

MicroRNA–Gene Interactions Impacted by Toxic Metal(oid)s during EMT and Carcinogenesis
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
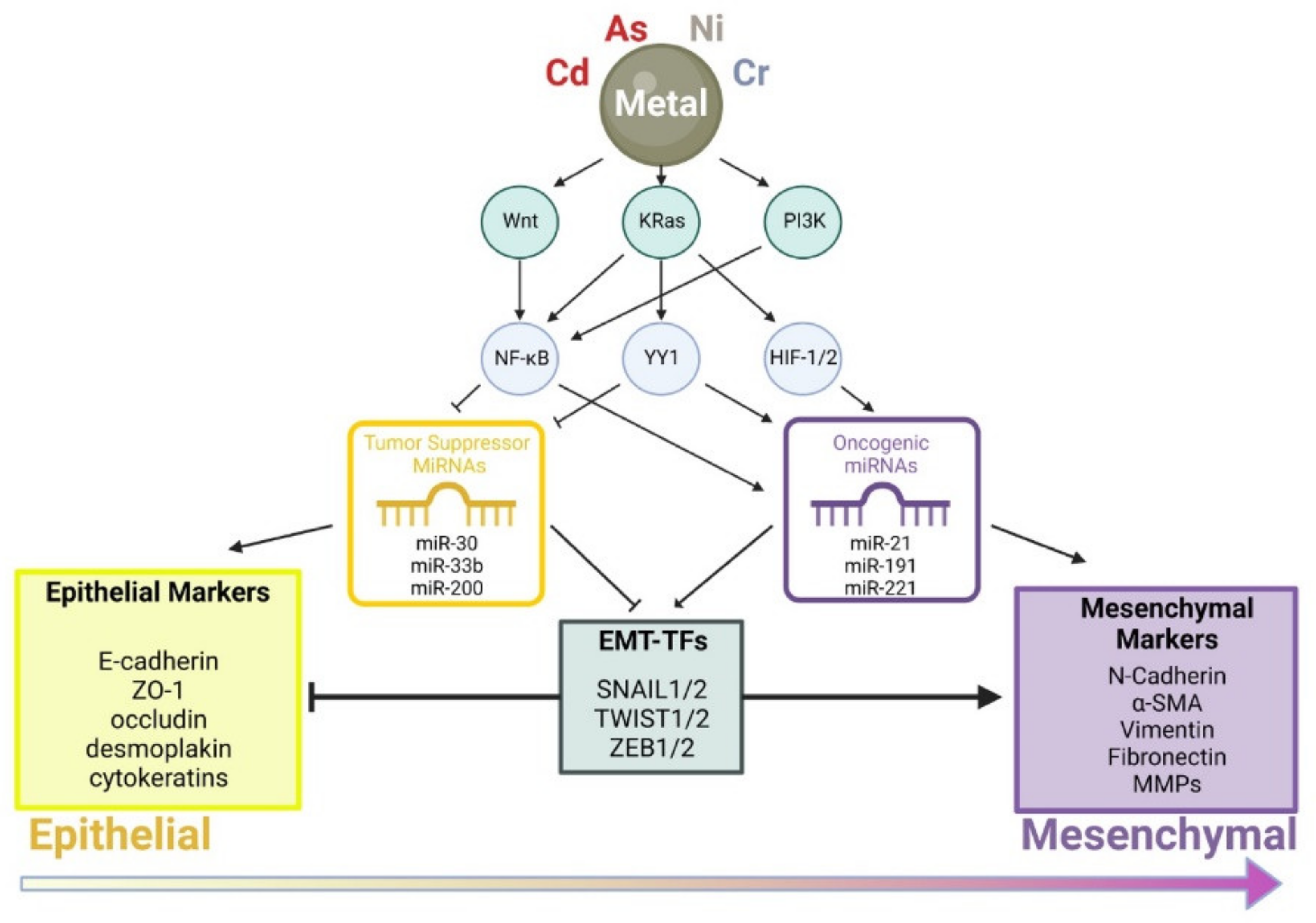
1. Introduction
2. Arsenic
2.1. Tumor Suppressing miRNAs Impacted by Arsenic Exposure
2.2. Oncogenic miRNAs Affected by Arsenic Exposure
2.3. MicroRNAs Impacted by Therapeutic Use of Arsenic
2.4. Emerging miRNA Targets in Arsenic Treated Cells
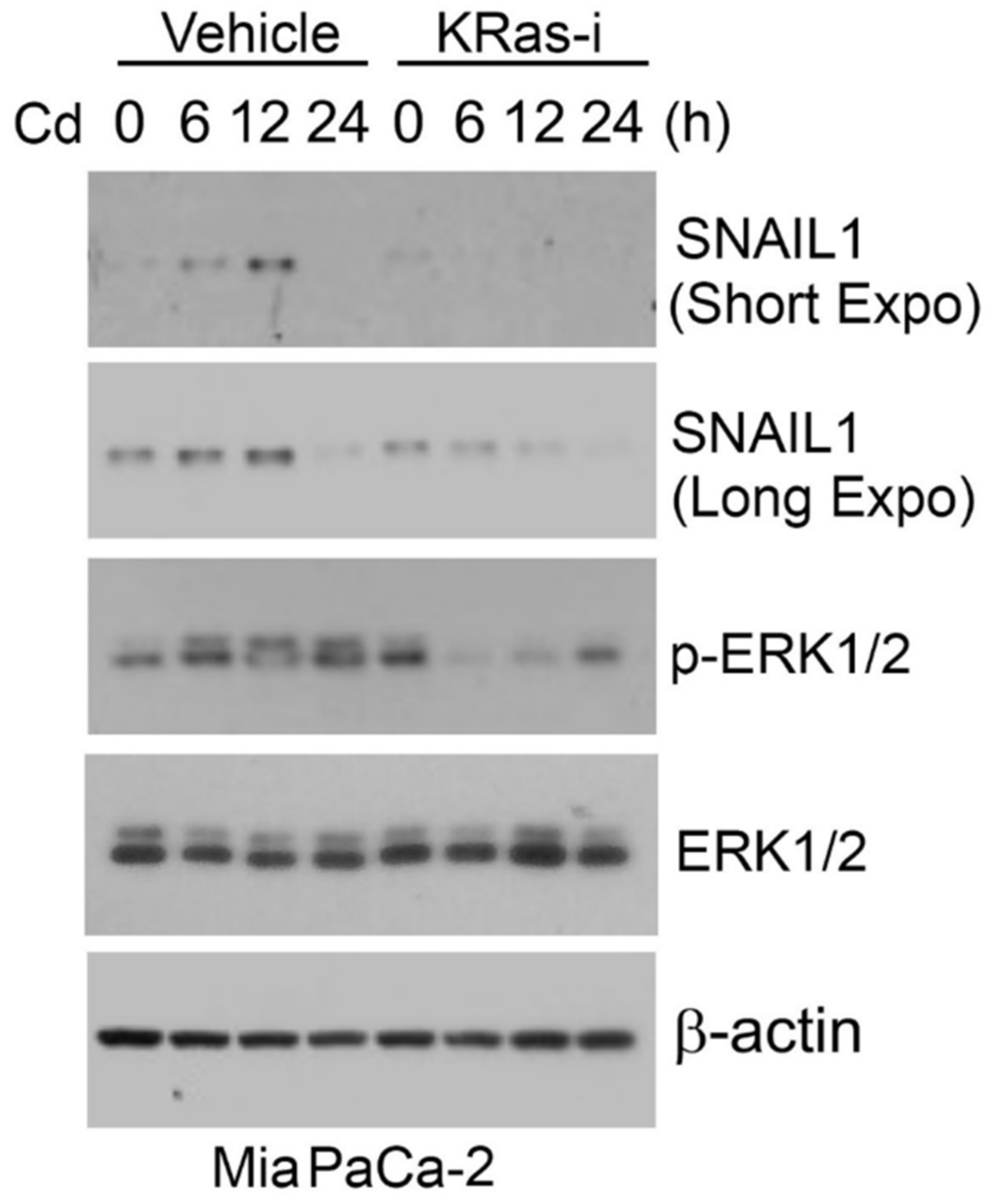
3. Cadmium
3.1. Tumor Suppressing miRNAs in Cd-Induced EMT
3.2. Oncogenic miRNAs Impacted by Cd Exposure
4. Nickel
4.1. Tumor Suppressing miRNAs Regulated by Ni
4.2. Oncogenic miRNAs Regulated by Ni
5. Chromium
5.1. Tumor Suppressing miRNAs Impacted by Cr(VI)
5.2. Oncogenic miRNAs Impacted by Cr(VI)
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fidler, I.J.; Poste, G. The “Seed and Soil” Hypothesis Revisited. Lancet Oncol. 2008, 9, 808. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular Mechanisms of Epithelial–Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Lecuit, T.; Le Goff, L. Orchestrating Size and Shape during Morphogenesis. Nature 2007, 450, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Marconi, G.D.; Fonticoli, L.; Rajan, T.S.; Pierdomenico, S.D.; Trubiani, O.; Pizzicannella, J.; Diomede, F. Epithelial-Mesenchymal Transition (EMT): The Type-2 EMT in Wound Healing, Tissue Regeneration and Organ Fibrosis. Cells 2021, 10, 1587. [Google Scholar] [CrossRef]
- Sauka-Spengler, T.; Bronner-Fraser, M. A Gene Regulatory Network Orchestrates Neural Crest Formation. Nat. Rev. Mol. Cell Biol. 2008, 9, 557–568. [Google Scholar] [CrossRef]
- Tiwari, N.; Gheldof, A.; Tatari, M.; Christofori, G. EMT as the Ultimate Survival Mechanism of Cancer Cells. Semin. Cancer Biol. 2012, 22, 194–207. [Google Scholar] [CrossRef]
- Ikenouchi, J.; Matsuda, M.; Furuse, M.; Tsukita, S. Regulation of Tight Junctions during the Epithelium-Mesenchyme Transition:Direct Repression of the Gene Expression of Claudins/Occludin by Snail. J. Cell Sci. 2003, 116, 1959–1967. [Google Scholar] [CrossRef]
- Savagner, P. The Epithelial–Mesenchymal Transition (EMT) Phenomenon. Ann. Oncol. 2010, 21, vii89–vii92. [Google Scholar] [CrossRef]
- Campbell, K.; Casanova, J. A Common Framework for EMT and Collective Cell Migration. Development 2016, 143, 4291–4300. [Google Scholar] [CrossRef]
- Evdokimova, V.; Tognon, C.; Ng, T.; Ruzanov, P.; Melnyk, N.; Fink, D.; Sorokin, A.; Ovchinnikov, L.P.; Davicioni, E.; Triche, T.J.; et al. Translational Activation of Snail1 and Other Developmentally Regulated Transcription Factors by YB-1 Promotes an Epithelial-Mesenchymal Transition. Cancer Cell 2009, 15, 402–415. [Google Scholar] [CrossRef]
- Chidgey, M.; Dawson, C. Desmosomes: A Role in Cancer? Br. J. Cancer 2007, 96, 1783–1787. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Christofori, G. EMT, the Cytoskeleton, and Cancer Cell Invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef]
- Hart, K.C.; Tan, J.; Siemers, K.A.; Sim, J.Y.; Pruitt, B.L.; Nelson, W.J.; Gloerich, M. E-Cadherin and LGN Align Epithelial Cell Divisions with Tissue Tension Independently of Cell Shape. Proc. Natl. Acad. Sci. USA 2017, 114, 3114. [Google Scholar] [CrossRef] [PubMed]
- Przybylo, J.A.; Radisky, D.C. Matrix Metalloproteinase-Induced Epithelial–Mesenchymal Transition: Tumor Progression at Snail’s Pace. Int. J. Biochem. Cell Biol. 2007, 39, 1082–1088. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, T.; Ibaragi, S.; Hu, G. Epithelial-Mesenchymal Transition and Cell Cooperativity in Metastasis. Cancer Res. 2009, 69, 7135–7139. [Google Scholar] [CrossRef] [PubMed]
