Simple Summary
VHL syndrome is an autosomal dominant hereditary cancer syndrome and one of the leading causes of death is ccRCC. Current target therapies may lead to stable disease or partial remission, as best response and scant evidence supports the therapeutic decision-making in metastatic ccRCC. Therefore, new genetic and epigenetics insights are needed, to guide the development of more effective target therapies and to promptly predict the presence and prognosis of ccRCC. Our review highlights all the new molecular perspectives based on research of genetic alterations, biological pathways and promising biomarkers in the VHL-associated hereditary ccRCC.
Abstract
Von Hippel-Lindau (VHL) disease is an autosomal dominant inherited cancer syndrome caused by germline mutations in the VHL tumor suppressor gene, characterized by the susceptibility to a wide array of benign and malign neoplasms, including clear-cell renal cell carcinoma. Moreover, VHL somatic inactivation is a crucial molecular event also in sporadic ccRCCs tumorigenesis. While systemic biomarkers in the VHL syndrome do not currently play a role in clinical practice, a new promising class of predictive biomarkers, microRNAs, has been increasingly studied. Lots of pan-genomic studies have deeply investigated the possible biological role of microRNAs in the development and progression of sporadic ccRCC; however, few studies have investigated the miRNA profile in VHL patients. Our review summarize all the new insights related to clinical and molecular features in VHL renal cancers, with a particular focus on the overlap with sporadic ccRCC.
1. Epidemiology, Clinical Features, Diagnosis and Screening of VHL Disease
1.1. Current Epidemiology and Clinical Spectrum
VHL disease is a rare inherited cancer syndrome, with a birth incidence between 1/27,300 and 1/45,500 live births, and a prevalence rate ranging between 1/39,000 and 1/91,000 individuals [1]. VHL-related tumors are usually diagnosed in adult age, although an earlier age of onset can be observed [2] depending on the VHL disease type and presenting clinical manifestations [3]. The most frequently reported presenting manifestation is central nervous system hemangioblastoma (CHB), followed by retinal hemangioblastoma (RHB), RCC, pancreatic cystic lesions, pheochromocytoma (Pheo), and genital system lesions (GSLs). CHB, RHB, and GSLs are associated with earlier clinical onset, at 29.5, 24.8, and 12.4 years of age, respectively [4].
1.2. VHL Disease Types, Clinical Spectrum
Von Hippel in 1904 and Lindau in 1927 were among the first to describe some features of VHL disease (retinal angiomatosis and cerebellar-spinal hemangioblastomas, respectively) [5,6]. VHL disease is characterized by a susceptibility to a wide array of tumors, including CHB (with cerebellar and spinal localization) and RHB, Pheo, endolymphatic sac tumors (ELSTs) of the middle ear, neuroendocrine tumors and serous cystadenomas of the pancreas, GSLs (such as papillary cystadenomas of the broad ligament and the epididymis) and, lastly, hereditary ccRCCs [7], with the latter being one of the main causes of death in this population, the leading one according to some studies [7,8,9].
VHL disease has diverse genotype and phenotype correlations, and variable intra- and inter-familial expressivity [10,11,12].
VHL disease can be classically divided into various types and subtypes according to the presence or absence of Pheo and ccRCC susceptibility: type 1 is the most common, it often displays deletions or truncating mutations and does not predispose to the development of Pheo; on the contrary, type 2 disease (7–20% of families) exhibits missense mutations and Pheo susceptibility [13,14,15]. Type 2 VHL disease can be further classified as type 2A (not including ccRCC), type 2C (Pheo is the only disease manifestation), and type 2B (featuring both ccRCCs and Pheo) [15].
Since affected families can shift between subtypes over time, the clinical utility of this classification is limited [15]. Liu et al. reported that VHL gene mutations affecting HIF1a binding (HM and TR) can increase the risk of developing ccRCC [16], furthermore, Gallou et al. found that mutations in the missense cluster regions were associated with an increased risk of RCC compared to those with missense mutations in other regions [17]. Kai R et al. and Liu et al. found, instead, that missense mutations in MCR are not associated with RCC risk, suggesting the possible contribution of a different mechanism from those involving VHL-HIF [16,18]. The organ involvement in VHL disease is represented in Figure 1.
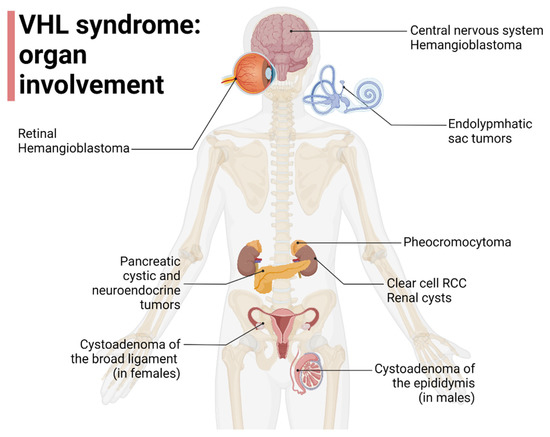
Figure 1.
Organ involvement in VHL disease, adapted from “Human Internal organs”, by BioRender.com (2022). Retrieved from https://app.biorender.com/biorender-templates (accessed on 1 October 2022).
1.3. Diagnostic Criteria and Screening
First diagnostic criteria—1964: patients with no family history of VHL disease and two related tumors (two CHBs alone or only one combined with a visceral tumor), or with family history and at least one VHL-related tumor [19]. Clinical and genetic findings are included in the current diagnostic criteria, including CHB, RHB, RCC, Pheo, paraganglioma, glomus tumor, ELST, pancreatic neuroendocrine tumors, and/or cysts. Even without a CHB or RHB, VHL disease can be diagnosed with at least one of the aforementioned manifestations and a positive VHL genetic test or with positive family history for VHL in at least one first-degree relative [20].
Careful family history and screening for other VHL-related tumors, in all patients who undergo hemangioblastoma genetic testing for VHL disease, is recommended [21].
Genetic testing on peripheral blood samples should be offered to all clinically suspected cases. Southern blotting (SB) is usually used to detect whole gene deletion and gene rearrangements, and fluorescence in situ hybridization has a consequent confirmatory role, but multiplex ligation-dependent probe amplification can be alternatively used instead of SB. In patients with a negative result on peripheral blood, genetic testing can be repeated by sampling other tissues [20].
In established cases, a comprehensive screening program is recommended, while with regard to ccRCC, an annual abdominal MRI starting from the ages of 10–16 years of age is of paramount importance. An annual abdominal ultrasound is a complementary technique, while abdominal CT is reserved for clinical scenarios in which MRI is contraindicated [2,22,23]. Screening programs vary across institutions, for example, Maher et al. proposed an annual ophthalmic examination for hemangioblastoma (starting from the age of 1), contrast-enhanced MRI of the brain and spine for hemangioblastomas of the central nervous system (starting at the age of 12), annual blood pressure evaluation and 24 h urinalysis for catecholamines and their metabolites or plasma free metanephrines for Pheo (starting at the age of 4 years), audiogram every two years for endolymphatic sac tumors (starting at the age of 16 years) and, finally, annual MRI of the abdomen for RCCs and pancreatic tumors (starting at the age of 12 years) [22].
2. Molecular Features of VHL Disease
2.1. Background and Overview of VHL Disease Genetics
In VHL disease, tumor susceptibility is underlain by genetic abnormalities behaving as tumor suppressor genes, causing a dysfunctional VHL-Hypoxia Inducible Factor (HIF) pathway, according to the 2-hit hypothesis for the development of cancer and Knudson’s theory of human carcinogenesis [24,25]. The first event is the inheritance of a germline pathogenic gene variant that results in a loss of function of one allele of the VHL-elongin C and elongin B (VCB) complex in all cells. This predisposing condition has to be followed by a second somatic event that, genetically or epigenetically, inactivates the wild-type allele in the cells involved by this acquired insult, finally leading to a loss of function of the VCB complex [24,26]. The inactivation of both alleles is crucial in carcinogenesis [27], and reinstatement of VHL function can partially reverse some effects of the biallelic inactivation [28]. High penetrance is observed, and up to 90% of patients will exhibit VHL-related clinical manifestations [1,7,15].
However, in about 20% of affected families, no VHL deletion or mutation is detectable [13]. VHL disease is associated with different molecular abnormalities besides the loss of the VHL gene, such as the loss of segments of the small arm of chromosome 3 (the VHL gene-harboring 3p25-26 [27], 3p12, 3p13-14.2, 3p21) [29], and several novel mutations involving elongin C-VHL binding (TCEB1 gene), BAP1 and SETD2 genes and several pathways (PI3K-AKT-mTOR, DNA methylation, p53-related and mRNA processing) [30,31].
Notably, VHL loss of function is not sufficient for ccRCCs to develop, [32,33] since this genetic anomaly is already present in benign renal cysts without foci of ccRCCs in VHL-deficient patients, as a consequence, mechanisms other than the VHL-HIF pathway are most likely involved in RCC progression [32,34].
A disrupted VHL pathway leads to dysfunctional ubiquitination of the VCB-Cullin 2-RBX1 (VCB-CR) complex and consequently to the constitutional activation of the HIF pathway and, finally, to subsequent alterations in downstream processes, mediated by abnormally expressed carcinogenic factors [35,36,37]. Alongside the HIF-related effect, several HIF-independent effects result from the loss of function of the VHL pathway [38]. A schematic of the VHL-HIF pathway is presented in Figure 2.
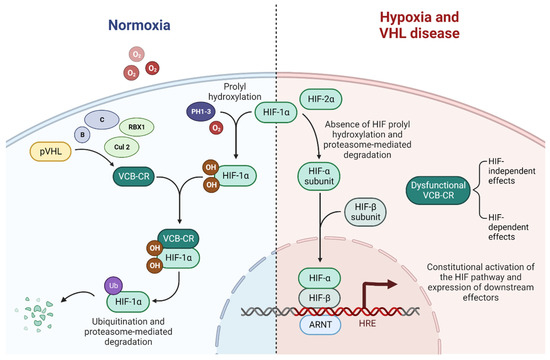
Figure 2.
The VHL-HIF pathway in normoxia, hypoxia, and VHL disease, adapted from “HIF signaling”, by BioRender.com (2022). Retrieved from https://app.biorender.com/biorender-templates (accessed on 1 October 2022).
2.2. VHL Gene, pVHL, HIF, and HIF-Independent Functions of pVHL
The VHL locus was first cloned and mapped between the late 80s and early 90s, while the VHL gene was identified in 1993 [39,40]. The coding sequence includes three exons, and based on the presence of exon 2, there are two mRNA isoforms [13]. The gene product is the VHL protein (pVHL), which consists of two isoforms, both of which have tumor-suppressing activity [41].
pVHL binds elongin C and elongin B to form the VCB complex [42], stabilizing its component and sheltering them from proteasomal degradation [43], and conferring a ubiquitin ligase activity, acting as an E3 ligase [34,44,45]. The VCB complex then binds to cullin 2 and RBX1 to form the VCB-CR complex [46].
