-4 and PD-1 immune checkpoint blocking monoclonal antibodies in patients with recurrent glioblastoma: A phase I clinical trial. J. Immunother. Cancer 2021, 9, e002296. [Google Scholar] [CrossRef]
- Reardon, D.A.; Kim, T.M.; Frenel, J.; Simonelli, M.; Lopez, J.; Subramaniam, D.S.; Siu, L.L.; Wang, H.; Krishnan, S.; Stein, K.; et al. Treatment with pembrolizumab in programmed death ligand 1–positive recurrent glioblastoma: Results from the multicohort phase 1 KEYNOTE-028 trial. Cancer 2021, 127, 1620–1629. [Google Scholar] [CrossRef]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef]
- Allen, E.; Jabouille, A.; Rivera, L.B.; Lodewijckx, I.; Missiaen, R.; Steri, V.; Feyen, K.; Tawney, J.; Hanahan, D.; Michael, I.P.; et al. Combined antiangiogenic and anti–PD-L1 therapy stimulates tumor immunity through HEV formation. Sci. Transl. Med. 2017, 9, eaak9679. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Yuan, J.; Righi, E.; Kamoun, W.S.; Ancukiewicz, M.; Nezivar, J.; Santosuosso, M.; Martin, J.D.; Martin, M.R.; Vianello, F.; et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 17561–17566. [Google Scholar] [CrossRef] [Green Version]
- Shrimali, R.K.; Yu, Z.; Theoret, M.R.; Chinnasamy, D.; Restifo, N.P.; Rosenberg, S.A. Antiangiogenic Agents Can Increase Lymphocyte Infiltration into Tumor and Enhance the Effectiveness of Adoptive Immunotherapy of Cancer. Cancer Res. 2010, 70, 6171–6180. [Google Scholar] [CrossRef] [Green Version]
- Nayak, L.; Molinaro, A.M.; Peters, K.B.; Clarke, J.L.; Jordan, J.T.; de Groot, J.F.; Nghiemphu, P.L.; Kaley, T.J.; Colman, H.; McCluskey, C.; et al. Randomized Phase II and Biomarker Study of Pembrolizumab plus Bevacizumab versus Pembrolizumab Alone for Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2021, 27, 1048–1057. [Google Scholar] [CrossRef]
- Lukas, R.V.; Rodon, J.; Becker, K.; Wong, E.T.; Shih, K.; Touat, M.; Fassò, M.; Osborne, S.; Molinero, L.; O’Hear, C.; et al. Clinical activity and safety of atezolizumab in patients with recurrent glioblastoma. J. Neuro Oncol. 2018, 140, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Dettmer, S.; Berberich, A.; Kessler, T.; Karapanagiotou-Schenkel, I.; Wick, A.; Winkler, F.; Pfaff, E.; Brors, B.; Debus, J.; et al. N2M2 (NOA-20) phase I/II trial of molecularly matched targeted therapies plus radiotherapy in patients with newly diagnosed non-MGMT hypermethylated glioblastoma. Neuro-Oncology 2018, 21, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Weathers, S.-P.S.; Zhu, H.; Knafl, M.; Damania, A.; Kamiya-Matsuoka, C.; Harrison, R.A.; Lyons, L.; Yun, C.; Darbonne, W.C.; Loghin, M.; et al. Baseline tumor genomic and gut microbiota association with clinical outcomes in newly diagnosed glioblastoma (GBM) treated with atezolizumab in combination with temozolomide (TMZ) and radiation. J. Clin. Oncol. 2022, 40, 2006. [Google Scholar] [CrossRef]
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; Dechant, C.; Farber, S.H.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C.; et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin. Cancer Res. 2018, 24, 4175–4186. [Google Scholar] [CrossRef] [Green Version]
- Ott, M.; Tomaszowski, K.-H.; Marisetty, A.; Kong, L.-Y.; Wei, J.; Duna, M.; Blumberg, K.; Ji, X.; Jacobs, C.; Fuller, G.N.; et al. Profiling of patients with glioma reveals the dominant immunosuppressive axis is refractory to immune function restoration. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Nduom, E.K.; Wei, J.; Yaghi, N.K.; Huang, N.; Kong, L.-Y.; Gabrusiewicz, K.; Ling, X.; Zhou, S.; Ivan, C.; Chen, J.Q.; et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro-Oncology 2015, 18, 195–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, M.; Prins, R.M.; Heimberger, A.B. The immune landscape of common CNS malignancies: Implications for immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 729–744. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; López-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inogés, S.; De Andrea, C.; De Cerio, A.L.-D.; Tejada, S.; et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef]
- Topalian, S.L.; Taube, J.M.; Pardoll, D.M. Neoadjuvant checkpoint blockade for cancer immunotherapy. Science 2020, 367, 6477. [Google Scholar] [CrossRef]
- Amaria, R.N.; Reddy, S.; Tawbi, H.A.; Davies, M.A.; Ross, M.I.; Glitza, I.C.; Cormier, J.N.; Lewis, C.; Hwu, W.-J.; Hanna, E.; et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat. Med. 2018, 24, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- El Andaloussi, A.; Lesniak, M.S. An increase in CD4+CD25+FOXP3+ regulatory T cells in tumor-infiltrating lymphocytes of human glioblastoma multiforme1. Neuro-Oncology 2006, 8, 234–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, M.D.; Baban, B.; Chandler, P.; Hou, D.-Y.; Singh, N.; Yagita, H.; Azuma, M.; Blazar, B.R.; Mellor, A.L.; Munn, D.H. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J. Clin. Investig. 2007, 117, 2570–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, L.; Ladomersky, E.; Lauing, K.L.; Wu, M.; Genet, M.; Gritsina, G.; Győrffy, B.; Brastianos, P.K.; Binder, D.C.; Sosman, J.A.; et al. Infiltrating T Cells Increase IDO1 Expression in Glioblastoma and Contribute to Decreased Patient Survival. Clin. Cancer Res. 2017, 23, 6650–6660. [Google Scholar] [CrossRef] [Green Version]
- Wainwright, D.A.; Balyasnikova, I.V.; Chang, A.L.; Ahmed, A.U.; Moon, K.S.; Auffinger, B.; Tobias, A.L.; Han, Y.; Lesniak, M.S. IDO Expression in Brain Tumors Increases the Recruitment of Regulatory T Cells and Negatively Impacts SurvivalIDO Regulates Treg Infiltration in Brain Tumors. Clin. Cancer Res. 2012, 18, 6110–6121. [Google Scholar] [CrossRef] [Green Version]
- Ladomersky, E.; Zhai, L.; Lenzen, A.; Lauing, K.L.; Qian, J.; Scholtens, D.M.; Gritsina, G.; Sun, X.; Liu, Y.; Yu, F.; et al. IDO1 Inhibition Synergizes with Radiation and PD-1 Blockade to Durably Increase Survival Against Advanced Glioblastoma. Clin. Cancer Res. 2018, 24, 2559–2573. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Ladomersky, E.; Mozny, A.; Kocherginsky, M.; O’Shea, K.; Reinstein, Z.Z.; Zhai, L.; Bell, A.; Lauing, K.L.; Bollu, L.; et al. Glioblastoma as an age-related neurological disorder in adults. Neuro-Oncology Adv. 2021, 3. [Google Scholar] [CrossRef]
