

Molecular Mechanisms of Anti-Estrogen Therapy Resistance and Novel Targeted Therapies

Abstract
Simple Summary
Abstract
1. Introduction
2. The Estrogen/ERα Axis Is the Major Oncogenic Signaling Source in ER+ Breast Cancer Cases
3. ERα Isoforms
4. Signaling Pathways Mediated by the Interaction of Estrogen with ERα and ERβ
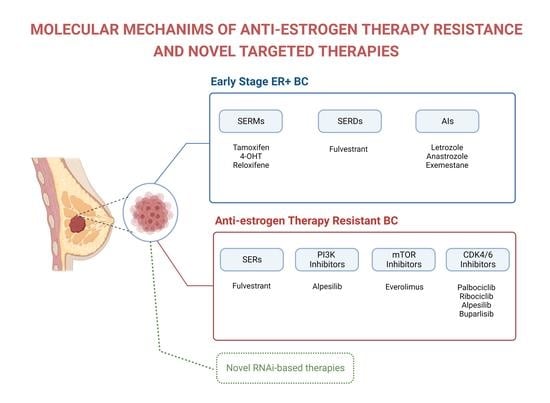
5. Anti-Estrogen Therapies
5.1. Selective ER Modulators (SERMs)
5.2. Selective ER Downregulators (SERDs)
5.3. Aromatase Inhibitors (AIs)
6. Mechanisms of Resistance to Anti-Estrogen Therapies
6.1. ERα Mutations
6.2. Loss of ERα Expression
6.3. ERα36 Overexpression
6.4. Loss of Progesterone Receptor (PR) Expression
6.5. Increased Tamoxifen Metabolism
6.6. Loss of the Cell-Cycle Regulator Retinoblastoma Protein
6.7. Increased Activity of Receptor Tyrosine Kinases (RTKs)
6.8. Breast Cancer Stem Cells (BCSCs)
6.9. Dysregulation of microRNA (miRNA) Expression
6.10. Dysregulation of Long Noncoding RNAs (LncRNAs)
7. Current and Potential Treatments to Improve Efficacy of Anti-Estrogen Treatments
7.1. SERDs
7.2. Targeting PI3K/AKT/mTOR Signaling
7.3. Targeting CDK4/CDK6
7.4. Combination of CDK4/6 and PI3K/AKT/mTOR Inhibitors
7.5. Novel RNA Interference-Based Therapies That Reverse Anti-Estrogen Therapy Resistance of ER+Breast Cancer
7.5.1. Therapeutic Targets That Potentially Reverse Anti-Estrogen Therapy Resistance
7.5.2. MicroRNA-Based Therapeutics That Potentially Reverse Anti-Estrogen Therapy Resistance
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Faldoni, F.L.C.; Rainho, C.A.; Rogatto, S.R. Epigenetics in Inflammatory Breast Cancer: Biological Features and Therapeutic Perspectives. Cells 2020, 9, 1164. [Google Scholar] [CrossRef] [PubMed]
- Effi, A.B.; Aman, N.A.; Koui, B.S.; Koffi, K.D.; Traoré, Z.C.; Kouyate, M. Immunohistochemical determination of estrogen and progesterone receptors in breast cancer: Relationship with clinicopathologic factors in 302 patients in Ivory Coast. BMC Cancer 2017, 17, 115. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Azad, K.A. Study of Receptor Status in Carcinoma Breast Patient. Chattagram Maa-O-Shishu Hosp. Med. Coll. J. 2017, 16, 48–50. [Google Scholar] [CrossRef]
- Ferreira Almeida, C.; Oliveira, A.; João Ramos, M.; Fernandes, P.A.; Teixeira, N.; Amaral, C. Estrogen receptor-positive (ER+) breast cancer treatment: Are multi-target compounds the next promising approach? Biochem. Pharmacol. 2020, 177, 113989. [Google Scholar] [CrossRef]
- Anderson, W.F.; Rosenberg, P.S.; Prat, A.; Perou, C.M.; Sherman, M.E. How Many Etiological Subtypes of Breast Cancer: Two, Three, Four, Or More? JNCI J. Natl. Cancer Inst. 2014, 106, dju165. [Google Scholar] [CrossRef] [PubMed]
- Kuran, D.; Pogorzelska, A.; Wiktorska, K. Breast Cancer Prevention-Is there a Future for Sulforaphane and Its Analogs? Nutrients 2020, 12, 1559. [Google Scholar] [CrossRef] [PubMed]
- Pearce, S.T.; Jordan, V.C. The biological role of estrogen receptors α and β in cancer. Crit. Rev. Oncol./Hematol. 2004, 50, 3–22. [Google Scholar] [CrossRef]
- Razandi, M.; Pedram, A.; Merchenthaler, I.; Greene, G.L.; Levin, E.R. Plasma Membrane Estrogen Receptors Exist and Functions as Dimers. Mol. Endocrinol. 2004, 18, 2854–2865. [Google Scholar] [CrossRef] [PubMed]
