

Targeting TET2 as a Therapeutic Approach for Angioimmunoblastic T Cell Lymphoma


Abstract
Simple Summary
Abstract
1. Introduction
2. AITL and Other Lymphomas with a TFH-Cell Phenotype
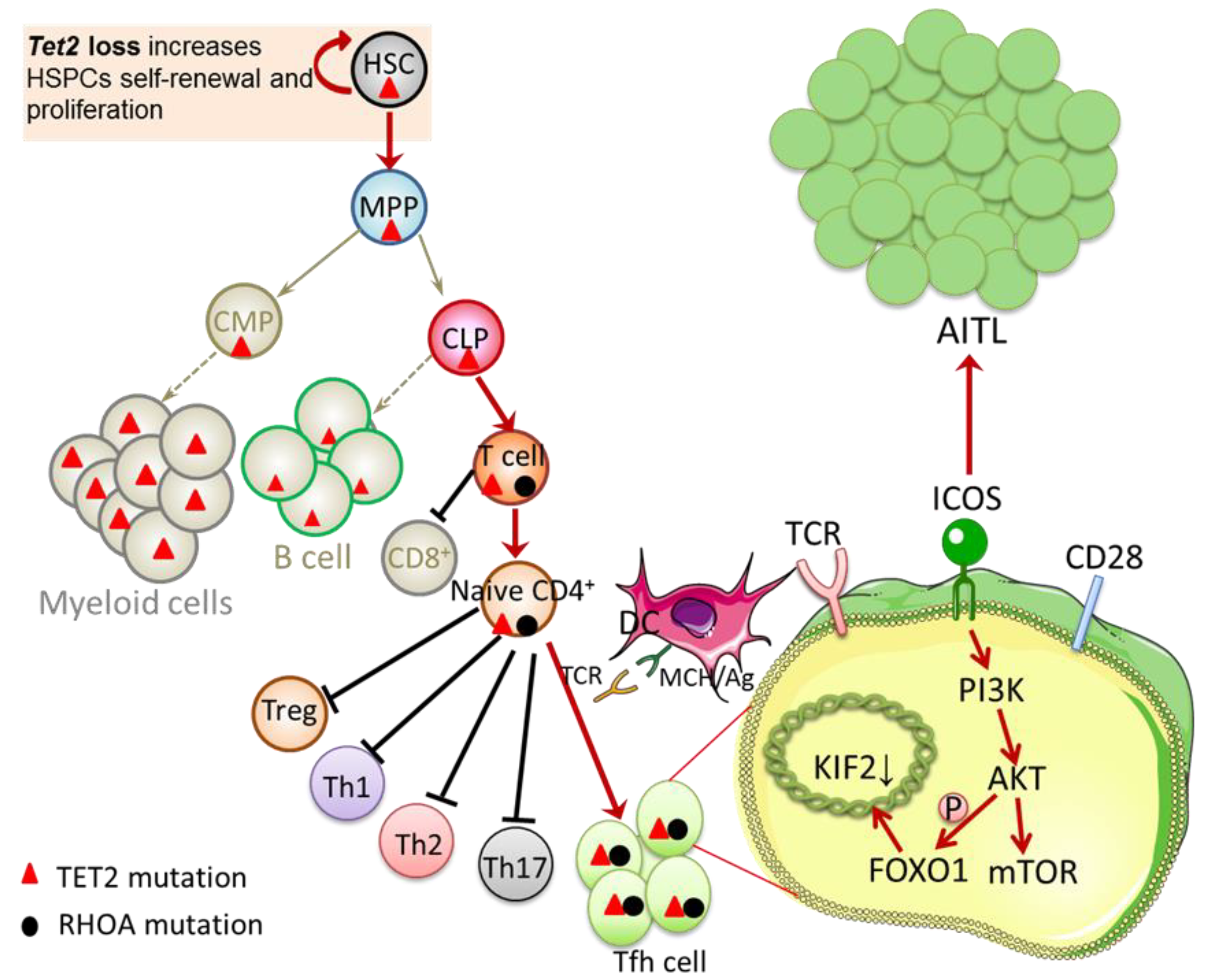
3. The Highly Recurrent TET2 Mutation in AITL
4. Crosstalk between TET2 and Other Genes in AITL
4.1. Interaction between TET2 and RHOA Mutations
4.2. Interaction between TET2 and DNMT3A Mutation
4.3. Interaction between TET2 and IDH2 Mutations
5. Role of TET2 Mutations in B-Cell Lymphoma Observed in AITL
5.1. Traditional View: AITL Precedes B-Cell Lymphoma
5.2. Recent View: AITL Occurs Concurrently with B Cell Lymphoma
6. New Drugs Targeting TET2 for Treatment of AITL
6.1. Histone Deacetylase Inhibitor (HDACis)
6.2. Hypomethylating Agents (HMAs)
6.3. Preclinical Trials of TET2 Targeting Agents
6.3.1. TET Enzyme Inhibitors
6.3.2. TET Enzyme Cofactors
6.3.3. Other Possibilities
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Grogg, K.L.; Attygalle, A.D.; Macon, W.R.; Remstein, E.D.; Kurtin, P.J.; Dogan, A. Angioimmunoblastic T-cell lymphoma: A neoplasm of germinal-center T-helper cells? Blood 2005, 106, 1501–1502. [Google Scholar] [CrossRef]
- Rob, A.C.; Javeed, I.; François, L.; Can, K.; de Laurence, L.; Jean-Philippe, J.; Marie, P.; Antoine, M.; Luc, X.; Pierre, B.; et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood 2012, 119, 1901–1903. [Google Scholar] [CrossRef]
- Lucile, C.; Christian, B.; Olivier, A.B. TET2 and DNMT3A mutations in human T-cell lymphoma. N. Engl. J. Med. 2012, 366, 95–96. [Google Scholar] [CrossRef]
- Quivoron, C.; Couronne, L.; Della Valle, V.; Lopez, C.K.; Plo, I.; Wagner-Ballon, O.; Do Cruzeiro, M.; Delhommeau, F.; Arnulf, B.; Stern, M.H.; et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 2011, 20, 25–38. [Google Scholar] [CrossRef]
- Scourzic, L.; Couronne, L.; Pedersen, M.T.; Della Valle, V.; Diop, M.; Mylonas, E.; Calvo, J.; Mouly, E.; Lopez, C.K.; Martin, N.; et al. DNMT3A(R882H) mutant and Tet2 inactivation cooperate in the deregulation of DNA methylation control to induce lymphoid malignancies in mice. Leukemia 2016, 30, 1388–1398. [Google Scholar] [CrossRef]
- Palomero, T.; Couronné, L.; Khiabanian, H.; Kim, M.-Y.; Ambesi-Impiombato, A.; Perez-Garcia, A.; Carpenter, Z.; Abate, F.; Allegretta, M.; Haydu, J.E.; et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat. Genet. 2014, 46, 166–170. [Google Scholar] [CrossRef]
- Sakata-Yanagimoto, M.; Enami, T.; Yoshida, K.; Shiraishi, Y.; Ishii, R.; Miyake, Y.; Muto, H.; Tsuyama, N.; Sato-Otsubo, A.; Okuno, Y.; et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat. Genet. 2014, 46, 171–175. [Google Scholar] [CrossRef]
- Nguyen, T.B.; Sakata-Yanagimoto, M.; Asabe, Y.; Matsubara, D.; Kano, J.; Yoshida, K.; Shiraishi, Y.; Chiba, K.; Tanaka, H.; Miyano, S.; et al. Identification of cell-type-specific mutations in nodal T-cell lymphomas. Blood Cancer J. 2017, 7, e516. [Google Scholar] [CrossRef]
- Schwartz, F.H.; Cai, Q.; Fellmann, E.; Hartmann, S.; Mayranpaa, M.I.; Karjalainen-Lindsberg, M.L.; Sundstrom, C.; Scholtysik, R.; Hansmann, M.L.; Kuppers, R. TET2 mutations in B cells of patients affected by angioimmunoblastic T-cell lymphoma. J. Pathol. 2017, 242, 129–133. [Google Scholar] [CrossRef]
- Dobay, M.P.; Lemonnier, F.; Missiaglia, E.; Bastard, C.; Vallois, D.; Jais, J.-P.; Scourzic, L.; Dupuy, A.; Fataccioli, V.; Pujals, A.; et al. Integrative clinicopathological and molecular analyses of angioimmunoblastic T-cell lymphoma and other nodal lymphomas of follicular helper T-cell origin. Haematologica 2017, 102, e148–e151. [Google Scholar] [CrossRef]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef]
- Byeong-Bae, P.; Baek-Yeol, R.; Jae, H.L.; Hyuck, C.K.; Sung, H.Y.; Hye, J.K.; Hyo, J.K.; Sung, Y.O.; Young, H.K.; Joo, R.H.; et al. Clinical features and treatment outcomes of angioimmunoblastic T-cell lymphoma. Leuk. Lymphoma 2007, 48, 716–722. [Google Scholar] [CrossRef]
- Chittima, S.; Kanchana, C.; Arnuparp, L.; Jakrawadee, J.; Kitsada, W.; Udomsak, B.; Lalita, N.; Weerasak, N.; Archrob, K.; Supachai, E.; et al. Clinical Features and Treatment Outcomes of Angioimmunoblastic T-Cell Lymphoma: An Analysis from a Nationwide Multicenter Registry, Thailand. Blood 2015, 126, 5064. [Google Scholar] [CrossRef]
- Jenni, K.; Pyry, U.; Kirsi-Maria, H.; Saila, K.; Hanna-Riikka, T.; Milla, E.L.K.; Siria, L.; Taina, T.-H.; Martine, V.; Mine, E.; et al. Incidence and clinicopathological features of Follicular T-cell lymphoma in Finland: A population-based immunohistochemical study. Hum. Pathol. 2021, 117, 79–87. [Google Scholar] [CrossRef]