- Lukas, R.; Sachdev, S.; Kumthekar, P.; Dixit, K.; Grimm, S.; Gondi, V.; Sharp, L.; Lezon, R.; James, D.; Lesniak, M.; et al. Ctim-12. a phase 1 trial of immunoradiotherapy with the ido enzyme inhibitor (bms-986205) and nivolumab in patients with newly diagnosed mgmt promoter unmethylated idhwt glioblastoma. Neuro-Oncology 2021, 23 (Suppl. 6), vi51–vi52. [Google Scholar] [CrossRef]
- Miska, J.; Rashidi, A.; Chang, A.L.; Muroski, M.E.; Han, Y.; Zhang, L.; Lesniak, M.S. Anti-GITR therapy promotes immunity against malignant glioma in a murine model. Cancer Immunol. Immunother. 2016, 65, 1555–1567. [Google Scholar] [CrossRef]
- Patel, M.A.; Kim, J.E.; Theodros, D.; Tam, A.; Velarde, E.; Kochel, C.M.; Francica, B.; Nirschl, T.R.; Ghasemzadeh, A.; Mathios, D.; et al. Agonist anti-GITR monoclonal antibody and stereotactic radiation induce immune-mediated survival advantage in murine intracranial glioma. J. Immunother. Cancer 2016, 4, 28. [Google Scholar] [CrossRef] [Green Version]
- Amoozgar, Z.; Kloepper, J.; Ren, J.; Tay, R.E.; Kazer, S.W.; Kiner, E.; Krishnan, S.; Posada, J.M.; Ghosh, M.; Mamessier, E.; et al. Targeting Treg cells with GITR activation alleviates resistance to immunotherapy in murine glioblastomas. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Heimberger, A.B.; Abou-Ghazal, M.; Reina-Ortiz, C.; Yang, D.S.; Sun, W.; Qiao, W.; Hiraoka, N.; Fuller, G.N. Incidence and Prognostic Impact of FoxP3+ Regulatory T Cells in Human Gliomas. Clin. Cancer Res. 2008, 14, 5166–5172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, E.E.W.; Pishvaian, M.J.; Shepard, D.R.; Wang, D.; Weiss, J.; Johnson, M.L.; Chung, C.H.; Chen, Y.; Huang, B.; Davis, C.B.; et al. A phase Ib study of utomilumab (PF-05082566) in combination with mogamulizumab in patients with advanced solid tumors. J. Immunother. Cancer 2019, 7, 342. [Google Scholar] [CrossRef] [PubMed]
- Segal, N.H.; He, A.R.; Doi, T.; Levy, R.; Bhatia, S.; Pishvaian, M.J.; Cesari, R.; Chen, Y.; Davis, C.B.; Huang, B.; et al. Phase I Study of Single-Agent Utomilumab (PF-05082566), a 4-1BB/CD137 Agonist, in Patients with Advanced Cancer. Clin. Cancer Res. 2018, 24, 1816–1823. [Google Scholar] [CrossRef] [Green Version]
- Desjardins, A.; Chandramohan, V.; Landi, D.B.; Johnson, M.O.; Khasraw, M.; Peters, K.B.; Low, J.; Herndon, J.E.; Threatt, S.; Bullock, C.A.; et al. A phase 1 trial of D2C7-it in combination with an Fc-engineered anti-CD40 monoclonal antibody (2141-V11) administered intratumorally via convection-enhanced delivery for adult patients with recurrent malignant glioma (MG). J. Clin. Oncol. 2022, 40, e14015. [Google Scholar] [CrossRef]
- Brock, C.S.; Newlands, E.S.; Wedge, S.R.; Bower, M.; Evans, H.; Colquhoun, I.; Roddie, M.; Glaser, M.; Brampton, M.H.; Rustin, G.J. Phase I trial of temozolomide using an extended continuous oral schedule. Cancer Res. 1998, 58, 4363–4367. [Google Scholar]
- Campian, J.L.; Piotrowski, A.F.; Ye, X.; Hakim, F.T.; Rose, J.; Yan, X.-Y.; Lu, Y.; Gress, R.; Grossman, S.A. Serial changes in lymphocyte subsets in patients with newly diagnosed high grade astrocytomas treated with standard radiation and temozolomide. J. Neuro-Oncology 2017, 135, 343–351. [Google Scholar] [CrossRef]
- Mathios, D.; Kim, J.E.; Mangraviti, A.; Phallen, J.; Park, C.-K.; Jackson, C.M.; Garzon-Muvdi, T.; Kim, E.; Theodros, D.; Polanczyk, M.; et al. Anti–PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Sci. Transl. Med. 2016, 8, 370ra180. [Google Scholar] [CrossRef] [Green Version]
- Karachi, A.; Yang, C.; Dastmalchi, F.; Sayour, E.J.; Huang, J.; Azari, H.; Long, Y.; Flores, C.; Mitchell, D.; Rahman, M. Modulation of temozolomide dose differentially affects T-cell response to immune checkpoint inhibition. Neuro-Oncology 2019, 21, 730–741. [Google Scholar] [CrossRef]
- Zhang, P.; Miska, J.; Lee-Chang, C.; Rashidi, A.; Panek, W.K.; An, S.; Zannikou, M.; Lopez-Rosas, A.; Han, Y.; Xiao, T.; et al. Therapeutic targeting of tumor-associated myeloid cells synergizes with radiation therapy for glioblastoma. Proc. Natl. Acad. Sci. USA 2019, 116, 23714–23723. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, W.; Shin, Y.S.; Xue, M.; Matsutani, T.; Masui, K.; Yang, H.; Ikegami, S.; Gu, Y.; Herrmann, K.; Johnson, D.; et al. Single-Cell Phosphoproteomics Resolves Adaptive Signaling Dynamics and Informs Targeted Combination Therapy in Glioblastoma. Cancer Cell 2016, 29, 563–573. [Google Scholar] [CrossRef] [Green Version]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational Analysis Reveals the Origin and Therapy-Driven Evolution of Recurrent Glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Zheng, S.; Amini, S.S.; Virk, S.M.; Mikkelsen, T.; Brat, D.J.; Grimsby, J.; Sougnez, C.; Muller, F.; Hu, J.; et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 2015, 25, 316–327. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Cazzato, E.; Ladewig, E.; Frattini, V.; Rosenbloom, D.I.S.; Zairis, S.; Abate, F.; Liu, Z.; Elliott, O.; Shin, Y.-J.; et al. Clonal evolution of glioblastoma under therapy. Nat. Genet. 2016, 48, 768–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, H.d.A.; Villar, R.C. Radiotherapy and immune response: The systemic effects of a local treatment. Clinics 2018, 73 (Suppl. S1), e557s. [Google Scholar] [CrossRef]
- Wu, Q.; Allouch, A.; Martins, I.; Brenner, C.; Modjtahedi, N.; Deutsch, E.; Perfettini, J.L. Modulating both tumor cell death and innate immunity is essential for improving radiation therapy effectiveness. Front. Immunol. 2017, 8, 613. [Google Scholar] [CrossRef]
- Cao, M.; Cabrera, R.; Xu, Y.; Liu, C.; Nelson, D. Different radiosensitivity of CD4+CD25+ regulatory T cells and effector T cells to low dose gamma irradiation in vitro. Int. J. Radiat. Biol. 2010, 87, 71–80. [Google Scholar] [CrossRef] [Green Version]