- Welsh, A.W.; Lannin, D.R.; Young, G.S.; Sherman, M.E.; Figueroa, J.D.; Henry, N.L.; Ryden, L.; Kim, C.; Love, R.R.; Schiff, R.; et al. Cytoplasmic estrogen receptor in breast cancer. Clin. Cancer Res. 2012, 18, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Enmark, E.; Pelto-Huikko, M.; Grandien, K.; Lagercrantz, S.; Lagercrantz, J.; Fried, G.; Nordenskjöld, M.; Gustafsson, J.A. Human estrogen receptor β-gene structure, chromosomal localization, and expression pattern. J. Clin. Endocrinol. Metab. 1997, 82, 4258–4265. [Google Scholar] [CrossRef] [PubMed]
- Rondón-Lagos, M.; Villegas, V.E.; Rangel, N.; Sánchez, M.C.; Zaphiropoulos, P.G. Tamoxifen Resistance: Emerging Molecular Targets. Int. J. Mol. Sci. 2016, 17, 1357. [Google Scholar] [CrossRef] [PubMed]
- Paterni, I.; Granchi, C.; Katzenellenbogen, J.A.; Minutolo, F. Estrogen receptors α (ERα) and β (ERβ): Subtype-selective ligands and clinical potential. Steroids 2014, 90, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Omoto, Y.; Iwase, H.; Yamashita, H.; Toyama, T.; Coombes, R.C.; Filipovic, A.; Warner, M.; Gustafsson, J. Differential expression of estrogen receptor α, β1, and β2 in lobular and ductal breast cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 1933–1938. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Butler, R.; Warner, M.; Gustafsson, J.; Wilczek, B.; Landgren, B.M. Effects of short-term estradiol and norethindrone acetate treatment on the breasts of normal postmenopausal women. Menopause 2013, 20, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Lattrich, C.; Juhasz-Boess, I.; Ortmann, O.; Treeck, O. Detection of an elevated HER2 expression in MCF-7 breast cancer cells overexpressing estrogen receptor β1. Oncol. Rep. 2008, 19, 811–817. [Google Scholar] [PubMed]
- Ruff, M.; Gangloff, M.; Wurtz, J.M.; Moras, D. Estrogen receptor transcription and transactivation: Structure-function relationship in DNA- and ligand-binding domains of estrogen receptors. Breast Cancer Res. BCR 2000, 2, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Chandra, V.; Rastinejad, F. Structural overview of the nuclear receptor superfamily: Insights into physiology and therapeutics. Annu. Rev. Physiol. 2010, 72, 247–272. [Google Scholar] [CrossRef] [PubMed]
- Augusto, T.V.; Correia-da-Silva, G.; Rodrigues, C.M.P.; Teixeira, N.; Amaral, C. Acquired resistance to aromatase inhibitors: Where we stand! Endocr.-Relat. Cancer 2018, 25, R283–R301. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, G.; Gallo, D.; Cossy, J.; Laïos, I.; Larsimont, D.; Laurent, G.; Jacquot, Y. Peptides targeting estrogen receptor α-potential applications for breast cancer treatment. Curr. Pharm. Des. 2011, 17, 2632–2653. [Google Scholar] [CrossRef]
- Yaşar, P.; Ayaz, G.; User, S.D.; Güpür, G.; Muyan, M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod. Med. Biol. 2016, 16, 4–20. [Google Scholar] [CrossRef]
- Ascenzi, P.; Bocedi, A.; Marino, M. Structure-function relationship of estrogen receptor α and β: Impact on human health. Mol. Asp. Med. 2006, 27, 299–402. [Google Scholar] [CrossRef] [PubMed]
- Thiebaut, C.; Konan, H.-P.; Guerquin, M.-J.; Chesnel, A.; Livera, G.; Le Romancer, M.; Dumond, H. The Role of ERα36 in Development and Tumor Malignancy. Int. J. Mol. Sci. 2020, 21, 4116. [Google Scholar] [CrossRef] [PubMed]
- Thornton, J.W. Evolution of vertebrate steroid receptors from an ancestral estrogen receptor by ligand exploitation and serial genome expansions. Proc. Natl. Acad. Sci. USA 2001, 98, 5671–5676. [Google Scholar] [CrossRef] [PubMed]
