
CIC-Rearranged Sarcomas: An Intriguing Entity That May Lead the Way to the Comprehension of More Common Cancers




Abstract
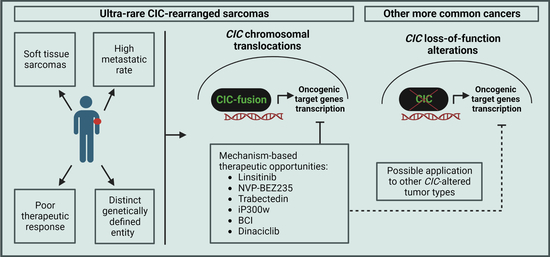
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Description of CIC-Rearranged Sarcoma
2.1. Epidemiology
2.2. Clinical Features and Treatment
2.3. Morphological and Genetic Features
3. Should CIC-Rearranged Sarcomas Be Studied Together with Other CIC-Deregulated Tumors?
4. Therapeutic Options and Perspectives
5. Critical Issues and Perspectives
- There is no consensus on how to treat patients with CIC-rearranged sarcomas. Prospective, multi-institutional clinical studies are highly recommended as well as the creation of a prospective registry that could be useful to better describe peculiarities of these tumors and to refine the best treatment.
- Treatment of rare cancers is complicated by recruitment difficulties, and it faces a lack of interest from Big Pharma in the development of new treatments. In this respect, the possibility to design basket trials where patients from different types of CIC-deregulated tumors are commonly treated based on the presence of a specific factor may represent an interesting perspective.
- A consensus regarding the workflow for the precise diagnosis of CIC sarcomas compared to other undifferentiated round cell sarcomas is still lacking. A multimodal approach combining immunohistochemistry and molecular biology techniques, including FISH, RT-PCR, and targeted RNA-seq, needs to be employed. Tumor specimens negative for EWS-specific fusion gene should be analyzed for the panel of known fusion transcripts including CIC-rearrangement, BCOR-rearrangement, EWSR1, or FUS fusion with no-ETS partner.
- The functional impact of CIC alterations is being explored through multiple cell-based and animal model systems, leading to the potential development of novel therapies to target CIC-altered cancers. Since the challenge for investigators focused on developing new therapies in the current era is to get the right therapy to the right patient at the right time, guidelines that govern the treatment decisions and precise roadmap should be created. Operational advances in the way we conduct clinical research will help us to identify promising therapies more quickly and to move them forward toward the clinic.
- The processes mediated by the CIC–ETV1/ETV4/ETV5 axis represent the majority of our knowledge on CIC functions. Additional CIC target genes have been discovered through molecular analyses of various cancer cells; however, their role in cancer still needs to be implemented. Furthermore, future studies should explore the still unclear mechanisms by which CIC regulates the expression of its target genes.
- Most of the studies have focused on the transcriptome comparison between CIC-rearranged sarcomas and other undifferentiated round cells sarcomas, mainly EWS. A comprehensive analysis of the genomic landscape of CIC-DUX4 sarcomas, including analysis of copy number alterations (CNAs), somatic point mutations, and insertions/deletions (indels) is still lacking.
- Experimental evidence demonstrates that epigenetic modulators represent a promising therapeutic opportunity in CIC-DUX4 cellular models. However, 1. the contribution of CIC-rearrangements to DNA methylation and histone modifications, and 2. epigenetic heterogeneity among CIC-rearranged sarcomas is still unexplored and should be addressed.
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Soft Tissue and Bone Tumors WHO Classification of Tumors, 5th ed.; WHO: Lyon, France, 2020; Volume 3.