- Massaous, J.; Hata, A. TGF-β Signalling through the Smad Pathway. Trends Cell Biol. 1997, 7, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K. Transforming Growth Factor-.BETA. Signaling in Epithelial-Mesenchymal Transition and Progression of Cancer. Proc. Jpn. Acad. Ser. B 2009, 85, 314–323. [Google Scholar] [CrossRef]
- Basu, S.; Cheriyamundath, S.; Ben-Ze’ev, A. Cell–Cell Adhesion: Linking Wnt/β-Catenin Signaling with Partial EMT and Stemness Traits in Tumorigenesis. F1000Res 2018, 7, 1488. [Google Scholar] [CrossRef]
- Secker, G.A.; Shortt, A.J.; Sampson, E.; Schwarz, Q.P.; Schultz, G.S.; Daniels, J.T. TGFβ Stimulated Re-Epithelialisation Is Regulated by CTGF and Ras/MEK/ERK Signalling. Exp. Cell Res. 2008, 314, 131–142. [Google Scholar] [CrossRef]
- Allen, C.E.; Du, J.; Jiang, B.; Huang, Q.; Yakovich, A.J.; Barnard, J.A. Transformation by Oncogenic Ras Expands the Early Genomic Response to Transforming Growth Factor β in Intestinal Epithelial Cells. Neoplasia 2008, 10, 1073–1082. [Google Scholar] [CrossRef]
- Janda, E.; Lehmann, K.; Killisch, I.; Jechlinger, M.; Herzig, M.; Downward, J.; Beug, H.; Grünert, S. Ras and TGFβ Cooperatively Regulate Epithelial Cell Plasticity and Metastasis. J. Cell Biol. 2002, 156, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Prasad, C.P.; Gupta, S.D.; Rath, G.; Ralhan, R. Wnt Signaling Pathway in Invasive Ductal Carcinoma of the Breast: Relationship between β-Catenin, Disheveled and Cyclin D1 Expression. Oncology 2007, 73, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Stemmer, V.; de Craene, B.; Berx, G.; Behrens, J. Snail Promotes Wnt Target Gene Expression and Interacts with β-Catenin. Oncogene 2008, 27, 5075–5080. [Google Scholar] [CrossRef] [PubMed]
- Gottardi, C.J.; Wong, E.; Gumbiner, B.M. E-Cadherin Suppresses Cellular Transformation by Inhibiting β-Catenin Signaling in an Adhesion-Independent Manner. J. Cell Biol. 2001, 153, 1049–1060. [Google Scholar] [CrossRef]
- Suarez-Carmona, M.; Lesage, J.; Cataldo, D.; Gilles, C. EMT and Inflammation: Inseparable Actors of Cancer Progression. Mol. Oncol. 2017, 11, 805–823. [Google Scholar] [CrossRef]
- Huang, C.; Yang, G.; Jiang, T.; Zhu, G.; Li, H.; Qiu, Z. The Effects and Mechanisms of Blockage of STAT3 Signaling Pathway on IL-6 Inducing EMT in Human Pancreatic Cancer Cells in Vitro. NEO 2011, 58, 396–405. [Google Scholar] [CrossRef]
- Huang, X.; Dai, S.; Dai, J.; Xiao, Y.; Bai, Y.; Chen, B.; Zhou, M. Luteolin Decreases Invasiveness, Deactivates STAT3 Signaling, and Reverses Interleukin-6 Induced Epithelial-Mesenchymal Transition and Matrix Metalloproteinase Secretion of Pancreatic Cancer Cells. Onco. Targets 2015, 8, 2989–3001. [Google Scholar] [CrossRef]
- Squarize, C.H.; Castilho, R.M.; Sriuranpong, V.; Pinto, D.S.; Gutkind, J.S. Molecular Cross-Talk between the NFκB and STAT3 Signaling Pathways in Head and Neck Squamous Cell Carcinoma. Neoplasia 2006, 8, 733–746. [Google Scholar] [CrossRef]
- Julien, S.; Puig, I.; Caretti, E.; Bonaventure, J.; Nelles, L.; van Roy, F.; Dargemont, C.; de Herreros, A.G.; Bellacosa, A.; Larue, L. Activation of NF-ΚB by Akt Upregulates Snail Expression and Induces Epithelium Mesenchyme Transition. Oncogene 2007, 26, 7445–7456. [Google Scholar] [CrossRef]
- Hobert, O. Gene Regulation by Transcription Factors and MicroRNAs. Science 2008, 319, 1785–1786. [Google Scholar] [CrossRef]