- Chantalat, E.; Boudou, F.; Laurell, H.; Palierne, G.; Houtman, R.; Melchers, D.; Rochaix, P.; Filleron, T.; Stella, A.; Burlet-Schiltz, O.; et al. The AF-1-deficient estrogen receptor ERα46 isoform is frequently expressed in human breast tumors. Breast Cancer Res. 2016, 18, 123. [Google Scholar] [CrossRef] [PubMed]
- Klinge, C.M.; Riggs, K.A.; Wickramasinghe, N.S.; Emberts, C.G.; McConda, D.B.; Barry, P.N.; Magnusen, J.E. Estrogen receptor α 46 is reduced in tamoxifen resistant breast cancer cells and re-expression inhibits cell proliferation and estrogen receptor α 66-regulated target gene transcription. Mol. Cell. Endocrinol. 2010, 323, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Jiang, J.; Ying, G.; Xie, X.Q.; Zhang, X.; Xu, W.; Zhang, X.; Song, E.; Bu, H.; Ping, Y.F.; et al. Tamoxifen enhances stemness and promotes metastasis of ERα36(+) breast cancer by upregulating ALDH1A1 in cancer cells. Cell Res. 2018, 28, 336–358. [Google Scholar] [CrossRef]
- Chamard-Jovenin, C.; Jung, A.C.; Chesnel, A.; Abecassis, J.; Flament, S.; Ledrappier, S.; Macabre, C.; Boukhobza, T.; Dumond, H. From ERα66 to ERα36: A generic method for validating a prognosis marker of breast tumor progression. BMC Syst. Biol. 2015, 9, 28. [Google Scholar] [CrossRef]
- Zhu, H.; Huang, Y.; Su, H.; Ma, Y.; Tao, Y.; Liao, D.J.; Liu, Y.; Feng, Z. Identification of a novel human estrogen receptor-α splice variant able to enhance malignant biological behaviors of breast cancer cells. Oncol. Lett. 2018, 15, 5339–5344. [Google Scholar] [CrossRef]
- Nardone, A.; De Angelis, C.; Trivedi, M.V.; Osborne, C.K.; Schiff, R. The changing role of ER in endocrine resistance. Breast 2015, 24 (Suppl. S2), S60–S66. [Google Scholar] [CrossRef]
- Brufsky, A.M.; Dickler, M.N. Estrogen Receptor-Positive Breast Cancer: Exploiting Signaling Pathways Implicated in Endocrine Resistance. Oncologist 2018, 23, 528–539. [Google Scholar] [CrossRef]
- Pietras, R.J.; Márquez-Garbán, D.C. Membrane-associated estrogen receptor signaling pathways in human cancers. Clin. Cancer Res. 2007, 13, 4672–4676. [Google Scholar] [CrossRef] [PubMed]
- Shupnik, M.A. Crosstalk between steroid receptors and the c-Src-receptor tyrosine kinase pathways: Implications for cell proliferation. Oncogene 2004, 23, 7979–7989. [Google Scholar] [CrossRef] [PubMed]
- Cheskis, B.J. Regulation of cell signalling cascades by steroid hormones. J. Cell. Biochem. 2004, 93, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Lumachi, F.; Santeufemia, D.A.; Basso, S.M. Current medical treatment of estrogen receptor-positive breast cancer. World J. Biol. Chem. 2015, 6, 231–239. [Google Scholar] [CrossRef]
- Patel, H.K.; Bihani, T. Selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs) in cancer treatment. Pharmacol. Ther. 2018, 186, 1–24. [Google Scholar] [CrossRef]
- Feng, Q.; O’Malley, B.W. Nuclear receptor modulation—Role of coregulators in selective estrogen receptor modulator (SERM) actions. Steroids 2014, 90, 39–43. [Google Scholar] [CrossRef]
- Pinkerton, J.V.; Conner, E.A. Beyond estrogen: Advances in tissue selective estrogen complexes and selective estrogen receptor modulators. Climacteric J. Int. Menopause Soc. 2019, 22, 140–147. [Google Scholar] [CrossRef]
- Gasco, M.; Argusti, A.; Bonanni, B.; Decensi, A. SERMs in chemoprevention of breast cancer. Eur. J. Cancer 2005, 41, 1980–1989. [Google Scholar] [CrossRef]
- Brauch, H.; Murdter, T.E.; Eichelbaum, M.; Schwab, M.J. Pharmacogenomics of tamoxifen therapy. Clin. Chem. 2009, 55, 1770–1782. [Google Scholar] [CrossRef]