In VHL-wild type patients in normoxic conditions, the VCB-CR complex is able to bind to prolyl hydroxylated hypoxia-induced factor 1α (HIF1α), while with hypoxia, this binding is prevented by the lack of hydroxylation of HIF1α by prolyl hydroxylase 1, 2 and 3, inactivated by the lack of their co-substrate: oxygen [47,48].
In VHL disease, the loss of function in the VCB-CR complex prevents its binding to the ubiquitous unstable subunit HIF1α, which subsequently heterodimerizes with the stable HIF1β to form HIF1, which translocates to the nucleus and acts as a transcription factor binding the hypoxia-response elements, leading to the constitutive activation of expressed downstream effectors that would physiologically be activated only in hypoxic conditions. HIF is a key driver of ccRCC progression and might be a tumor suppressor, directly contributing to tumorigenesis [34,49,50].
Some of the most relevant downstream effectors are TGFα, EGFR (involved in cell proliferation and survival), PDGFB (angiogenesis, lymphangiogenesis), the chemokine receptor CXCR4 and MMP2/9/14 and lysyl oxidase, UPAR, MMP2 (tumoral cell invasion and metastases), dysregulation of TWIST and activation of HGFR (epithelial-to-mesenchymal transition), VEGFA (angiogenesis) [50,51].
HIF2α is a subunit isoform peculiarly expressed only in the endothelium, lung, liver, and kidney cells, activated in hypoxia, that can heterodimerize with a β-subunit (HIF1β) to form HIF2, which displays a slightly different spectrum of downstream effects compared to HIF1, preferentially driving growth and angiogenetic processes [52]. HIF2, more than HIF1, appears to be a more prominent driver of RCC tumorigenesis [52,53,54]. It is known to activate mTOR complex 1 [55,56] and is increasingly being investigated as a target for direct inhibition in VHL-deficient ccRCCs [57,58]. Belzutifan, an HIF-2a inhibitor, was approved by the Food and Drug Administration on 13 August 2021 for the treatment of RCCs, CHB, and pancreatic neuroendocrine tumors not immediately requiring surgery [59].
pVHL also has HIF-independent functions (assembly and regulation of extracellular matrix, microtubule stabilization, maintenance of primary cilium, regulation of apoptosis, control of cell senescence-survival, transcriptional regulations, protein ubiquitination) that could, theoretically, influence tumorigenesis, however, its contribution is currently unknown [60,61,62,63]. An overview of VHL’s role in RCC tumorigenesis is provided in Figure 3.
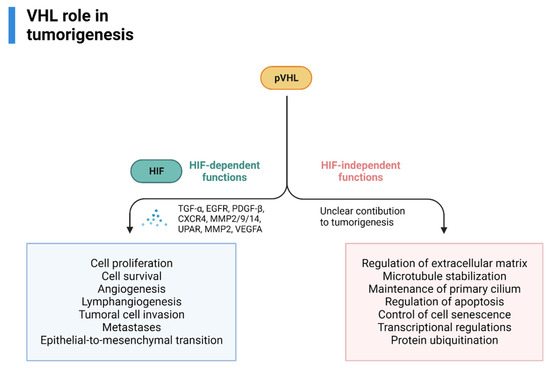
Figure 3.
VHL role in VHL-related RCC tumorigenesis, adapted from “Cell Differentiation Pathway (Layout, Vertical) 2”, by BioRender.com (2022). Retrieved from https://app.biorender.com/biorender-templates (accessed on 1 October 2022).
2.3. Difference between VHL-Related Sporadic and Hereditary ccRCC
The genetic abnormalities in sporadic ccRCC are similar to those observed in VHL disease, i.e., loss of 3p25, and VHL mutations are present in 60–90% of patients [64].
VHL mutations are typically frameshift or nonsense mutations, but missense mutations may be observed [65]. Epigenetic abnormalities have been described, and in particular, VHL silencing by methylation is present in 5–30% of sporadic ccRCCs, thus resulting in LOH in up to 98% of cases [66]. Biallelic inactivation of the VHL gene in sporadic ccRCCs is lower than expected, and Hamano et al. reported that only 44% of LOH ccRCCs showed this feature, implying the existence of a different 2nd hit mutational event [67].
Sporadic ccRCCs display mutation heterogeneity, with different subclones harboring different anomalies. In progressively smaller subclones, genetic abnormalities are named ubiquitous (such as VHL loss and chromosome 3p loss, early, truncal events, in every tumor cell), shared, and private [68].
VHL mutational status does not predict oncologic outcomes in sporadic ccRCCs since mixed results have been reported. Masahiro et al. reported a strong association between VHL alterations in sporadic ccRCC and better cancer-free survival and cancer-specific survival in non-metastatic disease treated with radical nephrectomy (RN), particularly in stage III patients [69]. On the contrary, Jung Han et al. could not find an association between somatic VHL alterations and prognosis in sporadic ccRCC, although patients with loss-of-function VHL mutations had decreased progression-free survival and overall survival [70]. Inactivation of the VHL gene in sporadic ccRCC is not associated with nuclear grade, disease stage, or overall survival, according to a recent meta-analysis by Kim et al. [71].
Gordan et al. reported an alternative classification of sporadic ccRCC based on the VHL allele, HIF1a, HIF2a, and MYC expression, recognizing three different molecular subgroups: VHL WT (wild-type VHL alleles, undetectable HIFα protein), H1H2 (VHL-deficient, expressing HIF1α and HIF2α proteins) and H2 tumors (VHL-deficient, expressing only HIF2α, enhanced MYC activity, higher proliferation rates) [72].
2.4. Clinical and Therapeutic Implications, Prognosis
The prognosis could be improved by performing a genetic diagnosis and careful follow-up [73]. Before the introduction of surveillance protocols, life expectancy was less than 50 years [13], but by adopting a regular screening program, earlier diagnosis and treatment of VHL-related tumors led to an extension of life expectancy to between 52.5 and 66 years [4,74,75].
Especially in advanced diseases, the prognosis remains poor. The incomplete understanding of VHL tumorigenesis slowed down the development of a rich target-therapy arsenal [38]. However, several steps forward have been made in understanding its pathogenesis, tailoring the surgical strategy, and enriching the arsenal of antineoplastic drugs with valid target-directed agents and immune-checkpoint inhibitors [35].
A poor prognosis is associated with advanced-stage disease, which implies a limited response to chemotherapy and radiotherapy, but a better response to immunotherapy, biologics, or target therapy [76].
Early-onset disease, a positive family history, and truncating VHL mutations are associated with reduced overall survival and disease-specific survival [75], and female sex is associated with decreased overall survival [77].
Risk factors for overall survival in VHL disease also include truncating mutations, type 1 disease, presence of CNB or RHB rather than abdominal lesions (RCCs, pancreatic neoplasms, Pheo) [4].
Several molecular pathways are involved in tumorigenesis and, consequently were investigated as possible therapeutic targets in this population, especially multiple tyrosine kinase inhibitors (TKis) linked to key downstream effectors. However, it was only recently that another class of drugs proved to be active and well tolerated in VHL patients, leading to FDA approval of the HIF-2a inhibitor Belzutifan [59].
3. Von Hippel-Lindau Disease and ccRCC
3.1. Clinical, Pathological, and Imaging Overview
ccRCC is acknowledged as a Renal Cell Tumor by 2016 WHO Classification of kidney tumors [78], representing the most common RCC histotype and accounting for 70% of cases [79].
VHL syndrome is the oldest and most common hereditary RCC syndrome, among at least twelve known entities [76].
Hereditary cancer syndrome of the kidney represents 5–8% of cases [80]. In a study by Ferstl et al., the prevalence of VHL RCC was 1.6% in a series of unselected RCCs [81].
VHL disease predisposes to the development of renal cysts and ccRCCs, and a single individual can present with multiple lesions [82]. A nationwide audit reported a median of 2–3 RCCs per individual [83,84]. In renal cysts, foci of dysplasia or RCC can be present, especially in complex cysts [82,84]. Foci of RCC can also be seen in the context of simple renal cysts [85], and VHL deficiency can also be observed in the cystic epithelium, supporting the hypothesis of cysts as precursor lesions of ccRCC in VHL disease [86].
ccRCC will develop in 30–70% of patients with VHL-disease [7,13,23,87]. The mean age at presentation is 39–44 years, the disease rarely presents before 20 years of age [37] but can occur between 15 and 75 years [73]. The probability of developing ccRCC by the age of 60 years is 0.69. Before the introduction of modern screening protocols, the reported median survival was 49 years and ccRCC was the leading cause of death [7,88]. VHL- and RCC-related deaths have decreased over time, with hemangioblastoma representing the first cause of death in this population [75].
In VHL patients with ccRCC, the 10-year disease-specific survival rate is 95% [73]. The overall survival in VHL-deficient individuals is progressively approaching that of unaffected siblings and of the general population [75].
Hereditary VHL-related ccRCCs display earlier onset and multifocal and bilateral tumors [88]: Neumann et al. described age at onset as being 25 years less than in sporadic ccRCCs, and an association with renal cyst and cystic organization, low histological grade, absence of metastasis in tumors smaller than 7 cm and better 10-year survival [81]. Poston et al. also documented that ccRCC in VHL disease tends to have a lower grade, with low local invasiveness [84]. Other authors, on the other hand, have reported a similar growth rate between sporadic and VHL disease ccRCCs [82,89,90].
The metastatic rate is around 11%, lower than for sporadic ccRCCs [73], and it is highly influenced by the tumor size and growth rate [91].
VHL disease displays a high rate of recurrence [76]; Ploussard et al. documented that among patients who underwent nephron-sparing surgery, 45.6% and 83.7% experienced a local recurrence at 5 and 10 years, respectively, with a 5-year and 10-year overall repeat surgery rate of 21.1% and 63.4%, respectively [92].
Diagnosis is often delayed due to the asymptomatic nature of the tumors, thus typical signs and symptoms of RCC, such as visible hematuria, flank pain, and palpable flank mass or masses are indicative of more advanced disease [88].
Unlike in the screening setting, the first-line diagnostic technique in a suspected case of RCC is contrast-enhanced abdominal CT to characterize the location, number, size, and appearance of solid lesions [88].