- Arrieta, V.A.; Iwamoto, F.; Lukas, R.V.; Sachdev, S.; Rabadan, R.; Sonabend, A.M. Can patient selection and neoadjuvant administration resuscitate PD-1 inhibitors for glioblastoma? J. Neurosurg. 2019, 132, 1667–1672. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Hodges, T.R.; Ott, M.; Xiu, J.; Gatalica, Z.; Swensen, J.; Zhou, S.; Huse, J.T.; De Groot, J.; Li, S.; Overwijk, W.W.; et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: Implications for immune checkpoint immunotherapy. Neuro-Oncology 2017, 19, 1047–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGrail, D.; Pilié, P.; Rashid, N.; Voorwerk, L.; Slagter, M.; Kok, M.; Jonasch, E.; Khasraw, M.; Heimberger, A.; Lim, B.; et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann. Oncol. 2021, 32, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chen, A.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 462–469. [Google Scholar] [CrossRef]
- Touat, M.; Li, Y.Y.; Boynton, A.N.; Spurr, L.F.; Iorgulescu, J.B.; Bohrson, C.L.; Cortes-Ciriano, I.; Birzu, C.; Geduldig, J.E.; Pelton, K.; et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 2020, 580, 517–523. [Google Scholar] [CrossRef]
- McGrail, D.J.; Pilié, P.G.; Rashid, N.U.; Voorwerk, L.; Slagter, M.; Kok, M.; Jonasch, E.; Khasraw, M.; Heimberger, A.B.; Ueno, N.T.; et al. Validation of cancer-type restricted benefit from immune checkpoint blockade in TMB-H tumors identified by the FoundationOne CDx assay. Ann. Oncol. 2022. in print. [Google Scholar] [CrossRef]
- Johanns, T.M.; Miller, C.A.; Dorward, I.G.; Tsien, C.; Chang, E.; Perry, A.; Uppaluri, R.; Ferguson, C.; Schmidt, R.E.; Dahiya, S.; et al. Immunogenomics of Hypermutated Glioblastoma: A Patient with Germline POLE Deficiency Treated with Checkpoint Blockade Immunotherapy. Cancer Discov. 2016, 6, 1230–1236. [Google Scholar] [CrossRef] [Green Version]
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; De Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J. Clin. Oncol. 2016, 34, 2206–2211. [Google Scholar] [CrossRef] [Green Version]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell–Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- I Wang, S.; Puc, J.; Li, J.; Bruce, J.N.; Cairns, P.; Sidransky, D.; Parsons, R. Somatic mutations of PTEN in glioblastoma multiforme. Cancer Res. 1997, 57, 4183–4186. [Google Scholar]
- Rasheed, B.K.; Stenzel, T.T.; McLendon, R.; Parsons, R.; Friedman, A.H.; Friedman, H.S.; Bigner, D.D.; Bigner, S.H. PTEN gene mutations are seen in high-grade but not in low-grade gliomas. Cancer Res. 1997, 57, 4187–4190. [Google Scholar] [PubMed]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrieta, V.A.; Chen, A.X.; Kane, J.R.; Kang, S.J.; Kassab, C.; Dmello, C.; Zhao, J.; Burdett, K.B.; Upadhyayula, P.S.; Lee-Chang, C.; et al. ERK1/2 phosphorylation predicts survival following anti-PD-1 immunotherapy in recurrent glioblastoma. Nat. Cancer 2021, 2, 1372–1386. [Google Scholar] [CrossRef] [PubMed]
- Kane, J.R.; Zhao, J.; Tsujiuchi, T.; Laffleur, B.; Arrieta, V.A.; Mahajan, A.; Rao, G.; Mela, A.; Dmello, C.; Chen, L.; et al. CD8+ T-cell–Mediated Immunoediting Influences Genomic Evolution and Immune Evasion in Murine Gliomas. Clin. Cancer Res. 2020, 26, 4390–4401. [Google Scholar] [CrossRef]
- Osborn, A.J.; Elledge, S.J.; Zou, L. Checking on the fork: The DNA-replication stress-response pathway. Trends Cell Biol. 2002, 12, 509–516. [Google Scholar] [CrossRef]
- McGrail, D.J.; Pilié, P.G.; Dai, H.; Lam, T.N.A.; Liang, Y.; Voorwerk, L.; Kok, M.; Zhang, X.H.-F.; Rosen, J.M.; Heimberger, A.B.; et al. Replication stress response defects are associated with response to immune checkpoint blockade in nonhypermutated cancers. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef]
- Adair, R.A.; Roulstone, V.; Scott, K.J.; Morgan, R.; Nuovo, G.J.; Fuller, M.; Beirne, D.; West, E.J.; Jennings, V.A.; Rose, A.; et al. Cell Carriage, Delivery, and Selective Replication of an Oncolytic Virus in Tumor in Patients. Sci. Transl. Med. 2012, 4, 138ra77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stojdl, D.F.; Lichty, B.D.; Knowles, S.; Marius, R.; Atkins, H.; Sonenberg, N.; Bell, J.C. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat. Med. 2000, 6, 821–825. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Song, X.; Wang, Y.; Liu, F.; Wei, J. Combining Oncolytic Viruses With Cancer Immunotherapy: Establishing a New Generation of Cancer Treatment. Front. Immunol. 2020, 11, 683. [Google Scholar] [CrossRef]
- Zeng, J.; Li, X.; Sander, M.; Zhang, H.; Yan, G.; Lin, Y. Oncolytic Viro-Immunotherapy: An Emerging Option in the Treatment of Gliomas. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef]
- Shoaf, M.L.; Peters, K.B. Clinical Trials of Oncolytic Viruses in Glioblastoma. Adv. Oncol. 2022, 2, 139–158. [Google Scholar] [CrossRef]
- Ager, C.R.; Reilley, M.J.; Nicholas, C.; Bartkowiak, T.; Jaiswal, A.R.; Curran, M.A. Intratumoral STING Activation with T-cell Checkpoint Modulation Generates Systemic Antitumor Immunity. Cancer Immunol. Res. 2017, 5, 676–684. [Google Scholar] [CrossRef] [Green Version]
- Sivick, K.E.; Desbien, A.L.; Glickman, L.H.; Reiner, G.L.; Corrales, L.; Surh, N.H.; Hudson, T.E.; Vu, U.T.; Francica, B.J.; Banda, T.; et al. Magnitude of therapeutic STING activation determines CD8(þ) T cell-mediated anti-tumor immunity. Cell Rep. 2018, 25, 3074–3085. [Google Scholar] [CrossRef] [Green Version]
- Corrales, L.; Gajewski, T.F. Molecular Pathways: Targeting the Stimulator of Interferon Genes (STING) in the Immunotherapy of CancerTargeting the STING Pathway in the Immunotherapy of Cancer. Clin. Cancer Res. 2015, 21, 4774–4779. [Google Scholar] [CrossRef] [Green Version]
- Ohkuri, T.; Ghosh, A.; Kosaka, A.; Zhu, J.; Ikeura, M.; David, M.; Watkins, S.C.; Sarkar, S.N.; Okada, H. STING Contributes to Antiglioma Immunity via Triggering Type I IFN Signals in the Tumor Microenvironment. Cancer Immunol. Res. 2014, 2, 1199–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudreau, C.E.; Najem, H.; Ott, M.; Horbinski, C.; Fang, D.; DeRay, C.M.; Levine, J.M.; Curran, M.A.; Heimberger, A.B. Intratumoral Delivery of STING Agonist Results in Clinical Responses in Canine Glioblastoma. Clin. Cancer Res. 2021, 27, 5528–5535. [Google Scholar] [CrossRef] [PubMed]
