
Chimeric Antigen Receptor Immunotherapy for Solid Tumors: Choosing the Right Ingredients for the Perfect Recipe

 ,
,
Abstract
Simple Summary
Abstract
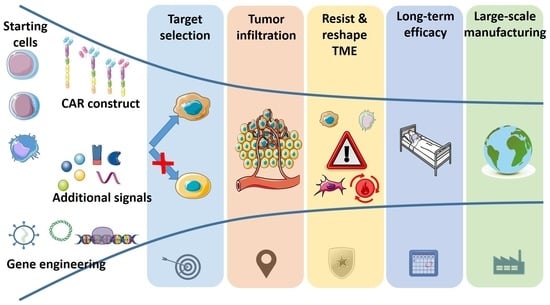
1. Introduction
2. The Challenges of Solid Tumors against CAR-Based Therapies (and Other Cell Immunotherapies)
3. The Experience with CAR-Based Therapies in Solid Tumors
4. The Cells to Engineer
4.1. Bulk T Cells
4.2. Specific T Cells Subsets
4.3. Natural Killer Cells
4.4. Macrophages
5. The Gene Engineering Tools
5.1. Cell Transduction Using Viral Vectors
5.2. Transposons for Gene Therapy of CAR
5.3. Gene Editing Using CRISPR/cas for CAR Engineering
6. Designing the Best CAR Construct against Solid Tumors
6.1. Tuning the Expression Level of CAR
6.2. Defining Which Antigen to Target and How
6.3. Choosing the Costimulation Signal
6.4. The Additional Signals to Overcome Solid Tumor Resistance
7. Manufacturing and Regulatory Aspects to Consider
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene Maraleucel for Patients with Relapsed or Refractory Large B-Cell Lymphomas (TRANSCEND NHL 001): A Multicentre Seamless Design Study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, L.D.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene Autoleucel, a B-Cell Maturation Antigen-Directed Chimeric Antigen Receptor T-Cell Therapy in Patients with Relapsed or Refractory Multiple Myeloma (CARTITUDE-1): A Phase 1b/2 Open-Label Study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef]
- Ivica, N.A.; Young, C.M. Tracking the Car-t Revolution: Analysis of Clinical Trials of Car-t and Tcr-t Therapies for the Treatment of Cancer (1997–2020). Healthcare 2021, 9, 1062. [Google Scholar] [CrossRef] [PubMed]
- Morotti, M.; Albukhari, A.; Alsaadi, A.; Artibani, M.; Brenton, J.D.; Curbishley, S.M.; Dong, T.; Dustin, M.L.; Hu, Z.; McGranahan, N.; et al. Promises and Challenges of Adoptive T-Cell Therapies for Solid Tumours. Br. J. Cancer 2021, 124, 1759–1776. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Gladney, W.L. Immune Escape Mechanisms as a Guide for Cancer Immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef]
- Rodriguez-Garcia, A.; Palazon, A.; Noguera-Ortega, E.; Powell, D.J.; Guedan, S. CAR-T Cells Hit the Tumor Microenvironment: Strategies to Overcome Tumor Escape. Front. Immunol. 2020, 11, 1109. [Google Scholar] [CrossRef]
- Hou, A.J.; Chen, L.C.; Chen, Y.Y. Navigating CAR-T Cells through the Solid-Tumour Microenvironment. Nat. Rev. Drug Discov. 2021, 20, 531–550. [Google Scholar] [CrossRef] [PubMed]
- Finn, O.J. Immuno-Oncology: Understanding the Function and Dysfunction of the Immune System in Cancer. Ann. Oncol. 2012, 23, viii6–viii9. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from Car T-Cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef]
- Lanitis, E.; Irving, M.; Coukos, G. Targeting the Tumor Vasculature to Enhance T Cell Activity. Curr. Opin. Immunol. 2015, 33, 55–63. [Google Scholar] [CrossRef]
- Kuczek, D.E.; Larsen, A.M.H.; Thorseth, M.L.; Carretta, M.; Kalvisa, A.; Siersbæk, M.S.; Simões, A.M.C.; Roslind, A.; Engelholm, L.H.; Noessner, E.; et al. Collagen Density Regulates the Activity of Tumor-Infiltrating T Cells. J. Immunother. Cancer 2019, 7, 68. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef]
- Hege, K.M.; Bergsland, E.K.; Fisher, G.A.; Nemunaitis, J.J.; Warren, R.S.; McArthur, J.G.; Lin, A.A.; Schlom, J.; June, C.H.; Sherwin, S.A. Safety, Tumor Trafficking and Immunogenicity of Chimeric Antigen Receptor (CAR)-T Cells Specific for TAG-72 in Colorectal Cancer. J. Immunother. Cancer 2017, 5, 22. [Google Scholar] [CrossRef]
- Pule, M.A.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-Specific T Cells Engineered to Coexpress Tumor-Specific Receptors: Persistence and Antitumor Activity in Individuals with Neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A Phase I Study on Adoptive Immunotherapy Using Gene-Modified T Cells for Ovarian Cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef]
- Park, J.R.; DiGiusto, D.L.; Slovak, M.; Wright, C.; Naranjo, A.; Wagner, J.; Meechoovet, H.B.; Bautista, C.; Chang, W.C.; Ostberg, J.R.; et al. Adoptive Transfer of Chimeric Antigen Receptor Re-Directed Cytolytic T Lymphocyte Clones in Patients with Neuroblastoma. Mol. Ther. 2007, 15, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C.H.J.; Sleijfer, S.; Vulto, A.G.; Kruit, W.H.J.; Kliffen, M.; Debets, R.; Gratama, J.W.; Stoter, G.; Oosterwijk, E. Treatment of Metastatic Renal Cell Carcinoma with Autologous T-Lymphocytes Genetically Retargeted against Carbonic Anhydrase IX: First Clinical Experience. J. Clin. Oncol. 2006, 24, e20-2. [Google Scholar] [CrossRef] [PubMed]
- Cappell, K.M.; Kochenderfer, J.N. A Comparison of Chimeric Antigen Receptors Containing CD28 versus 4-1BB Costimulatory Domains. Nat. Rev. Clin. Oncol. 2021, 18, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Barros, L.R.C.; Couto, S.C.F.; da Silva Santurio, D.; Paixão, E.A.; Cardoso, F.; da Silva, V.J.; Klinger, P.; Ribeiro, P.d.A.C.; Rós, F.A.; Oliveira, T.G.M.; et al. Systematic Review of Available CAR-T Cell Trials around the World. Cancers 2022, 14, 2667. [Google Scholar] [CrossRef]