The predominant histotype in VHL disease is ccRCC. A series on renal pathology in VHL disease showed that 91% of solid neoplasms in surgical specimens were ccRCCs, with different cytological features (pure clear cell features, clear cells with dispersed granular cells, and sarcomatoid RCC containing clear and granular cells in 53%, 45% and 1.5% of cases, respectively). The architectural pattern was typically trabecular, with a minority of cases displaying a microcystic pattern. The renal masses were all well-circumscribed, and a pseudocapsule (PSC) frequently surrounded the neoplasm. In this series, PSC microscopic invasion was present in 76% of cases, with no invasion of the surrounding parenchyma [84]. The neoplastic cells are large and have abundant clear cytoplasm that is rich in glycogen and lipids [93]. Wild-type sporadic ccRCCs may have different histological and clinical features, in a 10-year retrospective study, wild-type ccRCCs were associated with a higher nuclear grade, sarcomatoid component, dense lymphocyte infiltrate, nodal involvement, and presence of metastases compared to VHL-inactivated ccRCCs patients, with a markedly reduced disease-specific survival of 33 months versus 107 months, respectively [94]. A recent retrospective study by Wang et al. evaluated clinicopathological predictive markers in VHL-associated ccRCCs and in sporadic ccRCCs. The authors reported a higher histologic grade in the sporadic group, which exhibited a faster growth rate [95]. A kidney biopsy is seldom needed in patients with established VHL disease. Chahoud et al. recently published a report on the evaluation and diagnosis of limited renal masses in VHL disease, stating that a kidney biopsy can be indicated in selected clinical settings, i.e., in case of atypical imaging features suggesting a benign lesion (infections, foci of inflammation), in case of a positive history of extra-renal malignancy that could disseminate to the kidney prior to percutaneous ablation, or when post-interventional recurrence is suspected [96].
US is not a sensitive enough technique to characterize RCCs and, when used alone, is outperformed by MRI and CT [97].
The Bosniak system classification is useful for describing cystic lesions in VHL disease, but since small foci of dysplasia or RCC can harbor in these lesions, it should not be used to guide management in this population of patients, unlike in sporadic ccRCCs [98].
A national audit of VHL disease showed that MRI was the most common detection modality, followed by CT and ultrasonography, especially for bigger lesions [83]. MRI is better at detecting small renal masses than ultrasonography [83] and, although its sensitivity is inferior to that of abdominal CT, its use is preferred in screening protocols to spare patients from cumulative exposure due to ionizing radiations starting at a young age [83,99].
VHL has a saltatory growth pattern, with quiescent phases, that complicates the interpretation of imaging findings as treatment-response or natural quiescence [38]. A summary of the clinical key aspects of VHL-related ccRCC is presented in Figure 4.

Figure 4.
Clinical overview of VHL-related ccRCC, adapted from “4 Panels (Layout 2 × 2)”, by BioRender.com (2022). Retrieved from https://app.biorender.com/biorender-templates (accessed on 1 October 2022).
3.2. Peculiar Molecular Aspects of VHL-Related ccRCC
VHL mutations are ubiquitous, early, truncal events in ccRCCs [68].
Jonasch et al. found an association between the molecular abnormalities underlying ccRCCs and their timing and subdivided them into those that drive tumor initiation events (3p loss/5q gain, VHL mutation, VHL methylation), tumor progression (PBRM1/SETD2/BAP1 mutations, DNA repair defects, defects in mitosis) and those that confer lethality (PI3K pathway activation, 9p, 14q, 8q gain) [100].
ccRCCs are hypervascular tumors, and the main driving factor in tumor progression is represented by HIF overexpression and upregulated downstream effectors, above all VEGF [12]. The endothelium of VHL-deficient patients is highly dysfunctional, displaying altered cell adhesion, angiogenesis, migration, immune response, cell metabolism, and ROS homeostasis [101].
ccRCCs typically display large neoplastic cells, featuring an abundant, clear cytoplasm rich in glycogen and lipids, derived from a high glucose metabolism that, in VHL-null ccRCCs, can possibly be linked to the increased expression of an HIF downstream effector (the adipose differentiation-related peptide) acting as a lipid transporter [93]. A schematic of the genetic alterations, biological pathways involved, and investigated biomarkers in VHL syndrome RCC is synthetized in Table 1 and Table 2.

Table 1.
Genetic alterations, biological pathways of RCC in VHL disease.
Table 2.
Dysregulated miRNAs between VHL-associated and sporadic ccRCC samples, adapted from ref. 136.
4. Therapeutic Approach to VHL-Related ccRCC and Clinical Outcomes
4.1. Localized Disease
In the last decades, the implementation of genetic testing and the improvements in screening protocols have led to better clinical outcomes. The aim is to detect RCCs before the development of metastatic disease and to approach renal masses with nephron-sparing surgery so as to maximally preserve renal function. In view of the multifocal and bilateral disease presentation, the high recurrence rate, and the limited number of possible nephron-sparing surgical interventions because of their effects on renal function, tailored and careful surgical planning is of paramount importance.
Regarding target therapy, a recent phase II study of HIF-2a inhibitors in ccRCCs documented objective partial responses in 30 of the 61 enrolled patients, stable disease in 30 patients, and only one case of disease progression after a median follow-up time of 21.8 months [102]. The investigated drug, Belzutifan, proved to be active in renal and non-renal VHL-related cancer and well-tolerated by VHL patients, leading to its approval by the FDA on 13 August 2021 [59].
Lesions with a maximum diameter of less than 3 cm are usually treated with a surveillance protocol. The VHL alliance recommends repeating abdominal MRI every 3 to 6 months to assess tumor growth. In case of disease stability over 3 consecutive MRIs, the interval between scans can be extended to 2 years. On the contrary, if a lesion is greater than 3 cm, a urologic referral is mandatory [103]
Walther et al. investigated the 3 cm threshold as an indication for surgery; smaller tumors (group 1) underwent imaging surveillance and eventual surgery when the threshold was reached, while larger tumors (group 2) were immediately resected. After 60 months, no group 1 patients developed metastatic disease and only one required surgery, while after a 66-month follow-up, 12 of 44 group 2 patients required nephrectomy, and 11 of 44 progressed to the metastatic stage [104].
Surgery is the standard of care when dealing with >3 cm lesions, although the recent approval of Belzutifan in this setting has enriched the therapeutic arsenal and can be offered as an alternative choice. Belzutifan approval is based on the NCT03401788 trial, which included individuals with renal masses ranging from 10 to 61mm with no evidence of metastatic disease [102]. It is not currently approved for metastatic disease but can be offered to patients with <3 cm renal lesions. A summary of the drugs or compounds evaluated in a clinical trial for the treatment of VHL disease-ccRCC is provided in Table 3.
Table 3.
Summary of the clinical evidence on drugs or compounds evaluated in VHL disease ccRCC or with advanced ccRCCs.
A maximum renal mass diameter greater than 3 cm is associated with an increased risk of metastatic disease, although VHL-related ccRCCs tend to have limited local invasiveness and histologic grade [88,105]. Radical nephrectomy (RN) is not the first choice since nephron-sparing surgery (NSS) is effective and preserves renal function.
Steinbach et al. reported similar 5- and 10-year disease-specific survival rates for RN (95% and 77%, respectively) and NSS (100% and 81%, respectively). Moreover, the NSS group had 5- and 10-year recurrence-free survival rates of 71% and 15%, 51% displayed postoperative local recurrence, and only 2 of 49 patients progressed to metastatic disease. Finally, 23% developed ESKD, 6 of 15 were managed with renal transplantation, and 9 with dialysis [106]. Bratslavsky et al. investigated the outcomes of salvage partial nephrectomy (PN), intended as a 3rd or 4th PN on the same operated kidney. Twenty-three% of patients lost the renal unit, while the rest avoided the need for dialysis over a 25-month median follow-up [107].
Some investigators suggested adopting a 4 cm threshold to guide the treatment of renal masses in VHL disease, in an effort to maximally preserve the quality of life, renal survival, and overall survival [108].
A nationwide Japanese survey found that 203 VHL disease patients were affected by RCC (50.3%). Partial nephrectomy was performed in 46% of cases, radical nephrectomy in 31%, radiofrequency ablation (RFA) in 14%, and in 44% of cases two or more interventions were necessary. An increased number of surgeries correlated with a decrease in eGFR. The 10-year cancer-specific survival rate was higher than in non-VHL disease patients, reaching 95% [73]. In a UK national audit of VHL disease, reporting real-world data, clinical outcome was reported for 229 patients with RCC, of whom 22.2%, 58.5%, and 19.3% underwent clinical monitoring, surgery, and RFA or CB, respectively. Over a 5-year period, only 1.8% of patients had subsequent end-stage kidney disease (mean age 40.5 years), and 46.6% underwent kidney transplantation (mean age 37.3 years) [83].
When NSS is not feasible or does not prevent ESKD, renal replacement therapy is usually needed. Goldfarb et al. specifically investigated the clinical outcomes in VHL patients who underwent RN, developed ESKD, and received a renal transplantation, and compared the data with a control group of non-VHL transplanted patients. They reported that graft survival, patient survival, and renal function were similar between the two groups, supporting the safety of RT in VHL disease [109].
Surgery is usually not necessary for smaller than 3cm lesions due to the low grade and low metastatic rates [110], while larger masses, which are often multifocal and bilateral, should be treated with a nephron-sparing strategy to reduce the risk of inducing chronic kidney disease. The 10-year survival rate of the latter group of patients is 81% [20]. After a mean of 60 months follow-up following nephron sparing surgery for <3 cm tumors, Walther et al. reported no metastatic events and none of the patients needed renal replacement therapy [104].
Although smaller lesions are usually managed by close surveillance or Belzutifan, ablative techniques such as percutaneous RFA are increasingly being investigated for the treatment of <3 cm lesions, because of their efficacy and low complication rates [111]. Cryoablation has the advantage of being less painful and can ablate larger masses but, on account of its impact on renal function and technical feasibility, RFA is preferable in VHL disease [110]. RFA is not performed on <1 cm lesions because of the difficult imaging characterization leading to false positive and false negative results, and due to the low metastatic risk [110]. The most appropriate settings for nephron-sparing surgery are in patients with small renal masses of 2–3 cm diameter, not close to vessels or the bowel, or if unsuitable for more invasive surgery [112,113].
Ploussard et al. investigated clinical outcomes in VHL patients treated with NSS. At 5- and 10-year intervals, the respective local recurrence rates were 45.6% and 83.7%, while the respective overall repeat surgery rates were 23.1% and 63.4%. The mean time to local recurrence was 53 months. The 10-year disease-specific survival rate was 93.8% and none of the NSS-treated patients progressed to CKD or developed metastatic disease [92].