- Piperi, C.; Papavassiliou, K.A.; Papavassiliou, A.G. Pivotal Role of STAT3 in Shaping Glioblastoma Immune Microenvironment. Cells 2019, 8, 1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, S.D.; Srinivasan, V.M.; Heimberger, A.B. The role of STAT3 in tumor-mediated immune suppression. J. Neuro-Oncol. 2015, 123, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Ouedraogo, Z.G.; Biau, J.; Kemeny, J.-L.; Morel, L.; Verrelle, P.; Chautard, E. Role of STAT3 in Genesis and Progression of Human Malignant Gliomas. Mol. Neurobiol. 2016, 54, 5780–5797. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.; Ahn, S.H.; Kong, D.-S.; Lee, H.W.; Nam, D.-H. The role of STAT3 in glioblastoma progression through dual influences on tumor cells and the immune microenvironment. Mol. Cell. Endocrinol. 2017, 451, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.F.; Kong, L.-Y.; Jordan, J.; Conrad, C.; Madden, T.; Fokt, I.; Priebe, W.; Heimberger, A.B. A Novel Small Molecule Inhibitor of Signal Transducers and Activators of Transcription 3 Reverses Immune Tolerance in Malignant Glioma Patients. Cancer Res. 2007, 67, 9630–9636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Groot, J.; Ott, M.; Wei, J.; Kassab, C.; Fang, D.; Najem, H.; O’Brien, B.; Weathers, S.-P.; Matsouka, C.K.; Majd, N.K.; et al. A first-in-human Phase I trial of the oral p-STAT3 inhibitor WP1066 in patients with recurrent malignant glioma. CNS Oncol. 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Kassab, C.; Marisetty, A.; Hashimoto, Y.; Wei, J.; Zamler, D.B.; Leu, J.-S.; Tomaszowski, K.-H.; Sabbagh, A.; Fang, D.; et al. Radiation with STAT3 Blockade Triggers Dendritic Cell–T cell Interactions in the Glioma Microenvironment and Therapeutic Efficacy. Clin. Cancer Res. 2020, 26, 4983–4994. [Google Scholar] [CrossRef] [PubMed]
- Sonabend, A.M.; Velicu, S.; Ulasov, I.V.; Han, Y.; Tyler, B.; Brem, H.; Matar, M.M.; Fewell, J.G.; Anwer, K.; Lesniak, M.S. A safety and efficacy study of local delivery of interleukin-12 transgene by PPC polymer in a model of experimental glioma. Anti-Cancer Drugs 2008, 19, 133–142. [Google Scholar] [CrossRef] [Green Version]
- Chiocca, E.A.; Yu, J.S.; Lukas, R.V.; Solomon, I.H.; Ligon, K.L.; Nakashima, H.; Triggs, D.A.; Reardon, D.A.; Wen, P.; Stopa, B.M.; et al. Regulatable interleukin-12 gene therapy in patients with recurrent high-grade glioma: Results of a phase 1 trial. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Gelb, A.B.; Chen, C.C.; Rao, G.; Reardon, D.; Wen, P.Y.; Bi, W.L.; Peruzzi, P.; Amidei, C.; Triggs, D.; et al. Combined immunotherapy with controlled interleukin-12 gene therapy and immune checkpoint blockade in recurrent glioblastoma: An open-label, multi-institutional phase I trial. Neuro-Oncology 2021, 24, 951–963. [Google Scholar] [CrossRef]
- Lukas, R.; Oberheim-Bush, N.A.; Cavaliere, R.; Landolfi, J.; Yu, J.S.; Chen, C.; Cordova, C.; Amidei, C.; Buck, J.Y.; Hadar, N.; et al. CTIM-20. Final results of controlled il-12 monotherapy and in combination with pd-1 inhibitor in adult subjects with recurrent glioblastoma. Neuro-Oncology 2021, 23, vi54. [Google Scholar] [CrossRef]
- Lasek, W.; Zagożdżon, R.; Jakobisiak, M. Interleukin 12: Still a promising candidate for tumor immunotherapy? Cancer Immunol. Immunother. 2014, 63, 419–435. [Google Scholar] [CrossRef] [Green Version]
- Lotze, M.T. Interleukin 12: Cellular and molecular immunology of an important regulatory cytokine. Introd. Ann. N. Y. Acad. Sci. 1996, 795, xiii–xix. [Google Scholar] [CrossRef]
- Trinchieri, G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 2003, 3, 133–146. [Google Scholar] [CrossRef]
- Curtsinger, M.J.; Johnson, C.M.; Mescher, M.F. CD8 T cell clonal expansion and development of effector function require prolonged exposure to antigen, costimulation, and signal 3 cytokine. J. Immunol. 2003, 171, 5165–5171. [Google Scholar]
- Micallef, M.J.; Ohtsuki, T.; Kohno, K.; Tanabe, F.; Ushio, S.; Namba, M.; Tanimoto, T.; Torigoe, K.; Fujii, M.; Ikeda, M.; et al. Interferon-γ-inducing factor enhances T helper 1 cytokine production by stimulated human T cells: Synergism with interleukin-12 for interferon-γ production. Eur. J. Immunol. 1996, 26, 1647–1651. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Brem, S.; Desai, A.S.; Bagley, S.J.; Kurz, S.C.; De La Fuente, M.I.; Nagpal, S.; Welch, M.R.; Hormigo, A.; Carroll, N.; et al. INO-5401 and INO-9012 delivered intramuscularly (IM) with electroporation (EP) in combination with cemiplimab (REGN2810) in newly diagnosed glioblastoma (GBM): Interim results. J. Clin. Oncol. 2020, 38, 2514. [Google Scholar] [CrossRef]
- Van Vulpen, M.; Kal, H.B.; Taphoorn, M.J.; El Sharouni, S.Y. Changes in blood-brain barrier permeability induced by radiotherapy: Implications for timing of chemotherapy? (Review). Oncol. Rep. 2002, 9, 683–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, D.; Ma, J.; Xiao, J.; Tang, Z. Effect of Brain Irradiation on Blood-CSF Barrier Permeability of Chemotherapeutic Agents. Am. J. Clin. Oncol. 1997, 20, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, A.; Canney, M.; Vignot, A.; Reina, V.; Beccaria, K.; Horodyckid, C.; Karachi, C.; Leclercq, D.; Lafon, C.; Chapelon, J.-Y.; et al. Clinical trial of blood-brain barrier disruption by pulsed ultrasound. Sci. Transl. Med. 2016, 8, 343re2. [Google Scholar] [CrossRef]