- Antonescu, C. Round cell sarcomas beyond Ewing: Emerging entities. Histopathology 2014, 64, 26–37. [Google Scholar] [CrossRef]
- Grunewald, T.G.P.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; de Alava, E.; Kovar, H.; Sorensen, P.H.; Delattre, O.; Dirksen, U. Ewing sarcoma. Nat. Rev. Dis Primers 2018, 4, 5. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Owosho, A.A.; Zhang, L.; Chen, S.; Deniz, K.; Huryn, J.M.; Kao, Y.C.; Huang, S.C.; Singer, S.; Tap, W.; et al. Sarcomas With CIC-rearrangements Are a Distinct Pathologic Entity with Aggressive Outcome: A Clinicopathologic and Molecular Study of 115 Cases. Am. J. Surg. Pathol. 2017, 41, 941–949. [Google Scholar] [CrossRef]
- Gambarotti, M.; Benini, S.; Gamberi, G.; Cocchi, S.; Palmerini, E.; Sbaraglia, M.; Donati, D.; Picci, P.; Vanel, D.; Ferrari, S.; et al. CIC-DUX4 fusion-positive round-cell sarcomas of soft tissue and bone: A single-institution morphological and molecular analysis of seven cases. Histopathology 2016, 69, 624–634. [Google Scholar] [CrossRef]
- Italiano, A.; Sung, Y.S.; Zhang, L.; Singer, S.; Maki, R.G.; Coindre, J.M.; Antonescu, C.R. High prevalence of CIC fusion with double-homeobox (DUX4) transcription factors in EWSR1-negative undifferentiated small blue round cell sarcomas. Genes Chromosomes Cancer 2012, 51, 207–218. [Google Scholar] [CrossRef]
- Palmerini, E.; Righi, A.; Staals, E.L. Rare Primary Malignant Bone Sarcomas. Cancers 2020, 12, 3092. [Google Scholar] [CrossRef]
- Peters, T.L.; Kumar, V.; Polikepahad, S.; Lin, F.Y.; Sarabia, S.F.; Liang, Y.; Wang, W.L.; Lazar, A.J.; Doddapaneni, H.; Chao, H.; et al. BCOR-CCNB3 fusions are frequent in undifferentiated sarcomas of male children. Mod. Pathol. 2015, 28, 575–586. [Google Scholar] [CrossRef]
- Le Loarer, F.; Baud, J.; Azmani, R.; Michot, A.; Karanian, M.; Pissaloux, D. Advances in the classification of round cell sarcomas. Histopathology 2022, 80, 33–53. [Google Scholar] [CrossRef]
- Kao, Y.C.; Sung, Y.S.; Zhang, L.; Huang, S.C.; Argani, P.; Chung, C.T.; Graf, N.S.; Wright, D.C.; Kellie, S.J.; Agaram, N.P.; et al. Recurrent BCOR Internal Tandem Duplication and YWHAE-NUTM2B Fusions in Soft Tissue Undifferentiated Round Cell Sarcoma of Infancy: Overlapping Genetic Features With Clear Cell Sarcoma of Kidney. Am. J. Surg. Pathol. 2016, 40, 1009–1020. [Google Scholar] [CrossRef]
- WHO. Soft Tissue and Bone Tumors WHO Classification of Tumors, 4th ed.; WHO: Lyon, France, 2013; Volume 5.
- Yoshimoto, T.; Tanaka, M.; Homme, M.; Yamazaki, Y.; Takazawa, Y.; Antonescu, C.R.; Nakamura, T. CIC-DUX4 Induces Small Round Cell Sarcomas Distinct from Ewing Sarcoma. Cancer Res. 2017, 77, 2927–2937. [Google Scholar] [CrossRef]
- Le Loarer, F.; Pissaloux, D.; Watson, S.; Godfraind, C.; Galmiche-Rolland, L.; Silva, K.; Mayeur, L.; Italiano, A.; Michot, A.; Pierron, G.; et al. Clinicopathologic Features of CIC-NUTM1 Sarcomas, a New Molecular Variant of the Family of CIC-Fused Sarcomas. Am. J. Surg. Pathol. 2019, 43, 268–276. [Google Scholar] [CrossRef]
- Connolly, E.A.; Bhadri, V.A.; Wake, J.; Ingley, K.M.; Lewin, J.; Bae, S.; Wong, D.D.; Long, A.P.; Pryor, D.; Thompson, S.R.; et al. Systemic treatments and outcomes in CIC-rearranged Sarcoma: A national multi-centre clinicopathological series and literature review. Cancer Med. 2022, 11, 1805–1816. [Google Scholar] [CrossRef]