- Hirayama, A.V.; Gauthier, J.; Hay, K.A.; Voutsinas, J.M.; Wu, Q.; Gooley, T.; Li, D.; Cherian, S.; Chen, X.; Pender, B.S.; et al. The Response to Lymphodepletion Impacts PFS in Patients with Aggressive Non-Hodgkin Lymphoma Treated with CD19 CAR T Cells. Blood 2019, 133, 1876–1887. [Google Scholar] [CrossRef]
- Klebanoff, C.A.; Khong, H.T.; Antony, P.A.; Palmer, D.C.; Restifo, N.P. Sinks, Suppressors and Antigen Presenters: How Lymphodepletion Enhances T Cell-Mediated Tumor Immunotherapy. Trends Immunol. 2005, 26, 111–117. [Google Scholar] [CrossRef]
- Haas, A.R.; Tanyi, J.L.; O’Hara, M.H.; Gladney, W.L.; Lacey, S.F.; Torigian, D.A.; Soulen, M.C.; Tian, L.; McGarvey, M.; Nelson, A.M.; et al. Phase I Study of Lentiviral-Transduced Chimeric Antigen Receptor-Modified T Cells Recognizing Mesothelin in Advanced Solid Cancers. Mol. Ther. 2019, 27, 1919–1929. [Google Scholar] [CrossRef]
- Narayan, V.; Barber-Rotenberg, J.S.; Jung, I.Y.; Lacey, S.F.; Rech, A.J.; Davis, M.M.; Hwang, W.T.; Lal, P.; Carpenter, E.L.; Maude, S.L.; et al. PSMA-Targeting TGFβ-Insensitive Armored CAR T Cells in Metastatic Castration-Resistant Prostate Cancer: A Phase 1 Trial. Nat. Med. 2022, 28, 724–734. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2)–Specific Chimeric Antigen Receptor—Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef]
- Katz, S.C.; Burga, R.A.; McCormack, E.; Wang, L.J.; Mooring, W.; Point, G.R.; Khare, P.D.; Thorn, M.; Ma, Q.; Stainken, B.F.; et al. Phase I Hepatic Immunotherapy for Metastases Study of Intra-Arterial Chimeric Antigen Receptor-Modified T-Cell Therapy for CEA+ Liver Metastases. Clin. Cancer Res. 2015, 21, 3149–3159. [Google Scholar] [CrossRef]
- Feng, K.; Guo, Y.; Dai, H.; Wang, Y.; Li, X.; Jia, H.; Han, W. Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of Patients with EGFR-Expressing Advanced Relapsed/Refractory Non-Small Cell Lung Cancer. Sci. China Life Sci. 2016, 59, 468–479. [Google Scholar] [CrossRef]
- Guo, Y.; Feng, K.; Liu, Y.; Wu, Z.; Dai, H.; Yang, Q.; Wang, Y.; Jia, H.; Han, W. Phase I Study of Chimeric Antigen Receptor–Modified T Cells in Patients with EGFR-Positive Advanced Biliary Tract Cancers. Clin. Cancer Res. 2018, 24, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, Z.; Yang, Z.; Wang, M.; Li, S.; Li, Y.; Zhang, R.; Xiong, Z.; Wei, Z.; Shen, J.; et al. Phase I Escalating-Dose Trial of CAR-T Therapy Targeting CEA+ Metastatic Colorectal Cancers. Mol. Ther. 2017, 25, 1248–1258. [Google Scholar] [CrossRef] [PubMed]
- Tchou, J.; Zhao, Y.; Levine, B.L.; Zhang, P.J.; Davis, M.M.; Melenhorst, J.J.; Kulikovskaya, I.; Brennan, A.L.; Liu, X.; Lacey, S.F.; et al. Safety and Efficacy of Intratumoral Injections of Chimeric Antigen Receptor (CAR) T Cells in Metastatic Breast Cancer. Cancer Immunol. Res. 2017, 5, 1152–1161. [Google Scholar] [CrossRef] [PubMed]
- Heczey, A.; Louis, C.U.; Savoldo, B.; Dakhova, O.; Durett, A.; Grilley, B.; Liu, H.; Wu, M.F.; Mei, Z.; Gee, A.; et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol. Ther. 2017, 25, 2214–2224. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A Single Dose of Peripherally Infused EGFRvIII-Directed CAR T Cells Mediates Antigen Loss and Induces Adaptive Resistance in Patients with Recurrent Glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor–Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- Feng, K.; Liu, Y.; Guo, Y.; Qiu, J.; Wu, Z.; Dai, H.; Yang, Q.; Wang, Y.; Han, W. Phase I Study of Chimeric Antigen Receptor Modified T Cells in Treating HER2-Positive Advanced Biliary Tract Cancers and Pancreatic Cancers. Protein Cell 2018, 9, 838–847. [Google Scholar] [CrossRef]
- Beatty, G.L.; O’Hara, M.H.; Lacey, S.F.; Torigian, D.A.; Nazimuddin, F.; Chen, F.; Kulikovskaya, I.M.; Soulen, M.C.; McGarvey, M.; Nelson, A.M.; et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018, 155, 29–32. [Google Scholar] [CrossRef]
- Adusumilli, P.S.; Zauderer, M.G.; Rivière, I.; Solomon, S.B.; Rusch, V.W.; O’Cearbhaill, R.E.; Zhu, A.; Cheema, W.; Chintala, N.K.; Halton, E.; et al. A Phase i Trial of Regional Mesothelin-Targeted Car t-Cell Therapy in Patients with Malignant Pleural Disease, in Combination with the Anti–Pd-1 Agent Pembrolizumab. Cancer Discov. 2021, 11, 2748–2763. [Google Scholar] [CrossRef]
- Goff, S.L.; Morgan, R.A.; Yang, J.C.; Sherry, R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; Lu, Y.C.; Lu, L.; Zheng, Z.; et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor-Transduced t Cells Targeting Egfrviii in Patients with Glioblastoma. J. Immunother. 2019, 42, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.C.; Hardaway, J.; Prince, E.; Guha, P.; Cunetta, M.; Moody, A.; Wang, L.J.; Armenio, V.; Espat, N.J.; Junghans, R.P. HITM-SIR: Phase Ib Trial of Intraarterial Chimeric Antigen Receptor T-Cell Therapy and Selective Internal Radiation Therapy for CEA+ Liver Metastases. Cancer Gene Ther. 2020, 27, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Shi, Y.; Kaseb, A.O.; Qi, X.; Zhang, Y.; Chi, J.; Lu, Q.; Gao, H.; Jiang, H.; Wang, H.; et al. Chimeric Antigen Receptor-Glypican-3 T-Cell Therapy for Advanced Hepatocellular Carcinoma: Results of Phase I Trials. Clin. Cancer Res. 2020, 26, 3979–3989. [Google Scholar] [CrossRef] [PubMed]