4.2. Metastatic Disease
Currently, only scarce to moderate evidence guides therapeutic decision-making in the setting of metastatic VHL-related ccRCCs, and data are mostly extrapolated from the treatment approach to its sporadic counterpart. Metastatic RCCs are treated with target-therapy agents, mostly tyrosine-kinase inhibitors targeting VHL disease pathways, particularly the VEGF, PDGFR, and FGFR pathways. These agents include Semaxanib, Sunitinib, Pazopanib.
Complete radiologic and metabolic response of a metastatic RCC was reported after 11 administrations of Semaxanib, a tyrosine-kinase inhibitor that selectively targets VEGF [114].
Sunitinib is the most studied drug in VHL-related advanced RCC, and the main application is for palliative purposes, although complete remissions have been reported.
Several trials have demonstrated disease stability, partial response, and even complete regression [115] in metastatic RCCs treated with sunitinib [115,116,117,118,119,120,121]. Jonasch et al. documented a partial response rate of 33% in metastatic RCC patients treated with sunitinib, a better result than in hemangioblastomas. The authors speculated that the results could be explained by the greater expression of VEGFR2 in RCC cells [116].
Pazopanib is a 1st generation inhibitor of multiple tyrosine kinases (VEGFR-1/2/3, FGFR3, PDGFR α/β), approved for the palliative treatment of advanced RCC, seemingly with greater efficacy than sunitinib, albeit its use is supported by fewer and more inconsistent data [122,123].
Dovitinib is a multiple TKi; it is currently being studied for use in metastatic carcinoma, and the best response was found to be a stable disease for CNS hemangioblastomas [124].
Sorafenib is a multiple TKi; partial response in two patients was documented in patients with multiple small RCCs [125].
Although HIF2a inhibition has a strong biological rationale for the inhibition of an upstream abnormal target rather than one or more downstream effectors [38], to date, Belzutifan has not been evaluated in the metastatic stage of ccRCCs in VHL patients, thus other data are needed to support its use in this population. Clinical trials on Belzutifan in ccRCC, potentially also including VHL disease-ccRCC, are underway [126]. Interestingly, evidence is growing with regard to its use in advanced sporadic ccRCCs. A phase I/II trial on its use as monotherapy and an early analysis of a phase II trial on its combination with Cabozantinib both showed a favorable safety profile and promising antitumor activity [58,127].
5. MicroRNA Biomarkers of VHL-Associated Hereditary ccRCC
Even though the diagnosis of VHL syndrome is related to a well-known list of clinical and genetic features, as mentioned in the previous paragraph, the need for new molecular biomarkers able to predict the development and the aggressiveness of renal neoplasms in the VHL syndrome remains crucial in the oncological and nephrological panorama. In fact, one of the main problems related to VHL patients is represented by the multiple growths of ccRCC inside both kidneys, leading to multiple and consecutive surgical and radiological operations, when it is feasible [128]. As a result, VHL patients affected by ccRCC display an augmented risk of developing a moderate to an advanced stage of chronic kidney disease (CKD) due to the repetitive loss of nephrons mass [108], with a worse prognosis over time. Therefore, it would be fundamental for clinicians to better stratify VHL patients at risk of developing new ccRCC in order to avoid diagnostic delay leading to major surgical approaches such as radical nephrectomy in respect to conservative techniques such as nephron-sparing surgery [129]. In fact, the creation of a new molecular panel of circulating predictive biomarkers could help physicians to immediately highlight patients with a high predisposition of ccRCC development to follow a precise new medical algorithm able to detect and eliminate renal neoplasms at early stages with minimally invasive techniques, such as thermal or cryo-ablations, with the maximum saving of renal parenchyma over time [130]. In the molecular scenario, non-coding RNAs represent one of the most promising classes of predictive biomarkers in several types of malignancies and chronic conditions [131,132]. Non-coding RNAs are regulatory RNAs with no or little protein-coding potential [133]. However, in the last decade, several studies have highlighted their importance in the modulation of cell physiology and also cellular functions [134]. Among them, microRNAs are small non-coding single-stranded RNA molecules of 22 nucleotides in length that play a crucial role in the regulation of gene expression, highly involved in many functional processes [135]. Numerous studies both on solid and liquid biopsies have focused on microRNA expression profiles in sporadic ccRCC patients and have identified different miRNAs that act as oncogenes or tumor suppressors, regulating the main signaling pathways of ccRCC [136]. Moreover, different lines of evidence demonstrated that some panels of microRNAs could predict and determine the presence and aggressiveness of sporadic ccRCC, resulting in promising clinical biomarkers [135]. Surprisingly, only one report has so far studied the miRNA expression profile in VHL-associated hereditary ccRCC and compared that with the miRNA expression profile in sporadic ccRCC [137]. In this study, the authors characterized the mRNA and miRNA transcriptome (i) of several samples from multiple tumors occurring within the kidney of two patients affected with VHL disease and (ii) of 12 VHL-associated ccRCC, 22 sporadic ccRCC, and 17 normal adjacent tissues samples from patients with sporadic ccRCC [137]. They found that multiple kidney tumors in a patient affected with VHL disease show a very similar pattern of miRNAs ad mRNAs expression [137]. In addition, by using unsupervised hierarchical clustering of miRNA expression profiles, they were not able to discriminate between the two patients affected by VHL disease. However, the mRNA expression profile allows for the distinction between the two patients, maybe due to the different genetic background [137]. So, the VHL-associated hereditary ccRCC seems to be more homogeneous compared to the sporadic ccRCC. The high similarity in miRNA and mRNA expression indicated a similar molecular evolution of these synchronous tumors and suggested that the same molecular mechanisms underlie the pathogenesis of these hereditary tumors. As a matter of fact, reduced mutation burden and limited evidence of intra-tumor heterogeneity were detected in these tumors that exhibit complementarity of the evolutionary principles of contingency and convergence [138]. On the other hand, compared to normal reference samples, a total of 1377 genes and 51 miRNAs and a total of 1282 genes and 56 miRNAs were differentially expressed in the tumor samples of the two patients affected with VHL disease, respectively [137]. In addition, by using unsupervised hierarchical clustering analyses on the miRNA and mRNA expression profiles, the authors showed that tumor samples were well separated from the normal renal tissues. KEGG biological pathway analysis on dysregulated genes showed that three classes of pathways were overrepresented, and the most significant ones were similar in the two patients and were implicated in ‘immunity’ and ‘metabolism’ [137]. In addition, the authors found a total of 103 miRNAs differentially expressed in ccRCC samples compared to normal renal tissues. Sixty-eight are commonly dysregulated (12 upregulated and 56 downregulated) miRNAs in both VHL-associated and sporadic ccRCC. The authors identified also 18 differentially expressed miRNAs by directly comparing the VHL-associated and sporadic tumors (Table 2).
So, miRNA expression levels can distinguish VHL-associated tumors from sporadic ccRCC even though most differentially expressed miRNAs were similar between the two tumor groups [137]. Furthermore, by transcriptomic analysis, the authors showed that the mRNA expression profile was similar in VHL-associated and sporadic ccRCC. Among the dysregulated mRNAs in ccRCC samples compared to normal renal tissue samples, 2474 genes (959 up- and 1515 downregulated) were in common between VHL-associated and sporadic ccRCC, representing 71% and 81% of total dysregulated mRNAs in the two groups of samples, respectively [137]. So, even if VHL-associated and sporadic ccRCC are considered similar, more in-depth studies are needed to confirm this fact. The study of Gattolliat et al. showed similarities between the two entities, but also divergences in terms of miRNAs and mRNAs profiles helpful to distinguish and better characterize them [137]. One possible limitation of the above-mentioned study is represented by the fact that the relevant miRNA signatures were identified only in the tumor tissue of VHL patients, but not confirmed in the circulating plasma. From a precision medicine perspective, as described at the beginning of this paragraph, the use of a liquid biopsy instead of a tissue one could be fundamental for all VHL patients in order to avoid surgical or medical procedures (e.g., renal biopsy) which have the effect of damage or reduce the amount of nephron mass. Therefore, to find a promising tool for microRNAs in VHL patients, one should consider if the underlined tissue molecular signatures could reflect or not the circulating ones. However, different studies elucidated that tissue-derived microRNAs could be also completely different from the circulating ones, especially in the bloodstream where different organs can excrete different microRNAs and at different levels [139]. While new clinical trials will better investigate these aspects, a possible partial solution could be offered by the already studied circulating microRNAs in sporadic ccRCC, due to the overlap between the tumoral histology of sporadic and VHL ccRCC. As deeply described in a recent review of our group [136], different panels of circulating plasma and urine microRNAs in the ccRCC panorama have been investigated in both retrospective and prospective large studies, with promising results. Even though the absence of a reliable study in VHL patients concerning the evaluation of circulating microRNAs as predictive and prognostic biomarkers remains a gap to be filled, the confirmation or not of the already explored microRNAs in sporadic ccRCC in VHL patients could be a first step in the generation of a new molecular tool able to enter clinical practice.
6. Conclusions
Unlike research studies on the genetic bases of VHL disease, research studies on the transcriptome and non-coding RNA profiles on both liquid and solid biopsies are very scarce or absent in the context of VHL-associated hereditary ccRCC. The discovery of new biomarkers able to guide diagnosis, local or systemic therapy, and follow-up of VHL patients affected by ccRCC are urgent clinical needs. Given that non-coding RNA are emerging new molecular biomarkers for a plethora of diseases, including ccRCC, and could be promising therapeutic targets, preclinical and clinical research on the topic must be encouraged to improve the management of VHL patients.
Author Contributions
A.C., R.M., M.F., and F.T. have contributed to the writing, review, and editing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
The APC was funded by Biorek S.r.l.
Conflicts of Interest
Cinque is the founder and president of Biorek S.r.l.; Trevisani is the founderand CEO of Biorek S.r.l. The other co-authors declare no conflict of interest.