- Vieira, A.C.; Xavier, C.B.; Vieira, T.D.; Carvalho, F.M.; Scaranti, M.; Munhoz, R.R.; Carvalho, J.P. CIC-DUX4 rearranged uterine cervix round-cell sarcoma exhibiting near-complete pathologic response following radiation and neoadjuvant chemotherapy: A case report. Gynecol. Oncol. Rep. 2021, 36, 100745. [Google Scholar] [CrossRef]
- Brahmi, M.; Vanacker, H.; Macagno, N.; Tirode, F.; Dufresne, A. CIC-DUX4 sarcomas. Curr. Opin. Oncol. 2022, 34, 342–347. [Google Scholar] [CrossRef]
- Yoshida, A.; Goto, K.; Kodaira, M.; Kobayashi, E.; Kawamoto, H.; Mori, T.; Yoshimoto, S.; Endo, O.; Kodama, N.; Kushima, R.; et al. CIC-rearranged Sarcomas: A Study of 20 Cases and Comparisons With Ewing Sarcomas. Am. J. Surg. Pathol. 2016, 40, 313–323. [Google Scholar] [CrossRef]
- Ferrari, S.; Sundby Hall, K.; Luksch, R.; Tienghi, A.; Wiebe, T.; Fagioli, F.; Alvegard, T.A.; Brach Del Prever, A.; Tamburini, A.; Alberghini, M.; et al. Nonmetastatic Ewing family tumors: High-dose chemotherapy with stem cell rescue in poor responder patients. Results of the Italian Sarcoma Group/Scandinavian Sarcoma Group III protocol. Ann. Oncol. 2011, 22, 1221–1227. [Google Scholar] [CrossRef]
- Sbaraglia, M.; Righi, A.; Gambarotti, M.; Dei Tos, A.P. Ewing sarcoma and Ewing-like tumors. Virchows Arch. 2020, 476, 109–119. [Google Scholar] [CrossRef]
- Siegele, B.; Roberts, J.; Black, J.O.; Rudzinski, E.; Vargas, S.O.; Galambos, C. DUX4 Immunohistochemistry Is a Highly Sensitive and Specific Marker for CIC-DUX4 Fusion-positive Round Cell Tumor. Am. J. Surg. Pathol. 2017, 41, 423–429. [Google Scholar] [CrossRef]
- Smith, S.C.; Palanisamy, N.; Martin, E.; Almenara, J.; McHugh, J.B.; Choi, E.K.; Lucas, D.R.; Betz, B.L.; Thomas, D.; Patel, R.M. The utility of ETV1, ETV4 and ETV5 RNA in-situ hybridization in the diagnosis of CIC-DUX sarcomas. Histopathology 2017, 70, 657–663. [Google Scholar] [CrossRef]
- Le Guellec, S.; Velasco, V.; Perot, G.; Watson, S.; Tirode, F.; Coindre, J.M. ETV4 is a useful marker for the diagnosis of CIC-rearranged undifferentiated round-cell sarcomas: A study of 127 cases including mimicking lesions. Mod. Pathol. 2016, 29, 1523–1531. [Google Scholar] [CrossRef]
- Kawamura-Saito, M.; Yamazaki, Y.; Kaneko, K.; Kawaguchi, N.; Kanda, H.; Mukai, H.; Gotoh, T.; Motoi, T.; Fukayama, M.; Aburatani, H.; et al. Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Hum. Mol. Genet. 2006, 15, 2125–2137. [Google Scholar] [CrossRef] [PubMed]
- Oyama, R.; Takahashi, M.; Yoshida, A.; Sakumoto, M.; Takai, Y.; Kito, F.; Shiozawa, K.; Qiao, Z.; Arai, Y.; Shibata, T.; et al. Generation of novel patient-derived CIC- DUX4 sarcoma xenografts and cell lines. Sci. Rep. 2017, 7, 4712. [Google Scholar] [CrossRef]
- Hung, Y.P.; Fletcher, C.D.; Hornick, J.L. Evaluation of ETV4 and WT1 expression in CIC-rearranged sarcomas and histologic mimics. Mod. Pathol. 2016, 29, 1324–1334. [Google Scholar] [CrossRef]
- Sugita, S.; Arai, Y.; Aoyama, T.; Asanuma, H.; Mukai, W.; Hama, N.; Emori, M.; Shibata, T.; Hasegawa, T. NUTM2A-CIC fusion small round cell sarcoma: A genetically distinct variant of CIC-rearranged sarcoma. Hum. Pathol. 2017, 65, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Liu, L.; Yan, Y.; Jiang, L.; Han, S.; Shen, D.; Zhang, B. CIC-NUTM1 Sarcomas Affecting the Spine. Arch. Pathol. Lab. Med. 2022, 146, 735–741. [Google Scholar] [CrossRef]