- Hiroaki, M.; Mamiko, S.Y.; Joji, S.; Noriaki, Y.; Keiichiro, H.; Fumiko, A.; Eriko, Y.; Mai, T.; Kyohei, Y.; Takaharu, S.; et al. RHOA mutation in follicular T-cell lymphoma: Clinicopathological analysis of 16 cases. Pathol. Int. 2020, 70, 653–660. [Google Scholar] [CrossRef]
- Huang, Y.; Moreau, A.; Dupuis, J.; Streubel, B.; Petit, B.; Le Gouill, S.; Martin-Garcia, N.; Copie-Bergman, C.; Gaillard, F.; Qubaja, M.; et al. Peripheral T-cell lymphomas with a follicular growth pattern are derived from follicular helper T cells (TFH) and may show overlapping features with angioimmunoblastic T-cell lymphomas. Am. J. Surg. Pathol. 2009, 33, 682–690. [Google Scholar] [CrossRef]
- Liang, P.I.; Chang, S.T.; Lin, M.Y.; Hsieh, Y.C.; Chu, P.Y.; Chen, C.J.; Lin, K.J.; Jung, Y.C.; Hwang, W.S.; Huang, W.T.; et al. Angioimmunoblastic T-cell lymphoma in Taiwan shows a frequent gain of ITK gene. Int. J. Clin. Exp. Pathol. 2014, 7, 6097–6107. [Google Scholar]
- Lemonnier, F.; Couronne, L.; Parrens, M.; Jais, J.P.; Travert, M.; Lamant, L.; Tournillac, O.; Rousset, T.; Fabiani, B.; Cairns, R.A.; et al. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood 2012, 120, 1466–1469. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, S.; Chuang, S.; Ashton-Key, M.; Ochoa, E.; Bolli, N.; Vassiliou, G.; Gao, Z.; Du, M. Angioimmunoblastic T cell lymphoma: Novel molecular insights by mutation profiling. Oncotarget 2017, 8, 17763–17770. [Google Scholar] [CrossRef]
- Yao, W.Q.; Wu, F.; Zhang, W.; Chuang, S.S.; Thompson, J.S.; Chen, Z.; Zhang, S.W.; Clipson, A.; Wang, M.; Liu, H.; et al. Angioimmunoblastic T-cell lymphoma contains multiple clonal T-cell populations derived from a common TET2 mutant progenitor cell. J. Pathol. 2020, 250, 346–357. [Google Scholar] [CrossRef]
- Alexandra, B.; Kaushik, S.; Diwash, J.; Jyoti, K.; Malaya, K.S.; Nahid, S.; Roger, A.W.; Elumalai, R.; Benjamin, A.P.; Robert, S.O. A Comprehensive Analysis of Rhoa Mutation Positive and Negative Angioimmunoblastic T-Cell Lymphomas By Targeted Deep Sequencing, Expression Profiling, and Single Cell Digital Image Analysis. Blood 2019, 134, 5228. [Google Scholar] [CrossRef]
- Yingying, Y.; Ning, D.; Lan, M.; Yunfei, S.; Weiping, L.; Yuqin, S.; Shaokun, S.; Jun, Z. Correlation of mutational landscape and survival outcome of peripheral T-cell lymphomas. Exp. Hematol. Oncol. 2021, 10, 9. [Google Scholar] [CrossRef]
- Nguyen, T.B.; Sakata-Yanagimoto, M.; Fujisawa, M.; Nuhat, S.T.; Miyoshi, H.; Nannya, Y.; Hashimoto, K.; Fukumoto, K.; Bernard, O.A.; Kiyoki, Y.; et al. Dasatinib Is an Effective Treatment for Angioimmunoblastic T-cell Lymphoma. Cancer Res. 2020, 80, 1875–1884. [Google Scholar] [CrossRef]
- Wang, C.; McKeithan, T.W.; Gong, Q.; Zhang, W.; Bouska, A.; Rosenwald, A.; Gascoyne, R.D.; Wu, X.; Wang, J.; Muhammad, Z.; et al. IDH2R172 mutations define a unique subgroup of patients with angioimmunoblastic T-cell lymphoma. Blood 2015, 126, 1741–1752. [Google Scholar] [CrossRef]
- François, L.; Violaine, S.; Asma, B.-F.; Anne-Ségolène, C.; Emmanuel, B.; Guillaume, C.; Virginie, F.; Laura, P.; Cyrielle, R.; Audrey, L.; et al. Integrative analysis of a phase 2 trial combining lenalidomide with CHOP in angioimmunoblastic T-cell lymphoma. Blood Adv. 2021, 5, 539–548. [Google Scholar] [CrossRef]
- Steinhilber, J.; Mederake, M.; Bonzheim, I.; Serinsoz-Linke, E.; Muller, I.; Fallier-Becker, P.; Lemonnier, F.; Gaulard, P.; Fend, F.; Quintanilla-Martinez, L. The pathological features of angioimmunoblastic T-cell lymphomas with IDH2(R172) mutations. Mod. Pathol. 2019, 32, 1123–1134. [Google Scholar] [CrossRef]
- Marta, R.; Ruth, A.-A.; Laura, T.-R.; Socorro, M.R.-P.; Rebeca, M.-A.; Laura, C.; Jennifer, B.; Teresa, V.; Raúl, C.; Margarita, S.-B.; et al. Peripheral T-cell lymphoma: Molecular profiling recognizes subclasses and identifies prognostic markers. Blood Adv. 2021, 5, 5588–5598. [Google Scholar] [CrossRef]
- Saillard, C.; Guermouche, H.; Derrieux, C.; Bruneau, J.; Frenzel, L.; Couronne, L.; Asnafi, V.; Macintyre, E.; Trinquand, A.; Lhermitte, L.; et al. Response to 5-azacytidine in a patient with TET2-mutated angioimmunoblastic T-cell lymphoma and chronic myelomonocytic leukaemia preceded by an EBV-positive large B-cell lymphoma. Hematol. Oncol. 2017, 35, 864–868. [Google Scholar] [CrossRef]
- Fadela, B.; Jocelyne, D.; Jacques, B.; Catherine, T.; Daniel, E.; Gilles, S.; Bertrand, C. Profiles and prognostic values of serum LDH isoenzymes in patients with haematopoietic malignancies. Bull. Cancer 2004, 91, E229–E240. [Google Scholar]