- Straathof, K.; Flutter, B.; Wallace, R.; Jain, N.; Loka, T.; Depani, S.; Wright, G.; Thomas, S.; Cheung, G.W.K.; Gileadi, T.; et al. Antitumor Activity without On-Target off-Tumor Toxicity of GD2-Chimeric Antigen Receptor T Cells in Patients with Neuroblastoma. Sci. Transl. Med. 2020, 12, eabd6169. [Google Scholar] [CrossRef]
- Wang, Z.; Li, N.; Feng, K.; Chen, M.; Zhang, Y.; Liu, Y.; Yang, Q.; Nie, J.; Tang, N.; Zhang, X.; et al. Phase I Study of CAR-T Cells with PD-1 and TCR Disruption in Mesothelin-Positive Solid Tumors. Cell. Mol. Immunol. 2021, 18, 2188–2198. [Google Scholar] [CrossRef]
- Heczey, A.; Courtney, A.N.; Montalbano, A.; Robinson, S.; Liu, K.; Li, M.; Ghatwai, N.; Dakhova, O.; Liu, B.; Raveh-Sadka, T.; et al. Anti-GD2 CAR-NKT Cells in Patients with Relapsed or Refractory Neuroblastoma: An Interim Analysis. Nat. Med. 2020, 26, 1686–1690. [Google Scholar] [CrossRef]
- Heczey, A.; Courtney, A.; Ghatwai, N.; Thakkar, S.; Rathi, P.; Xu, X.; Ngai, H.; Di Piero, E.J.; Grilley, B.; Sher, A.; et al. Anti-GD2 CAR NKT Cells Are Safe and Produce Antitumor Responses in Patients with Relapsed/Refractory Neuroblastoma. In Proceedings of the 25th American Society of Gene and Cell Therapy’s Annual Meeting, Washington, DC, USA, 16–19 May 2022. [Google Scholar]
- Abou-el-Enein, M.; Elsallab, M.; Feldman, S.A.; Fesnak, A.D.; Heslop, H.E.; Marks, P.; Till, B.G.; Bauer, G.; Savoldo, B. Scalable Manufacturing of CAR T Cells for Cancer Immunotherapy. Blood Cancer Discov. 2021, 2, 408–422. [Google Scholar] [CrossRef]
- Wagner, D.L.; Koehl, U.; Chmielewski, M.; Scheid, C.; Stripecke, R. Review: Sustainable Clinical Development of CAR-T Cells—Switching from Viral Transduction Towards CRISPR-Cas Gene Editing. Front. Immunol. 2022, 13, 865424. [Google Scholar] [CrossRef]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef]
- Yu, W.-L.; Hua, Z.-C. Chimeric Antigen Receptor T-Cell (CAR T) Therapy for Hematologic and Solid Malignancies: Efficacy and Safety—A Systematic Review with Meta-Analysis. Cancers 2019, 11, 47. [Google Scholar] [CrossRef]
- McKean, M.; Carabasi, M.H.; Stein, M.N.; Schweizer, M.T.; Luke, J.J.; Narayan, V.; Parikh, R.A.; Pachynski, R.K.; Zhang, J.; Peddareddigari, V.G.R.; et al. Safety and Early Efficacy Results from a Phase 1, Multicenter Trial of PSMA-Targeted Armored CAR T Cells in Patients with Advanced MCRPC. J. Clin. Oncol. 2022, 40, 94. [Google Scholar] [CrossRef]
- Lian, J.; Yue, Y.; Yu, W.; Zhang, Y. Immunosenescence: A Key Player in Cancer Development. J. Hematol. Oncol. 2020, 13, 151. [Google Scholar] [CrossRef]
- Das, R.K.; Vernau, L.; Grupp, S.A.; Barrett, D.M. Naïve T-Cell Deficits at Diagnosis and after Chemotherapy Impair Cell Therapy Potential in Pediatric Cancers. Cancer Discov. 2019, 9, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Dhakal, B.; Chhabra, S.; Savani, B.N.; Hamadani, M. Promise and Pitfalls of Allogeneic Chimeric Antigen Receptor Therapy in Plasma Cell and Lymphoid Malignancies. Br. J. Haematol. 2022, 197, 28–40. [Google Scholar] [CrossRef]
- Heczey, A.; Liu, D.; Tian, G.; Courtney, A.N.; Wei, J.; Marinova, E.; Gao, X.; Guo, L.; Yvon, E.; Hicks, J.; et al. Invariant NKT Cells with Chimeric Antigen Receptor Provide a Novel Platform for Safe and Effective Cancer Immunotherapy. Blood 2014, 124, 2824–2833. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, M.; Hu, J.; Sommariva, M.; Gautam, S.; Fellowes, V.; Hocker, J.D.; Dougherty, S.; Qin, H.; Klebanoff, C.A.; Fry, T.J.; et al. Generation of Clinical-Grade CD19-Specific CAR-Modified CD81 Memory Stem Cells for the Treatment of Human B-Cell Malignancies. Blood 2016, 128, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Rezvani, K.; Rouce, R.; Liu, E.; Shpall, E. Engineering Natural Killer Cells for Cancer Immunotherapy. Mol. Ther. 2017, 25, 1769–1781. [Google Scholar] [CrossRef] [PubMed]
- Sebestyen, Z.; Prinz, I.; Déchanet-Merville, J.; Silva-Santos, B.; Kuball, J. Translating Gammadelta (Γδ) T Cells and Their Receptors into Cancer Cell Therapies. Nat. Rev. Drug Discov. 2020, 19, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, M.; Ramos, C.A.; Durett, A.; Liu, E.; Dakhova, O.; Liu, H.; Creighton, C.J.; Gee, A.P.; Heslop, H.E.; et al. Closely Related T-Memory Stem Cells Correlate with in Vivo Expansion of CAR.CD19-T Cells and Are Preserved by IL-7 and IL-15. Blood 2014, 123, 3750–3759. [Google Scholar] [CrossRef]
- Gattinoni, L.; Lugli, E.; Ji, Y.; Pos, Z.; Paulos, C.M.; Quigley, M.F.; Almeida, J.R.; Gostick, E.; Yu, Z.; Carpenito, C.; et al. A Human Memory T Cell Subset with Stem Cell–like Properties. Nat. Med. 2011, 17, 1290–1297. [Google Scholar] [CrossRef]