- Smith, S.C.; Buehler, D.; Choi, E.Y.; McHugh, J.B.; Rubin, B.P.; Billings, S.D.; Balzer, B.; Thomas, D.G.; Lucas, D.R.; Goldblum, J.R.; et al. CIC-DUX sarcomas demonstrate frequent MYC amplification and ETS-family transcription factor expression. Mod. Pathol. 2015, 28, 57–68. [Google Scholar] [CrossRef]
- Simon-Carrasco, L.; Jimenez, G.; Barbacid, M.; Drosten, M. The Capicua tumor suppressor: A gatekeeper of Ras signaling in development and cancer. Cell Cycle 2018, 17, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Mocciaro, E.; Runfola, V.; Ghezzi, P.; Pannese, M.; Gabellini, D. DUX4 Role in Normal Physiology and in FSHD Muscular Dystrophy. Cells 2021, 10, 3322. [Google Scholar] [CrossRef]
- Okimoto, R.A.; Wu, W.; Nanjo, S.; Olivas, V.; Lin, Y.K.; Ponce, R.K.; Oyama, R.; Kondo, T.; Bivona, T.G. CIC-DUX4 oncoprotein drives sarcoma metastasis and tumorigenesis via distinct regulatory programs. J. Clin. Investig. 2019, 129, 3401–3406. [Google Scholar] [CrossRef]
- Choi, S.H.; Gearhart, M.D.; Cui, Z.; Bosnakovski, D.; Kim, M.; Schennum, N.; Kyba, M. DUX4 recruits p300/CBP through its C-terminus and induces global H3K27 acetylation changes. Nucleic Acids Res. 2016, 44, 5161–5173. [Google Scholar] [CrossRef]
- Xu, H.; Wang, Z.; Jin, S.; Hao, H.; Zheng, L.; Zhou, B.; Zhang, W.; Lv, H.; Yuan, Y. Dux4 induces cell cycle arrest at G1 phase through upregulation of p21 expression. Biochem. Biophys. Res. Commun 2014, 446, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Shadle, S.C.; Zhong, J.W.; Campbell, A.E.; Conerly, M.L.; Jagannathan, S.; Wong, C.J.; Morello, T.D.; van der Maarel, S.M.; Tapscott, S.J. DUX4-induced dsRNA and MYC mRNA stabilization activate apoptotic pathways in human cell models of facioscapulohumeral dystrophy. PLoS Genet. 2017, 13, e1006658. [Google Scholar] [CrossRef] [PubMed]
- Chew, G.L.; Campbell, A.E.; De Neef, E.; Sutliff, N.A.; Shadle, S.C.; Tapscott, S.J.; Bradley, R.K. DUX4 Suppresses MHC Class I to Promote Cancer Immune Evasion and Resistance to Checkpoint Blockade. Dev. Cell 2019, 50, 658–671.e657. [Google Scholar] [CrossRef]
- Lee, C.J.; Chan, W.I.; Cheung, M.; Cheng, Y.C.; Appleby, V.J.; Orme, A.T.; Scotting, P.J. CIC, a member of a novel subfamily of the HMG-box superfamily, is transiently expressed in developing granule neurons. Brain Res. Mol. Brain Res. 2002, 106, 151–156. [Google Scholar] [CrossRef]
- Fores, M.; Simon-Carrasco, L.; Ajuria, L.; Samper, N.; Gonzalez-Crespo, S.; Drosten, M.; Barbacid, M.; Jimenez, G. A new mode of DNA binding distinguishes Capicua from other HMG-box factors and explains its mutation patterns in cancer. PLoS Genet. 2017, 13, e1006622. [Google Scholar] [CrossRef]
- Lee, Y. Regulation and function of capicua in mammals. Exp. Mol. Med. 2020, 52, 531–537. [Google Scholar] [CrossRef]
- Carrabotta, M.; Laginestra, M.A.; Durante, G.; Mancarella, C.; Landuzzi, L.; Parra, A.; Ruzzi, F.; Toracchio, L.; De Feo, A.; Giusti, V.; et al. Integrated Molecular Characterization of Patient-Derived Models Reveals Therapeutic Strategies for Treating CIC-DUX4 Sarcoma. Cancer Res. 2022, 82, 708–720. [Google Scholar] [CrossRef]