- Kiyotani, K.; Mushiroda, T.; Nakamura, Y.; Zembutsu, H. Pharmacogenomics of tamoxifen: Roles of drug metabolizing enzymes and transporters. Drug Metab. Pharmacokinet. 2012, 27, 122–131. [Google Scholar] [CrossRef]
- Fisher, B.; Costantino, J.P.; Wickerham, D.L.; Redmond, C.K.; Kavanah, M.; Cronin, W.M.; Vogel, V.; Robidoux, A.; Dimitrov, N.; Atkins, J.; et al. Tamoxifen for prevention of breast cancer: Report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J. Natl. Cancer Inst. 1998, 90, 1371–1388. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V.C.; Murphy, C.S. Endocrine pharmacology of antiestrogens as antitumor agents. Endocr. Rev. 1990, 11, 578–610. [Google Scholar] [CrossRef] [PubMed]
- Viedma-Rodríguez, R.; Baiza-Gutman, L.; Salamanca-Gómez, F.; Diaz-Zaragoza, M.; Martínez-Hernández, G.; Ruiz Esparza-Garrido, R.; Velázquez-Flores, M.A.; Arenas-Aranda, D. Mechanisms associated with resistance to tamoxifen in estrogen receptor-positive breast cancer (Review). Oncol. Rep. 2014, 32, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Serrano, D.; Lazzeroni, M.; Zambon, C.F.; Macis, D.; Maisonneuve, P.; Johansson, H.; Guerrieri-Gonzaga, A.; Plebani, M.; Basso, D.; Gjerde, J.; et al. Efficacy of tamoxifen based on cytochrome P450 2D6, CYP2C19 and SULT1A1 genotype in the Italian Tamoxifen Prevention Trial. Pharm. J. 2011, 11, 100–107. [Google Scholar] [CrossRef]
- Goetz, M.P.; Suman, V.J.; Reid, J.M.; Northfelt, D.W.; Mahr, M.A.; Ralya, A.T.; Kuffel, M.; Buhrow, S.A.; Safgren, S.L.; McGovern, R.M.; et al. First-in-Human Phase I Study of the Tamoxifen Metabolite Z-Endoxifen in Women with Endocrine-Refractory Metastatic Breast Cancer. J. Clin. Oncol. 2017, 35, 3391–3400. [Google Scholar] [CrossRef]
- Gennari, L.; Merlotti, D.; Paola, V.D.; Nuti, R. Raloxifene in breast cancer prevention. Expert Opin. Drug Saf. 2008, 7, 259–270. [Google Scholar] [CrossRef]
- Thompson, E.W.; Reich, R.; Shima, T.B.; Albini, A.; Graf, J.; Martin, G.R.; Dickson, R.B.; Lippman, M.E. Differential regulation of growth and invasiveness of MCF-7 breast cancer cells by antiestrogens. Cancer Res. 1988, 48, 6764–6768. [Google Scholar]
- Freiss, G.; Galtier, F.; Puech, C.; Aknin, C.; Maudelonde, T.; Chalbos, D.; Vignon, F. Anti-growth factor activities of benzothiophenes in human breast cancer cells. J. Steroid Biochem. Mol. Biol. 2005, 94, 451–460. [Google Scholar] [CrossRef]
- Saha, T.; Makar, S.; Swetha, R.; Gutti, G.; Singh, S.K. Estrogen signaling: An emanating therapeutic target for breast cancer treatment. Eur. J. Med. Chem. 2019, 177, 116–143. [Google Scholar] [CrossRef]
- Baumann, C.K.; Castiglione-Gertsch, M. Estrogen Receptor Modulators and Down Regulators. Drugs 2007, 67, 2335–2353. [Google Scholar] [CrossRef]
- Kieser, K.J.; Kim, D.W.; Carlson, K.E.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A. Characterization of the pharmacophore properties of novel selective estrogen receptor downregulators (SERDs). J. Med. Chem. 2010, 53, 3320–3329. [Google Scholar] [CrossRef] [PubMed]
- Dauvois, S.; Danielian, P.S.; White, R.; Parker, M.G. Antiestrogen ICI 164,384 reduces cellular estrogen receptor content by increasing its turnover. Proc. Natl. Acad. Sci. USA 1992, 89, 4037–4041. [Google Scholar] [CrossRef] [PubMed]
- Vergote, I.; Abram, P. Fulvestrant, a new treatment option for advanced breast cancer: Tolerability versus existing agents. Ann. Oncol. 2006, 17, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Tyson, J.J.; Dixon, J.M. Endocrine resistance in breast cancer—An overview and update. Mol. Cell. Endocrinol. 2015, 418 Pt 3, 220–234. [Google Scholar] [CrossRef]