- Arcangeli, S.; Bove, C.; Mezzanotte, C.; Camisa, B.; Falcone, L.; Manfredi, F.; Bezzecchi, E.; El Khoury, R.; Norata, R.; Sanvito, F.; et al. CAR T Cell Manufacturing from Naive/Stem Memory T Lymphocytes Enhances Antitumor Responses While Curtailing Cytokine Release Syndrome. J. Clin. Investig. 2022, 132, e150807. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Fernández, C.; Escribà-Garcia, L.; Caballero, A.; Escudero-López, E.; Ujaldón-Miró, C.; Montserrat-Torres, R.; Pujol-Fernández, P.; Sierra, J.; Briones, J. Memory Stem T Cells Modified with a Redesigned CD30-chimeric Antigen Receptor Show an Enhanced Antitumor Effect in Hodgkin Lymphoma. Clin. Transl. Immunol. 2021, 10, e1268. [Google Scholar] [CrossRef] [PubMed]
- Saura-Esteller, J.; de Jong, M.; King, L.A.; Ensing, E.; Winograd, B.; de Gruijl, T.D.; Parren, P.W.H.I.; van der Vliet, H.J. Gamma Delta T-Cell Based Cancer Immunotherapy: Past-Present-Future. Front. Immunol. 2022, 13, 2649. [Google Scholar] [CrossRef] [PubMed]
- Makkouk, A.; Yang, X.; Barca, T.; Lucas, A.; Turkoz, M.; Wong, J.T.S.; Nishimoto, K.P.; Brodey, M.M.; Tabrizizad, M.; Gundurao, S.R.Y.; et al. Off-the-Shelf Vδ1 Gamma Delta T Cells Engineered with Glypican-3 (GPC-3)-Specific Chimeric Antigen Receptor (CAR) and Soluble IL-15 Display Robust Antitumor Efficacy against Hepatocellular Carcinoma. J. Immunother. Cancer 2021, 9, e003441. [Google Scholar] [CrossRef]
- Nelson, A.; Lukacs, J.D.; Johnston, B. The Current Landscape of NKT Cell Immunotherapy and the Hills Ahead. Cancers 2021, 13, 5174. [Google Scholar] [CrossRef] [PubMed]
- Metelitsa, L.S.; Wu, H.W.; Wang, H.; Yang, Y.; Warsi, Z.; Asgharzadeh, S.; Groshen, S.; Wilson, S.B.; Seeger, R.C. Natural Killer T Cells Infiltrate Neuroblastomas Expressing the Chemokine CCL2. J. Exp. Med. 2004, 199, 1213–1221. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.; Courtney, A.N.; Jena, B.; Heczey, A.; Liu, D.; Marinova, E.; Guo, L.; Xu, X.; Torikai, H.; Mo, Q.; et al. CD62L+ NKT Cells Have Prolonged Persistence and Antitumor Activity in Vivo. J. Clin. Investig. 2016, 126, 2341–2355. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Huang, W.; Heczey, A.; Liu, D.; Guo, L.; Wood, M.; Jin, J.; Courtney, A.N.; Liu, B.; Di Pierro, E.J.; et al. NKT Cells Coexpressing a GD2-Specific Chimeric Antigen Receptor and IL15 Show Enhanced in Vivo Persistence and Antitumor Activity against Neuroblastoma. Clin. Cancer Res. 2019, 25, 7126–7138. [Google Scholar] [CrossRef]
- Rotolo, A.; Caputo, V.S.; Holubova, M.; Baxan, N.; Dubois, O.; Chaudhry, M.S.; Xiao, X.; Goudevenou, K.; Pitcher, D.S.; Petevi, K.; et al. Enhanced Anti-Lymphoma Activity of CAR19-INKT Cells Underpinned by Dual CD19 and CD1d Targeting. Cancer Cell 2018, 34, 596–610.E11. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Gang, M.; Marin, N.D.; Wong, P.; Neal, C.C.; Marsala, L.; Foster, M.; Schappe, T.; Meng, W.; Tran, J.; Schaettler, M.; et al. CAR-Modified Memory-like NK Cells Exhibit Potent Responses to NK-Resistant Lymphomas. Blood 2020, 136, 2308–2318. [Google Scholar] [CrossRef]
- Tang, X.; Yang, L.; Li, Z.; Nalin, A.P.; Dai, H.; Xu, T.; Yin, J.; You, F.; Zhu, M.; Shen, W.; et al. First-in-Man Clinical Trial of CAR NK-92 Cells: Safety Test of CD33-CAR NK-92 Cells in Patients with Relapsed and Refractory Acute Myeloid Leukemia. Am. J. Cancer Res. 2018, 8, 1083–1089. [Google Scholar] [PubMed]
- Torelli, G.F.; Rozera, C.; Santodonato, L.; Peragine, N.; D’Agostino, G.; Montefiore, E.; Napolitano, M.R.; Monque, D.M.; Carlei, D.; Mariglia, P.; et al. A Good Manufacturing Practice Method to Ex Vivo Expand Natural Killer Cells for Clinical Use. Blood Transfus. 2015, 13, 464–471. [Google Scholar] [CrossRef]
- Quintarelli, C.; Sivori, S.; Caruso, S.; Carlomagno, S.; Falco, M.; Boffa, I.; Orlando, D.; Guercio, M.; Abbaszadeh, Z.; Sinibaldi, M.; et al. Efficacy of Third-Party Chimeric Antigen Receptor Modified Peripheral Blood Natural Killer Cells for Adoptive Cell Therapy of B-Cell Precursor Acute Lymphoblastic Leukemia. Leukemia 2020, 34, 1102–1115. [Google Scholar] [CrossRef]
- Morrissey, M.A.; Williamson, A.P.; Steinbach, A.M.; Roberts, E.W.; Kern, N.; Headley, M.B.; Vale, R.D. Chimeric Antigen Receptors That Trigger Phagocytosis. eLife 2018, 7, e36688. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, L.; Su, H.F.; Liu, Q.; Shen, J.; Dai, H.; Zheng, W.; Lu, Y.; Zhang, W.; Bei, Y.; et al. Chimeric Antigen Receptor Macrophage Therapy for Breast Tumours Mediated by Targeting the Tumour Extracellular Matrix. Br. J. Cancer 2019, 121, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human Chimeric Antigen Receptor Macrophages for Cancer Immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef]
- Irving, M.; Lanitis, E.; Migliorini, D.; Ivics, Z.; Guedan, S. Choosing the Right Tool for Genetic Engineering: Clinical Lessons from Chimeric Antigen Receptor-T Cells. Hum. Gene Ther. 2021, 32, 1044–1058. [Google Scholar] [CrossRef]
- Sloas, C.; Gill, S.; Klichinsky, M. Engineered CAR-Macrophages as Adoptive Immunotherapies for Solid Tumors. Front. Immunol. 2021, 12, 783305. [Google Scholar] [CrossRef]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Bartholomae, C.C.; Ranzani, M.; Benedicenti, F.; Sergi, L.S.; Ambrosi, A.; Ponzoni, M.; et al. The Genotoxic Potential of Retroviral Vectors Is Strongly Modulated by Vector Design and Integration Site Selection in a Mouse Model of HSC Gene Therapy. J. Clin. Investig. 2009, 119, 964–975. [Google Scholar] [CrossRef]