- Odejide, O.; Weigert, O.; Lane, A.A.; Toscano, D.; Lunning, M.A.; Kopp, N.; Kim, S.; van Bodegom, D.; Bolla, S.; Schatz, J.H.; et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood 2014, 123, 1293–1296. [Google Scholar] [CrossRef]
- François, L.; Jehan, D.; Pierre, S.; Olivier, T.; Morgane, C.; Clémentine, S.; Laura, P.; Ambroise, M.; Cyrielle, R.; Virginie, F.; et al. Treatment with 5-azacytidine induces a sustained response in patients with angioimmunoblastic T-cell lymphoma. Blood 2018, 132, 2305–2309. [Google Scholar] [CrossRef]
- Sebastian, F.-P.; Lisa, M.; Rohan, P.J.; Daniel, A.A. A Survey of Somatic Mutations in 41 Genes in a Cohort of T-Cell Lymphomas Identifies Frequent Mutations in Genes Involved in Epigenetic Modification. Appl. Immunohistochem. Mol. Morphol. 2019, 27, 416–422. [Google Scholar] [CrossRef]
- Bar-Sagi, D.; Hall, A. Ras and Rho GTPases: A family reunion. Cell 2000, 103, 227–238. [Google Scholar] [CrossRef]
- Sandrine, E.-M.; Alan, H. Rho GTPases in cell biology. Nature 2002, 420, 629–635. [Google Scholar] [CrossRef]
- Vallois, D.; Dobay, M.P.; Morin, R.D.; Lemonnier, F.; Missiaglia, E.; Juilland, M.; Iwaszkiewicz, J.; Fataccioli, V.; Bisig, B.; Roberti, A.; et al. Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T-cell-derived lymphomas. Blood 2016, 128, 1490–1502. [Google Scholar] [CrossRef]
- Hae Yong, Y.; Min Kyung, S.; Seung Ho, L.; Sangok, K.; Haeseung, L.; Seongjin, P.; Sang Cheol, K.; Byungwook, L.; Kyoohyoung, R.; Jong-Eun, L.; et al. A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat. Genet. 2014, 46, 371–375. [Google Scholar] [CrossRef]
- Fujisawa, M.; Sakata-Yanagimoto, M.; Nishizawa, S.; Komori, D.; Gershon, P.; Kiryu, M.; Tanzima, S.; Fukumoto, K.; Enami, T.; Muratani, M.; et al. Activation of RHOA-VAV1 signaling in angioimmunoblastic T-cell lymphoma. Leukemia 2018, 32, 694–702. [Google Scholar] [CrossRef]
- Tybulewicz, V.L. Vav-family proteins in T-cell signalling. Curr. Opin. Immunol. 2005, 17, 267–274. [Google Scholar] [CrossRef]
- Komori, D.; Sakata-Yanagimoto, M.; Nuhat, S.T.; Fukumoto, K.; Fujisawa, M.; Nishizawa, S.; Matsue, K.; Izutsu, K.; Nakamura, N.; Yoshida, K.; et al. Recurrent VAV1 Abnormalities in Angioimmunoblastic T Cell Lymphoma. Blood 2016, 128, 4104. [Google Scholar] [CrossRef]
- Samuel, Y.N.; Leon, B.; Kristen, S.; Tiffany, d.; Jon, C.A.; Abner, L.; David, M.W. RhoA G17V is sufficient to induce autoimmunity and promotes T cell lymphomagenesis in mice. Blood 2018, 132, 935–947. [Google Scholar] [CrossRef]
- Horwitz, S.M.; Moskowitz, A.J.; Jacobsen, E.D.; Mehta-Shah, N.; Khodadoust, M.S.; Fisher, D.C.; Myskowski, P.; Wang, E.B.K.; Tawa, M.; Davey, T.; et al. The Combination of Duvelisib, a PI3K-δ,γ Inhibitor, and Romidepsin Is Highly Active in Relapsed/Refractory Peripheral T-Cell Lymphoma with Low Rates of Transaminitis: Results of Parallel Multicenter, Phase 1 Combination Studies with Expansion Cohorts. Blood 2018, 132, 683. [Google Scholar] [CrossRef]
- Stone, E.L.; Pepper, M.; Katayama, C.D.; Kerdiles, Y.M.; Lai, C.Y.; Emslie, E.; Lin, Y.C.; Yang, E.; Goldrath, A.W.; Li, M.O.; et al. ICOS coreceptor signaling inactivates the transcription factor FOXO1 to promote Tfh cell differentiation. Immunity 2015, 42, 239–251. [Google Scholar] [CrossRef]
- Zang, S.; Li, J.; Yang, H.; Zeng, H.; Han, W.; Zhang, J.; Lee, M.; Moczygemba, M.; Isgandarova, S.; Yang, Y.; et al. Mutations in 5-methylcytosine oxidase TET2 and RhoA cooperatively disrupt T cell homeostasis. J. Clin. Investig. 2017, 127, 2998–3012. [Google Scholar] [CrossRef]
- Cortes, J.R.; Ambesi-Impiombato, A.; Couronne, L.; Quinn, S.A.; Kim, C.S.; da Silva Almeida, A.C.; West, Z.; Belver, L.; Martin, M.S.; Scourzic, L.; et al. RHOA G17V Induces T Follicular Helper Cell Specification and Promotes Lymphomagenesis. Cancer Cell 2018, 33, 259–273. [Google Scholar] [CrossRef]
- Mhaidly, R.; Krug, A.; Gaulard, P.; Lemonnier, F.; Ricci, J.-E.; Verhoeyen, E. New preclinical models for angioimmunoblastic T-cell lymphoma: Filling the GAP. Oncogenesis 2020, 9, 73. [Google Scholar] [CrossRef]
- Challen, G.A.; Sun, D.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.; Xi, Y.; et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 2011, 44, 23–31. [Google Scholar] [CrossRef]
- Allison, M.; Liubin, Y.; Benjamin, R.; Ting, Z.; Edmund, C.; Choladda, V.C.; Grant, A.C.; Wei, L.; David, W.; Vivienne, I.R.; et al. Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood 2015, 125, 629–638. [Google Scholar] [CrossRef]