References
- Binderup, M.L.M. Von Hippel-Lindau Disease: Diagnosis and Factors Influencing Disease Outcome. Dan. Med. J. 2018, 65, 1–29. [Google Scholar]
- Rednam, S.P.; Erez, A.; Druker, H.; Janeway, K.A.; Kamihara, J.; Kohlmann, W.K.; Nathanson, K.L.; States, L.J.; Tomlinson, G.E.; Villani, A.; et al. Von Hippel-Lindau and Hereditary Pheochromocytoma/Paraganglioma Syndromes: Clinical Features, Genetics, and Surveillance Recommendations in Childhood. Clin. Cancer Res. 2017, 23, e68–e75. [Google Scholar] [CrossRef] [PubMed]
- Maher, E.R.; Yates, J.R.W.; Ferguson-Smith, M.A. Statistical Analysis of the Two Stage Mutation Model in von Hippel-Lindau Disease, and in Sporadic Cerebellar Haemangioblastoma and Renal Cell Carcinoma. J. Med. Genet. 1990, 27, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Qiu, J.; Yang, W.; Ma, K.; Li, L.; Xie, H.; Xu, Y.; Gong, Y.; Zhou, J.; Cai, L.; et al. Clinical Characteristics and Risk Factors for Survival in Affected Offspring of von Hippel-Lindau Disease Patients. J. Med. Genet. 2021, 59, 951–956. [Google Scholar] [CrossRef]
- v. Hippel, E. Über Eine Sehr Seltene Erkrankung Der Netzhaut. Albrecht Von Graefes Arch. Für Ophthalmol. 1904, 59, 83–106. [Google Scholar] [CrossRef]
- Lindau, A. Zur Frage der Angiomatosis retinæ und ihrer hirnkomplikationen. Acta Ophthalmol. 1926, 4, 193–226. [Google Scholar] [CrossRef]
- Maher, E.R.; Yates, J.R.W.; Harries, R.; Benjamin, C.; Harris, R.; Moore, A.T.; Ferguson-Smith, M.A. Clinical Features and Natural History of von Hippel-Lindau Disease. QJM 1990, 77, 1151–1163. [Google Scholar] [CrossRef]
- Horton, W.A.; Wong, V.; Eldridge, R. Von Hippel-Lindau Disease: Clinical and Pathological Manifestations in Nine Families with 50 Affected Members. Arch. Intern. Med. 1976, 136, 769–777. [Google Scholar] [CrossRef]
- Lamiell, J.M.; Salazar, F.G.; Edward Hsia, Y. Von Hippel-Lindau Disease Affecting 43 Members of a Single Kindred. Medicine 1989, 68, 1–29. [Google Scholar] [CrossRef]
- Shanbhogue, K.P.; Hoch, M.; Fatterpaker, G.; Chandarana, H. Von Hippel-Lindau Disease: Review of Genetics and Imaging. Radiol. Clin. N. Am. 2016, 54, 409–422. [Google Scholar] [CrossRef]
- Neumann, H.P.H.; Wiestler, O.D. Clustering of Features of von Hippel-Lindau Syndrome: Evidence for a Complex Genetic Locus. The Lancet 1991, 337, 1052–1054. [Google Scholar] [CrossRef]
- Nordstrom-O’Brien, M.; van der Luijt, R.B.; van Rooijen, E.; van den Ouweland, A.M.; Majoor-Krakauer, D.F.; Lolkema, M.P.; van Brussel, A.; Voest, E.E.; Giles, R.H. Genetic Analysis of von Hippel-Lindau Disease. Hum. Mutat. 2010, 31, 521–537. [Google Scholar] [CrossRef] [PubMed]
- Von Hippel-Lindau Syndrome. A Pleomorphic Condition. Available online: https://pubmed.ncbi.nlm.nih.gov/10630173/ (accessed on 21 May 2022).
- Friedrich, C.A. Genotype-Phenotype Correlation in von Hippel-Lindau Syndrome. Hum. Mol. Genet. 2001, 10, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Gossage, L.; Eisen, T.; Maher, E.R. VHL, the Story of a Tumour Suppressor Gene. Nat. Rev. Cancer 2015, 15, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.J.; Wang, J.Y.; Peng, S.H.; Li, T.; Ning, X.H.; Hong, B.A.; Liu, J.Y.; Wu, P.J.; Zhou, B.W.; Zhou, J.C.; et al. Genotype and Phenotype Correlation in von Hippel–Lindau Disease Based on Alteration of the HIF-α Binding Site in VHL Protein. Genet. Med. 2018, 20, 1266–1273. [Google Scholar] [CrossRef]
- Gallou, C.; Chauveau, D.; Richard, S.; Joly, D.; Giraud, S.; Olschwang, S.; Martin, N.; Saquet, C.; Chrétien, Y.; Méjean, A.; et al. Genotype-Phenotype Correlation in von Hippel-Lindau Families with Renal Lesions. Hum. Mutat. 2004, 24, 215–224. [Google Scholar] [CrossRef]
- Kai, R.O.; Woodward, E.R.; Killick, P.; Lim, C.; Macdonald, F.; Maher, E.R. Genotype-Phenotype Correlations in von Hippel-Lindau Disease. Hum. Mutat. 2007, 28, 143–149. [Google Scholar] [CrossRef]
- Melmon, K.L.; Rosen, S.W. Lindau’s disease. Review of the literature and study of a large kindred. Am. J. Med. 1964, 36, 595–617. [Google Scholar] [CrossRef]
- Chittiboina, P.; Lonser, R.R. Von Hippel-Lindau Disease. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2015; Volume 132. [Google Scholar]
- Gläsker, S.; Bender, B.U.; Apel, T.W.; Natt, E.; van Velthoven, V.; Scheremet, R.; Zentner, J.; Neumann, H.P.H. The Impact of Molecular Genetic Analysis of the VHL Gene in Patients with Haemangioblastomas of the Central Nervous System. J. Neurol. Neurosurg. Psychiatry 1999, 67, 758–762. [Google Scholar] [CrossRef]
- Maher, E. Von Hippel-Lindau Disease. Curr. Mol. Med. 2004, 4, 833–842. [Google Scholar] [CrossRef]
- Varshney, N.; Kebede, A.A.; Owusu-Dapaah, H.; Lather, J.; Kaushik, M.; Bhullar, J.S. A Review of Von Hippel-Lindau Syndrome. J. Kidney Cancer VHL 2017, 4, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.Y.; Kaelin, W.G. Role of VHL Gene Mutation in Human Cancer. J. Clin. Oncol. 2004, 22, 4991–5004. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G. Mutation and Cancer: Statistical Study of Retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Somatic Inactivation of the VHL Gene in Von Hippel-Lindau Disease Tumors. Available online: https://pubmed.ncbi.nlm.nih.gov/9106522/ (accessed on 22 May 2022).
- Crossey, P.A.; Foster, K.; Richards, F.M.; Phipps, M.E.; Latif, F.; Tory, K.; Jones, M.H.; Bentley, E.; Kumar, R.; Lerman, M.I.; et al. Molecular Genetic Investigations of the Mechanism of Tumourigenesis in von Hippel-Lindau Disease: Analysis of Allele Loss in VHL Tumours. Hum. Genet. 1994, 93, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, O.; Kibel, A.; Gray, S.; Kaelin, W.G. Tumour Suppression by the Human von Hippel-Lindau Gene Product. Nat. Med. 1995, 1, 822–826. [Google Scholar] [CrossRef]
- Inactivation of the von Hippel-Lindau (VHL) Tumour Suppressor Gene and Allelic Losses at Chromosome Arm 3p in Primary Renal Cell Carcinoma: Evidence for a VHL-Independent Pathway in Clear Cell Renal Tumourigenesis. Available online: https://pubmed.ncbi.nlm.nih.gov/9624531/ (accessed on 25 May 2022).
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H.; et al. Integrated Molecular Analysis of Clear-Cell Renal Cell Carcinoma. Nat. Genet. 2013, 45, 860–867. [Google Scholar] [CrossRef]
- Li, J.; Guo, L.; Ai, Z. An Integrated Analysis of Cancer Genes in Clear Cell Renal Cell Carcinoma. Future Oncol. 2017, 13, 715–725. [Google Scholar] [CrossRef]
- Rankin, E.B.; Tomaszewski, J.E.; Haase, V.H. Renal Cyst Development in Mice with Conditional Inactivation of the von Hippel-Lindau Tumor Suppressor. Cancer Res. 2006, 66, 2576–2583. [Google Scholar] [CrossRef]
- Vortmeyer, A.O.; Yuan, Q.; Lee, Y.S.; Zhuang, Z.; Oldfield, E.H. Developmental Effects of von Hippel-Lindau Gene Deficiency. Ann. Neurol. 2004, 55, 721–728. [Google Scholar] [CrossRef]
- Kaelin, W.G. Molecular Basis of the VHL Hereditary Cancer Syndrome. Nat. Rev. Cancer 2002, 2, 673–682. [Google Scholar] [CrossRef]
- Schanza, L.M.; Seles, M.; Stotz, M.; Fosselteder, J.; Hutterer, G.C.; Pichler, M.; Stiegelbauer, V. MicroRNAs Associated with Von Hippel-Lindau Pathway in Renal Cell Carcinoma: A Comprehensive Review. Int. J. Mol. Sci. 2017, 18, 2495. [Google Scholar] [CrossRef] [PubMed]
- White, N.M.A.; Yousef, G.M. MicroRNAs: Exploring a New Dimension in the Pathogenesis of Kidney Cancer. BMC Med. 2010, 8, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Shuin, T.; Yamasaki, I.; Tamura, K.; Okuda, H.; Furihata, M.; Ashida, S. Von Hippel-Lindau Disease: Molecular Pathological Basis, Clinical Criteria, Genetic Testing, Clinical Features of Tumors and Treatment. Jpn. J. Clin. Oncol. 2006, 36, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Gläsker, S.; Vergauwen, E.; Koch, C.A.; Kutikov, A.; Vortmeyer, A.O. Von Hippel-Lindau Disease: Current Challenges and Future Prospects. OncoTargets Ther. 2020, 13, 5669–5690. [Google Scholar] [CrossRef] [PubMed]
- Seizinger, B.R.; Rouleau, G.A.; Ozelius, L.J.; Lane, A.H.; Farmer, G.E.; Lamiell, J.M.; Haines, J.; Yuen, J.W.M.; Collins, D.; Majoor-Krakauer, D.; et al. Von Hippel-Lindau Disease Maps to the Region of Chromosome 3 Associated with Renal Cell Carcinoma. Nature 1988, 332, 268–269. [Google Scholar] [CrossRef] [PubMed]
- Latif, F.; Tory, K.; Gnarra, J.; Yao, M.; Duh, F.M.; Orcutt, M.; Stackhouse, T.; Kuzmin, I.; Modi, W.; Geil, L.; et al. Identification of the von Hippel-Lindau Disease Tumor Suppressor Gene. Science 1993, 260, 1317–1320. [Google Scholar] [CrossRef]