- Shenouda, S.K.; Alahari, S.K. MicroRNA Function in Cancer: Oncogene or a Tumor Suppressor? Cancer Metastasis Rev. 2009, 28, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.-Q.; Chen, Z.-Q.; Cao, X.-X.; Xu, J.-D.; Xu, J.-W.; Chen, Y.-Y.; Wang, W.-J.; Chen, Q.; Tang, F.; Liu, X.-P.; et al. Involvement of NF-ΚB/MiR-448 Regulatory Feedback Loop in Chemotherapy-Induced Epithelial–Mesenchymal Transition of Breast Cancer Cells. Cell Death Differ. 2011, 18, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Gautam, N.; Mishra, A.; Gupta, R. Heavy Metals and Living Systems: An Overview. Indian J. Pharm. 2011, 43, 246. [Google Scholar] [CrossRef] [PubMed]
- Popoola, O.E.; Popoola, A.O.; Purchase, D. Levels of Awareness and Concentrations of Heavy Metals in the Blood of Electronic Waste Scavengers in Nigeria. J. Health Pollut. 2019, 9, 190311. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Kim, Y.J.; Seo, Y.R. An Overview of Carcinogenic Heavy Metal: Molecular Toxicity Mechanism and Prevention. J. Cancer Prev. 2015, 20, 232–240. [Google Scholar] [CrossRef]
- Ng, J.C. Environmental Contamination of Arsenic and Its Toxicological Impact on Humans. Environ. Chem. 2005, 2, 146. [Google Scholar] [CrossRef]
- Smith, A.H.; Smith, M.M.H. Arsenic Drinking Water Regulations in Developing Countries with Extensive Exposure. Toxicology 2004, 198, 39–44. [Google Scholar] [CrossRef]
- Kitchin, K.T. Recent Advances in Arsenic Carcinogenesis: Modes of Action, Animal Model Systems, and Methylated Arsenic Metabolites. Toxicol. Appl. Pharmacol. 2001, 172, 249–261. [Google Scholar] [CrossRef]
- Vahter, M.; Concha, G. Role of Metabolism in Arsenic Toxicity. Pharmacol. Toxicol. 2008, 89, 1–5. [Google Scholar] [CrossRef]
- Benbrahim-Tallaa, L.; Webber, M.M.; Waalkes, M.P. Mechanisms of Acquired Androgen Independence during Arsenic-Induced Malignant Transformation of Human Prostate Epithelial Cells. Environ. Health Perspect. 2007, 115, 243–247. [Google Scholar] [CrossRef]
- Chen, Q.Y.; Costa, M. PI3K/Akt/MTOR Signaling Pathway and the Biphasic Effect of Arsenic in Carcinogenesis. Mol. Pharm. 2018, 94, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Duan, Q.; Komissarova, E.; Dai, W. Arsenic Trioxide Suppresses Paclitaxel-Induced Mitotic Arrest. Cell Prolif. 2009, 42, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Soignet, S.L.; Maslak, P.; Wang, Z.-G.; Jhanwar, S.; Calleja, E.; Dardashti, L.J.; Corso, D.; DeBlasio, A.; Gabrilove, J.; Scheinberg, D.A.; et al. Complete Remission after Treatment of Acute Promyelocytic Leukemia with Arsenic Trioxide. N. Engl. J. Med. 1998, 339, 1341–1348. [Google Scholar] [CrossRef]
- Cui, X.; Wakai, T.; Shirai, Y.; Yokoyama, N.; Hatakeyama, K.; Hirano, S. Arsenic Trioxide Inhibits DNA Methyltransferase and Restores Methylation-Silenced Genes in Human Liver Cancer Cells. Hum. Pathol. 2006, 37, 298–311. [Google Scholar] [CrossRef] [PubMed]
- Michailidi, C.; Hayashi, M.; Datta, S.; Sen, T.; Zenner, K.; Oladeru, O.; Brait, M.; Izumchenko, E.; Baras, A.; VandenBussche, C.; et al. Involvement of Epigenetics and EMT-Related MiRNA in Arsenic-Induced Neoplastic Transformation and Their Potential Clinical Use. Cancer Prev. Res. 2015, 8, 208–221. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Humphries, B.; Xiao, H.; Jiang, Y.; Yang, C. Epithelial to Mesenchymal Transition in Arsenic-Transformed Cells Promotes Angiogenesis through Activating β-Catenin–Vascular Endothelial Growth Factor Pathway. Toxicol. Appl. Pharmacol. 2013, 271, 20–29. [Google Scholar] [CrossRef][Green Version]