- Bergamino, M.A.; López-Knowles, E.; Morani, G.; Tovey, H.; Kilburn, L.; Schuster, E.F.; Alataki, A.; Hills, M.; Xiao, H.; Holcombe, C.; et al. HER2-enriched subtype and novel molecular subgroups drive aromatase inhibitor resistance and an increased risk of relapse in early ER+/HER2+ breast cancer. EBioMedicine 2022, 83, 104205. [Google Scholar] [CrossRef] [PubMed]
- Cuzick, J.; Sestak, I.; Baum, M.; Buzdar, A.; Howell, A.; Dowsett, M.; Forbes, J.F. Effect of anastrozole and tamoxifen as adjuvant treatment for early-stage breast cancer: 10-year analysis of the ATAC trial. Lancet. Oncol. 2010, 11, 1135–1141. [Google Scholar] [CrossRef]
- Dowsett, M.; Cuzick, J.; Ingle, J.; Coates, A.; Forbes, J.; Bliss, J.; Buyse, M.; Baum, M.; Buzdar, A.; Colleoni, M.; et al. Meta-analysis of breast cancer outcomes in adjuvant trials of aromatase inhibitors versus tamoxifen. J. Clin. Oncol. 2010, 28, 509–518. [Google Scholar] [CrossRef]
- Rani, A.; Stebbing, J.; Giamas, G.; Murphy, J. Endocrine Resistance in Hormone Receptor Positive Breast Cancer-From Mechanism to Therapy. Front. Endocrinol. 2019, 10, 245. [Google Scholar] [CrossRef]
- Szostakowska, M.; Trębińska-Stryjewska, A.; Grzybowska, E.A.; Fabisiewicz, A. Resistance to endocrine therapy in breast cancer: Molecular mechanisms and future goals. Breast Cancer Res. Treat. 2019, 173, 489–497. [Google Scholar] [CrossRef]
- Li, S.; Shen, D.; Shao, J.; Crowder, R.; Liu, W.; Prat, A.; He, X.; Liu, S.; Hoog, J.; Lu, C.; et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013, 4, 1116–1130. [Google Scholar] [CrossRef]
- Jeselsohn, R.; Yelensky, R.; Buchwalter, G.; Frampton, G.; Meric-Bernstam, F.; Gonzalez-Angulo, A.M.; Ferrer-Lozano, J.; Perez-Fidalgo, J.A.; Cristofanilli, M.; Gómez, H.; et al. Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin. Cancer Res. 2014, 20, 1757–1767. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Wu, Y.M.; Vats, P.; Su, F.; Lonigro, R.J.; Cao, X.; Kalyana-Sundaram, S.; Wang, R.; Ning, Y.; Hodges, L.; et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 2013, 45, 1446–1451. [Google Scholar] [CrossRef]
- Jeselsohn, R.; Buchwalter, G.; De Angelis, C.; Brown, M.; Schiff, R. ESR1 mutations—A mechanism for acquired endocrine resistance in breast cancer. Nat. Rev. Clin. Oncol. 2015, 12, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Toy, W.; Shen, Y.; Won, H.; Green, B.; Sakr, R.A.; Will, M.; Li, Z.; Gala, K.; Fanning, S.; King, T.A.; et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 2013, 45, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Yudt, M.R.; Vorojeikina, D.; Zhong, L.; Skafar, D.F.; Sasson, S.; Gasiewicz, T.A.; Notides, A.C. Function of estrogen receptor tyrosine 537 in hormone binding, DNA binding, and transactivation. Biochemistry 1999, 38, 14146–14156. [Google Scholar] [CrossRef] [PubMed]
- Harrod, A.; Fulton, J.; Nguyen, V.T.M.; Periyasamy, M.; Ramos-Garcia, L.; Lai, C.F.; Metodieva, G.; de Giorgio, A.; Williams, R.L.; Santos, D.B.; et al. Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer. Oncogene 2017, 36, 2286–2296. [Google Scholar] [CrossRef] [PubMed]
- Fanning, S.W.; Mayne, C.G.; Dharmarajan, V.; Carlson, K.E.; Martin, T.A.; Novick, S.J.; Toy, W.; Green, B.; Panchamukhi, S.; Katzenellenbogen, B.S.; et al. Estrogen receptor α somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. eLife 2016, 5, e12792. [Google Scholar] [CrossRef] [PubMed]