- Mainprize, T.; Lipsman, N.; Huang, Y.; Meng, Y.; Bethune, A.; Ironside, S.; Heyn, C.; Alkins, R.; Trudeau, M.; Sahgal, A.; et al. Blood-Brain Barrier Opening in Primary Brain Tumors with Non-invasive MR-Guided Focused Ultrasound: A Clinical Safety and Feasibility Study. Sci. Rep. 2019, 9, 321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dréan, A.; Lemaire, N.; Bouchoux, G.; Goldwirt, L.; Canney, M.; Goli, L.; Bouzidi, A.; Schmitt, C.; Guehennec, J.; Verreault, M.; et al. Temporary blood–brain barrier disruption by low intensity pulsed ultrasound increases carboplatin delivery and efficacy in preclinical models of glioblastoma. J. Neuro-Oncol. 2019, 144, 33–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idbaih, A.; Canney, M.; Belin, L.; Desseaux, C.; Vignot, A.; Bouchoux, G.; Asquier, N.; Law-Ye, B.; Leclercq, D.; Bissery, A.; et al. Safety and Feasibility of Repeated and Transient Blood–Brain Barrier Disruption by Pulsed Ultrasound in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2019, 25, 3793–3801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonabend, A.M.; Gould, A.; Luan, Y.; Hou, Y.; Chen, L.; Kobayashi, M.A.; Castro, B.A.; Zhang, D.Y.; Korobova, F.V.; Amidei, C.; et al. Repeated opening of the blood-brain barrier with the skull-implantable SonoCloud-9 (SC9) device: Phase 1 trial of nab-paclitaxel and SC9 in recurrent glioblastoma. J. Clin. Oncol. 2022, 40, 2016. [Google Scholar] [CrossRef]
- Zhang, D.Y.; Dmello, C.; Chen, L.; Arrieta, V.A.; Gonzalez-Buendia, E.; Kane, J.R.; Magnusson, L.P.; Baran, A.; James, C.D.; Horbinski, C.; et al. Ultrasound-mediated Delivery of Paclitaxel for Glioma: A Comparative Study of Distribution, Toxicity, and Efficacy of Albumin-bound Versus Cremophor FormulationsUS-Delivered ABX Extends Survival in GBM PDX Mouse Model. Clin. Cancer Res. 2020, 26, 477–486. [Google Scholar] [CrossRef]
- Lau, T.S.; Chan, L.K.Y.; Man, G.C.W.; Wong, C.H.; Lee, J.H.S.; Yim, S.F.; Cheung, T.H.; McNeish, I.A.; Kwong, J. Paclitaxel induces immunogenic cell death in ovarian cancer via TLR4/IKK2/SNARE-dependent exocytosis. Cancer Immunol. Res. 2020, 8, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Decraene, B.; Yang, Y.; De Smet, F.; Garg, A.D.; Agostinis, P.; De Vleeschouwer, S. Immunogenic cell death and its therapeutic or prognostic potential in high-grade glioma. Genes Immun. 2022, 23, 1–11. [Google Scholar] [CrossRef]
- Sabbagh, A.; Beccaria, K.; Ling, X.; Marisetty, A.; Ott, M.; Caruso, H.; Barton, E.; Kong, L.-Y.; Fang, D.; Latha, K.; et al. Opening of the Blood–Brain Barrier Using Low-Intensity Pulsed Ultrasound Enhances Responses to Immunotherapy in Preclinical Glioma Models. Clin. Cancer Res. 2021, 27, 4325–4337. [Google Scholar] [CrossRef] [PubMed]
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.-P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 2020, 181, 1643–1660. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [Green Version]
- Alban, T.J.; Alvarado, A.G.; Sorensen, M.D.; Bayik, D.; Volovetz, J.; Serbinowski, E.; Mulkearns-Hubert, E.E.; Sinyuk, M.; Hale, J.S.; Onzi, G.R.; et al. Global immune fingerprinting in glioblastoma patient peripheral blood reveals immune-suppression signatures associated with prognosis. JCI Insight 2018, 3, e122264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raychaudhuri, B.; Rayman, P.; Ireland, J.; Ko, J.; Rini, B.; Borden, E.C.; Garcia, J.; Vogelbaum, M.A.; Finke, J. Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro-Oncology 2011, 13, 591–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, A.L.; Miska, J.; Wainwright, D.A.; Dey, M.; Rivetta, C.V.; Yu, D.; Kanojia, D.; Pituch, K.C.; Qiao, J.; Pytel, P.; et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor CellsCCL2 in Treg and MDSC Trafficking to Glioma. Cancer Res. 2016, 76, 5671–5682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayik, D.; Zhou, Y.; Park, C.; Hong, C.; Vail, D.; Silver, D.J.; Lauko, A.; Roversi, G.; Watson, D.C.; Lo, A.; et al. Myeloid-Derived Suppressor Cell Subsets Drive Glioblastoma Growth in a Sex-Specific MannerMDSC Subset Sexual Dimorphism Informs GBM Immunotherapy. Cancer Discov. 2020, 10, 1210–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores-Toro, J.A.; Luo, D.; Gopinath, A.; Sarkisian, M.R.; Campbell, J.J.; Charo, I.F.; Singh, R.; Schall, T.J.; Datta, M.; Jain, R.K.; et al. CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. Proc. Natl. Acad. Sci. USA 2020, 117, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Miska, J.; Rashidi, A.; Lee-Chang, C.; Gao, P.; Lopez-Rosas, A.; Zhang, P.; Burga, R.; Castro, B.; Xiao, T.; Han, Y.; et al. Polyamines drive myeloid cell survival by buffering intracellular pH to promote immunosuppression in glioblastoma. Sci. Adv. 2021, 7, eabc8929. [Google Scholar] [CrossRef]
- Peereboom, D.M.; Alban, T.J.; Grabowski, M.M.; Alvarado, A.G.; Otvos, B.; Bayik, D.; Roversi, G.; McGraw, M.; Huang, P.; Mohammadi, A.M.; et al. Metronomic capecitabine as an immune modulator in glioblastoma patients reduces myeloid-derived suppressor cells. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [Green Version]
- Akkari, L.; Bowman, R.L.; Tessier, J.; Klemm, F.; Handgraaf, S.M.; de Groot, M.; Quail, D.F.; Tillard, L.; Gadiot, J.; Huse, J.T.; et al. Dynamic changes in glioma macrophage populations after radiotherapy reveal CSF-1R inhibition as a strategy to overcome resistance. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Lin, C.-C.; Gil-Martin, M.; Bauer, T.M.; Naing, A.; Lim, D.W.-T.; Sarantopoulos, J.; Geva, R.; Ando, Y.; Fan, L.; Choudhury, S.; et al. Abstract CT171: Phase I study of BLZ945 alone and with spartalizumab (PDR001) in patients (pts) with advanced solid tumors. Cancer Res. 2020, 80, CT171. [Google Scholar] [CrossRef]
- Butowski, N.; Colman, H.; De Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro-Oncology 2016, 18, 557–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S.; Yang, R.; Mirzaei, R.; Rawji, K.; Poon, C.; Mishra, M.K.; Zemp, F.J.; Bose, P.; Kelly, J.; Dunn, J.F.; et al. Control of brain tumor growth by reactivating myeloid cells with niacin. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Daher, M.; Rezvani, K. Next generation natural killer cells for cancer immunotherapy: The promise of genetic engineering. Curr. Opin. Immunol. 2018, 51, 146–153. [Google Scholar] [CrossRef]
- Shaim, H.; Shanley, M.; Basar, R.; Daher, M.; Gumin, J.; Zamler, D.B.; Uprety, N.; Wang, F.; Huang, Y.; Gabrusiewicz, K.; et al. Targeting the αv integrin/TGF-β axis improves natural killer cell function against glioblastoma stem cells. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Kabelitz, D.; Serrano, R.; Kouakanou, L.; Peters, C.; Kalyan, S. Cancer immunotherapy with γδ T cells: Many paths ahead of us. Cell. Mol. Immunol. 2020, 17, 925–939. [Google Scholar] [CrossRef]