- Zhang, X.; Su, J.; Jeong, M.; Ko, M.; Huang, Y.; Park, H.J.; Guzman, A.; Lei, Y.; Huang, Y.H.; Rao, A.; et al. DNMT3A and TET2 compete and cooperate to repress lineage-specific transcription factors in hematopoietic stem cells. Nat. Genet. 2016, 48, 1014–1023. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef]
- Sakata-Yanagimoto, M. Multistep tumorigenesis in peripheral T cell lymphoma. Int. J. Hematol. 2015, 102, 523–527. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef]
- Maria, E.F.; Omar, A.-W.; Chao, L.; Patrick, S.W.; Jay, P.; Alan, S.; Yushan, L.; Neha, B.; Aparna, V.; Hugo, F.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Cairns, R.A.; Mak, T.W. Oncogenic isocitrate dehydrogenase mutations: Mechanisms, models, and clinical opportunities. Cancer Discov. 2013, 3, 730–741. [Google Scholar] [CrossRef]
- François, L.; Rob, A.C.; Satoshi, I.; Wanda, Y.L.; Aurélie, D.; Sophie, B.; Nadine, M.; Virginie, F.; Romain, P.; Andrew, W.; et al. The IDH2 R172K mutation associated with angioimmunoblastic T-cell lymphoma produces 2HG in T cells and impacts lymphoid development [Medical Sciences]. Proc. Natl. Acad. Sci. USA 2016, 113, 15084–15089. [Google Scholar] [CrossRef]
- Tayla, B.H.; Alyssa, B.; Jiayu, Y.; Waseem, L.; Catalina, A.; Qiang, G.; Weiwei, Z.; Yuping, L.; Bhavana, J.D.; Maarja-Liisa, N.; et al. Genetic drivers of oncogenic pathways in molecular subgroups of peripheral T-cell lymphoma. Blood 2019, 133, 1664–1676. [Google Scholar] [CrossRef]
- Mario, L.M.-P.; Yessenia, I.S.; Carlos, P.; Renato, B.-H.; Francisco, V.; Roberto, N.M. Epstein–Barr virus-associated B-cell lymphoproliferative disorders and lymphomas: A review. Pathology 2020, 52, 40–52. [Google Scholar] [CrossRef]
- Ho, J.W.; Ho, F.C.; Chan, A.C.; Liang, R.H.; Srivastava, G. Frequent detection of Epstein-Barr virus-infected B cells in peripheral T-cell lymphomas. J. Pathol. 1998, 185, 79–85. [Google Scholar] [CrossRef]
- Willenbrock, K.; Brauninger, A.; Hansmann, M.L. Frequent occurrence of B-cell lymphomas in angioimmunoblastic T-cell lymphoma and proliferation of Epstein-Barr virus-infected cells in early cases. Br. J. Haematol. 2007, 138, 733–739. [Google Scholar] [CrossRef]
- Bräuninger, A.; Spieker, T.; Willenbrock, K.; Gaulard, P.; Wacker, H.H.; Rajewsky, K.; Hansmann, M.L.; Küppers, R. Survival and clonal expansion of mutating "forbidden" (immunoglobulin receptor-deficient) epstein-barr virus-infected b cells in angioimmunoblastic t cell lymphoma. J. Exp. Med. 2001, 194, 927–940. [Google Scholar] [CrossRef]
- Marie, P.; Antoine, M.; Laurence, L.; Richard, D.; Corinne, H.; Olivier, T.; Francoise, B.; Céline, B.; Brigitte, B.h.; Josette, B.; et al. Angioimmunoblastic T-Cell Lymphoma (AITL) Is the Most Prevalent T-Cell Lymphoma Entity in Western Europe. Blood 2012, 120, 1607. [Google Scholar] [CrossRef]
- Lemonnier, F.; Mak, T.W. Angioimmunoblastic T-cell lymphoma: More than a disease of T follicular helper cells. J. Pathol. 2017, 242, 387–390. [Google Scholar] [CrossRef]
- Nicolae, A.; Pittaluga, S.; Venkataraman, G.; Vijnovich-Baron, A.; Xi, L.; Raffeld, M.; Jaffe, E.S. Peripheral T-cell lymphomas of follicular T-helper cell derivation with Hodgkin/Reed-Sternberg cells of B-cell lineage: Both EBV-positive and EBV-negative variants exist. Am. J. Surg. Pathol. 2013, 37, 816–826. [Google Scholar] [CrossRef]
- Suefuji, N.; Niino, D.; Arakawa, F.; Karube, K.; Kimura, Y.; Kiyasu, J.; Takeuchi, M.; Miyoshi, H.; Yoshida, M.; Ichikawa, A.; et al. Clinicopathological analysis of a composite lymphoma containing both T- and B-cell lymphomas. Pathol. Int. 2012, 62, 690–698. [Google Scholar] [CrossRef]
- Ayoma Deepthi, A.; Charalampia, K.; Jehan, D.; Karen Lynne, G.; Timothy Charles, D.; Andrew Charles, W.; Shih Sung, C.; José, C.; Peter Gershon, I.; Ming-Qing, D.; et al. Histologic evolution of angioimmunoblastic T-cell lymphoma in consecutive biopsies: Clinical correlation and insights into natural history and disease progression. Am. J. Surg. Pathol. 2007, 31, 1077–1088. [Google Scholar] [CrossRef]
- Krug, A.; Tari, G.; Saidane, A.; Gaulard, P.; Ricci, J.-E.; Lemonnier, F.; Verhoeyen, E. Novel T Follicular Helper-like T-Cell Lymphoma Therapies: From Preclinical Evaluation to Clinical Reality. Cancers 2022, 14, 2392. [Google Scholar] [CrossRef]
- James, O.A. The aggressive peripheral T-cell lymphomas: 2017. Am. J. Hematol. 2017, 92, 706–715. [Google Scholar] [CrossRef]
- Horwitz, S.M.; Ansell, S.; Ai, W.Z.; Barnes, J.; Barta, S.K.; Brammer, J.; Clemens, M.W.; Dogan, A.; Foss, F.; Ghione, P.; et al. T-Cell Lymphomas, Version 2.2022, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. JNCCN 2022, 20, 285–308. [Google Scholar] [CrossRef]