- Encarnación, C.A.; Ciocca, D.R.; McGuire, W.L.; Clark, G.M.; Fuqua, S.A.; Osborne, C.K. Measurement of steroid hormone receptors in breast cancer patients on tamoxifen. Breast Cancer Res. Treat. 1993, 26, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.R.; Saccani-Jotti, G.; Smith, I.E.; Salter, J.; Newby, J.; Coppen, M.; Ebbs, S.R.; Dowsett, M. Changes in estrogen receptor, progesterone receptor, and pS2 expression in tamoxifen-resistant human breast cancer. Cancer Res. 1995, 55, 3331–3338. [Google Scholar] [PubMed]
- Liang, Y.K.; Zeng, D.; Xiao, Y.S.; Wu, Y.; Ouyang, Y.X.; Chen, M.; Li, Y.C.; Lin, H.Y.; Wei, X.L.; Zhang, Y.Q.; et al. MCAM/CD146 promotes tamoxifen resistance in breast cancer cells through induction of epithelial-mesenchymal transition, decreased ERα expression and AKT activation. Cancer Lett. 2017, 386, 65–76. [Google Scholar] [CrossRef]
- De Kruijff, I.E.; Timmermans, A.M.; den Bakker, M.A.; Trapman-Jansen, A.; Foekens, R.; Meijer-Van Gelder, M.E.; Oomen-de Hoop, E.; Smid, M.; Hollestelle, A.; van Deurzen, C.H.M.; et al. The Prevalence of CD146 Expression in Breast Cancer Subtypes and Its Relation to Outcome. Cancers 2018, 10, 134. [Google Scholar] [CrossRef]
- Yu, S.; Gong, X.; Ma, Z.; Zhang, M.; Huang, L.; Zhang, J.; Zhao, S.; Zhu, T.; Yu, Z.; Chen, L. Endocrine resistant breast cancer cells with loss of ERα expression retain proliferative ability by reducing caspase7-mediated HDAC3 cleavage. Cell. Oncol. 2020, 43, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, Z.; Hao, Q.; Li, W.; Xu, Y.; Zhang, J.; Zhang, W.; Wang, S.; Liu, S.; Li, M.; et al. Loss of ERα induces amoeboid-like migration of breast cancer cells by downregulating vinculin. Nat. Commun. 2017, 8, 14483. [Google Scholar] [CrossRef]
- Pagano, M.T.; Ortona, E.; Dupuis, M.L. A Role for Estrogen Receptor α36 in Cancer Progression. Front. Endocrinol. 2020, 11, 506. [Google Scholar] [CrossRef] [PubMed]
- Blows, F.M.; Driver, K.E.; Schmidt, M.K.; Broeks, A.; van Leeuwen, F.E.; Wesseling, J.; Cheang, M.C.; Gelmon, K.; Nielsen, T.O.; Blomqvist, C.; et al. Subtyping of breast cancer by immunohistochemistry to investigate a relationship between subtype and short and long term survival: A collaborative analysis of data for 10,159 cases from 12 studies. PLoS Med. 2010, 7, e1000279. [Google Scholar] [CrossRef] [PubMed]
- Thike, A.A.; Chng, M.J.; Tan, P.H.; Fook-Chong, S. Immunohistochemical expression of hormone receptors in invasive breast carcinoma: Correlation of results of H-score with pathological parameters. Pathology 2001, 33, 21–25. [Google Scholar] [CrossRef]
- Cui, X.; Schiff, R.; Arpino, G.; Osborne, C.K.; Lee, A.V. Biology of progesterone receptor loss in breast cancer and its implications for endocrine therapy. J. Clin. Oncol. 2005, 23, 7721–7735. [Google Scholar] [CrossRef] [PubMed]
- Skinner, L.; Barnes, D.M.; Ribeiro, G. The clinical value of multiple steroid receptor assays in breast cancer management. Cancer 1980, 46, 2939–2945. [Google Scholar] [CrossRef]
