
Computational Analysis Reveals the Temporal Acquisition of Pathway Alterations during the Evolution of Cancer

 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
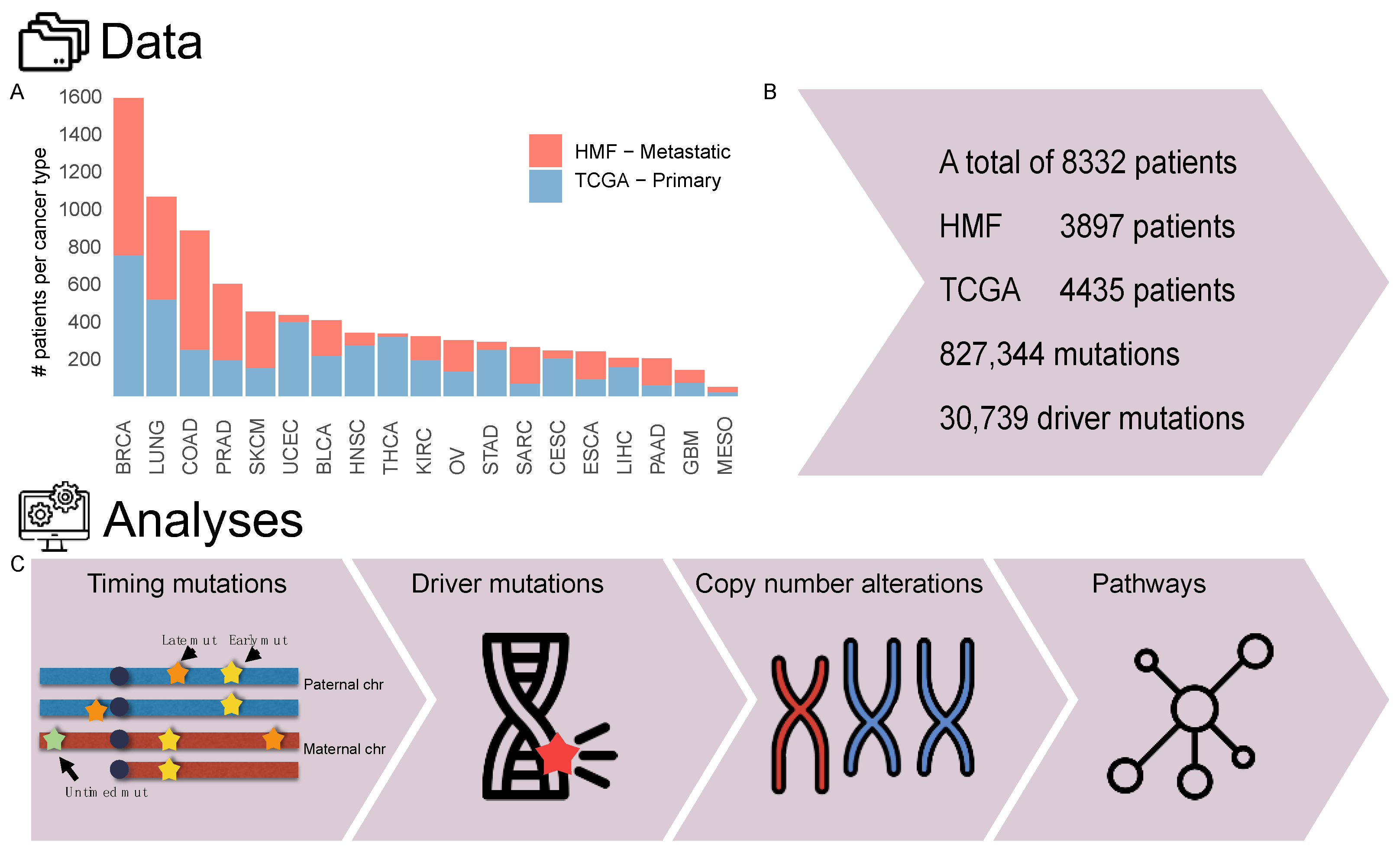
2.1. Cohort Overview
2.2. Relative Timing of Mutations
2.3. Annotation of Driver Events
2.4. Copy Number Alterations
2.5. Enriched and Depleted Genes and Pathways
2.6. Hotspot Mutations
2.7. Statistical Analysis
3. Results
3.1. Metastatic Tumours Have a Higher Number of Driver Mutations
3.2. Most Cancer Genes Are Affected by Driver Mutations at the Same Frequency in Primary and Metastatic Cancer
3.3. Metastatic Tumours Have a Higher Level of Somatic Copy Number Alterations
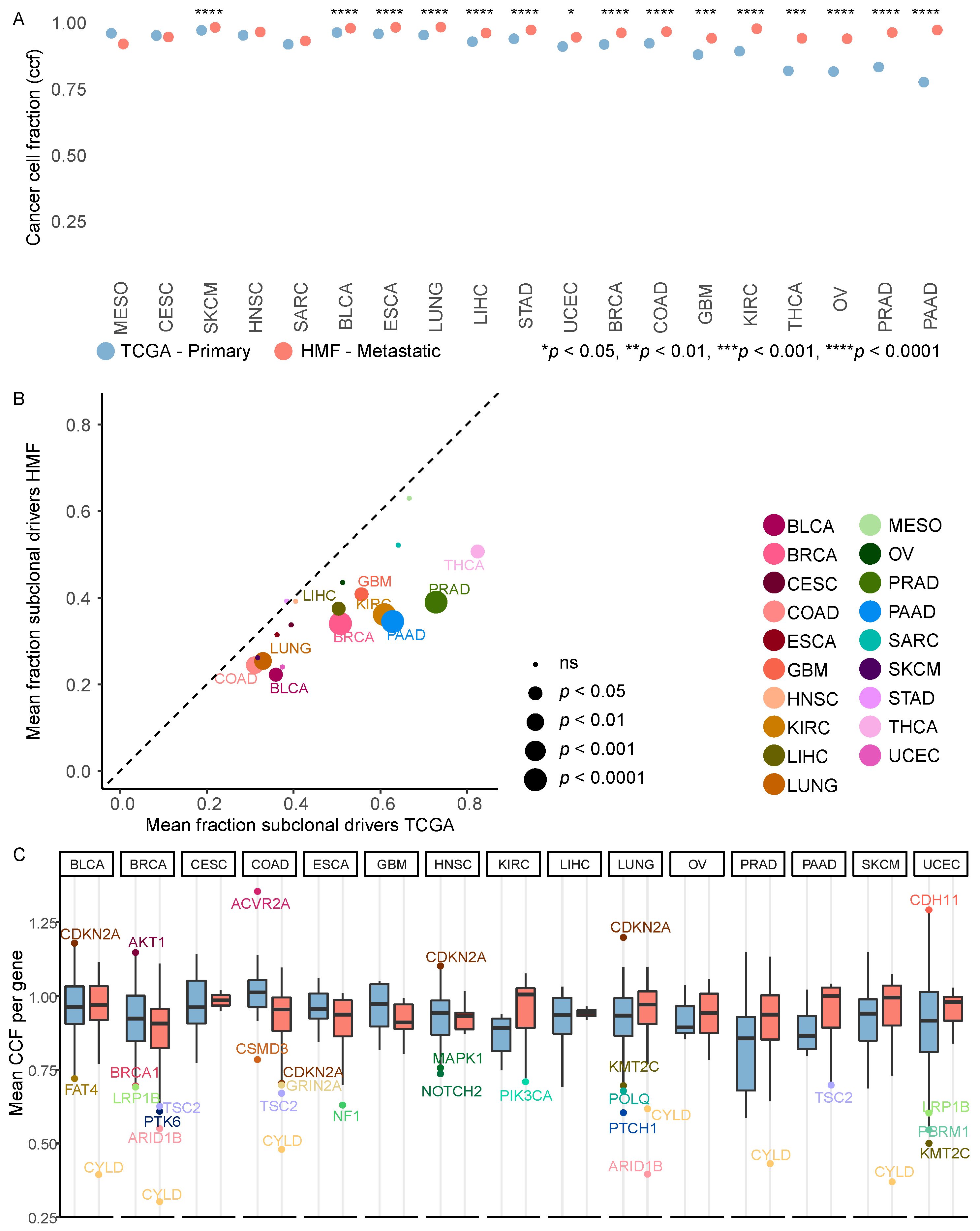