- Nakai, S.; Yamada, S.; Outani, H.; Nakai, T.; Yasuda, N.; Mae, H.; Imura, Y.; Wakamatsu, T.; Tamiya, H.; Tanaka, T.; et al. Establishment of a novel human CIC-DUX4 sarcoma cell line, Kitra-SRS, with autocrine IGF-1R activation and metastatic potential to the lungs. Sci. Rep. 2019, 9, 15812. [Google Scholar] [CrossRef] [PubMed]
- Simon-Carrasco, L.; Grana, O.; Salmon, M.; Jacob, H.K.C.; Gutierrez, A.; Jimenez, G.; Drosten, M.; Barbacid, M. Inactivation of Capicua in adult mice causes T-cell lymphoblastic lymphoma. Genes Dev. 2017, 31, 1456–1468. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Brunetti, L.; Rousseaux, M.W.C.; Lu, H.C.; Wan, Y.W.; Revelli, J.P.; Liu, Z.; Goodell, M.A.; Zoghbi, H.Y. Loss of Capicua alters early T cell development and predisposes mice to T cell lymphoblastic leukemia/lymphoma. Proc. Natl. Acad. Sci. USA 2018, 115, E1511–E1519. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Agrawal, N.; Jiao, Y.; Sausen, M.; Wood, L.D.; Hruban, R.H.; Rodriguez, F.J.; Cahill, D.P.; McLendon, R.; Riggins, G.; et al. Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science 2011, 333, 1453–1455. [Google Scholar] [CrossRef] [PubMed]
- Gleize, V.; Alentorn, A.; Connen de Kerillis, L.; Labussiere, M.; Nadaradjane, A.A.; Mundwiller, E.; Ottolenghi, C.; Mangesius, S.; Rahimian, A.; Ducray, F.; et al. CIC inactivating mutations identify aggressive subset of 1p19q codeleted gliomas. Ann. Neurol. 2015, 78, 355–374. [Google Scholar] [CrossRef] [PubMed]
- Okimoto, R.A.; Breitenbuecher, F.; Olivas, V.R.; Wu, W.; Gini, B.; Hofree, M.; Asthana, S.; Hrustanovic, G.; Flanagan, J.; Tulpule, A.; et al. Inactivation of Capicua drives cancer metastasis. Nat. Genet. 2017, 49, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Seim, I.; Jeffery, P.L.; Thomas, P.B.; Nelson, C.C.; Chopin, L.K. Whole-Genome Sequence of the Metastatic PC3 and LNCaP Human Prostate Cancer Cell Lines. G3 Genes Genomes Genet. 2017, 7, 1731–1741. [Google Scholar] [CrossRef]
- Kim, E.; Kim, D.; Lee, J.S.; Yoe, J.; Park, J.; Kim, C.J.; Jeong, D.; Kim, S.; Lee, Y. Capicua suppresses hepatocellular carcinoma progression by controlling the ETV4-MMP1 axis. Hepatology 2018, 67, 2287–2301. [Google Scholar] [CrossRef]
- Kim, J.W.; Ponce, R.K.; Okimoto, R.A. Capicua in Human Cancer. Trends Cancer 2021, 7, 77–86. [Google Scholar] [CrossRef]
- Huang, S.C.; Zhang, L.; Sung, Y.S.; Chen, C.L.; Kao, Y.C.; Agaram, N.P.; Singer, S.; Tap, W.D.; D’Angelo, S.; Antonescu, C.R. Recurrent CIC Gene Abnormalities in Angiosarcomas: A Molecular Study of 120 Cases With Concurrent Investigation of PLCG1, KDR, MYC, and FLT4 Gene Alterations. Am. J. Surg. Pathol. 2016, 40, 645–655. [Google Scholar] [CrossRef]
- Jimenez, G.; Shvartsman, S.Y.; Paroush, Z. The Capicua repressor--a general sensor of RTK signaling in development and disease. J. Cell Sci. 2012, 125, 1383–1391. [Google Scholar] [CrossRef]
- Astigarraga, S.; Grossman, R.; Diaz-Delfin, J.; Caelles, C.; Paroush, Z.; Jimenez, G. A MAPK docking site is critical for downregulation of Capicua by Torso and EGFR RTK signaling. EMBO J. 2007, 26, 668–677. [Google Scholar] [CrossRef]
- Ajuria, L.; Nieva, C.; Winkler, C.; Kuo, D.; Samper, N.; Andreu, M.J.; Helman, A.; Gonzalez-Crespo, S.; Paroush, Z.; Courey, A.J.; et al. Capicua DNA-binding sites are general response elements for RTK signaling in Drosophila. Development 2011, 138, 915–924. [Google Scholar] [CrossRef]