- Yang, D.; Tang, S.; Yang, Y.; Yang, F.; Jiang, W.; Liu, Y.; Zhang, F.; Fang, H.; Wang, S.; Zhang, Y. Generation and Validation of MiR-100 Hepatocyte-Specific Knock-Out Mice. Front. Oncol. 2019, 9, 535. [Google Scholar] [CrossRef]
- Yang, J.; Chen, Z.; Wang, X.; Xu, M.; Fang, H.; Li, F.; Liu, Y.; Jiang, Y.; Ding, Y.; Li, J.; et al. Inactivation of MiR-100 Combined with Arsenic Treatment Enhances the Malignant Transformation of BEAS-2B Cells via Stimulating Epithelial-Mesenchymal Transition. Cancer Biol. Ther. 2017, 18, 965–973. [Google Scholar] [CrossRef]
- Wang, Q.; Yang, H.-S. The Role of Pdcd4 in Tumour Suppression and Protein Translation: Pdcd4 in Tumour Suppression and Protein Translation. Biol. Cell 2018, 110, 169–177. [Google Scholar] [CrossRef]
- Luo, F.; Ji, J.; Liu, Y.; Xu, Y.; Zheng, G.; Jing, J.; Wang, B.; Xu, W.; Shi, L.; Lu, X.; et al. MicroRNA-21, up-Regulated by Arsenite, Directs the Epithelial–Mesenchymal Transition and Enhances the Invasive Potential of Transformed Human Bronchial Epithelial Cells by Targeting PDCD4. Toxicol. Lett. 2015, 232, 301–309. [Google Scholar] [CrossRef]
- Luo, F.; Xu, Y.; Ling, M.; Zhao, Y.; Xu, W.; Liang, X.; Jiang, R.; Wang, B.; Bian, Q.; Liu, Q. Arsenite Evokes IL-6 Secretion, Autocrine Regulation of STAT3 Signaling, and MiR-21 Expression, Processes Involved in the EMT and Malignant Transformation of Human Bronchial Epithelial Cells. Toxicol. Appl. Pharmacol. 2013, 273, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Luo, F.; Liu, Y.; Zhang, A.; Li, J.; Wang, B.; Xu, W.; Shi, L.; Liu, X.; Lu, L.; et al. The IL-6/STAT3 Pathway via MiR-21 Is Involved in the Neoplastic and Metastatic Properties of Arsenite-Transformed Human Keratinocytes. Toxicol. Lett. 2015, 237, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Yang, Q.; Wang, D.; Luo, F.; Liu, X.; Xue, J.; Yang, P.; Xu, H.; Lu, J.; Zhang, A.; et al. MicroRNA-191, Regulated by HIF-2α, Is Involved in EMT and Acquisition of a Stem Cell-like Phenotype in Arsenite-Transformed Human Liver Epithelial Cells. Toxicol. Vitr. 2018, 48, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Tapeh, B.E.-G.; Alivand, M.R.; Solalii, S. Potential Interactions between MiRNAs and Hypoxia: A New Layer in Cancer Hypoxia. Anti-Cancer Agents Med. Chem. 2021, 21, 2315–2326. [Google Scholar] [CrossRef]
- Xue, J.; Chen, C.; Luo, F.; Pan, X.; Xu, H.; Yang, P.; Sun, Q.; Liu, X.; Lu, L.; Yang, Q.; et al. CircLRP6 Regulation of ZEB1 via MiR-455 Is Involved in the Epithelial-Mesenchymal Transition During Arsenite-Induced Malignant Transformation of Human Keratinocytes. Toxicol. Sci. 2018, 162, 450–461. [Google Scholar] [CrossRef]
- Xiao, T.; Xue, J.; Shi, M.; Chen, C.; Luo, F.; Xu, H.; Chen, X.; Sun, B.; Sun, Q.; Yang, Q.; et al. Circ008913, via MiR-889 Regulation of DAB2IP/ZEB1, Is Involved in the Arsenite-Induced Acquisition of CSC-like Properties by Human Keratinocytes in Carcinogenesis. Metallomics 2018, 10, 1328–1338. [Google Scholar] [CrossRef]
- Yun, E.-J.; Baek, S.T.; Xie, D.; Tseng, S.-F.; Dobin, T.; Hernandez, E.; Zhou, J.; Zhang, L.; Yang, J.; Sun, H.; et al. DAB2IP Regulates Cancer Stem Cell Phenotypes through Modulating Stem Cell Factor Receptor and ZEB1. Oncogene 2015, 34, 2741–2752. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, F.; Mu, J.; Ye, X.; Si, L.; Ning, S.; Li, Z.; Li, Y. Arsenic Trioxide Attenuates the Invasion Potential of Human Liver Cancer Cells through the Demethylation-Activated MicroRNA-491. Toxicol. Lett. 2014, 227, 75–83. [Google Scholar] [CrossRef]