3.4. Metastatic Tumours Are More Clonal Than Primary Tumours
3.5. Driver Mutations Outside Common Cancer Genes
3.6. Timed Driver Mutations Show Similar Patterns in Primary and Metastatic Tumours
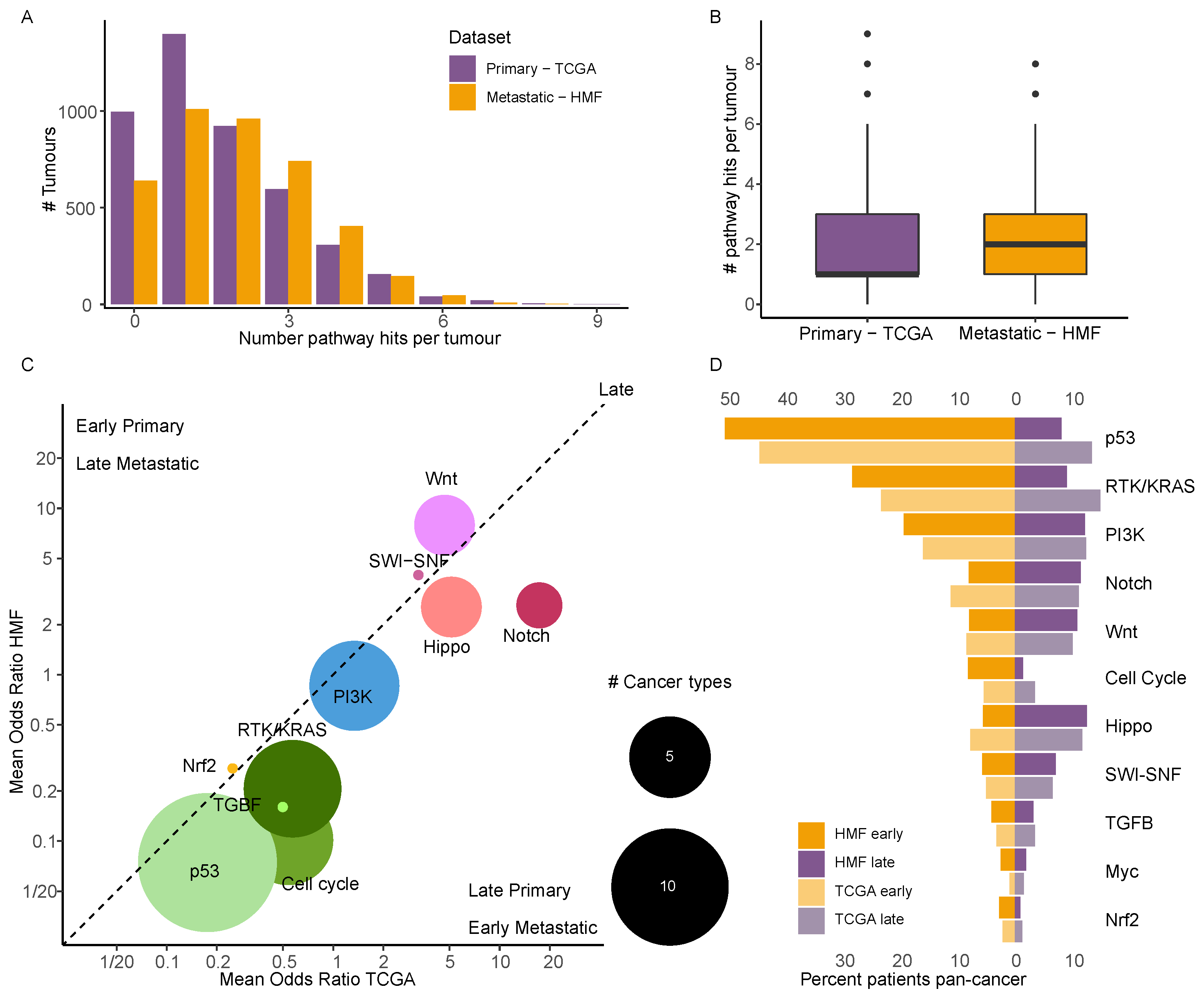
3.7. Driver Events in Cancer Specific Pathways Show near Identical Timing in Primary and Metastatic Cancer
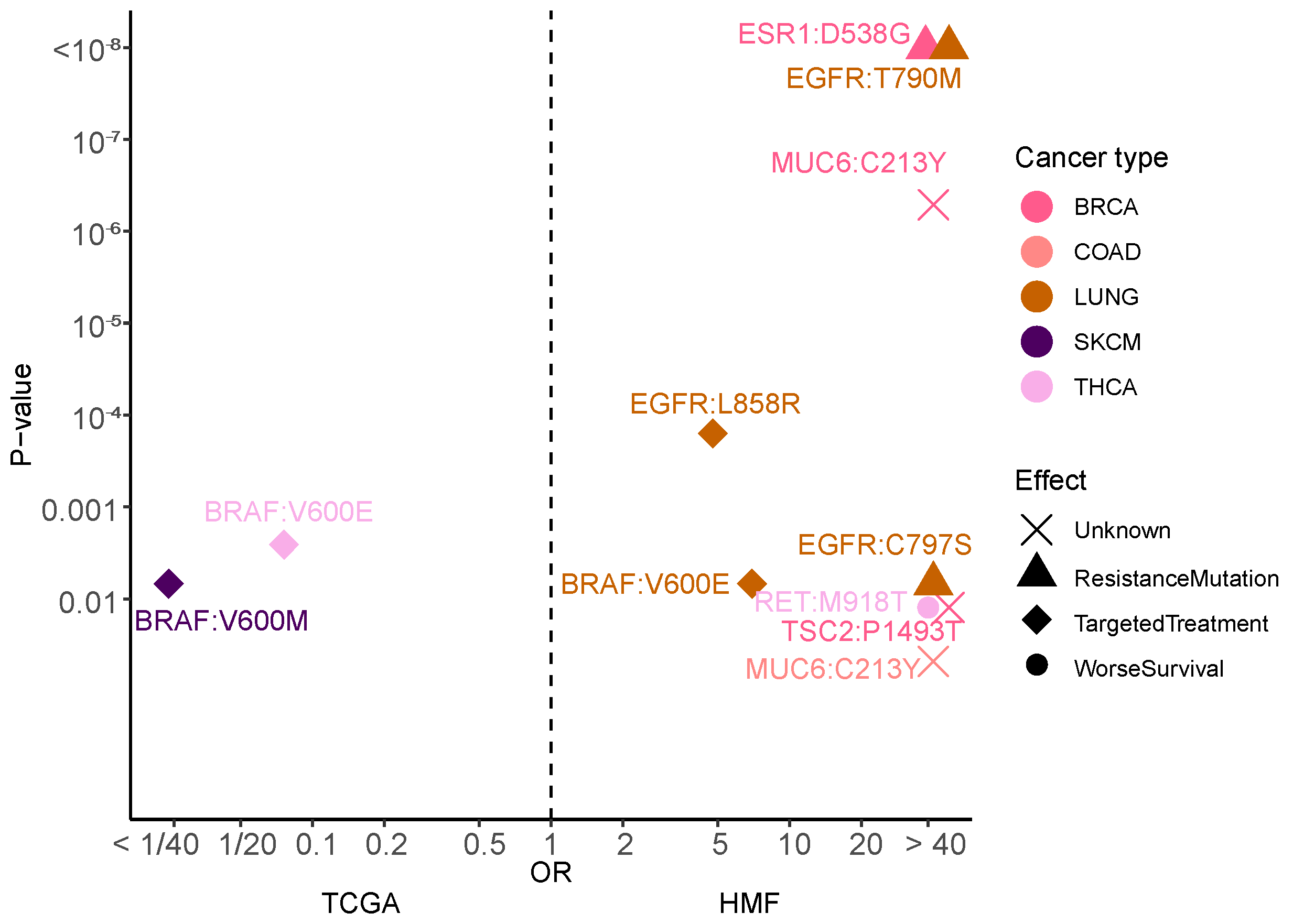
3.8. Mutational Hotspots Indicate Treatment Resistance as Driver for Metastatic Evolution
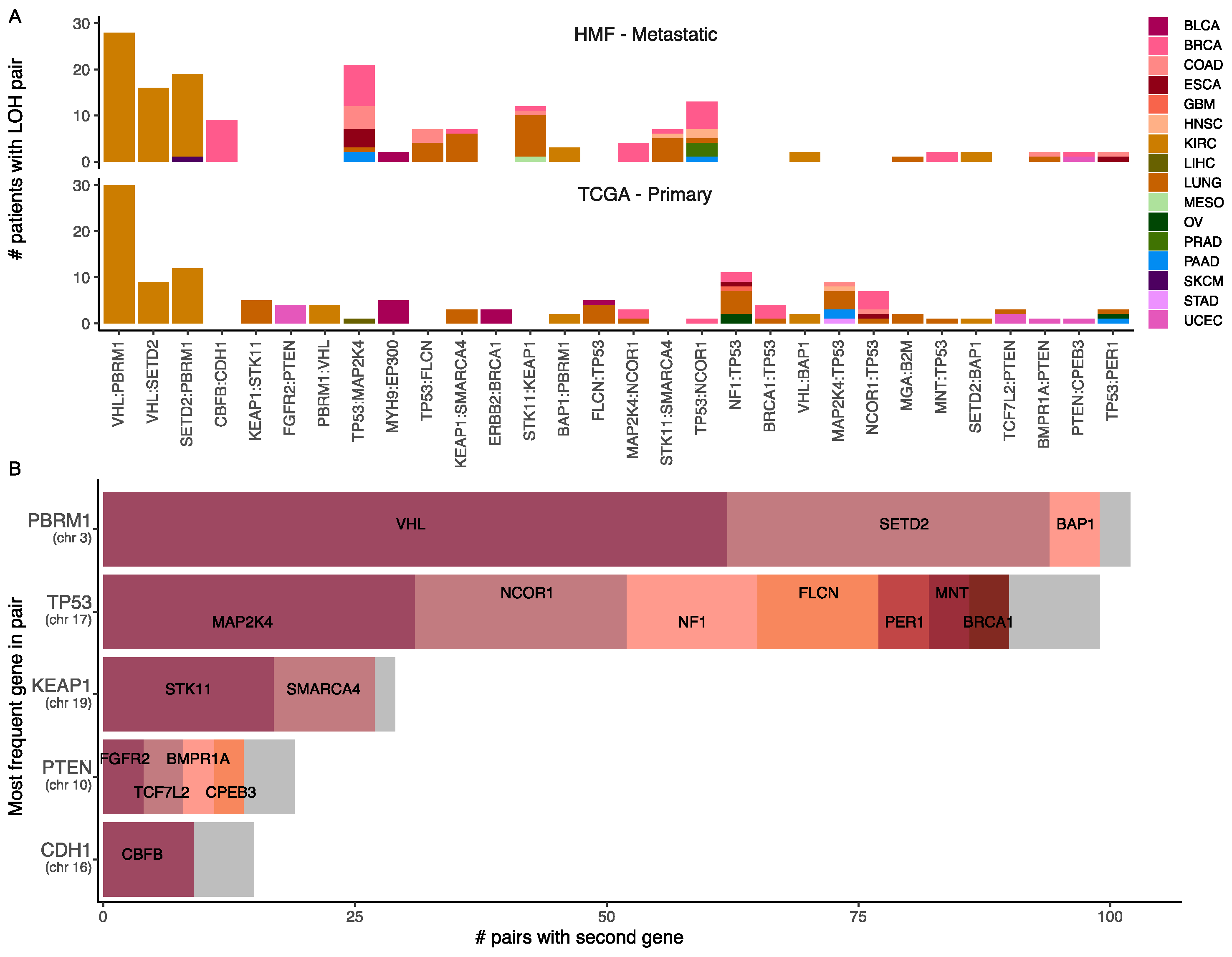
3.9. Loss of Heterozygosity and Driver Mutations Primes for Additional Mutations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hu, Z.; Li, Z.; Ma, Z.; Curtis, C. Multi-Cancer Analysis of Clonality and the Timing of Systemic Spread in Paired Primary Tumors and Metastases. Nat. Genet. 