- Tari, G.; Lemonnier, F.; Morschhauser, F. Epigenetic focus on angioimmunoblastic T-cell lymphoma: Pathogenesis and treatment. Curr. Opin. Oncol. 2021, 33, 400–405. [Google Scholar] [CrossRef]
- Coiffier, B.; Pro, B.; Prince, H.M.; Foss, F.; Sokol, L.; Greenwood, M.; Caballero, D.; Borchmann, P.; Morschhauser, F.; Wilhelm, M.; et al. Results From a Pivotal, Open-Label, Phase II Study of Romidepsin in Relapsed or Refractory Peripheral T-Cell Lymphoma After Prior Systemic Therapy. J. Clin. Oncol. 2012, 30, 631–636. [Google Scholar] [CrossRef]
- Foss, F.; Advani, R.; Duvic, M.; Hymes, K.B.; Intragumtornchai, T.; Lekhakula, A.; Shpilberg, O.; Lerner, A.; Belt, R.J.; Jacobsen, E.D.; et al. A Phase II trial of Belinostat (PXD101) in patients with relapsed or refractory peripheral or cutaneous T-cell lymphoma. Br. J. Haematol. 2014, 168, 811–819. [Google Scholar] [CrossRef]
- O’Connor, O.A.; Horwitz, S.; Masszi, T.; Van Hoof, A.; Brown, P.; Doorduijn, J.; Hess, G.; Jurczak, W.; Knoblauch, P.; Chawla, S.; et al. Belinostat in Patients With Relapsed or Refractory Peripheral T-Cell Lymphoma: Results of the Pivotal Phase II BELIEF (CLN-19) Study. J. Clin. Oncol. 2015, 33, 2492–2499. [Google Scholar] [CrossRef]
- Shi, Y.; Dong, M.; Hong, X.; Zhang, W.; Feng, J.; Zhu, J.; Yu, L.; Ke, X.; Huang, H.; Shen, Z.; et al. Results from a multicenter, open-label, pivotal phase II study of chidamide in relapsed or refractory peripheral T-cell lymphoma. Ann. Oncol. 2015, 26, 1766–1771. [Google Scholar] [CrossRef]
- Shi, Y.; Jia, B.; Xu, W.; Li, W.; Liu, T.; Liu, P.; Zhao, W.; Zhang, H.; Sun, X.; Yang, H.; et al. Chidamide in relapsed or refractory peripheral T cell lymphoma: A multicenter real-world study in China. J. Hematol. Oncol. 2017, 10, 69. [Google Scholar] [CrossRef]
- Dupuis, J.; Morschhauser, F.; Ghesquières, H.; Tilly, H.; Casasnovas, O.; Thieblemont, C.; Ribrag, V.; Bossard, C.; Bras, F.L.; Bachy, E.; et al. Combination of romidepsin with cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated patients with peripheral T-cell lymphoma: A non-randomised, phase 1b/2 study. Lancet Haematol. 2015, 2, e160–e165. [Google Scholar] [CrossRef]
- Moskowitz, A.J.; Koch, R.; Mehta-Shah, N.; Myskowski, P.; Kheterpal, M.; Dogan, A.; Davey, T.; Galasso, N.; Evan, M.; Shah, M.; et al. In Vitro, In Vivo, and Parallel Phase I Evidence Support the Safety and Activity of Duvelisib, a PI3K-δ,γ Inhibitor, in Combination with Romidepsin or Bortezomib in Relapsed/Refractory T-Cell Lymphoma. Blood 2017, 130, 819. [Google Scholar] [CrossRef]
- Bachy, E.; Camus, V.; Thieblemont, C.; Sibon, D.; Casasnovas, R.-O.; Ysebaert, L.; Damaj, G.; Guidez, S.; Pica, G.M.; Kim, W.S.; et al. Romidepsin Plus CHOP Versus CHOP in Patients With Previously Untreated Peripheral T-Cell Lymphoma: Results of the Ro-CHOP Phase III Study (Conducted by LYSA). J. Clin. Oncol. 2021, 40, 242–251. [Google Scholar] [CrossRef]
- Falchi, L.; Ma, H.; Klein, S.; Lue, J.K.; Montanari, F.; Marchi, E.; Deng, C.; Kim, H.A.; Rada, A.; Jacob, A.T.; et al. Combined oral 5-azacytidine and romidepsin are highly effective in patients with PTCL: A multicenter phase 2 study. Blood 2021, 137, 2161–2170. [Google Scholar] [CrossRef]
- Ding, K.; Shi, X.; Yang, H.; Cao, L.; Zhao, X.; Liu, H.; Wu, W.; Zhang, X.; Wang, L.; Xu, W.; et al. The Interim Efficacy of Epigenetic Priming Regimen with Azacytidine and Chidamide in Patients with Relapsed or Refractory Peripheral T Cell Lymphoma. Blood 2021, 138, 2461. [Google Scholar] [CrossRef]
- Jessica, E.B.; Melissa, J.P.; Ricky, W.J. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Ricky, W.J.; Jonathan, D.L. Histone deacetylase inhibitors in cancer therapy: Is transcription the primary target? Cancer Cell 2003, 4, 13–18. [Google Scholar] [CrossRef]
- Peart, M.J.; Smyth, G.K.; Laar, R.K.v.; Bowtell, D.D.; Richon, V.M.; Marks, P.A.; Holloway, A.J.; Johnstone, R.W. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 3697–3702. [Google Scholar] [CrossRef]
- Walid, R.; Mark, B.; Ricky, W.J.; Prince, H.M. Histone deacetylase inhibitors in lymphoma and solid malignancies. Expert Rev. Anticancer Ther. 2008, 8, 413–432. [Google Scholar] [CrossRef]
- Yun, L.; Zhijuan, L.; Kun, C.; Zhihong, F.; Zhifeng, L.; Yiming, L.; Bing, X. Prognostic role of TET2 deficiency in myelodysplastic syndromes: A meta-analysis. Oncotarget 2017, 8, 43295–43305. [Google Scholar] [CrossRef]
- Cedena, M.T.; Inmaculada, R.; Alejandro, S.-L.; Rosa, A.; Esther, O.; María, A.; Esperanza, S.; Fernando, R.; José, C.; María, D.-C.; et al. Mutations in the DNA methylation pathway and number of driver mutations predict response to azacitidine in myelodysplastic syndromes. Oncotarget 2017, 8, 106948–106961. [Google Scholar] [CrossRef]