- Rivière, I.; Sadelain, M. Chimeric Antigen Receptors: A Cell and Gene Therapy Perspective. Mol. Ther. 2017, 25, 1117–1124. [Google Scholar] [CrossRef]
- Ellis, G.I.; Sheppard, N.C.; Riley, J.L. Genetic Engineering of T Cells for Immunotherapy. Nat. Rev. Genet. 2021, 22, 427–447. [Google Scholar] [CrossRef]
- Kumar, M.; Keller, B.; Makalou, N.; Sutton, R.E. Systematic Determination of the Packaging Limit of Lentiviral Vectors. Hum. Gene Ther. 2001, 12, 1893–1905. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Lopez, M.; Garcia-Perez, J. DNA Transposons: Nature and Applications in Genomics. Curr. Genom. 2010, 11, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Harris, E.; Elmer, J.J. Optimization of Electroporation and Other Non-Viral Gene Delivery Strategies for T Cells. Biotechnol. Prog. 2021, 37, e3066. [Google Scholar] [CrossRef]
- Kebriaei, P.; Singh, H.; Huls, M.H.; Figliola, M.J.; Bassett, R.; Olivares, S.; Jena, B.; Dawson, M.J.; Kumaresan, P.R.; Su, S.; et al. Phase i Trials Using Sleeping Beauty to Generate CD19-Specific CAR T Cells. J. Clin. Investig. 2016, 126, 3363–3376. [Google Scholar] [CrossRef] [PubMed]
- Magnani, C.F.; Gaipa, G.; Lussana, F.; Belotti, D.; Gritti, G.; Napolitano, S.; Matera, G.; Cabiati, B.; Buracchi, C.; Borleri, G.; et al. Sleeping Beauty–Engineered CAR T Cells Achieve Antileukemic Activity without Severe Toxicities. J. Clin. Investig. 2020, 130, 6021–6033. [Google Scholar] [CrossRef] [PubMed]
- Amberger, M.; Ivics, Z. Latest Advances for the Sleeping Beauty Transposon System: 23 Years of Insomnia but Prettier than Ever: Refinement and Recent Innovations of the Sleeping Beauty Transposon System Enabling Novel, Nonviral Genetic Engineering Applications. BioEssays 2020, 42, 2000136. [Google Scholar] [CrossRef]
- Prommersberger, S.; Reiser, M.; Beckmann, J.; Danhof, S.; Amberger, M.; Quade-Lyssy, P.; Einsele, H.; Hudecek, M.; Bonig, H.; Ivics, Z. CARAMBA: A First-in-Human Clinical Trial with SLAMF7 CAR-T Cells Prepared by Virus-Free Sleeping Beauty Gene Transfer to Treat Multiple Myeloma. Gene Ther. 2021, 28, 560–571. [Google Scholar] [CrossRef]
- Pickar-Oliver, A.; Gersbach, C.A. The next Generation of CRISPR–Cas Technologies and Applications. Nat. Rev. Mol. Cell Biol. 2019, 20, 490–507. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, R.; Graham, C.; Yallop, D.; Jozwik, A.; Mirci-Danicar, O.C.; Lucchini, G.; Pinner, D.; Jain, N.; Kantarjian, H.; Boissel, N.; et al. Genome-Edited, Donor-Derived Allogeneic Anti-CD19 Chimeric Antigen Receptor T Cells in Paediatric and Adult B-Cell Acute Lymphoblastic Leukaemia: Results of Two Phase 1 Studies. Lancet 2020, 396, 1885–1894. [Google Scholar] [CrossRef]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; Van Der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC Locus with CRISPR/Cas9 Enhances Tumour Rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Harbottle, J.A. Immunotherapy to Get on Point with Base Editing. Drug Discov. Today 2021, 26, 2350–2357. [Google Scholar] [CrossRef]
- Alabanza, L.; Pegues, M.; Geldres, C.; Shi, V.; Wiltzius, J.J.W.; Sievers, S.A.; Yang, S.; Kochenderfer, J.N. Function of Novel Anti-CD19 Chimeric Antigen Receptors with Human Variable Regions Is Affected by Hinge and Transmembrane Domains. Mol. Ther. 2017, 25, 2452–2465. [Google Scholar] [CrossRef]
- Frigault, M.J.; Lee, J.; Basil, M.C.; Carpenito, C.; Motohashi, S.; Scholler, J.; Kawalekar, O.U.; Guedan, S.; McGettigan, S.E.; Posey, A.D.; et al. Identification of Chimeric Antigen Receptors That Mediate Constitutive or Inducible Proliferation of T Cells. Cancer Immunol. Res. 2015, 3, 356–367. [Google Scholar] [CrossRef]
- Arcangeli, S.; Rotiroti, M.C.; Bardelli, M.; Simonelli, L.; Magnani, C.F.; Biondi, A.; Biagi, E.; Tettamanti, S.; Varani, L. Balance of Anti-CD123 Chimeric Antigen Receptor Binding Affinity and Density for the Targeting of Acute Myeloid Leukemia. Mol. Ther. 2017, 25, 1933–1945. [Google Scholar] [CrossRef] [PubMed]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB Costimulation Ameliorates T Cell Exhaustion Induced by Tonic Signaling of Chimeric Antigen Receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef]
- Gomes-Silva, D.; Mukherjee, M.; Srinivasan, M.; Krenciute, G.; Dakhova, O.; Zheng, Y.; Cabral, J.M.S.; Rooney, C.M.; Orange, J.S.; Brenner, M.K.; et al. Tonic 4-1BB Costimulation in Chimeric Antigen Receptors Impedes T Cell Survival and Is Vector-Dependent. Cell Rep. 2017, 21, 17–26. [Google Scholar] [CrossRef]
- Ho, J.Y.; Wang, L.; Liu, Y.; Ba, M.; Yang, J.; Zhang, X.; Chen, D.; Lu, P.; Li, J. Promoter Usage Regulating the Surface Density of CAR Molecules May Modulate the Kinetics of CAR-T Cells In Vivo. Mol. Ther.—Methods Clin. Dev. 2021, 21, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Kosti, P.; Opzoomer, J.W.; Larios-Martinez, K.I.; Henley-Smith, R.; Scudamore, C.L.; Okesola, M.; Taher, M.Y.M.; Davies, D.M.; Muliaditan, T.; Larcombe-Young, D.; et al. Hypoxia-Sensing CAR T Cells Provide Safety and Efficacy in Treating Solid Tumors. Cell Rep. Med. 2021, 2, 100227. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Liao, Q.; Zhao, C.; Zhu, C.; Feng, M.; Liu, Z.; Jiang, L.; Zhang, L.; Ding, X.; Yuan, M.; et al. Conditioned CAR-T Cells by Hypoxia-Inducible Transcription Amplification (HiTA) System Significantly Enhances Systemic Safety and Retains Antitumor Efficacy. J. Immunother. Cancer 2021, 9, e002755. [Google Scholar] [CrossRef] [PubMed]