- Blankenship, C.; Naglich, J.G.; Whaley, J.M.; Seizinger, B.; Kley, N. Alternate Choice of Initiation Codon Produces a Biologically Active Product of the von Hippel Lindau Gene with Tumor Suppressor Activity. Oncogene 1999, 18, 1529–1535. [Google Scholar] [CrossRef]
- Takagi, Y.; Pause, A.; Conaway, R.C.; Conaway, J.W. Identification of Elongin C Sequences Required for Interaction with the von Hippel-Lindau Tumor Suppressor Protein. J. Biol. Chem. 1997, 272, 27444–27449. [Google Scholar] [CrossRef]
- Schoenfeld, A.R.; Davidowitz, E.J.; Burk, R.D. Elongin BC Complex Prevents Degradation of von Hippel-Lindau Tumor Suppressor Gene Products. Proc. Natl. Acad. Sci. USA 2000, 97, 8507–8512. [Google Scholar] [CrossRef]
- Iwai, K.; Yamanaka, K.; Kamura, T.; Minato, N.; Conaway, R.C.; Conaway, J.W.; Klausner, R.D.; Pause, A. Identification of the von Hippel-Lindau Tumor-Suppressor Protein as Part of an Active E3 Ubiquitin Ligase Complex. Proc. Natl. Acad. Sci. USA 1999, 96, 12436–12441. [Google Scholar] [CrossRef]
- Kuo, C.-Y.; Lin, C.-H.; Hsu, T. VHL Inactivation in Precancerous Kidney Cells Induces an Inflammatory Response via ER Stress–Activated IRE1α Signaling. Cancer Res. 2017, 77, 13–3406. [Google Scholar] [CrossRef] [PubMed]
- Clifford, S.C.; Astuti, D.; Hooper, L.; Maxwell, P.H.; Ratcliffe, P.J.; Maher, E.R. The PVHL-Associated SCF Ubiquitin Ligase Complex: Molecular Genetic Analysis of Elongin B and C, Rbx1 and HIF-1alpha in Renal Cell Carcinoma. Oncogene 2001, 20, 5067–5074. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-α to the von Hippel-Lindau Ubiquitylation Complex by O2-Regulated Prolyl Hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.; Kentsis, A.; Osman, R.; Pan, Z.Q. Inactivation of VHL by Tumorigenic Mutations That Disrupt Dynamic Coupling of the PVHL.Hypoxia-Inducible Transcription Factor-1alpha Complex. J. Biol. Chem. 2005, 280, 7985–7996. [Google Scholar] [CrossRef]
- Li, L.; Zhang, L.; Zhang, X.; Yan, Q.; Minamishima, Y.A.; Olumi, A.F.; Mao, M.; Bartz, S.; Kaelin, W.G. Hypoxia-Inducible Factor Linked to Differential Kidney Cancer Risk Seen with Type 2A and Type 2B VHL Mutations. Mol. Cell Biol. 2007, 27, 5381–5392. [Google Scholar] [CrossRef]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for Cancer Therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Carroll, V.A.; Ashcroft, M. Role of Hypoxia-Inducible Factor (HIF)-1alpha versus HIF-2alpha in the Regulation of HIF Target Genes in Response to Hypoxia, Insulin-like Growth Factor-I, or Loss of von Hippel-Lindau Function: Implications for Targeting the HIF Pathway. Cancer Res. 2006, 66, 6264–6270. [Google Scholar] [CrossRef]
- Baldewijns, M.M.; van Vlodrop, I.J.H.; Vermeulen, P.B.; Soetekouw, P.M.M.B.; van Engeland, M.; de Bruïne, A.P. VHL and HIF Signalling in Renal Cell Carcinogenesis. J. Pathol. 2010, 221, 125–138. [Google Scholar] [CrossRef]
- Schödel, J.; Grampp, S.; Maher, E.R.; Moch, H.; Ratcliffe, P.J.; Russo, P.; Mole, D.R. Hypoxia, Hypoxia-Inducible Transcription Factors, and Renal Cancer. Eur. Urol. 2016, 69, 646–657. [Google Scholar] [CrossRef]
- Elorza, A.; Soro-Arnáiz, I.; Meléndez-Rodríguez, F.; Rodríguez-Vaello, V.; Marsboom, G.; de Cárcer, G.; Acosta-Iborra, B.; Albacete-Albacete, L.; Ordóñez, A.; Serrano-Oviedo, L.; et al. HIF2α Acts as an MTORC1 Activator through the Amino Acid Carrier SLC7A5. Mol. Cell 2012, 48, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Ganner, A.; Gehrke, C.; Klein, M.; Thegtmeier, L.; Matulenski, T.; Wingendorf, L.; Wang, L.; Pilz, F.; Greidl, L.; Meid, L.; et al. VHL Suppresses RAPTOR and Inhibits MTORC1 Signaling in Clear Cell Renal Cell Carcinoma. Sci. Rep. 2021, 11, 14827. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Kaelin, W.G. Targeting the HIF2-VEGF Axis in Renal Cell Carcinoma. Nat. Med. 2020, 26, 1519–1530. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.M.; Choueiri, T.K.; Papadopoulos, K.P.; Plimack, E.R.; Merchan, J.R.; McDermott, D.F.; Michaelson, M.D.; Appleman, L.J.; Thamake, S.; Perini, R.F.; et al. The Oral HIF-2 α Inhibitor MK-6482 in Patients with Advanced Clear Cell Renal Cell Carcinoma (RCC): Updated Follow-up of a Phase I/II Study. J. Clin. Oncol. 2021, 39, 273. [Google Scholar] [CrossRef]
- FDA Approves Belzutifan for Cancers Associated with von Hippel-Lindau Disease|FDA. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-belzutifan-cancers-associated-von-hippel-lindau-disease (accessed on 29 May 2022).
- Zhang, J.; Zhang, Q. VHL and Hypoxia Signaling: Beyond HIF in Cancer. Biomedicines 2018, 6, 35. [Google Scholar] [CrossRef]
- Haase, V.H. The VHL Tumor Suppressor: Master Regulator of HIF. Curr. Pharm. Des. 2009, 15, 3895. [Google Scholar] [CrossRef]
- Kapitsinou, P.P.; Haase, V.H. The VHL Tumor Suppressor and HIF: Insights from Genetic Studies in Mice. Cell Death Differ. 2008, 15, 650–659. [Google Scholar] [CrossRef]
- Tarade, D.; Ohh, M. The HIF and Other Quandaries in VHL Disease. Oncogene 2018, 37, 139–147. [Google Scholar] [CrossRef]
- Rathmell, W.K.; Chen, S. VHL Inactivation in Renal Cell Carcinoma: Implications for Diagnosis, Prognosis and Treatment. Expert Rev. Anticancer Ther. 2008, 8, 63–73. [Google Scholar] [CrossRef][Green Version]
- Gnarra, J.R.; Tory, K.; Weng, Y.; Schmidt, L.; Wei, M.H.; Li, H.; Latif, F.; Liu, S.; Chen, F.; Duh, F.M.; et al. Mutations of the VHL Tumour Suppressor Gene in Renal Carcinoma. Nat. Genet. 1994, 7, 85–90. [Google Scholar] [CrossRef]
- Young, A.C.; Craven, R.A.; Cohen, D.; Taylor, C.; Booth, C.; Harnden, P.; Cairns, D.A.; Astuti, D.; Gregory, W.; Maher, E.R.; et al. Analysis of VHL Gene Alterations and Their Relationship to Clinical Parameters in Sporadic Conventional Renal Cell Carcinoma. Clin. Cancer Res. 2009, 15, 7582–7592. [Google Scholar] [CrossRef] [PubMed]
- Hamano, K.; Esumi, M.; Igarashi, H.; Chino, K.; Mochida, J.; Ishida, H.; Okada, K. Biallelic Inactivation of the von Hippel-Lindau Tumor Suppressor Gene in Sporadic Renal Cell Carcinoma. J. Urol. 2002, 167, 713–717. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Yoshida, M.; Kishida, T.; Nakaigawa, N.; Baba, M.; Kobayashi, K.; Miura, T.; Moriyama, M.; Nagashima, Y.; Nakatani, Y.; et al. VHL Tumor Suppressor Gene Alterations Associated with Good Prognosis in Sporadic Clear-Cell Renal Carcinoma. J. Natl. Cancer Inst. 2002, 94, 1569–1575. [Google Scholar] [CrossRef] [PubMed]
- Somatic VHL Alteration and Its Impact on Prognosis in Patients with Clear Cell Renal Cell Carcinoma. Available online: https://pubmed.ncbi.nlm.nih.gov/15809750/ (accessed on 22 May 2022).
- Kim, H.S.; Kim, J.H.; Jang, H.J.; Han, B.; Zang, D.Y. Clinicopathologic Significance of Vhl Gene Alteration in Clear-Cell Renal Cell Carcinoma: An Updated Meta-Analysis and Review. Int. J. Mol. Sci. 2018, 19, 2529. [Google Scholar] [CrossRef]
- Gordan, J.D.; Lal, P.; Dondeti, V.R.; Letrero, R.; Parekh, K.N.; Oquendo, C.E.; Greenberg, R.A.; Flaherty, K.T.; Rathmell, W.K.; Keith, B.; et al. HIF-Alpha Effects on c-Myc Distinguish Two Subtypes of Sporadic VHL-Deficient Clear Cell Renal Carcinoma. Cancer Cell 2008, 14, 435–446. [Google Scholar] [CrossRef]
- Takayanagi, S.; Mukasa, A.; Nakatomi, H.; Kanno, H.; Kuratsu, J.I.; Nishikawa, R.; Mishima, K.; Natsume, A.; Wakabayashi, T.; Houkin, K.; et al. Development of Database and Genomic Medicine for von Hippel-Lindau Disease in Japan. Neurol. Med. Chir. 2017, 57, 59–65. [Google Scholar] [CrossRef][Green Version]
- Wilding, A.; Ingham, S.L.; Lalloo, F.; Clancy, T.; Huson, S.M.; Moran, A.; Evans, D.G. Life Expectancy in Hereditary Cancer Predisposing Diseases: An Observational Study. J. Med. Genet. 2012, 49, 264–269. [Google Scholar] [CrossRef]
- Wang, J.Y.; Peng, S.H.; Li, T.; Ning, X.H.; Liu, S.J.; Hong, B.A.; Liu, J.Y.; Wu, P.J.; Zhou, B.W.; Zhou, J.C.; et al. Risk Factors for Survival in Patients with von Hippel-Lindau Disease. J. Med. Genet. 2018, 55, 322–328. [Google Scholar] [CrossRef]
- Bausch, B.; Jilg, C.; Gläsker, S.; Vortmeyer, A.; Lützen, N.; Anton, A.; Eng, C.; Neumann, H.P.H. Renal Cancer in von Hippel-Lindau Disease and Related Syndromes. Nat. Rev. Nephrol. 2013, 9, 529–538. [Google Scholar] [CrossRef]
- Binderup, M.L.M.; Jensen, A.M.; Budtz-Jørgensen, E.; Bisgaard, M.L. Survival and Causes of Death in Patients with von Hippel-Lindau Disease. J. Med. Genet. 2017, 54, 11–18. [Google Scholar] [CrossRef]
- IARC Publications Website—Urinary and Male Genital Tumours. Available online: https://publications.iarc.fr/Book-And-Report-Series/Who-Classification-Of-Tumours/Urinary-And-Male-Genital-Tumours-2022 (accessed on 15 August 2022).