- Morgane, C.; Julie, B.; Olivier, K.; François, L.; Richard, D.; Philippe, G.; Isabelle, R.; Coralie, D.; Olivier, H.; François, L. Efficacy of 5-azacytidine in a TET2 mutated angioimmunoblastic T cell lymphoma. Br. J. Haematol. 2014, 168, 913–916. [Google Scholar] [CrossRef]
- O’Connor, O.A.; Falchi, L.; Lue, J.K.; Marchi, E.; Kinahan, C.; Sawas, A.; Deng, C.; Montanari, F.; Amengual, J.E.; Kim, H.A.; et al. Oral 5-azacytidine and romidepsin exhibit marked activity in patients with PTCL: A multicenter phase 1 study. Blood 2019, 134, 1395–1405. [Google Scholar] [CrossRef]
- Elizabeth, E.C.; Kurtis, E.B.; Sanna, M.; James, G.H.; Stephen, B.B. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet. 1999, 21, 103–107. [Google Scholar] [CrossRef]
- Melanie, R.H.; Aleksandra, K.; Karoline, K.; Irene, S.; Martin, B.; Ana-Iris, S.; Veronika, S.; Gerda, E. Antineoplastic activity of the DNA methyltransferase inhibitor 5-aza-2’-deoxycytidine in anaplastic large cell lymphoma. Biochimie 2012, 94, 2297–2307. [Google Scholar] [CrossRef]
- Jabbour, E.; Issa, J.P.; Garcia-Manero, G.; Kantarjian, H. Evolution of decitabine development: Accomplishments, ongoing investigations, and future strategies. Cancer 2008, 112, 2341–2351. [Google Scholar] [CrossRef]
- Benigno, C.V.; Yang, L.; David, M.; Yan, L.; Yago, N.; Richard, E.C.; Borje, S.A. Combination of a hypomethylating agent and inhibitors of PARP and HDAC traps PARP1 and DNMT1 to chromatin, acetylates DNA repair proteins, down-regulates NuRD and induces apoptosis in human leukemia and lymphoma cells. Oncotarget 2017, 9, 3908–3921. [Google Scholar] [CrossRef]
- Chua, G.N.L.; Wassarman, K.L.; Sun, H.; Alp, J.A.; Jarczyk, E.I.; Kuzio, N.J.; Bennett, M.J.; Malachowsky, B.G.; Kruse, M.; Kennedy, A.J. Cytosine-Based TET Enzyme Inhibitors. ACS Med. Chem. Lett. 2019, 10, 180–185. [Google Scholar] [CrossRef]
- Guan, Y.; Tiwari, A.D.; Phillips, J.G.; Hasipek, M.; Grabowski, D.R.; Pagliuca, S.; Gopal, P.; Kerr, C.M.; Adema, V.; Radivoyevitch, T.; et al. A Therapeutic Strategy for Preferential Targeting of TET2 Mutant and TET-dioxygenase Deficient Cells in Myeloid Neoplasms. Blood Cancer Discov. 2020, 2, 146–161. [Google Scholar] [CrossRef]
- Shenoy, N.; Bhagat, T.; Nieves, E.; Stenson, M.; Lawson, J.; Choudhary, G.S.; Habermann, T.; Nowakowski, G.; Singh, R.; Wu, X.; et al. Upregulation of TET activity with ascorbic acid induces epigenetic modulation of lymphoma cells. Blood Cancer J. 2017, 7, e587. [Google Scholar] [CrossRef]
- Smith-Díaz, C.C.; Magon, N.J.; McKenzie, J.L.; Hampton, M.B.; Vissers, M.C.M.; Das, A.B. Ascorbate Inhibits Proliferation and Promotes Myeloid Differentiation in TP53-Mutant Leukemia. Front. Oncol. 2021, 11, 709543. [Google Scholar] [CrossRef]
- Guan, Y.; Greenberg, E.F.; Hasipek, M.; Chen, S.; Liu, X.; Kerr, C.M.; Gackowski, D.; Zarakowska, E.; Radivoyevitch, T.; Gu, X.; et al. Context dependent effects of ascorbic acid treatment in TET2 mutant myeloid neoplasia. Commun. Biol. 2020, 3, 493. [Google Scholar] [CrossRef]
- Bensberg, M.; Rundquist, O.; Selimović, A.; Lagerwall, C.; Benson, M.; Gustafsson, M.; Vogt, H.; Lentini, A.; Nestor, C.E. TET2 as a tumor suppressor and therapeutic target in T-cell acute lymphoblastic leukemia [Genetics]. Proc. Natl. Acad. Sci. USA 2021, 118, e2110758118. [Google Scholar] [CrossRef]
- Gerecke, C.; Schumacher, F.; Edlich, A.; Wetzel, A.; Yealland, G.; Neubert, L.K.; Scholtka, B.; Homann, T.; Kleuser, B. Vitamin C promotes decitabine or azacytidine induced DNA hydroxymethylation and subsequent reactivation of the epigenetically silenced tumour suppressor CDKN1A in colon cancer cells. Oncotarget 2018, 9, 32822–32840. [Google Scholar] [CrossRef]
- Zeng, H.; He, H.; Guo, L.; Li, J.; Lee, M.; Han, W.; Guzman, A.G.; Zang, S.; Zhou, Y.; Zhang, X.; et al. Antibiotic treatment ameliorates Ten-eleven translocation 2 (TET2) loss-of-function associated hematological malignancies. Cancer Lett. 2019, 467, 1–8. [Google Scholar] [CrossRef]
- Li, C.; Peng, C.; Jiang, Z.; Hu, H.; Lin, C.; Gao, Y.; Liu, D.; Sun, B.; Wang, D. Ginkgo biloba Extract Inhibited Cell Proliferation and Invasion by Stimulating TET2 Expression Through miR-29a in Colorectal Carcinoma Cells. DNA Cell Biol. 2022, 41. [Google Scholar] [CrossRef]
- Kawahori, K.; Kondo, Y.; Yuan, X.; Kawasaki, Y.; Hanzawa, N.; Tsujimoto, K.; Wada, F.; Kohda, T.; Ishigami, A.; Yamada, T.; et al. Ascorbic acid during the suckling period is required for proper DNA demethylation in the liver. Sci. Rep. 2020, 10, 21228. [Google Scholar] [CrossRef]