- Tahiri, A.; Tekpli, X.; Satheesh, S.V.; DeWijn, R.; Lüders, T.; Bukholm, I.R.; Hurtado, A.; Geisler, J.; Kristensen, V.N. Loss of progesterone receptor is associated with distinct tyrosine kinase profiles in breast cancer. Breast Cancer Res. Treat. 2020, 183, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, H.; Russell, I.A.; Stark, R.; Rueda, O.M.; Hickey, T.E.; Tarulli, G.A.; Serandour, A.A.; Birrell, S.N.; Bruna, A.; Saadi, A.; et al. Progesterone receptor modulates ERα action in breast cancer. Nature 2015, 523, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Petruolo, O.A.; Pilewskie, M.; Patil, S.; Barrio, A.V.; Stempel, M.; Wen, H.Y.; Morrow, M. Standard Pathologic Features Can Be Used to Identify a Subset of Estrogen Receptor-Positive, HER2 Negative Patients Likely to Benefit from Neoadjuvant Chemotherapy. Ann. Surg. Oncol. 2017, 24, 2556–2562. [Google Scholar] [CrossRef] [PubMed]
- Van Mackelenbergh, M.T.; Denkert, C.; Nekljudova, V.; Karn, T.; Schem, C.; Marmé, F.; Stickeler, E.; Jackisch, C.; Hanusch, C.; Huober, J.; et al. Outcome after neoadjuvant chemotherapy in estrogen receptor-positive and progesterone receptor-negative breast cancer patients: A pooled analysis of individual patient data from ten prospectively randomized controlled neoadjuvant trials. Breast Cancer Res. Treat. 2018, 167, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Kunc, M.; Popęda, M.; Biernat, W.; Senkus, E. Lost but Not Least-Novel Insights into Progesterone Receptor Loss in Estrogen Receptor-Positive Breast Cancer. Cancers 2021, 13, 4755. [Google Scholar] [CrossRef] [PubMed]
- Stearns, V.; Johnson, M.D.; Rae, J.M.; Morocho, A.; Novielli, A.; Bhargava, P.; Hayes, D.F.; Desta, Z.; Flockhart, D.A. Active tamoxifen metabolite plasma concentrations after coadministration of tamoxifen and the selective serotonin reuptake inhibitor paroxetine. J. Natl. Cancer Inst. 2003, 95, 1758–1764. [Google Scholar] [CrossRef] [PubMed]
- Machuca, T.N.; Hsin, M.K.; Ott, H.C.; Chen, M.; Hwang, D.M.; Cypel, M.; Waddell, T.K.; Keshavjee, S. Injury-specific ex vivo treatment of the donor lung: Pulmonary thrombolysis followed by successful lung transplantation. Am. J. Respir. Crit. Care Med. 2013, 188, 878–880. [Google Scholar] [CrossRef]
- Boocock, D.J.; Brown, K.; Gibbs, A.H.; Sanchez, E.; Turteltaub, K.W.; White, I.N. Identification of human CYP forms involved in the activation of tamoxifen and irreversible binding to DNA. Carcinogenesis 2002, 23, 1897–1902. [Google Scholar] [CrossRef]
- Justenhoven, C.; Hamann, U.; Pierl, C.B.; Baisch, C.; Harth, V.; Rabstein, S.; Spickenheuer, A.; Pesch, B.; Brüning, T.; Winter, S.; et al. CYP2C19* 17 is associated with decreased breast cancer risk. Breast Cancer Res. Treat. 2009, 115, 391–396. [Google Scholar] [CrossRef]
- Schroth, W.; Antoniadou, L.; Fritz, P.; Schwab, M.; Muerdter, T.; Zanger, U.M.; Simon, W.; Eichelbaum, M.; Brauch, H. Breast cancer treatment outcome with adjuvant tamoxifen relative to patient CYP2D6 and CYP2C19 genotypes. J. Clin. Oncol. 2007, 25, 5187–5193. [Google Scholar] [CrossRef]
- Wu, X.; Hawse, J.R.; Subramaniam, M.; Goetz, M.P.; Ingle, J.N.; Spelsberg, T.C. The tamoxifen metabolite, endoxifen, is a potent antiestrogen that targets estrogen receptor α for degradation in breast cancer cells. Cancer Res. 2009, 69, 1722–1727. [Google Scholar] [CrossRef]
- Borges, S.; Desta, Z.; Li, L.; Skaar, T.C.; Ward, B.A.; Nguyen, A.; Jin, Y.; Storniolo, A.M.; Nikoloff, D.M.; Wu, L.; et al. Quantitative effect of CYP2D6 genotype and inhibitors on tamoxifen metabolism: Implication for optimization of breast cancer treatment. Clin. Pharmacol. Ther. 2006, 80, 61–74. [Google Scholar] [CrossRef]