2020, 52, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.S.; Ahrenfeldt, J.; Sokač, M.; Kisistók, J.; Thomsen, M.K.; Maretty, L.; McGranahan, N.; Birkbak, N.J. Treatment Represents a Key Driver of Metastatic Cancer Evolution. Cancer Res. 2022, 82, 2918–2927. [Google Scholar] [CrossRef]
- Priestley, P.; Baber, J.; Lolkema, M.P.; Steeghs, N.; de Bruijn, E.; Shale, C.; Duyvesteyn, K.; Haidari, S.; van Hoeck, A.; Onstenk, W.; et al. Pan-Cancer Whole-Genome Analyses of Metastatic Solid Tumours. Nature 2019, 575, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Wu, Y.-M.; Lonigro, R.J.; Vats, P.; Cobain, E.; Everett, J.; Cao, X.; Rabban, E.; Kumar-Sinha, C.; Raymond, V.; et al. Integrative Clinical Genomics of Metastatic Cancer. Nature 2017, 548, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.; Fong, C.; Luthra, A.; Smith, S.A.; DiNatale, R.G.; Nandakumar, S.; Walch, H.; Chatila, W.K.; Madupuri, R.; Kundra, R.; et al. Genomic Characterization of Metastatic Patterns from Prospective Clinical Sequencing of 25,000 Patients. Cell 2022, 185, 563–575.e11. [Google Scholar] [CrossRef] [PubMed]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular Principles of Metastasis: A Hallmark of Cancer Revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J.; Obenauf, A.C. Metastatic Colonization by Circulating Tumour Cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The