- Bunda, S.; Heir, P.; Metcalf, J.; Li, A.S.C.; Agnihotri, S.; Pusch, S.; Yasin, M.; Li, M.; Burrell, K.; Mansouri, S.; et al. CIC protein instability contributes to tumorigenesis in glioblastoma. Nat. Commun. 2019, 10, 661. [Google Scholar] [CrossRef] [PubMed]
- Weissmann, S.; Cloos, P.A.; Sidoli, S.; Jensen, O.N.; Pollard, S.; Helin, K. The Tumor Suppressor CIC Directly Regulates MAPK Pathway Genes via Histone Deacetylation. Cancer Res. 2018, 78, 4114–4125. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Ouyang, Z.; Hou, Z.; Yan, Y.; Zhi, Z.; Shi, M.; Du, M.; Liu, H.; Wen, Y.; Shao, Y. CIC Is a Mediator of the ERK1/2-DUSP6 Negative Feedback Loop. iScience 2020, 23, 101635. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Tan, T.H. DUSPs, to MAP kinases and beyond. Cell Biosci. 2012, 2, 24. [Google Scholar] [CrossRef] [PubMed]
- Dissanayake, K.; Toth, R.; Blakey, J.; Olsson, O.; Campbell, D.G.; Prescott, A.R.; MacKintosh, C. ERK/p90(RSK)/14-3-3 signalling has an impact on expression of PEA3 Ets transcription factors via the transcriptional repressor capicua. Biochem. J. 2011, 433, 515–525. [Google Scholar] [CrossRef]
- Kim, E.; Lu, H.C.; Zoghbi, H.Y.; Song, J.J. Structural basis of protein complex formation and reconfiguration by polyglutamine disease protein Ataxin-1 and Capicua. Genes Dev. 2013, 27, 590–595. [Google Scholar] [CrossRef]
- Wong, D.; Lounsbury, K.; Lum, A.; Song, J.; Chan, S.; LeBlanc, V.; Chittaranjan, S.; Marra, M.; Yip, S. Transcriptomic analysis of CIC and ATXN1L reveal a functional relationship exploited by cancer. Oncogene 2019, 38, 273–290. [Google Scholar] [CrossRef]
- Ni, C.; Jiang, W.; Wang, Z.; Wang, Z.; Zhang, J.; Zheng, X.; Liu, Z.; Ou, H.; Jiang, T.; Liang, W.; et al. LncRNA-AC006129.1 reactivates a SOCS3-mediated anti-inflammatory response through DNA methylation-mediated CIC downregulation in schizophrenia. Mol. Psychiatry 2021, 26, 4511–4528. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, M.; Shuang, T.; Liu, Y.; Zhang, Y.; Shi, C. MiR-1307 influences the chemotherapeutic sensitivity in ovarian cancer cells through the regulation of the CIC transcriptional repressor. Pathol. Res. Pract. 2019, 215, 152606. [Google Scholar] [CrossRef]
- Choi, N.; Park, J.; Lee, J.S.; Yoe, J.; Park, G.Y.; Kim, E.; Jeon, H.; Cho, Y.M.; Roh, T.Y.; Lee, Y. miR-93/miR-106b/miR-375-CIC-CRABP1: A novel regulatory axis in prostate cancer progression. Oncotarget 2015, 6, 23533–23547. [Google Scholar] [CrossRef]
- Miao, L.J.; Yan, S.; Zhuang, Q.F.; Mao, Q.Y.; Xue, D.; He, X.Z.; Chen, J.P. miR-106b promotes proliferation and invasion by targeting Capicua through MAPK signaling in renal carcinoma cancer. Onco Targets Ther. 2019, 12, 3595–3607. [Google Scholar] [CrossRef]
- Fryer, J.D.; Yu, P.; Kang, H.; Mandel-Brehm, C.; Carter, A.N.; Crespo-Barreto, J.; Gao, Y.; Flora, A.; Shaw, C.; Orr, H.T.; et al. Exercise and genetic rescue of SCA1 via the transcriptional repressor Capicua. Science 2011, 334, 690–693. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Chen, L.H.; Hansen, L.J.; Carpenter, A.B.; Moure, C.J.; Liu, H.; Pirozzi, C.J.; Diplas, B.H.; Waitkus, M.S.; Greer, P.K.; et al. Cic Loss Promotes Gliomagenesis via Aberrant Neural Stem Cell Proliferation and Differentiation. Cancer Res. 2017, 77, 6097–6108. [Google Scholar] [CrossRef] [PubMed]