- Yin, R.; Mao, S.-Q.; Zhao, B.; Chong, Z.; Yang, Y.; Zhao, C.; Zhang, D.; Huang, H.; Gao, J.; Li, Z.; et al. Ascorbic Acid Enhances Tet-Mediated 5-Methylcytosine Oxidation and Promotes DNA Demethylation in Mammals. J. Am. Chem. Soc. 2013, 35, 10396–10403. [Google Scholar] [CrossRef]
- Blaschke, K.; Ebata, K.T.; Karimi, M.M.; Zepeda-Martínez, J.A.; Goyal, P.; Mahapatra, S.; Tam, A.; Laird, D.J.; Hirst, M.; Rao, A.; et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature 2013, 500, 222–226. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Diagnosis | Experiment Type | AITL Cases | Mutation Cases | Mutation Rate | Mutation Sites | Mutation Type (Rate) | Amino Acid Change | Mult-Mutation Rate | Reference |
|---|---|---|---|---|---|---|---|---|---|
| AITL& PTCL-NOS | Microarray | 30 | 10 | AITL: 33.3%; PTCL-NOS: 20% | NA | NA | NA | NA | Cyril Quivoron [4] et al. (2011) |
| AITL& PTCL-NOS | direct sequencing | 86 | 40 | AITL: 47%; PTCL-NOS: 38% | NA | NA | NA | NA | Lemonnier [18] et al. (2012) |
| AITL | NGS | 85 | 65 | 76% | 115 | Missense: 20/115 (17%); Nonsense: 38/115 (33%); Splice site: 3/115 (3%); Frameshift: 54/115 (47%); | p.Q673*, p.Q727*, p.Q765*, p.R1486*, p.Y1148* | 43/65 (66%) | Odejide [30] et al. (2014) |
| AITL& PTCL-NOS | Targeted resequencing | 46 | 38 | AITL: 82.6%; PTCL-NOS: 48.5% | 70 | Frameshift: 29/70 (42%); Missense: 19/70 (27%); Nonframeshift: 1/70 (1%); Nonsense: 16/70 (23%); Splice site: 5/70 (7%) | p.E1318 splice; | 28/38 (74%) | Sakata-Yanagimoto [7] et al. (2014) |
| AITL& PTCL-NOS | Targeted resequencing | 39 | 32 | AITL: 82.1%; PTCL-NOS: 46.3% | 48 | NA | NA | 13/32 (41%) | Wang [24] et al. (2015) |
| AITL | WES | 9 | 9 | 100% | 15 | frameshift or nonsense changes: 14/15 (93%) | NA | 6/9 (67%) | Wang [19] et al. (2017) |
| AITL | Sanger | 13 | 12 | 92% | 15 | premature stop codons or deletions:11/15 (73%);replacement:4/15 (27%) | NA | NA | Schwartz [9] et al. (2017) |
| AITL& Nodal PTCL with TFH phenotype& PTCL-NOS | Targeted resequencing | 48 | 36 | AITL:75%; Nodal PTCL with TFH phenotype:100% PTCL-NOS: 55.9% | NA | NA | NA | NA | T B Nguyen [8] et al. (2017) |
| AITL& PTCL-NOS & PTCL-TFH&FTCL | NA | 64 | 31 | AITL: 48%; PTCL-NOS:17% PTCL-TFH: 64% FTCL:75% | NA | NA | NA | NA | Dobay [10] et al. (2017) |
| AITL | Targeted resequencing | 12 | 12 | 100% | 17 | NA | NA | 7/12 (58%) | Lemonnier [31] et al. (2018) |
| AITL& PTCL-NOS | Targeted Exon Sequencing | 13 | 5 | AITL:38%; PTCL-NOS:31% | 65 | NA | R126C, G1869W(2/13); N202K; D302Y; Y620; A893T; W1291; | NA | Fernandez-Pol [32] et al. (2019) |
| AITL | NGS | 44 | 38 | 86% | 60 | NA | NA | 23/38 (61%) | Julia Steinhilber [26] et al. (2019) |
| AITL& PTCL-TFH | Fluidigm Access Array& Illumina MiSeq | 94 | NA | AITL: 72%; PTCL-TFH: 73% | 154 | frameshift indels or Nonsense changes: 118 (77%) | NA | 57% | Yao [20] et al. (2020) |
| AITL | Targeted sequencing | 10 | 6 | 60% | 6 | NA | R550; Q1274; G422Efs’ 5; L34F; Q909; G422Efs’ 5 | 0/6 | Butzmann [21] et al. (2020) |
| AITL | Targeted sequencing | 5 | 4 | 80% | 8 | Frameshift insertion: 2/8 (25%); Nonsilent: 3/8 (37.5%);Frameshift deletion:3/8 (37.5%) | NA | 4/5 (80%) | Tran B. Nguyen [23] et al. (2020) |
| AITL | NGS | 28 | 25 | 85% | 75 | Missense: 26/75 (34.7%);Nosens: 22/75 (29.3%);Frameshift: 22/75 (29.3%);Splice: 4/75 (5.3%);CDS-indel: 1/75 (1.3%) | NA | 22/28 (79%) | Ye [22] et al. (2021) |
| AITL | NGS | 44 | NA | NA | 49 | Frameshift: 18/49 (36.7%);Missense: 12/49 (24.5%);Splice: 3/49 (6%);Stop_gained: 12/49 (24.5%);Synonymous: 3/49 (6%);3_prime_UTR: 1/49 (2%) | NA | NA | Marta Rodríguez [27] et al. (2021) |
| AITL& PTCL-TFH | Targeted resequencing | 63 | 49 | AITL: 78%; PTCL-TFH: 58% | NA | NA | NA | 28/49 (57%) | Lemonnier [25] et al. (2021) |
| 5 | PTCL Subtype | Design | Primary Endpoint | ORR | CR | PR | Median PFS (Months) | Median OS (Months) | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Romidepsin | PTCL n = 130 (PTCL-NOS n = 67, AITL n = 27) | Phase II, Open-Label | CR/Cru | 25% | 15% | 11% | 4 | NA | Coiffier [72] et al. (2012) |
| Belinostat | PTCL n = 24 (PTCL-NOS n = 13, AITL n = 3) | Phase II | ORR | 25% | 8.30% | 16.70% | NA | NA | Foss [73] et al. (2015) |
| Belinostat | PTCL n = 120 (PTCL-NOS n = 77, AITL n = 22) | Phase II, Open-Label, multicenter | ORR | 26% | 11% | 15% | 1.6 | 7.9 | O’Connor [74] et al. (2015) |
| Chidamide | PTCL n = 79 (PTCL-NOS n = 27, AITL n = 10) | Phase II, Open-Label, multicenter | ORR | 28% | 9% | 14% | 2.1 | 21.4 | Shi [75] et al. (2015) |
| Chidamide | PTCL n = 256 | Phase II, multicenter | ORR | 39.06% (PTCL 37.3% AITL 49.23%) | PTCL 8.73% AITL 9.23% | PTCL 28.57% AITL 40% | 4.3 | NA | Shi [76] et al. (2017) |