- Liao, Y.; Wei, Y.; Zhou, X.; Yang, J.-Y.; Dai, C.; Chen, Y.-J.; Agarwal, N.K.; Sarbassov, D.; Shi, D.; Yu, D.; et al. Peptidyl-Prolyl Cis/Trans Isomerase Pin1 Is Critical for the Regulation of PKB/Akt Stability and Activation Phosphorylation. Oncogene 2009, 28, 2436–2445. [Google Scholar] [CrossRef]
- Liou, Y.-C.; Ryo, A.; Huang, H.-K.; Lu, P.-J.; Bronson, R.; Fujimori, F.; Uchida, T.; Hunter, T.; Lu, K.P. Loss of Pin1 Function in the Mouse Causes Phenotypes Resembling Cyclin D1-Null Phenotypes. Proc. Natl. Acad. Sci. USA 2002, 99, 1335–1340. [Google Scholar] [CrossRef]
- Ryo, A.; Nakamura, M.; Wulf, G.; Liou, Y.-C.; Lu, K.P. Pin1 Regulates Turnover and Subcellular Localization of β-Catenin by Inhibiting Its Interaction with APC. Nat. Cell Biol. 2001, 3, 793–801. [Google Scholar] [CrossRef]
- Kozono, S.; Lin, Y.-M.; Seo, H.-S.; Pinch, B.; Lian, X.; Qiu, C.; Herbert, M.K.; Chen, C.-H.; Tan, L.; Gao, Z.J.; et al. Arsenic Targets Pin1 and Cooperates with Retinoic Acid to Inhibit Cancer-Driving Pathways and Tumor-Initiating Cells. Nat. Commun. 2018, 9, 3069. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Sun, D.; Li, X.; Zhang, Z. Alterations of MiRNAs and Their Potential Roles in Arsenite-Induced Transformation of Human Bronchial Epithelial Cells. Genes 2017, 8, 254. [Google Scholar] [CrossRef]
- Qu, J.; Li, M.; An, J.; Zhao, B.; Zhong, W.; Gu, Q.; Cao, L.; Yang, H.; Hu, C. MicroRNA-33b Inhibits Lung Adenocarcinoma Cell Growth, Invasion, and Epithelial-Mesenchymal Transition by Suppressing Wnt/β-Catenin/ZEB1 Signaling. Int. J. Oncol. 2015, 47, 2141–2152. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhao, Y.; Smith, E.; Goodall, G.J.; Drew, P.A.; Brabletz, T.; Yang, C. Reversal and Prevention of Arsenic-Induced Human Bronchial Epithelial Cell Malignant Transformation by MicroRNA-200b. Toxicol. Sci. 2011, 121, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Seelan, R.S.; Greene, R.M.; Pisano, M.M. Impact of Prenatal Arsenate Exposure on Gene Expression in a Pure Population of Migratory Cranial Neural Crest Cells. Reprod. Toxicol. 2019, 86, 76–85. [Google Scholar] [CrossRef]
- Zhao, Q.; Jiang, F.; Zhuang, H.; Chu, Y.; Zhang, F.; Wang, C. MicroRNA MiR-124-3p Suppresses Proliferation and Epithelial–Mesenchymal Transition of Hepatocellular Carcinoma via ARRDC1 (Arrestin Domain Containing 1). Bioengineered 2022, 13, 8255–8265. [Google Scholar] [CrossRef]
- Chen, L.; Wang, Q.; Wang, G.; Wang, H.; Huang, Y.; Liu, X.; Cai, X. MiR-16 Inhibits Cell Proliferation by Targeting IGF1R and the Raf1–MEK1/2–ERK1/2 Pathway in Osteosarcoma. FEBS Lett. 2013, 587, 1366–1372. [Google Scholar] [CrossRef] [PubMed]
- Palus, J.; Rydzynski, K.; Dziubaltowska, E.; Wyszynska, K.; Natarajan, A.T.; Nilsson, R. Genotoxic Effects of Occupational Exposure to Lead and Cadmium. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2003, 540, 19–28. [Google Scholar] [CrossRef]
- Hayat, M.T.; Nauman, M.; Nazir, N.; Ali, S.; Bangash, N. Environmental Hazards of Cadmium: Past, Present, and Future. In Cadmium Toxicity and Tolerance in Plants; Elsevier: Amsterdam, The Netherlands, 2019; pp. 163–183. ISBN 9780128148648. [Google Scholar]
- Satarug, S.; Garrett, S.H.; Sens, M.A.; Sens, D.A. Cadmium, Environmental Exposure, and Health Outcomes. Environ. Health Perspect. 2010, 118, 182–190. [Google Scholar] [CrossRef]
- Rani, A.; Kumar, A.; Lal, A.; Pant, M. Cellular Mechanisms of Cadmium-Induced Toxicity: A Review. Int. J. Environ. Health Res. 2014, 24, 378–399. [Google Scholar] [CrossRef]