- Yoshimatsu, Y.; Noguchi, R.; Tsuchiya, R.; Kito, F.; Sei, A.; Sugaya, J.; Nakagawa, M.; Yoshida, A.; Iwata, S.; Kawai, A.; et al. Establishment and characterization of NCC-CDS2-C1: A novel patient-derived cell line of CIC-DUX4 sarcoma. Hum. Cell 2020, 33, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, A.; Matsumoto, K.; Yoshimatsu, Y.; Sin, Y.; Kubota, A.; Saito, T.; Mizumoto, A.; Ohashi, S.; Muto, M.; Noguchi, R.; et al. The CAM Model for CIC-DUX4 Sarcoma and Its Potential Use for Precision Medicine. Cells 2021, 10, 2613. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.K.; Wu, W.; Ponce, R.K.; Kim, J.W.; Okimoto, R.A. Negative MAPK-ERK regulation sustains CIC-DUX4 oncoprotein expression in undifferentiated sarcoma. Proc. Natl. Acad. Sci. USA 2020, 117, 20776–20784. [Google Scholar] [CrossRef] [PubMed]
- Unni, A.M.; Harbourne, B.; Oh, M.H.; Wild, S.; Ferrarone, J.R.; Lockwood, W.W.; Varmus, H. Hyperactivation of ERK by multiple mechanisms is toxic to RTK-RAS mutation-driven lung adenocarcinoma cells. Elife 2018, 7, e33718. [Google Scholar] [CrossRef]
- Wang, B.; Krall, E.B.; Aguirre, A.J.; Kim, M.; Widlund, H.R.; Doshi, M.B.; Sicinska, E.; Sulahian, R.; Goodale, A.; Cowley, G.S.; et al. ATXN1L, CIC, and ETS Transcription Factors Modulate Sensitivity to MAPK Pathway Inhibition. Cell Rep. 2017, 18, 1543–1557. [Google Scholar] [CrossRef]
- Liao, S.; Davoli, T.; Leng, Y.; Li, M.Z.; Xu, Q.; Elledge, S.J. A genetic interaction analysis identifies cancer drivers that modify EGFR dependency. Genes Dev. 2017, 31, 184–196. [Google Scholar] [CrossRef]
- Bosnakovski, D.; Ener, E.T.; Cooper, M.S.; Gearhart, M.D.; Knights, K.A.; Xu, N.C.; Palumbo, C.A.; Toso, E.A.; Marsh, G.P.; Maple, H.J.; et al. Inactivation of the CIC-DUX4 oncogene through P300/CBP inhibition, a therapeutic approach for CIC-DUX4 sarcoma. Oncogenesis 2021, 10, 68. [Google Scholar] [CrossRef]
- Ponce, R.K.M.; Thomas, N.J.; Bui, N.Q.; Kondo, T.; Okimoto, R.A. WEE1 kinase is a therapeutic vulnerability in CIC-DUX4 undifferentiated sarcoma. JCI Insight 2022, 7, e152293. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mancarella, C.; Carrabotta, M.; Toracchio, L.; Scotlandi, K. CIC-Rearranged Sarcomas: An Intriguing Entity That May Lead the Way to the Comprehension of More Common Cancers. Cancers 2022, 14, 5411. https://doi.org/10.3390/cancers14215411
Mancarella C, Carrabotta M, Toracchio L, Scotlandi K. CIC-Rearranged Sarcomas: An Intriguing Entity That May Lead the Way to the Comprehension of More Common Cancers. Cancers. 2022; 14(21):5411. https://doi.org/10.3390/cancers14215411
Chicago/Turabian StyleMancarella, Caterina, Marianna Carrabotta, Lisa Toracchio, and Katia Scotlandi. 2022. "CIC-Rearranged Sarcomas: An Intriguing Entity That May Lead the Way to the Comprehension of More Common Cancers" Cancers 14, no. 21: 5411. https://doi.org/10.3390/cancers14215411
APA StyleMancarella, C., Carrabotta, M., Toracchio, L., & Scotlandi, K. (2022). CIC-Rearranged Sarcomas: An Intriguing Entity That May Lead the Way to the Comprehension of More Common Cancers. Cancers, 14(21), 5411. https://doi.org/10.3390/cancers14215411