- Groh, V.; Rhinehart, R.; Secrist, H.; Bauer, S.; Grabstein, K.H.; Spies, T. Broad tumor-associated expression and recognition by tumor-derived γδ T cells of MICA and MICB. Proc. Natl. Acad. Sci. USA 1999, 96, 6879–6884. [Google Scholar] [CrossRef] [Green Version]
- Fujimiya, Y.; Suzuki, Y.; Katakura, R.; Miyagi, T.; Yamaguchi, T.; Yoshimoto, T.; Ebina, T. In vitro interleukin 12 activation of peripheral blood CD3(+)CD56(+) and CD3(+)CD56(-) gammadelta T cells from glioblastoma patients. Clin. Cancer Res. 1997, 3, 633–643. [Google Scholar] [PubMed]
- Yamaguchi, T.; Fujimiya, Y.; Suzuki, Y.; Katakura, R.; Ebina, T. A simple method for the propagation and purification of γδT cells from the peripheral blood of glioblastoma patients using solid-phase anti-CD3 antibody and soluble IL-2. J. Immunol. Methods 1997, 205, 19–28. [Google Scholar] [CrossRef]
- Nabors, L.B.; Lamb, L.S.; Beelen, M.J.; Pillay, T.; ter Haak, M.; Youngblood, S.; Vaickus, L.; Lobbous, M. Phase 1 trial of drug resistant immunotherapy: A first-in-class combination of MGMT-modified γδ t cells and temozolomide chemotherapy in newly diagnosed glioblastoma. J. Clin. Oncol. 2021, 39, 2057. [Google Scholar] [CrossRef]
- Nabors, L.B.; Lobbous, M.; Lamb, L.S.; Rochlin, K.; Pillay, T.; Youngblood, S.; Haak, M.t.; Goswami, T. “Phase I study of drug-resistant immunotherapy (DRI) with gene-modified autologous γδ T cells in patients with newly diagnosed glioblastoma multiforme (GBM) receiving maintenance temozolomide (TMZ). J. Clin. Oncol. 2022, 40, 2044. [Google Scholar] [CrossRef]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Larsen, M.S.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef]
- Garg, K.; Maurer, M.; Griss, J.; Brüggen, M.-C.; Wolf, I.H.; Wagner, C.; Willi, N.; Mertz, K.D.; Wagner, S.N. Tumor-associated B cells in cutaneous primary melanoma and improved clinical outcome. Hum. Pathol. 2016, 54, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, S.; Lee, A.H.S.; Paish, E.C.; Macmillan, R.D.; Ellis, I.; Green, A.R. The prognostic significance of B lymphocytes in invasive carcinoma of the breast. Breast Cancer Res. Treat. 2011, 132, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Edin, S.; Kaprio, T.; Hagström, J.; Larsson, P.; Mustonen, H.K.; Böckelman, C.; Strigård, K.; Gunnarsson, U.; Haglund, C.; Palmqvist, R. The Prognostic Importance of CD20+ B lymphocytes in Colorectal Cancer and the Relation to Other Immune Cell subsets. Sci. Rep. 2019, 9, 19997–19999. [Google Scholar] [CrossRef] [PubMed]
- Griss, J.; Bauer, W.; Wagner, C.; Simon, M.; Chen, M.; Grabmeier-Pfistershammer, K.; Maurer-Granofszky, M.; Roka, F.; Penz, T.; Bock, C.; et al. B cells sustain inflammation and predict response to immune checkpoint blockade in human melanoma. Nat. Commun. 2019, 10, 4186. [Google Scholar] [CrossRef] [Green Version]
- van Herpen, C.M.; van der Voort, R.; van der Laak, J.A.; Klasen, I.S.; de Graaf, A.O.; van Kempen, L.C.; de Vries, I.J.; Boer, T.D.; Dolstra, H.; Torensma, R.; et al. Intratumoral rhIL-12 administration in head and neck squamous cell carcinoma patients induces B cell activation. Int. J. Cancer 2008, 123, 2354–2361. [Google Scholar] [CrossRef]
- Santoiemma, P.P.; Reyes, C.; Wang, L.-P.; McLane, M.W.; Feldman, M.D.; Tanyi, J.L.; Powell, D.J. Systematic evaluation of multiple immune markers reveals prognostic factors in ovarian cancer. Gynecol. Oncol. 2016, 143, 120–127. [Google Scholar] [CrossRef]
- Goeppert, B.; Frauenschuh, L.; Zucknick, M.; Stenzinger, A.; Andrulis, M.; Klauschen, F.; Joehrens, K.; Warth, A.; Renner, M.; Mehrabi, A.; et al. Prognostic impact of tumour-infiltrating immune cells on biliary tract cancer. Br. J. Cancer 2013, 109, 2665–2674. [Google Scholar] [CrossRef] [Green Version]
- Lee-Chang, C.; Bodogai, M.; Moritoh, K.; Olkhanud, P.B.; Chan, A.C.; Croft, M.; Mattison, J.A.; Holst, P.J.; Gress, R.E.; Ferrucci, L.; et al. Accumulation of 4-1BBL+ B cells in the elderly induces the generation of granzyme-B+ CD8+ T cells with potential antitumor activity. Blood 2014, 124, 1450–1459. [Google Scholar] [CrossRef]
- Lee-Chang, C.; Miska, J.; Hou, D.; Rashidi, A.; Zhang, P.; Burga, R.A.; Jusué-Torres, I.; Xiao, T.; Arrieta, V.A.; Zhang, D.Y.; et al. Activation of 4-1BBL+ B cells with CD40 agonism and IFNγ elicits potent immunity against glioblastoma. J. Exp. Med. 2021, 218, e20200913. [Google Scholar] [CrossRef]


| Completed Trials | ||||||
| Vaccine Type/Target | Study Title | Phase | Size | Intervention Details | Primary Endpoint | Reference |
| EGFRvIII | Phase III Study of Rindopepimut/GM-CSF in Patients With Newly Diagnosed Glioblastoma (ACT IV) | 3 | 745 | Rindopepimut + TMZ vs. TMZ | OS = 20.1 (95% CI 18.5–22.1) vs. 20.0 mo (95% CI 18.1–21.9) | NCT01480479 |
| A Study of Rindopepimut/GM-CSF in Patients with Relapsed EGFRvIII Positive Glioblastoma (ReACT) | 2 | 73 | Rindopepimut + bevacizumab vs. Bevacizumab | PFS at 6 mo = 28% vs. 16% (HR = 0.72, 95% CI 0.42–1.21) | NCT01498328 | |
| WT1 | Phase II clinical trial of Wilms tumor 1 peptide vaccination for patients with recurrent glioblastoma multiforme | 2 | 21 | WT1 peptide | mPFS = 20 weeks 6 mo PFS rate = 33.3% | N/A |
| A phase I study of the WT2725 dosing emulsion in patients with advanced malignancies | 1 | 62 | WT1 peptide (WT2725) | Maximum tolerated dose. * Response rate in GBM = 20% * OS12 in GBM = 33% | NCT01621542 | |
| IMT-03 Clinical Trial for Newly Diagnosed Malignant Glioma with WT1-W10 Vaccination | 1/2 | 27 | WT peptide (W10) | PFS = 12.7 mo OS = 21.9 mo | N/A | |
| IDH1 | Targeting IDH1R132H in WHO Grade III-IV IDH1R132H-mutated Gliomas by a Peptide Vaccine—a Phase I Safety, Tolerability and Immunogenicity Multicenter Trial (NOA-16) | 1 | 33 | IDH1 peptide vaccine | Safety and immunogenicity. 93.3% with vaccine-induced immune response | NCT02454634 |