- Jonasch, E.; Gao, J.; Rathmell, W.K. Renal Cell Carcinoma. BMJ 2014, 349, g4797. [Google Scholar] [CrossRef] [PubMed]
- Freifeld, Y.; Ananthakrishnan, L.; Margulis, V. Imaging for Screening and Surveillance of Patients with Hereditary Forms of Renal Cell Carcinoma. Curr. Urol. Rep. 2018, 19, 82. [Google Scholar] [CrossRef] [PubMed]
- Prevalence, Morphology and Biology of Renal Cell Carcinoma in von Hippel-Lindau Disease Compared to Sporadic Renal Cell Carcinoma. Available online: https://pubmed.ncbi.nlm.nih.gov/9751329/ (accessed on 21 May 2022).
- Choyke, P.L.; Glenn, G.M.; Walther, M.M.; Zbar, B.; Weiss, G.H.; Alexander, R.B.; Hayes, W.S.; Long, J.P.; Thakore, K.N.; Linehan, W.M. The Natural History of Renal Lesions in von Hippel-Lindau Disease: A Serial CT Study in 28 Patients. Am. J. Roentgenol. 1992, 159, 1229–1234. [Google Scholar] [CrossRef] [PubMed]
- Maher, E.R.; Adlard, J.; Barwell, J.; Brady, A.F.; Brennan, P.; Cook, J.; Crawford, G.S.; Dabir, T.; Davidson, R.; Dyer, R.; et al. Evaluation of Tumour Surveillance Protocols and Outcomes in von Hippel-Lindau Disease in a National Health Service. Br. J. Cancer 2022, 126, 1339–1345. [Google Scholar] [CrossRef]
- Poston, C.D.; Jaffe, G.S.; Lubensky, I.A.; Solomon, D.; Zbar, B.; Linehan, W.M.; Walther, M.M. Characterization of the Renal Pathology of a Familial Form of Renal Cell Carcinoma Associated with von Hippel-Lindau Disease: Clinical and Molecular Genetic Implications. J. Urol. 1995, 153, 22–26. [Google Scholar] [CrossRef]
- Solomon, D.; Schwartz, A. Renal Pathology in von Hippel-Lindau Disease. Hum. Pathol. 1988, 19, 1072–1079. [Google Scholar] [CrossRef]
- Montani, M.; Heinimann, K.; von Teichman, A.; Rudolph, T.; Perren, A.; Moch, H. VHL-Gene Deletion in Single Renal Tubular Epithelial Cells and Renal Tubular Cysts: Further Evidence for a Cyst-Dependent Progression Pathway of Clear Cell Renal Carcinoma in von Hippel-Lindau Disease. Am. J. Surg. Pathol. 2010, 34, 806–815. [Google Scholar] [CrossRef]
- Peng, S.; Shepard, M.J.; Wang, J.; Li, T.; Ning, X.; Cai, L.; Zhuang, Z.; Gong, K. Genotype-Phenotype Correlations in Chinese von Hippel-Lindau Disease Patients. Oncotarget 2017, 8, 38456–38465. [Google Scholar] [CrossRef]
- Lonser, R.R.; Glenn, G.M.; Walther, M.; Chew, E.Y.; Libutti, S.K.; Linehan, W.M.; Oldfield, E.H. Von Hippel-Lindau Disease. Lancet 2003, 361, 2059–2067. [Google Scholar] [CrossRef]
- Jilg, C.A.; Neumann, H.P.; Gläsker, S.; Schäfer, O.; Ardelt, P.U.; Schwardt, M.; Schultze-Seemann, W. Growth Kinetics in von Hippel-Lindau-Associated Renal Cell Carcinoma. Urol. Int. 2012, 88, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, J.; Zhang, L.; Yao, L.; Zhang, C.; He, Z.; Li, X.; Zhou, L. Natural History and Growth Kinetics of Clear Cell Renal Cell Carcinoma in Sporadic and von Hippel-Lindau Disease. Transl. Androl. Urol. 2021, 10, 1064. [Google Scholar] [CrossRef] [PubMed]
- Gumundsson, E.; Hellborg, H.; Lundstam, S.; Erikson, S.; Ljungberg, B. Metastatic Potential in Renal Cell Carcinomas ≤7 Cm: Swedish Kidney Cancer Quality Register Data. Eur. Urol. 2011, 60, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Ploussard, G.; Droupy, S.; Ferlicot, S.; Ples, R.; Rocher, L.; Richard, S.; Benoit, G. Local Recurrence after Nephron-Sparing Surgery in von Hippel-Lindau Disease. Urology 2007, 70, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Tabuchi, H.; Nagashima, Y.; Baba, M.; Nakaigawa, N.; Ishiguro, H.; Hamada, K.; Inayama, Y.; Kishida, T.; Hattori, K.; et al. Gene Expression Analysis of Renal Carcinoma: Adipose Differentiation-Related Protein as a Potential Diagnostic and Prognostic Biomarker for Clear-Cell Renal Carcinoma. J. Pathol. 2005, 205, 377–387. [Google Scholar] [CrossRef]
- Dagher, J.; Kammerer-Jacquet, S.F.; Brunot, A.; Pladys, A.; Patard, J.J.; Bensalah, K.; Perrin, C.; Verhoest, G.; Mosser, J.; Lespagnol, A.; et al. Wild-Type VHL Clear Cell Renal Cell Carcinomas Are a Distinct Clinical and Histologic Entity: A 10-Year Follow-Up. Eur. Urol. Focus 2016, 1, 284–290. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, L.; Qiu, J.; Li, Z.; Wu, Y.; Zhang, C.; Yao, L.; Gong, K.; Li, X.; Zhou, L. Natural History of Von Hippel–Lindau Disease-Associated and Sporadic Clear Cell Renal Cell Carcinoma: A Comparative Study. J. Cancer Res. Clin. Oncol. 2021, 148, 2631–2641. [Google Scholar] [CrossRef]
- Chahoud, J.; McGettigan, M.; Parikh, N.; Boris, R.S.; Iliopoulos, O.; Rathmell, W.K.; Daniels, A.B.; Jonasch, E.; Spiess, P.E. Evaluation, Diagnosis and Surveillance of Renal Masses in the Setting of VHL Disease. World J. Urol. 2021, 39, 2409–2415. [Google Scholar] [CrossRef]
- Meister, M.; Choyke, P.; Anderson, C.; Patel, U. Radiological Evaluation, Management, and Surveillance of Renal Masses in Von Hippel-Lindau Disease. Clin. Radiol. 2009, 64, 589–600. [Google Scholar] [CrossRef]
- Ashouri, K.; Mohseni, S.; Tourtelot, J.; Sharma, P.; Spiess, P.E. Implications of Von Hippel-Lindau Syndrome and Renal Cell Carcinoma. J. Kidney Cancer VHL 2015, 2, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Maher, E.R.; Neumann, H.P.H.; Richard, S. Von Hippel-Lindau Disease: A Clinical and Scientific Review. Eur. J. Hum. Genet. 2011, 19, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Jonasch, E.; Walker, C.L.; Rathmell, W.K. Clear Cell Renal Cell Carcinoma Ontogeny and Mechanisms of Lethality. Nat. Rev. Nephrol. 2021, 17, 245–261. [Google Scholar] [CrossRef] [PubMed]
- de Rojas-P, I.; Albiñana, V.; Taranets, L.; Recio-Poveda, L.; Cuesta, A.M.; Popov, N.; Kronenberger, T.; Botella, L.M. The Endothelial Landscape and Its Role in Von Hippel-Lindau Disease. Cells 2021, 10, 2313. [Google Scholar] [CrossRef] [PubMed]
- Jonasch, E.; Donskov, F.; Iliopoulos, O.; Rathmell, W.K.; Narayan, V.K.; Maughan, B.L.; Oudard, S.; Else, T.; Maranchie, J.K.; Welsh, S.J.; et al. Belzutifan for Renal Cell Carcinoma in von Hippel-Lindau Disease. N. Engl. J. Med. 2021, 385, 2036–2046. [Google Scholar] [CrossRef]
- VHLA Suggested Active Surveillance Guidelines. Available online: https://www.vhl.org/wp-content/uploads/2020/10/Active-Surveillance-Guidelines-2020.pdf (accessed on 1 October 2022).
- Walther, M.M.; Choyke, P.L.; Glenn, G.; Lyne, J.C.; Rayford, W.; Venzon, D.; Linehan, W.M. Renal Cancer in Families with Hereditary Renal Cancer: Prospective Analysis of a Tumor Size Threshold for Renal Parenchymal Sparing Surgery. J. Urol. 1999, 161, 1475–1479. [Google Scholar] [CrossRef]
- Duffey, B.G.; Choyke, P.L.; Glenn, G.; Grubb, R.L.; Venzon, D.; Linehan, W.M.; Walther, M.M. The Relationship between Renal Tumor Size and Metastases in Patients with von Hippel-Lindau Disease. J. Urol. 2004, 172, 63–65. [Google Scholar] [CrossRef]
- Treatment of Renal Cell Carcinoma in von Hippel-Lindau Disease: A Multicenter Study. Available online: https://pubmed.ncbi.nlm.nih.gov/7752324/ (accessed on 29 May 2022).