Cancer Genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Reiter, J.G.; Bozic, I.; Allen, B.; Chatterjee, K.; Nowak, M.A. The Effect of One Additional Driver Mutation on Tumor Progression. Evol. Appl. 2013, 6, 34–45. [Google Scholar] [CrossRef]
- Birkbak, N.J.; McGranahan, N. Cancer Genome Evolutionary Trajectories in Metastasis. Cancer Cell 2020, 37, 8–19. [Google Scholar] [CrossRef]
- AACR Project GENIE Consortium AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [CrossRef]
- McGranahan, N.; Favero, F.; de Bruin, E.C.; Birkbak, N.J.; Szallasi, Z.; Swanton, C. Clonal Status of Actionable Driver Events and the Timing of Mutational Processes in Cancer Evolution. Sci. Transl. Med. 2015, 7, 283ra54. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. Predicting Deleterious Amino Acid Substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The Causes and Consequences of Genetic Heterogeneity in Cancer Evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Kassambara, A. Ggpubr: “ggplot2” Based Publication Ready Plots, R Package Version 0.4.0; 2020. Available online: https://CRAN.R-project.org/package=ggpubr (accessed on 26 October 2022).
- Hadley, W.; Seidel, D. Scale Functions for Visualization; Version 1.2.1; RStudio: Vienna, Austria, 2019. [Google Scholar]
- Slowikowski, K. Ggrepel: Automatically Position Non-Overlapping Text Labels with “ggplot2” 2021. Available online: https://rdrr.io/cran/ggrepel/ (accessed on 26 October 2022).