| CMV | Vaccine Therapy in Treating Patients with Newly Diagnosed Glioblastoma Multiforme (ATTAC) | 1/2 | 12 | pp65-DC vaccine + Td preconditioning | Safety and feasability * OS = 20.6–47.3 mo vs. 13.8–41.3 mo (p = 0.013) | NCT00639639 |
| Long-term Survival in Glioblastoma with Cytomegalovirus pp65-Targeted Vaccination (ATTC-GM) | 1 | 11 | pp65 DC vaccine + GM-CSF | Safety and feasibility * mPFS = 25.3 mo vs. 8 mo (p = 0.0001), * mOS = 41.1 vs. 19.2 mo (p = 0.0001) | NCT00639639 | |
| Evaluation of Overcoming Limited Migration and Enhancing Cytomegalovirus-specific Dendritic Cell Vaccines with Adjuvant TEtanus Pre-conditioning in Patients With Newly-diagnosed Glioblastoma (ELEVATE) | 2 | 43 | pp65 DC vaccine + TMZ vs. Pp65 DC vaccine + TMZ + preconditioning | 3-year OS = 34% (95% CI 19–63%) vs. 6% (95% CI 1–42%) | NCT02366728 | |
| Multipeptide Vaccines | Efficacy finding cohort of a cancer peptide vaccine, TAS0313, in treating recurrent glioblastoma | 1/2 | 9 | TAS0313 | Safety and efficacy. ORR = 11.1% (95% CI = 0.3–48.2%) | JapicCTI-183824 |
| A Randomized, Double-blind, Controlled Phase IIb Study of the Safety and Efficacy of ICT-107 in Newly Diagnosed Patients with Glioblastoma Multiforme (GBM) Following Resection and Chemoradiation | 2 | 124 | ICT-107 vs. placebo | OS = 17 vs. 15 mo (HR = 0.87, p = 0.58). * PFS = 11.2 vs. 9.0 mo (HR = 0.57, p = 0.011) | NCT01280552 | |
| Neoantigens | A Phase I Study of a Personalized NeoAntigen Cancer Vaccine With Radiotherapy Plus Pembrolizumab/MK-3475 Among Newly Diagnosed Glioblastoma Patients | 1/1b | 8 | NeoVax + RT vs. Neovax + RT + Pembrolizumab | Safety and tolerability | NCT02287428 |
| Ongoing Trials | ||||||
| Vaccine Type/Target | Study Title | Phase | Size | Intervention details | Primary endpoint | Reference |
| Survivin | A Phase II Study of the Safety and Efficacy of SVN53-67/M57-KLH (SurVaxM) in Survivin-Positive Newly Diagnosed Glioblastoma | 2 | 66 | SurVaxM + TMZ | PFS at 6 mo | NCT02455557 |
| Phase II Study of Pembrolizumab Plus SurVaxM for Glioblastoma at First Recurrence | 2 | 40 | SurVaxM + pembrolizumab | PFS at 6 mo | NCT04013672 | |
| TERT | Anticancer Therapeutic Vaccination Using Telomerase-derived Universal Cancer Peptides in Glioblastoma (UCPVax-Glio) | 1/2 | 56 | UCPVax vs. UCPVax + TMZ | Anti-TERT T-cell response | NCT04280848 |
| Multipeptide Vaccines | First-in-Human, Phase 1b/2a Trial of a Multipeptide Therapeutic Vaccine in Patients with Progressive Glioblastoma (ROSALIE) | 1/2 | 52 | EO2041 + nivolumab vs. EO2041 + nivolumab + bevacizumab | Safety and tolerability | NCT04116658 |
| Autologous Vaccine | Study Title | Phase | Size | Intervention | Primary Endpoint | Reference |
|---|---|---|---|---|---|---|
| Dendritic cells | A Phase III Clinical Trial Evaluating DCVax®-L, Autologous Dendritic Cells Pulsed With Tumor Lysate Antigen For The Treatment Of Glioblastoma Multiforme (GBM) | 3 | 331 | TMZ + DCVax-L vs. TMZ + placebo | mOS = 23.1 mo (95% CI 21.2–25.4) | NCT00045968 |
| Phase 1 Study of a Dendritic Cell Vaccine for Patients with Either Newly Diagnosed or Recurrent Glioblastoma | 1 | 36 | DC vaccine + GBM stem-like cell lysate | Safety and tolerability * mPFS = 8.75 mo in new GBM, 3.23 mo in recurrent GBM * mOS = 20.36 mo in new GBM, 11.97 mo in recurrent GBM | NCT02010606 | |
| HSP | Phase I/II Trial of Heat Shock Protein Peptide Complex-96 (HSPPC-96) Vaccine for Patients With Recurrent High Grade Glioma | 1/2 | 41 | HSPPC-96 vaccine | OS at 6 mo = 90.2% (95% CI 75.9–96.8%) mOS = 42.6 weeks (95% CI = 34.7–50.5) | NCT00293423 |
| A Phase II Randomized Trial Comparing the Efficacy of Heat Shock Protein Peptide Complex-96 (HSPPC-96) Vaccine Given with Bevacizumab versus Bevacizumab Alone in the Treatment of Surgically Resectable Recurrent Glioblastoma | 2 | 90 | HSPPC-96 vaccine + bevacicumab vs. Bevacizumab alone | OS = 7.5 vs. 10.7 mo (HR = 2.06) | NCT01814813 |
| Study Title | Phase | Size | Intervention Details | Primary Endpoint | Reference | |
|---|---|---|---|---|---|---|
| Newly diagnosed GBM | An Investigational Immuno-therapy Study of Nivolumab Compared to Temozolomide, Each Given With Radiation Therapy, for Newly-diagnosed Patients With Glioblastoma (GBM, a Malignant Brain Cancer) (Checkmate 498) | 3 | 560 | Nivolumab + RT vs. TMZ + RT | OS (endpoint not met) mOS = 13.4 vs. 14.9 mo (HR = 1.31, 95% CI = 1.09–1.58, p = 0.0037) | NCT02617589 |
| An Investigational Immuno-therapy Study of Temozolomide Plus Radiation Therapy With Nivolumab or Placebo, for Newly Diagnosed Patients With Glioblastoma (GBM, a Malignant Brain Cancer) (Checkmate 548) | 3 | 716 | Nivolumab + RT + TMZ vs. Placebo + RT + TMZ | PFS = 10.6 vs. 10.3 mo (HR = 1.06) mOS = 28.9 vs. 32.1 mo (HR = 1.10) | NCT02667587 | |
| Phase I/II Study to Evaluate the Safety and Clinical Efficacy of Atezolizumab (aPDL1) in Combination With Temozolomide and Radiation in Patients With Newly Diagnosed Glioblastoma (GBM) | 1/2 | 60 | Atezolizumab + RT + TMZ vs. adjuvant atezolizumab + TMZ | MTD, mOS = 17.1 mo | NCT03174197 | |
| Recurrent GBM | A Study of the Effectiveness and Safety of Nivolumab Compared to Bevacizumab and of Nivolumab With or Without Ipilimumab in Glioblastoma Patients (Checkmate 143) | 3 | 369 | Nivolumab vs. Bevacizumab | mOS = 9.8 vs. 10.0 mo (HR = 1.04, 95% CI = 0.83–1.30) | NCT02017717 |
| Study of Pembrolizumab (MK-3475) in Participants With Advanced Solid Tumors (MK-3475-028/KEYNOTE-28) | 1 | 26 | Pembrolizumab | mOS = 13.1 mo | NCT02054806 | |
| Pembrolizumab +/− Bevacizumab for Recurrent GBM | 2 | 80 | Pembrolizumab + bevacizumab vs. Pembrolizumab | PFS at 6 mo = 26.0% (95% CI 16.3–41.5) vs. 6.7% (95% CI 1.7–25.4) | NCT02337491 | |
| A Study of Atezolizumab (an Engineered Anti-Programmed Death-Ligand 1 [PDL1] Antibody) to Evaluate Safety, Tolerability and Pharmacokinetics in Participants With Locally Advanced or Metastatic Solid Tumors | 1 | 16 | Atezolizumab | Safety profile, mOS = 4.2 mo | NCT01375842 |
| Target | Study Title | Phase | Size | Intervention | Primary Outcome | Reference | |
|---|---|---|---|---|---|---|---|