- Tanwar, V.S.; Zhang, X.; Jagannathan, L.; Jose, C.C.; Cuddapah, S. Cadmium Exposure Upregulates SNAIL through MiR-30 Repression in Human Lung Epithelial Cells. Toxicol. Appl. Pharmacol. 2019, 373, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Jiang, Y.-L.; Fei, J.; Cao, P.; Zhang, C.; Xie, G.-F.; Wang, L.-X.; Cao, W.; Fu, L.; Zhao, H. Circulatory Cadmium Positively Correlates with Epithelial-Mesenchymal Transition in Patients with Chronic Obstructive Pulmonary Disease. Ecotoxicol. Environ. Saf. 2021, 215, 112164. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Wang, L.; Lu, H.; Yang, G.; Zhang, Z.; Fu, H.; Lu, X.; Wei, M.; Sun, J.; Zhao, Q.; et al. Downregulation of Tumor Suppressor QKI in Gastric Cancer and Its Implication in Cancer Prognosis. Biochem. Biophys. Res. Commun. 2012, 422, 187–193. [Google Scholar] [CrossRef]
- Zhou, M.; Li, L.; Chen, B.; Pan, S.; Tu, W.; Hou, Y.; Chen, P.; Hernández, R.R.; Zhou, X. Circ-SHPRH Suppresses Cadmium-Induced Transformation of Human Bronchial Epithelial Cells by Regulating QKI Expression via MiR-224–5p. Ecotoxicol. Environ. Saf. 2021, 220, 112378. [Google Scholar] [CrossRef] [PubMed]
- Mortoglou, M.; Buha Djordjevic, A.; Djordjevic, V.; Collins, H.; York, L.; Mani, K.; Valle, E.; Wallace, D.; Uysal-Onganer, P. Role of MicroRNAs in Response to Cadmium Chloride in Pancreatic Ductal Adenocarcinoma. Arch. Toxicol. 2022, 96, 467–485. [Google Scholar] [CrossRef]
- Choi, B.H.; Philips, M.R.; Chen, Y.; Lu, L.; Dai, W. K-Ras Lys-42 Is Crucial for Its Signaling, Cell Migration, and Invasion. J. Biol. Chem. 2018, 293, 17574–17581. [Google Scholar] [CrossRef]
- Choi, B.H.; Kou, Z.; Colon, T.M.; Chen, C.-H.; Chen, Y.; Dai, W. Identification of Radil as a Ras Binding Partner and Putative Activator. J. Biol. Chem. 2021, 296, 100314. [Google Scholar] [CrossRef]
- Shao, D.D.; Xue, W.; Krall, E.B.; Bhutkar, A.; Piccioni, F.; Wang, X.; Schinzel, A.C.; Sood, S.; Rosenbluh, J.; Kim, J.W.; et al. KRAS and YAP1 Converge to Regulate EMT and Tumor Survival. Cell 2014, 158, 171–184. [Google Scholar] [CrossRef]
- Ngalame, N.N.O.; Waalkes, M.P.; Tokar, E.J. Silencing KRAS Overexpression in Cadmium-Transformed Prostate Epithelial Cells Mitigates Malignant Phenotype. Chem. Res. Toxicol. 2016, 29, 1458–1467. [Google Scholar] [CrossRef]
- Lin, X.; Chen, L.; Yao, Y.; Zhao, R.; Cui, X.; Chen, J.; Hou, K.; Zhang, M.; Su, F.; Chen, J.; et al. CCL18-Mediated down-Regulation of MiR98 and MiR27b Promotes Breast Cancer Metastasis. Oncotarget 2015, 6, 20485–20499. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.; Woo, Y.; Valiathan, R.R.; Jung, H.Y.; Park, S.Y.; Kim, Y.N.; Kim, H.-R.C.; Fridman, R.; Moon, A. Discoidin Domain Receptor 1 Is a Novel Transcriptional Target of ZEB1 in Breast Epithelial Cells Undergoing H-Ras-Induced Epithelial to Mesenchymal Transition: Discoidin Domain Receptor 1 Is a Target of ZEB1. Int. J. Cancer 2015, 136, E508–E520. [Google Scholar] [CrossRef] [PubMed]
- Sarvagalla, S.; Kolapalli, S.P.; Vallabhapurapu, S. The Two Sides of YY1 in Cancer: A Friend and a Foe. Front. Oncol. 2019, 9, 1230. [Google Scholar] [CrossRef] [PubMed]
- Verheul, T.C.J.; van Hijfte, L.; Perenthaler, E.; Barakat, T.S. The Why of YY1: Mechanisms of Transcriptional Regulation by Yin Yang 1. Front. Cell Dev. Biol. 2020, 8, 592164. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-C.; Kuo, I.-Y.; Wu, L.-T.; Kuan, W.-H.; Liao, S.-Y.; Jen, J.; Yang, Y.-E.; Tang, C.-W.; Chen, Y.-R.; Wang, Y.-C. Dysregulated Kras/YY1/ZNF322A/Shh Transcriptional Axis Enhances Neo-Angiogenesis to Promote Lung Cancer Progression. Theranostics 2020, 10, 10001–10015. [Google Scholar] [CrossRef]