- Juillerat, A.; Marechal, A.; Filhol, J.M.; Valogne, Y.; Valton, J.; Duclert, A.; Duchateau, P.; Poirot, L. An Oxygen Sensitive Self-Decision Making Engineered CAR T-Cell. Sci. Rep. 2017, 7, 39833. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case Report of a Serious Adverse Event Following the Administration of t Cells Transduced with a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C.H.J.; Sleijfer, S.; Van Steenbergen, S.; Van Elzakker, P.; Van Krimpen, B.; Groot, C.; Vulto, A.; Den Bakker, M.; Oosterwijk, E.; Debets, R.; et al. Treatment of Metastatic Renal Cell Carcinoma with CAIX CAR-Engineered T Cells: Clinical Evaluation and Management of on-Target Toxicity. Mol. Ther. 2013, 21, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Castellarin, M.; Sands, C.; Da, T.; Scholler, J.; Graham, K.; Buza, E.; Fraietta, J.A.; Zhao, Y.; June, C.H. A Rational Mouse Model to Detect On-Target, off-Tumor CAR T Cell Toxicity. JCI Insight 2020, 5, e136012. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, S.; Fang, C.; Yang, S.; Olalere, D.; Pequignot, E.C.; Cogdill, A.P.; Li, N.; Ramones, M.; Granda, B.; et al. Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res. 2015, 75, 3596–3607. [Google Scholar] [CrossRef]
- Park, S.; Shevlin, E.; Vedvyas, Y.; Zaman, M.; Park, S.; Hsu, Y.M.S.; Min, I.M.; Jin, M.M. Micromolar Affinity CAR T Cells to ICAM-1 Achieves Rapid Tumor Elimination While Avoiding Systemic Toxicity. Sci. Rep. 2017, 7, 14366. [Google Scholar] [CrossRef]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.H.; et al. Tandem CAR T Cells Targeting HER2 and IL13Rα2 Mitigate Tumor Antigen Escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef]
- Bielamowicz, K.; Fousek, K.; Byrd, T.T.; Samaha, H.; Mukherjee, M.; Aware, N.; Wu, M.F.; Orange, J.S.; Sumazin, P.; Man, T.K.; et al. Trivalent CAR T Cells Overcome Interpatient Antigenic Variability in Glioblastoma. Neuro-Oncology 2018, 20, 506–518. [Google Scholar] [CrossRef]
- Tong, C.; Zhang, Y.; Liu, Y.; Ji, X.; Zhang, W.; Guo, Y.; Han, X.; Ti, D.; Dai, H.; Wang, C.; et al. Optimized Tandem CD19/CD20 CAR-Engineered T Cells in Refractory/Relapsed B-Cell Lymphoma. Blood 2020, 136, 1632–1644. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Y.; Liu, Y.; Tong, C.; Wang, C.; Guo, Y.; Ti, D.; Yang, Q.; Qiao, S.; Wu, Z.; et al. Long-Term Activity of Tandem CD19/CD20 CAR Therapy in Refractory/Relapsed B-Cell Lymphoma: A Single-Arm, Phase 1–2 Trial. Leukemia 2022, 36, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Katsarou, A.; Sjöstrand, M.; Naik, J.; Mansilla-Soto, J.; Kefala, D.; Kladis, G.; Nianias, A.; Ruiter, R.; Poels, R.; Sarkar, I.; et al. Combining a CAR and a Chimeric Costimulatory Receptor Enhances T Cell Sensitivity to Low Antigen Density and Promotes Persistence. Sci. Transl. Med. 2021, 13, eabh1962. [Google Scholar] [CrossRef] [PubMed]
- Wilkie, S.; Van Schalkwyk, M.C.I.; Hobbs, S.; Davies, D.M.; Van Der Stegen, S.J.C.; Pereira, A.C.P.; Burbridge, S.E.; Box, C.; Eccles, S.A.; Maher, J. Dual Targeting of ErbB2 and MUC1 in Breast Cancer Using Chimeric Antigen Receptors Engineered to Provide Complementary Signaling. J. Clin. Immunol. 2012, 32, 1059–1070. [Google Scholar] [CrossRef] [PubMed]
- Lanitis, E.; Poussin, M.; Klattenhoff, A.W.; Song, D.; Sandaltzopoulos, R.; June, C.H.; Powell, D.J. Chimeric Antigen Receptor T Cells with Dissociated Signaling Domains Exhibit Focused Antitumor Activity with Reduced Potential for Toxicity In Vivo. Cancer Immunol. Res. 2013, 1, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Kloss, C.C.; Condomines, M.; Cartellieri, M.; Bachmann, M.; Sadelain, M. Combinatorial Antigen Recognition with Balanced Signaling Promotes Selective Tumor Eradication by Engineered T Cells. Nat. Biotechnol. 2013, 31, 71–75. [Google Scholar] [CrossRef]
- Chen, L.; Flies, D.B. Molecular Mechanisms of T Cell Co-Stimulation and Co-Inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Cantoni, C.; Cristina, M.; Biassoni, R.; Moretta, L. Activating Receptors and Coreceptors Involved in Human Natural Killer Cell-Mediated Cytolysis. Annu. Rev. Immunol. 2001, 19, 197–223. [Google Scholar] [CrossRef]
- Wang, N.; Liang, H.; Zen, K. Molecular Mechanisms That Influence the Macrophage M1-M2 Polarization Balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef]
- Finney, H.M.; Akbar, A.N.; Lawson, A.D.G. Activation of Resting Human Primary T Cells with Chimeric Receptors: Costimulation from CD28, Inducible Costimulator, CD134, and CD137 in Series with Signals from the TCRζ Chain. J. Immunol. 2004, 172, 104–113. [Google Scholar] [CrossRef]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Rivière, I.; Sadelain, M. Human T-Lymphocyte Cytotoxicity and Proliferation Directed by a Single Chimeric TCRζ/CD28 Receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef]