| TReg Targeting | IDO1 | A Phase 1/2a Study of BMS-986205 Administered in Combination With Nivolumab (Anti-PD-1 Monoclonal Antibody) and in Combination With Both Nivolumab and Ipilimumab (Anti-CTLA-4 Monoclonal Antibody) in Advanced Malignant Tumors | 1 | 30 | IDO1 inhibitor (BMS 986205) + RT + nivolumab with vs. without TMZ | MTD | NCT04047706 |
| GITR | A Phase II Study of the Anti-GITR Agonist INCAGN1876 and the PD-1 Inhibitor INCMGA00012 in Combination With Stereotactic Radiosurgery in Recurrent Glioblastoma | 2 | 32 | Anti-GITR agonist (INCAGN1876) + PD1 inhibitor (INCMGA00012) + SRS with vs. without surgery | Objective radiographic response | NCT04225039 | |
| T-Cell Activating | 41BB | A Phase 1b study of utomilumab (PF-05082566) in combination with mogamulizumab in patients with advanced solid tumors | 1/1b | 24 | Utomilumab (PF-05082566) + CCR4 mAb | MTD | NCT02444793 |
| OX-40L | Phase I Trial of DNX-2440 Oncolytic Adenovirus in Patients With Recurrent Glioblastoma | 1 | 24 | DNX-2440 virus | MTD | NCT03714334 | |
| CD40L | A Phase 1 Trial of D2C7-IT in Combination With an Fc-engineered Anti-CD40 Monoclonal Antibody (2141-V11) Administered Intratumorally Via Convection-Enhanced Delivery for Adult Patients With Recurrent Malignant Glioma | 1 | 8 | CD40 agonist antibody (2141-V11) + EGFRvIII immunotoxin (D2C7-IT) | MTD | NCT04547777 |
| Primary Mechanism | Study Title | Phase | Size | Intervention | Primary Outcome | Reference |
|---|---|---|---|---|---|---|
| STING agonism | Intratumoral Delivery of STING Agonist Results in Clinical Responses in Canine Glioblastoma | Preclincial | 6 | STING agonist IACS-8779 | MTD and radiographic response | N/A |
| STAT3 inhibition | A first-in-human Phase I trial of the oral p-STAT3 inhibitor WP1066 in patients with recurrent malignant glioma | 1 | 8 | p-STAT3 inhibitor WP1066 | MTD. * mOS = 25 mo. | NCT02977780 |
| IL-12 therapy | An Open-Label, Multi-Center Trial of INO-5401 and INO-9012 Delivered by Electroporation (EP) in Combination With REGN2810 in Subjects With Newly-Diagnosed Glioblastoma (GBM) | 1/2 | 52 | INO-9012 (IL-12 plasmid) and INO-5401 + cemiplimab + TMZ + radiation | Safety (ongoing) | NCT03491683 |
| A Phase I Study of Ad-RTS-hIL-12, an Inducible Adenoviral Vector Engineered to Express hIL-12 in the Presence of the Activator Ligand Veledimex in Subjects With Recurrent or Progressive Glioblastoma or Grade III Malignant Glioma | 1 | 31 | Veledimex + Ad-RTS-hIL-12 | Safety and tolerability. * mOS = 12.7 mo | NCT02026271 | |
| BBB disruption | A Study to Evaluate the Safety of Transient Opening of the Blood-Brain Barrier by Low Intensity Pulsed Ultrasound With the SonoCloud Implantable Device in Patients With Recurrent Glioblastoma Before Chemotherapy Administration | 1/2 | 21 | SonoCloud + carboplatin | Safety. * mPFS = 4.11 mo, mOS = 12.94 mo | NCT02253212 |
| A Study to Evaluate the Safety and Feasibility of Blood-Brain Barrier Disruption Using Transcranial MRI-Guided Focused Ultrasound With Intravenous Ultrasound Contrast Agents in the Treatment of Brain Tumours With Doxorubicin | 1 | 5 | ExABlate | Safety | NCT02343991 | |
| A Study to Evaluate the Safety and the Efficacy of Transient Opening of the Blood-brain Barrier (BBB) by Low Intensity Pulsed Ultrasound With the SonoCloud-9 Implantable Device in Recurrent Glioblastoma Patients Eligible for Surgery and for Carboplatin Chemotherapy | 1/2a | 33 | Sonocloud-9 device + carboplatin | MTD and BBB opening (ongoing) | NCT03744026 | |
| Phase 1/2 Trial of Blood-brain Barrier Opening With an Implantable Ultrasound Device SonoCloud-9 and Treatment With Albumin-bound Paclitaxel in Patients With Recurrent Glioblastoma | 1/2 | 17 | Sonocloud-9 device + paclitaxel + carboplatin | Maximum tolerated dose and 1-year survival rate (ongoing) | NCT04528680 |
| Target | Study Title | Phase | Size | Intervention | Primary Outcome | Reference |
|---|---|---|---|---|---|---|
| MDSC | Targeting Myeloid Derived Suppressor Cells in Recurrent Glioblastoma: Phase 0/1 Trial of Low Dose Capecitabine + Bevacizumab in Patients With Recurrent Glioblastoma | 0/1 | 4 | Capecitabine + bevacizumab | MDSC reduction ranging between 20% and 79% from baseline | NCT02669173 |
| A Phase I/II, Open-label, Multi-center Study of the Safety and Efficacy of BLZ945 as Single Agent and in Combination With PDR001 in Adults Patients With Advanced Solid Tumors | 1/2 | 146 | CSF1R inhibitor (BLZ945) with vs. without PD1 blockade (PDR001) | MTD, 6 mo PFS | NCT02829723 | |
| A Phase 2 Study of Orally Administered PLX3397 in Patients With Recurrent Glioblastoma | 2 | 38 | CSF1R inhibitor (PLX3397) | 6 mo PFS = 8.6% | NCT01349036 | |
| Niacin (ongoing) | A Phase I-II Study of Niacin in Patients With Newly Diagnosed Glioblastoma Receiving Concurrent Radiotherapy and Temozolomide Followed by Monthly Temozolomide | 1/2 | 59 | Niacin + RT + TMZ | MTD, 6mo PFS | NCT04677049 |
| Gamma delta cells (ongoing) | A Phase I Study of Drug Resistant Immunotherapy (DRI) With Activated, Gene Modified γδ T Cells in Patients With Newly Diagnosed Glioblastoma Multiforme Receiving Maintenance Temozolomide Chemotherapy | 1 | 12 | Gene-modified gamma delta T cells | MTD | NCT04165941 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, T.Q.; Wainwright, D.A.; Lee-Chang, C.; Miska, J.; Sonabend, A.M.; Heimberger, A.B.; Lukas, R.V. Next Steps for Immunotherapy in Glioblastoma. Cancers 2022, 14, 4023. https://doi.org/10.3390/cancers14164023
Cao TQ, Wainwright DA, Lee-Chang C, Miska J, Sonabend AM, Heimberger AB, Lukas RV. Next Steps for Immunotherapy in Glioblastoma. Cancers. 2022; 14(16):4023. https://doi.org/10.3390/cancers14164023
Chicago/Turabian StyleCao, Toni Q., Derek A. Wainwright, Catalina Lee-Chang, Jason Miska, Adam M. Sonabend, Amy B. Heimberger, and Rimas V. Lukas. 2022. "Next Steps for Immunotherapy in Glioblastoma" Cancers 14, no. 16: 4023. https://doi.org/10.3390/cancers14164023
APA StyleCao, T. Q., Wainwright, D. A., Lee-Chang, C., Miska, J., Sonabend, A. M., Heimberger, A. B., & Lukas, R. V. (2022). Next Steps for Immunotherapy in Glioblastoma. Cancers, 14(16), 4023. https://doi.org/10.3390/cancers14164023