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational Heterogeneity in Cancer and the Search for New Cancer-Associated Genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Elisei, R.; Cosci, B.; Romei, C.; Bottici, V.; Renzini, G.; Molinaro, E.; Agate, L.; Vivaldi, A.; Faviana, P.; Basolo, F.; et al. Prognostic Significance of Somatic RET Oncogene Mutations in Sporadic Medullary Thyroid Cancer: A 10-Year Follow-up Study. J. Clin. Endocrinol. Metab. 2008, 93, 682–687. [Google Scholar] [CrossRef]
- Turajlic, S.; Xu, H.; Litchfield, K.; Rowan, A.; Horswell, S.; Chambers, T.; O’Brien, T.; Lopez, J.I.; Watkins, T.B.K.; Nicol, D.; et al. Deterministic Evolutionary Trajectories Influence Primary Tumor Growth: TRACERx Renal. Cell 2018, 173, 595–610. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Watkins, T.B.K.; Lim, E.L.; Petkovic, M.; Elizalde, S.; Birkbak, N.J.; Wilson, G.A.; Moore, D.A.; Grönroos, E.; Rowan, A.; Dewhurst, S.M.; et al. Pervasive Chromosomal Instability and Karyotype Order in Tumour Evolution. Nature 2020, 587, 126–132. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- De Mattos-Arruda, L.; Sammut, S.-J.; Ross, E.M.; Bashford-Rogers, R.; Greenstein, E.; Markus, H.; Morganella, S.; Teng, Y.; Maruvka, Y.; Pereira, B.; et al. The Genomic and Immune Landscapes of Lethal Metastatic Breast Cancer. Cell Rep. 2019, 27, 2690–2708.e10. [Google Scholar] [CrossRef]
- Nguyen, K.-S.H.; Kobayashi, S.; Costa, D.B. Acquired Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Non–Small-Cell Lung Cancers Dependent on the Epidermal Growth Factor Receptor Pathway. Clin. Lung Cancer 2009, 10, 281–289. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.-J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The Role of BRAF V600 Mutation in Melanoma. J. Transl. Med. 2012, 10, 85. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Cancer Type |
|---|---|
| BLCA | Bladder Urothelial Carcinoma |
| BRCA | Breast invasive carcinoma |
| CESC | Cervical squamous cell carcinoma and endocervical adenocarcinoma |
| COAD | Colon adenocarcinoma |
| ESCA | Esophageal carcinoma |
| GBM | Glioblastoma multiforme |
| HNSC | Head and Neck squamous cell carcinoma |
| KIRC | Kidney renal clear cell carcinoma |
| LIHC | Liver hepatocellular carcinoma |
| LUNG | Non small cell lung cancer |
| MESO | Mesothelioma |
| OV | Ovarian serous cystadenocarcinoma |
| PAAD | Pancreatic adenocarcinoma |
| PRAD | Prostate adenocarcinoma |
| SARC | Sarcoma |
| SKCM | Skin Cutaneous Melanoma |
| STAD | Stomach adenocarcinoma |
| THCA | Thyroid carcinoma |
| UCEC | Uterine Corpus Endometrial Carcinoma |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahrenfeldt, J.; Christensen, D.S.; Sokač, M.; Kisistók, J.; McGranahan, N.; Birkbak, N.J. Computational Analysis Reveals the Temporal Acquisition of Pathway Alterations during the Evolution of Cancer. Cancers 2022, 14, 5817. https://doi.org/10.3390/cancers14235817
Ahrenfeldt J, Christensen DS, Sokač M, Kisistók J, McGranahan N, Birkbak NJ. Computational Analysis Reveals the Temporal Acquisition of Pathway Alterations during the Evolution of Cancer. Cancers. 2022; 14(23):5817. https://doi.org/10.3390/cancers14235817
Chicago/Turabian StyleAhrenfeldt, Johanne, Ditte S. Christensen, Mateo Sokač, Judit Kisistók, Nicholas McGranahan, and Nicolai J. Birkbak. 2022. "Computational Analysis Reveals the Temporal Acquisition of Pathway Alterations during the Evolution of Cancer" Cancers 14, no. 23: 5817. https://doi.org/10.3390/cancers14235817
APA StyleAhrenfeldt, J., Christensen, D. S., Sokač, M., Kisistók, J., McGranahan, N., & Birkbak, N. J. (2022). Computational Analysis Reveals the Temporal Acquisition of Pathway Alterations during the Evolution of Cancer. Cancers, 14(23), 5817. https://doi.org/10.3390/cancers14235817