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.H.; Geiger, T.L.; Campana, D. Chimeric Receptors with 4-1BB Signaling Capacity Provoke Potent Cytotoxicity against Acute Lymphoblastic Leukemia. Leukemia 2004, 18, 676–684. [Google Scholar] [CrossRef]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef]
- Sabbagh, L.; Snell, L.M.; Watts, T.H. TNF Family Ligands Define Niches for T Cell Memory. Trends Immunol. 2007, 28, 333–339. [Google Scholar] [CrossRef]
- Van Der Stegen, S.J.C.; Hamieh, M.; Sadelain, M. The Pharmacology of Second-Generation Chimeric Antigen Receptors. Nat. Rev. Drug Discov. 2015, 14, 499–509. [Google Scholar] [CrossRef]
- Kegler, A.; Koristka, S.; Bergmann, R.; Berndt, N.; Arndt, C.; Feldmann, A.; Hoffmann, A.; Bornhäuser, M.; Schmitz, M.; Bachmann, M.P. T Cells Engrafted with a UniCAR 28/z Outperform UniCAR BB/z-Transduced T Cells in the Face of Regulatory T Cell-Mediated Immunosuppression. Oncoimmunology 2019, 8, e1621676. [Google Scholar] [CrossRef]
- Lindner, S.E.; Johnson, S.M.; Brown, C.E.; Wang, L.D. Chimeric Antigen Receptor Signaling: Functional Consequences and Design Implications. Sci. Adv. 2020, 6, eaaz3223. [Google Scholar] [CrossRef]
- Roselli, E.; Boucher, J.C.; Li, G.; Kotani, H.; Spitler, K.; Reid, K.; Cervantes, E.V.; Bulliard, Y.; Tu, N.; Lee, S.B.; et al. 4-1BB and Optimized CD28 Co-Stimulation Enhances Function of Human Mono-Specific and Bi-Specific Third-Generation CAR T Cells. J. Immunother. Cancer 2021, 9, e003354. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liu, Q.; Zhong, M.; Wang, Z.; Chen, Z.; Zhang, Y.; Xing, H.; Tian, Z.; Tang, K.; Liao, X.; et al. 2B4 Costimulatory Domain Enhancing Cytotoxic Ability of Anti-CD5 Chimeric Antigen Receptor Engineered Natural Killer Cells against T Cell Malignancies. J. Hematol. Oncol. 2019, 12, 49. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zeng, J.; Liu, T.; Xu, Q.; Song, X.; Zeng, J. Dnam1 and 2b4 Costimulatory Domains Enhance the Cytotoxicity of Anti-Gpc3 Chimeric Antigen Receptor-Modified Natural Killer Cells against Hepatocellular Cancer Cells In Vitro. Cancer Manag. Res. 2020, 12, 3247–3255. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human IPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-Tumor Activity. Cell Stem Cell 2018, 23, 181–192.E5. [Google Scholar] [CrossRef] [PubMed]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T Cells with Cell-Intrinsic PD-1 Checkpoint Blockade Resist Tumor-Mediated Inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef] [PubMed]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-Mediated PD-1 Disruption Enhances Anti-Tumor Efficacy of Human Chimeric Antigen Receptor T Cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted Delivery of a PD-1-Blocking ScFV by CAR-T Cells Enhances Anti-Tumor Efficacy In Vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ranganathan, R.; Jiang, S.; Fang, C.; Sun, J.; Kim, S.; Newick, K.; Lo, A.; June, C.H.; Zhao, Y.; et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res. 2016, 76, 1578–1590. [Google Scholar] [CrossRef]
- Zou, F.; Lu, L.; Liu, J.; Xia, B.; Zhang, W.; Hu, Q.; Liu, W.; Zhang, Y.; Lin, Y.; Jing, S.; et al. Engineered Triple Inhibitory Receptor Resistance Improves Anti-Tumor CAR-T Cell Performance via CD56. Nat. Commun. 2019, 10, 4109. [Google Scholar] [CrossRef]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase Promotes Tumor Infiltration and Antitumor Activity of CAR-Redirected T Lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef]
- Foeng, J.; Comerford, I.; McColl, S.R. Harnessing the Chemokine System to Home CAR-T Cells into Solid Tumors. Cell Rep. Med. 2022, 3, 100543. [Google Scholar] [CrossRef]
- Sagnella, S.M.; White, A.L.; Yeo, D.; Saxena, P.; van Zandwijk, N.; Rasko, J.E.J. Locoregional Delivery of CAR-T Cells in the Clinic. Pharmacol. Res. 2022, 182, 106329. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKS, the Fourth-generation CAR T Cells: Current Developments and Clinical Translation. Adv. Cell Gene Ther. 2020, 3, e84. [Google Scholar] [CrossRef]
- Koneru, M.; Purdon, T.J.; Spriggs, D.; Koneru, S.; Brentjens, R.J. IL-12 Secreting Tumor-Targeted Chimeric Antigen Receptor T Cells Eradicate Ovarian Tumors In Vivo. Oncoimmunology 2015, 4, e994446. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. IL-12 Release by Engineered T Cells Expressing Chimeric Antigen Receptors Can Effectively Muster an Antigen-Independent Macrophage Response on Tumor Cells That Have Shut down Tumor Antigen Expression. Cancer Res. 2011, 71, 5697–5706. [Google Scholar] [CrossRef] [PubMed]
- Adachi, K.; Kano, Y.; Nagai, T.; Okuyama, N.; Sakoda, Y.; Tamada, K. IL-7 and CCL19 Expression in CAR-T Cells Improves Immune Cell Infiltration and CAR-T Cell Survival in the Tumor. Nat. Biotechnol. 2018, 36, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Su, J.; Sun, R.; Sun, Y.; Wang, Y.; Dong, Y.; Shi, B.; Jiang, H.; Li, Z. Coexpression of IL7 and CCL21 Increases Efficacy of CAR-T Cells in Solid Tumors without Requiring Preconditioned Lymphodepletion. Clin. Cancer Res. 2020, 26, 5494–5505. [Google Scholar] [CrossRef]