- Bratslavsky, G.; Liu, J.J.; Johnson, A.D.; Sudarshan, S.; Choyke, P.L.; Linehan, W.M.; Pinto, P.A. Salvage Partial Nephrectomy for Hereditary Renal Cancer: Feasibility and Outcomes. J. Urol. 2008, 179, 67–70. [Google Scholar] [CrossRef]
- Jilg, C.A.; Neumann, H.P.H.; Gläsker, S.; Schäfer, O.; Leiber, C.; Ardelt, P.U.; Schwardt, M.; Schultze-Seemann, W. Nephron Sparing Surgery in von Hippel-Lindau Associated Renal Cell Carcinoma; Clinicopathological Long-Term Follow-Up. Fam. Cancer 2012, 11, 387–394. [Google Scholar] [CrossRef]
- Goldfarb, D.A.; Neumann, H.P.H.; Penn, I.; Novick, A.C. Results of Renal Transplantation in Patients with Renal Cell Carcinoma and von Hippel-Lindau Disease. Transplantation 1997, 64, 1726–1729. [Google Scholar] [CrossRef]
- Park, B.K.; Kim, C.K.; Park, S.Y.; Shen, S.H. Percutaneous Radiofrequency Ablation of Renal Cell Carcinomas in Patients with von Hippel Lindau Disease: Indications, Techniques, Complications, and Outcomes. Acta Radiol. 2013, 54, 418–427. [Google Scholar] [CrossRef]
- Allasia, M.; Soria, F.; Battaglia, A.; Gazzera, C.; Calandri, M.; Caprino, M.P.; Lucatello, B.; Velrti, A.; Maccario, M.; Pasini, B.; et al. Radiofrequency Ablation for Renal Cancer in Von Hippel-Lindau Syndrome Patients: A Prospective Cohort Analysis. Clin. Genitourin. Cancer 2017, 16, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Park, B.K.; Kim, C.K.; LeeLee, H.M.; Jeon, S.S.; Seo, S.I.; Jeong, B.C.; Choi, H.Y. Percutaneous Radiofrequency Ablation of Renal Cell Carcinomas in Patients with von Hippel Lindau Disease Previously Undergoing a Radical Nephrectomy or Repeated Nephron-Sparing Surgery. Acta Radiol. 2011, 52, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Park, B.K.; Kim, C.K. Percutaneous Radio Frequency Ablation of Renal Tumors in Patients with von Hippel-Lindau Disease: Preliminary Results. J. Urol. 2010, 183, 1703–1707. [Google Scholar] [CrossRef] [PubMed]
- Jennens, R.R.; Rosenthal, M.A.; Lindeman, G.J.; Michael, M. Complete Radiological and Metabolic Response of Metastatic Renal Cell Carcinoma to SU5416 (Semaxanib) in a Patient with Probable von Hippel-Lindau Syndrome. Urol. Oncol. 2004, 22, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Tsimafeyeu, I. Sunitinib Treatment of Metastatic Renal Cell Carcinoma in von Hippel-Lindau Disease. J. Cancer Res. Ther. 2015, 11, 920–922. [Google Scholar] [CrossRef]
- Jonasch, E.; McCutcheon, I.E.; Waguespack, S.G.; Wen, S.; Davis, D.W.; Smith, L.A.; Tannir, N.M.; Gombos, D.S.; Fuller, G.N.; Matin, S.F. Pilot Trial of Sunitinib Therapy in Patients with von Hippel-Lindau Disease. Ann. Oncol. 2011, 22, 2661–2666. [Google Scholar] [CrossRef]
- Roma, A.; Maruzzo, M.; Basso, U.; Brunello, A.; Zamarchi, R.; Bezzon, E.; Pomerri, F.; Zovato, S.; Opocher, G.; Zagonel, V. First-Line Sunitinib in Patients with Renal Cell Carcinoma (RCC) in von Hippel–Lindau (VHL) Disease: Clinical Outcome and Patterns of Radiological Response. Fam. Cancer 2015, 14, 309–316. [Google Scholar] [CrossRef]
- Kobayashi, A.; Takahashi, M.; Imai, H.; Akiyama, S.; Sugiyama, S.; Komine, K.; Saijo, K.; Takahashi, M.; Takahashi, S.; Shirota, H.; et al. Attainment of a Long-Term Favorable Outcome by Sunitinib Treatment for Pancreatic Neuroendocrine Tumor and Renal Cell Carcinoma Associated with von Hippel-Lindau Disease. Intern. Med. 2016, 55, 629–634. [Google Scholar] [CrossRef][Green Version]
- Kim, H.C.; Lee, J.S.; Kim, S.H.; So, H.S.; Woo, C.Y.; Lee, J.L. Sunitinib Treatment for Metastatic Renal Cell Carcinoma in Patients with von Hippel-Lindau Disease. Cancer Res. Treat. 2013, 45, 349–353. [Google Scholar] [CrossRef]
- Sunitinib Treatment for Multifocal Renal Cell Carcinoma (RCC) and Pancreatic Neuroendocrine Tumor (NET) in Patient with Von Hippel-Lindau Disease. Case Report. Available online: https://pubmed.ncbi.nlm.nih.gov/26812297/ (accessed on 28 May 2022).
- Oudard, S.; Elaidi, R.; Brizard, M.; le Rest, C.; Caillet, V.; Deveaux, S.; Benoit, G.; Corréas, J.M.; Benoudiba, F.; David, P.; et al. Sunitinib for the Treatment of Benign and Malignant Neoplasms from von Hippel-Lindau Disease: A Single-Arm, Prospective Phase II Clinical Study from the PREDIR Group. Oncotarget 2016, 7, 85306–85317. [Google Scholar] [CrossRef][Green Version]
- Kim, B.Y.S.; Jonasch, E.; McCutcheon, I.E. Pazopanib Therapy for Cerebellar Hemangioblastomas in von Hippel-Lindau Disease: Case Report. Target. Oncol. 2012, 7, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Jonasch, E.; McCutcheon, I.E.; Gombos, D.S.; Ahrar, K.; Perrier, N.D.; Liu, D.; Robichaux, C.C.; Villarreal, M.F.; Weldon, J.A.; Woodson, A.H.; et al. Pazopanib in Patients with von Hippel-Lindau Disease: A Single-Arm, Single-Centre, Phase 2 Trial. Lancet Oncol. 2018, 19, 1351–1359. [Google Scholar] [CrossRef]
- Pilié, P.; Hasanov, E.; Matin, S.F.; Woodson, A.H.H.; Marcott, V.D.; Bird, S.; Slack, R.S.; Fuller, G.N.; McCutcheon, I.E.; Jonasch, E. Pilot Study of Dovitinib in Patients with von Hippel-Lindau Disease. Oncotarget 2018, 9, 23390–23395. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.H.; Yu, Y.D.; Kang, M.H.; Park, D.S. Sorafenib Treatment for Recurrent Stage T1 Bilateral Renal Cell Carcinoma in Patients with Von Hippel- Lindau Disease: A Case Report and Literature Review. Can. Urol. Assoc. J. 2015, 9, E651–E653. [Google Scholar] [CrossRef] [PubMed]
- A Study of Belzutifan (MK-6482) in Participants with Advanced Clear Cell Renal Cell Carcinoma (MK-6482-018)—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04846920?term=belzutifan&draw=2&rank=2 (accessed on 29 May 2022).
- Choueiri, T.K.; Bauer, T.M.; McDermott, D.F.; Arrowsmith, E.; Roy, A.; Perini, R.F.; Vickery, D.; Tykodi, S.S. Phase 2 Study of the Oral Hypoxia-Inducible Factor 2α (HIF-2α) Inhibitor MK-6482 in Combination with Cabozantinib in Patients with Advanced Clear Cell Renal Cell Carcinoma (CcRCC). J. Clin. Oncol. 2021, 39, 272. [Google Scholar] [CrossRef]
- Renal Cell Carcinoma in von Hippel-Lindau Disease. Nephron Sparing Surgery. Available online: https://pubmed.ncbi.nlm.nih.gov/30403378/ (accessed on 24 June 2022).
- Metwalli, A.R.; Linehan, W.M. Nephron-Sparing Surgery for Multifocal and Hereditary Renal Tumors. Curr. Opin. Urol. 2014, 24, 466–473. [Google Scholar] [CrossRef]
- Bianchi, L.; Bianchi, F.M.; Chessa, F.; Barbaresi, U.; Casablanca, C.; Piazza, P.; Mottaran, A.; Droghetti, M.; Roveroni, C.; Balestrazzi, E.; et al. Percutaneous Tumor Ablation versus Partial Nephrectomy for Small Renal Mass: The Impact of Histologic Variant and Tumor Size. Minerva Urol. Nephrol. 2021, 73, 581–590. [Google Scholar] [CrossRef]
- Trevisani, F.; Ghidini, M.; Larcher, A.; Lampis, A.; Lote, H.; Manunta, P.; Alibrandi, M.T.S.; Zagato, L.; Citterio, L.; Dell’Antonio, G.; et al. MicroRNA 193b-3p as a Predictive Biomarker of Chronic Kidney Disease in Patients Undergoing Radical Nephrectomy for Renal Cell Carcinoma. Br. J. Cancer 2016, 115, 1343–1350. [Google Scholar] [CrossRef]
- Trevisani, F.; Floris, M.; Vago, R.; Minnei, R.; Cinque, A. Long Non-Coding RNAs as Novel Biomarkers in the Clinical Management of Papillary Renal Cell Carcinoma Patients: A Promise or a Pledge? Cells 2022, 11, 1658. [Google Scholar] [CrossRef]
- Cinque, A.; Capasso, A.; Vago, R.; Floris, M.; Lee, M.W.; Minnei, R.; Trevisani, F. MicroRNA Signatures in the Upper Urinary Tract Urothelial Carcinoma Scenario: Ready for the Game Changer? Int. J. Mol. Sci. 2022, 23, 2602. [Google Scholar] [CrossRef]
- Ghidini, M.; Hahne, J.C.; Frizziero, M.; Tomasello, G.; Trevisani, F.; Lampis, A.; Passalacqua, R.; Valeri, N. MicroRNAs as Mediators of Resistance Mechanisms to Small-Molecule Tyrosine Kinase Inhibitors in Solid Tumours. Target. Oncol. 2018, 13, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Trevisani, F.; Floris, M.; Minnei, R.; Cinque, A. Renal Oncocytoma: The Diagnostic Challenge to Unmask the Double of Renal Cancer. Int. J. Mol. Sci. 2022, 23, 2603. [Google Scholar] [CrossRef] [PubMed]
- Cinque, A.; Vago, R.; Trevisani, F. Circulating RNA in Kidney Cancer: What We Know and What We Still Suppose. Genes 2021, 12, 835. [Google Scholar] [CrossRef] [PubMed]
- Gattolliat, C.H.; Couvé, S.; Meurice, G.; Oréar, C.; Droin, N.; Chiquet, M.; Ferlicot, S.; Verkarre, V.; Vasiliu, V.; Molinié, V.; et al. Integrative Analysis of Dysregulated MicroRNAs and MRNAs in Multiple Recurrent Synchronized Renal Tumors from Patients with von Hippel-Lindau Disease. Int. J. Oncol. 2018, 53, 1455–1468. [Google Scholar] [CrossRef]
- Fisher, R.; Horswell, S.; Rowan, A.; Salm, M.P.; de Bruin, E.C.; Gulati, S.; McGranahan, N.; Stares, M.; Gerlinger, M.; Varela, I.; et al. Development of Synchronous VHL Syndrome Tumors Reveals Contingencies and Constraints to Tumor Evolution. Genome Biol. 2014, 15, 433. [Google Scholar] [CrossRef]
- Shi, L.; Wang, M.; Li, H.; You, P. MicroRNAs in Body Fluids: A More Promising Biomarker for Clear Cell Renal Cell Carcinoma. Cancer Manag. Res. 2021, 13, 7663–7675. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).