- Yuan, P.; He, X.-H.; Rong, Y.-F.; Cao, J.; Li, Y.; Hu, Y.-P.; Liu, Y.; Li, D.; Lou, W.; Liu, M.-F. KRAS/NF-ΚB/YY1/MiR-489 Signaling Axis Controls Pancreatic Cancer Metastasis. Cancer Res. 2017, 77, 100–111. [Google Scholar] [CrossRef]
- Huang, Y.; Tao, T.; Liu, C.; Guan, H.; Zhang, G.; Ling, Z.; Zhang, L.; Lu, K.; Chen, S.; Xu, B.; et al. Upregulation of MiR-146a by YY1 Depletion Correlates with Delayed Progression of Prostate Cancer. Int. J. Oncol. 2017, 50, 421–431. [Google Scholar] [CrossRef]
- Yi, C.; Li, G.; Wang, W.; Sun, Y.; Zhang, Y.; Zhong, C.; Stovall, D.B.; Li, D.; Shi, J.; Sui, G. Disruption of YY1-EZH2 Interaction Using Synthetic Peptides Inhibits Breast Cancer Development. Cancers 2021, 13, 2402. [Google Scholar] [CrossRef]
- Genchi, G.; Carocci, A.; Lauria, G.; Sinicropi, M.S.; Catalano, A. Nickel: Human Health and Environmental Toxicology. Int. J. Environ. Res. Public Health 2020, 17, 679. [Google Scholar] [CrossRef]
- De Brouwere, K.; Buekers, J.; Cornelis, C.; Schlekat, C.E.; Oller, A.R. Assessment of Indirect Human Exposure to Environmental Sources of Nickel: Oral Exposure and Risk Characterization for Systemic Effects. Sci. Total Environ. 2012, 419, 25–36. [Google Scholar] [CrossRef]
- Kasprzak, K. Nickel Carcinogenesis. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2003, 533, 67–97. [Google Scholar] [CrossRef] [PubMed]
- Pietruska, J.R.; Liu, X.; Smith, A.; McNeil, K.; Weston, P.; Zhitkovich, A.; Hurt, R.; Kane, A.B. Bioavailability, Intracellular Mobilization of Nickel, and HIF-1α Activation in Human Lung Epithelial Cells Exposed to Metallic Nickel and Nickel Oxide Nanoparticles. Toxicol. Sci. 2011, 124, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Cruz, M.T.; Goncalo, M.; Figueiredo, A.; Carvalho, A.P.; Duarte, C.B.; Lopes, M.C. Contact Sensitizer Nickel Sulfate Activates the Transcription Factors NF-KB and AP-1 and Increases the Expression of Nitric Oxide Synthase in a Skin Dendritic Cell Line. Exp. Derm. 2004, 13, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Jose, C.C.; Jagannathan, L.; Tanwar, V.S.; Zhang, X.; Zang, C.; Cuddapah, S. Nickel Exposure Induces Persistent Mesenchymal Phenotype in Human Lung Epithelial Cells through Epigenetic Activation of ZEB1. Mol. Carcinog. 2018, 57, 794–806. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-H.; Tang, S.-C.; Wang, P.-H.; Lee, H.; Ko, J.-L. Nickel-Induced Epithelial-Mesenchymal Transition by Reactive Oxygen Species Generation and E-Cadherin Promoter Hypermethylation. J. Biol. Chem. 2012, 287, 25292–25302. [Google Scholar] [CrossRef]
- Chiou, Y.-H.; Liou, S.-H.; Wong, R.-H.; Chen, C.-Y.; Lee, H. Nickel May Contribute to EGFR Mutation and Synergistically Promotes Tumor Invasion in EGFR-Mutated Lung Cancer via Nickel-Induced MicroRNA-21 Expression. Toxicol. Lett. 2015, 237, 46–54. [Google Scholar] [CrossRef] [PubMed]
- To, W.S.; Midwood, K.S. Plasma and Cellular Fibronectin: Distinct and Independent Functions during Tissue Repair. Fibrogenesis Tissue Repair 2011, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Golubnitschaja, O.; Yeghiazaryan, K. Opinion Controversy to Chromium Picolinate Therapy’s Safety and Efficacy: Ignoring ‘Anecdotes’ of Case Reports or Recognising Individual Risks and New Guidelines Urgency to Introduce Innovation by Predictive Diagnostics? EPMA J. 2012, 3, 11. [Google Scholar] [CrossRef]