- Fultang, L.; Booth, S.; Yogev, O.; da Costa, B.M.; Tubb, V.; Panetti, S.; Stavrou, V.; Scarpa, U.; Jankevics, A.; Lloyd, G.; et al. Metabolic Engineering against the Arginine Microenvironment Enhances CAR-T Cell Proliferation and Therapeutic Activity. Blood 2020, 136, 1155–1160. [Google Scholar] [CrossRef]
- Ligtenberg, M.A.; Mougiakakos, D.; Mukhopadhyay, M.; Witt, K.; Lladser, A.; Chmielewski, M.; Riet, T.; Abken, H.; Kiessling, R. Coexpressed Catalase Protects Chimeric Antigen Receptor–Redirected T Cells as Well as Bystander Cells from Oxidative Stress–Induced Loss of Antitumor Activity. J. Immunol. 2016, 196, 759–766. [Google Scholar] [CrossRef]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-Shelf’ Allogeneic CAR T Cells: Development and Challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- Salmikangas, P.; Kinsella, N.; Chamberlain, P. Chimeric Antigen Receptor T-Cells (CAR T-Cells) for Cancer Immunotherapy—Moving Target for Industry? Pharm. Res. 2018, 35, 152. [Google Scholar] [CrossRef]
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular Remission of Infant B-ALL after Infusion of Universal TALEN Gene-Edited CAR T Cells. Sci. Transl. Med. 2017, 9, eaaj2013. [Google Scholar] [CrossRef]
- Stroncek, D.F.; Somerville, R.P.T.; Highfill, S.L. Point-of-Care Cell Therapy Manufacturing; It’s Not for Everyone. J. Transl. Med. 2022, 20, 34. [Google Scholar] [CrossRef]
- Consultation on Point of Care Manufacturing. Available online: https://www.gov.uk/government/consultations/point-of-care-consultation/consultation-on-point-of-care-manufacturing (accessed on 29 October 2022).
- Draft EMA Regulatory Science to 2025. Available online: https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/draft-ema-regulatory-science-2025-strategic-reflection_en.pdf (accessed on 29 October 2022).



{kind=link}
{kind=link}
{kind=link}
| Disease | Trial Phase | No. of pt | Target | CAR Construct | Additional Strategies | Ref. |
|---|---|---|---|---|---|---|
| HER2+ solid tumors | I/II | 19 | HER2 | CD28, CD3z | None | [30] |
| Liver metastases | I | 6 | CEA | CD28, CD3z | None | [31] |
| Non-Small Cell Lung Cancer | I | 11 | EGFR | 4-1BB, CD3z | None, Cy * alone or Cy with additional cytotoxic drugs | [32] |
| Biliary tract cancer | I | 19 | EGFR | 4-1BB, CD3z | Cy/nab-paclitaxel | [33] |
| Metastatic colorectal cancer | I | 10 | CEA | CD28, CD3z | Cy | [34] |
| Breast cancer | 0 | 6 | MET | 4-1BB, CD3z | None | [35] |
| Neuroblastoma | I | 11 | GD2 | CD28, OX40, CD3z | None, Flu/Cy, Flu/Cy+PD-1 inhibitor | [36] |
| Glioblastoma | I | 10 | EGFR | 4-1BB, CD3z | None | [37] |
| Glioblastoma | I | 17 | HER2 | CD28, CD3z | None | [38] |
| Biliary tract cancer and pancreatic carcinoma | I | 11 | HER2 | 4-1BB, CD3z | Cy/nab-paclitaxel | [39] |
| Pancreatic ductal adenocarcinoma | I | 6 | mesothelin | 4-1BB, CD3z | None | [40] |
| Mesothelioma, Ovarian carcinoma, Pancreatic ductal carcinoma | I | 15 | mesothelin | 4-1BB, CD3z | None or Cy | [28] |
| Prostate carcinoma, pancreatic carcinoma | I | 13 | PSMA | 4-1BB, CD3z + TGFBDN | None or Flu/Cy | [29] |
| Mesothelin+ Solid tumor | I | 27 | mesothelin | CD28, CD3z | None, Cy, Cy+ PD-1 inhibitor | [41] |
| Glioblastoma | I | 18 | EGFR | CD28, 4-1BB, CD3z | Flu/Cy + IL-2 | [42] |
| Liver metastases | Ib | 6 | CEA | CD28, CD3z | Selective intra-arterial radiation with SIR spheres | [43] |
| Hepatocellular carcinoma | I | 13 | GPC3 | CD28, CD3z | Cy alone or Flu/Cy | [44] |
| Neuroblastoma | I | 17 | GD2 | CD28, CD3z | None, Cy, Flu/Cy | [45] |
| Mesothelin+ Solid tumor | I | 15 | mesothelin | CD28, CD3z | None | [46] |
| Neuroblastoma | I | 3 | GD2 | CD28, CD3z + IL15 | Flu/C | [47,48] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castiello, L.; Santodonato, L.; Napolitano, M.; Carlei, D.; Montefiore, E.; Monque, D.M.; D’Agostino, G.; Aricò, E. Chimeric Antigen Receptor Immunotherapy for Solid Tumors: Choosing the Right Ingredients for the Perfect Recipe. Cancers 2022, 14, 5351. https://doi.org/10.3390/cancers14215351
Castiello L, Santodonato L, Napolitano M, Carlei D, Montefiore E, Monque DM, D’Agostino G, Aricò E. Chimeric Antigen Receptor Immunotherapy for Solid Tumors: Choosing the Right Ingredients for the Perfect Recipe. Cancers. 2022; 14(21):5351. https://doi.org/10.3390/cancers14215351
Chicago/Turabian StyleCastiello, Luciano, Laura Santodonato, Mariarosaria Napolitano, Davide Carlei, Enrica Montefiore, Domenica Maria Monque, Giuseppina D’Agostino, and Eleonora Aricò. 2022. "Chimeric Antigen Receptor Immunotherapy for Solid Tumors: Choosing the Right Ingredients for the Perfect Recipe" Cancers 14, no. 21: 5351. https://doi.org/10.3390/cancers14215351
APA StyleCastiello, L., Santodonato, L., Napolitano, M., Carlei, D., Montefiore, E., Monque, D. M., D’Agostino, G., & Aricò, E. (2022). Chimeric Antigen Receptor Immunotherapy for Solid Tumors: Choosing the Right Ingredients for the Perfect Recipe. Cancers, 14(21), 5351. https://doi.org/10.3390/cancers14215351