| Romidepsin + CHOP | PTCL n = 37 (PTCL-NOS n = 9, AITL n = 15) | phase 1b/2 | ORR | 69% | 51% | 17% | 21.3 | NA | Dupuis [77] et al. (2015) |
| Panobinostat + bortezomib | PTCL n = 25 (PTCL-NOS n = 9, AITL n = 8) | Phase II, Open-Label, multicenter | ORR | 43% (PTCL 22% AITL 50%) | 21.5% (PTCL 11% AITL 25%) | 21.5% (PTCL 11% AITL 25%) | NA | NA | Tan [76] et al. (2015) |
| Chidamide + chemotherapy | PTCL n = 127 | Phase II, multicenter | ORR | 51.18% | NA | NA | 5.4 | NA | Shi [76] et al. (2017) |
| Duvelisib + Romidepsin | T-cell lymphoma n = 12 | Phase I | ORR | 50% | NA | NA | NA | NA | Moskowitz [78] et al. (2017) |
| Duvelisib + bortezomib | T-cell lymphoma n = 17 | Phase I | ORR | 53% | 20% | 23% | NA | NA | Moskowitz [78] et al. (2017) |
| 5-Azacytidine | AITL n = 12 PTCL n = 37 | Clinic trial | ORR | 75% | 50% | 25% | 15 | 21 | Lemonnier [31] et al. (2018) |
| Duvelisib + Romidepsin | T-cell lymphoma n = 39 (PTCL n = 22) | Phase I, Parallel Multicenter | ORR | 51% (PTCL 55%) | 17% (PTCL 27%) | 34% | 8.8 (PTCL) | NA | Horwitz [41] et al. (2018) |
| Romidepsin + CHOP | PTCL n = 421 (Ro-CHOP n = 211) | Phase III | PFS | 63% | 41% | 22% | 12 | 51.8 | Bachy [79] et al. (2021) |
| 5-Azacytidine + romidepsin | PTCL n = 25 (PTCL-NOS n = 4, AITL n = 14) | Phase II, multicenter | ORR | 61% | 48% | 13% | 8 | Not reached | Falchi [80] et al. (2021) |
| 5-Azacytidine + Chidamide | PTCL n = 24 (PTCL-NOS n = 4, AITL n = 15) | Phase II | ORR | 68.8% (AITL 72.7%) | 31.2% (AITL 36.4%) | 37.5% (AITL 36.4%) | NA | NA | Ding [81] et al. (2021) |
| Drugs | Disease of Study | Models | Mechanism | Limitations | Reference |
|---|---|---|---|---|---|
| Bobcat339 (TET enzyme inhibitors) | NA | HT-22 cells | Reduce DNA 5hmC levels in hippocampal | No testing in animal model and clinical trial | Gabriella [94] et al. (2019) |
| TET-specific inhibitors (TETi76) | MDS | Cell-permeable diethyl ester of TETi76 and different human leukemia cell lines (K562, MEG-01, SIG-M5, OCI-AML5, and MOLM13) | Decrease cytosine hydroxymethylation and restrict clonal out-growth of TET2 mutant | Potential to replicate the TET2 mutation | Guan [95] et al. (2020) |
| Ascorbic acid (AA) | DLBCL | Lymphoma cell lines LY-1 (DLBCL), Karpas 299 (T-cell NHL), and Jeko (mantle cell NHL) | Enhance TET activity and an increase in the hydroxymethylcytosine fraction; reactivate SMAD1 | The target, route of administration, and dose are unclear | N Shenoy [96] et al. (2017) |
| Ascorbate | AML | SKM-1 cells | Increase TET activity | The target, route of administration, and dose are unclear | Carlos [97] et al. (2021) |
| Ascorbic acid (AA) | Myeloid neoplasia (MN) | TET2−/− mice | facilitate Fe(III)/Fe(II) redox reaction | The target, route of administration, and dose are unclear | Guan [98] et al. (2020) |
| Ascorbic acid (AA) + 5-Azacytidine (5-aza) | Pediatric T-cell acute lymphoblastic leukemia (T-ALL) | TET2-silenced T-ALL cells | Stable re-expression of the TET2 gene; up-regulation of methylated genes and human endogenous retroviruses (HERVs) | The target, route of administration, and dose are unclear | Maike [99] et al. (2021) |
| Vitamin C | Colorectal cancer (CRC) | HCT 116 cells | Increase expression of CDKN1A | The target, route of administration, and dose are unclear | Christian [100] et al. (2018) |
| Four-antibiotic cocktail | CMML | TET2 KO mice | Supress TNF-α signaling | How antibiotics inhibit the pathways associated with TNF is unclear | Zeng [101] et al. (2019) |
| Ginkgo biloba extract (GBE) | Colorectal cancer (CRC) | SW480 cells | Reduce expression of miR-29a | No testing in the clinical trial | Li [102] et al. (2022) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, L.; Zhang, X.; Li, H.; Lin, S.; Zang, S. Targeting TET2 as a Therapeutic Approach for Angioimmunoblastic T Cell Lymphoma. Cancers 2022, 14, 5699. https://doi.org/10.3390/cancers14225699
Hu L, Zhang X, Li H, Lin S, Zang S. Targeting TET2 as a Therapeutic Approach for Angioimmunoblastic T Cell Lymphoma. Cancers. 2022; 14(22):5699. https://doi.org/10.3390/cancers14225699
Chicago/Turabian StyleHu, Lina, Xuanye Zhang, Huifeng Li, Suxia Lin, and Shengbing Zang. 2022. "Targeting TET2 as a Therapeutic Approach for Angioimmunoblastic T Cell Lymphoma" Cancers 14, no. 22: 5699. https://doi.org/10.3390/cancers14225699
APA StyleHu, L., Zhang, X., Li, H., Lin, S., & Zang, S. (2022). Targeting TET2 as a Therapeutic Approach for Angioimmunoblastic T Cell Lymphoma. Cancers, 14(22), 5699. https://doi.org/10.3390/cancers14225699