- Seidler, A.; Jähnichen, S.; Hegewald, J.; Fishta, A.; Krug, O.; Rüter, L.; Strik, C.; Hallier, E.; Straube, S. Systematic Review and Quantification of Respiratory Cancer Risk for Occupational Exposure to Hexavalent Chromium. Int. Arch. Occup. Environ. Health 2013, 86, 943–955. [Google Scholar] [CrossRef]
- Hu, L.; Liu, X.; Chervona, Y.; Yang, F.; Tang, M.; Darzynkiewicz, Z.; Dai, W. Chromium Induces Chromosomal Instability, Which Is Partly Due to Deregulation of BubR1 and Emi1, Two APC/C Inhibitors. Cell Cycle 2011, 10, 2373–2379. [Google Scholar] [CrossRef]
- Nickens, K.P.; Patierno, S.R.; Ceryak, S. Chromium Genotoxicity: A Double-Edged Sword. Chem. Biol. Interact. 2010, 188, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Chuang, S.-M.; Liou, G.-Y.; Yang, J.-L. Activation of JNK, P38 and ERK Mitogen-Activated Protein Kinases by Chromium(VI) Is Mediated through Oxidative Stress but Does Not Affect Cytotoxicity. Carcinogenesis 2000, 21, 1491–1500. [Google Scholar] [CrossRef] [PubMed]
- Qie, Y.; Zhou, D.; Wu, Z.; Liu, S.; Shen, C.; Hu, H.; Zhang, C.; Xu, Y. Low-Dose Hexavalent Chromium(VI) Exposure Promotes Prostate Cancer Cell Proliferation by Activating MAGEB2-AR Signal Pathway. Ecotoxicol. Environ. Saf. 2022, 241, 113724. [Google Scholar] [CrossRef]
- Wang, L.; Bayanbold, K.; Zhao, L.; Wang, Y.; Adamcakova-Dodd, A.; Thorne, P.S.; Yang, H.; Jiang, B.-H.; Liu, L.-Z. Redox Sensitive MiR-27a/b/Nrf2 Signaling in Cr(VI)-Induced Carcinogenesis. Sci. Total Environ. 2022, 809, 151118. [Google Scholar] [CrossRef] [PubMed]
- Bocci, F.; Tripathi, S.C.; Vilchez Mercedes, S.A.; George, J.T.; Casabar, J.P.; Wong, P.K.; Hanash, S.M.; Levine, H.; Onuchic, J.N.; Jolly, M.K. NRF2 Activates a Partial Epithelial-Mesenchymal Transition and Is Maximally Present in a Hybrid Epithelial/Mesenchymal Phenotype. Integr. Biol. 2019, 11, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Yazaki, K.; Matsuno, Y.; Yoshida, K.; Sherpa, M.; Nakajima, M.; Matsuyama, M.; Kiwamoto, T.; Morishima, Y.; Ishii, Y.; Hizawa, N. ROS-Nrf2 Pathway Mediates the Development of TGF-Β1-Induced Epithelial-Mesenchymal Transition through the Activation of Notch Signaling. Eur. J. Cell Biol. 2021, 100, 151181. [Google Scholar] [CrossRef]
- Pratheeshkumar, P.; Son, Y.-O.; Divya, S.P.; Turcios, L.; Roy, R.V.; Hitron, J.A.; Wang, L.; Kim, D.; Dai, J.; Asha, P.; et al. Hexavalent Chromium Induces Malignant Transformation of Human Lung Bronchial Epithelial Cells via ROS-Dependent Activation of MiR-21-PDCD4 Signaling. Oncotarget 2016, 7, 51193–51210. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, F.; Lee, E.; Cuddapah, S.; Choi, B.H.; Dai, W. MicroRNA–Gene Interactions Impacted by Toxic Metal(oid)s during EMT and Carcinogenesis. Cancers 2022, 14, 5818. https://doi.org/10.3390/cancers14235818
Tran F, Lee E, Cuddapah S, Choi BH, Dai W. MicroRNA–Gene Interactions Impacted by Toxic Metal(oid)s during EMT and Carcinogenesis. Cancers. 2022; 14(23):5818. https://doi.org/10.3390/cancers14235818
Chicago/Turabian StyleTran, Franklin, Eunji Lee, Suresh Cuddapah, Byeong Hyeok Choi, and Wei Dai. 2022. "MicroRNA–Gene Interactions Impacted by Toxic Metal(oid)s during EMT and Carcinogenesis" Cancers 14, no. 23: 5818. https://doi.org/10.3390/cancers14235818
APA StyleTran, F., Lee, E., Cuddapah, S., Choi, B. H., & Dai, W. (2022). MicroRNA–Gene Interactions Impacted by Toxic Metal(oid)s during EMT and Carcinogenesis. Cancers, 14(23), 5818. https://doi.org/10.3390/cancers14235818