- Gjerde, J.; Hauglid, M.; Breilid, H.; Lundgren, S.; Varhaug, J.; Kisanga, E.; Mellgren, G.; Steen, V.; Lien, E.A. Effects of CYP2D6 and SULT1A1 genotypes including SULT1A1 gene copy number on tamoxifen metabolism. Ann. Oncol. 2008, 19, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Goetz, M.P.; Rae, J.M.; Suman, V.J.; Safgren, S.L.; Ames, M.M.; Visscher, D.W.; Reynolds, C.; Couch, F.J.; Lingle, W.L.; Flockhart, D.A. Pharmacogenetics of tamoxifen biotransformation is associated with clinical outcomes of efficacy and hot flashes. J. Clin. Oncol. 2005, 23, 9312–9318. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.A.; Yee, D. Killing the second messenger: Targeting loss of cell cycle control in endocrine-resistant breast cancer. Endocr.-Relat. Cancer 2011, 18, C19–C24. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.; Thijssen, B.; McDermott, U.; Garnett, M.; Wessels, L.F.; Bernards, R. Targeting the RB-E2F pathway in breast cancer. Oncogene 2016, 35, 4829–4835. [Google Scholar] [CrossRef]
- Finn, R.S.; Dering, J.; Conklin, D.; Kalous, O.; Cohen, D.J.; Desai, A.J.; Ginther, C.; Atefi, M.; Chen, I.; Fowst, C.; et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009, 11, R77. [Google Scholar] [CrossRef]
- Bollard, J.; Miguela, V.; Ruiz de Galarreta, M.; Venkatesh, A.; Bian, C.B.; Roberto, M.P.; Tovar, V.; Sia, D.; Molina-Sánchez, P.; Nguyen, C.B.; et al. Palbociclib (PD-0332991), a selective CDK4/6 inhibitor, restricts tumour growth in preclinical models of hepatocellular carcinoma. Gut 2017, 66, 1286–1296. [Google Scholar] [CrossRef]
- Gopalan, P.K.; Villegas, A.G.; Cao, C.; Pinder-Schenck, M.; Chiappori, A.; Hou, W.; Zajac-Kaye, M.; Ivey, A.M.; Kaye, F.J. CDK4/6 inhibition stabilizes disease in patients with p16-null non-small cell lung cancer and is synergistic with mTOR inhibition. Oncotarget 2018, 9, 37352–37366. [Google Scholar] [CrossRef]
- Wander, S.A.; Cohen, O.; Gong, X.; Johnson, G.N.; Buendia-Buendia, J.E.; Lloyd, M.R.; Kim, D.; Luo, F.; Mao, P.; Helvie, K.; et al. The Genomic Landscape of Intrinsic and Acquired Resistance to Cyclin-Dependent Kinase 4/6 Inhibitors in Patients with Hormone Receptor-Positive Metastatic Breast Cancer. Cancer Discov. 2020, 10, 1174–1193. [Google Scholar] [CrossRef]
- Wei, J.; Sun, H.; Zhang, A.; Wu, X.; Li, Y.; Liu, J.; Duan, Y.; Xiao, F.; Wang, H.; Lv, M.; et al. A novel AXL chimeric antigen receptor endows T cells with anti-tumor effects against triple negative breast cancers. Cell. Immunol. 2018, 331, 49–58. [Google Scholar] [CrossRef]
- Roberts, P.J.; Bisi, J.E.; Strum, J.C.; Combest, A.J.; Darr, D.B.; Usary, J.E.; Zamboni, W.C.; Wong, K.-K.; Perou, C.M.; Sharpless, N.E. Multiple roles of cyclin-dependent kinase 4/6 inhibitors in cancer therapy. J. Natl. Cancer Inst. 2012, 104, 476–487. [Google Scholar] [CrossRef]
- Templeton, A.J.; Diez-Gonzalez, L.; Ace, O.; Vera-Badillo, F.; Šeruga, B.; Jordán, J.; Amir, E.; Pandiella, A.; Ocaña, A. Prognostic relevance of receptor tyrosine kinase expression in breast cancer: A meta-analysis. Cancer Treat. Rev. 2014, 40, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, E.A.; Sutherland, R.L. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer 2009, 9, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, S.; Joh, J.; Remick, S.C.; Miller, D.M.; Yan, J.; Kanaan, Z.; Chao, J.-H.; Krem, M.M.; Basu, S.K. MLLT11/AF1q boosts oncogenic STAT3 activity through Src-PDGFR tyrosine kinase signaling. Oncotarget 2016, 7, 43960–43973. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.-Z.; Zhang, H.; Li, J.; He, T.; Yeo, E.-J.; Soong, D.Y.; Carragher, N.O.; Munro, A.; Chang, A.; Bresnick, A.R.; et al. FLT1 signaling in metastasis-associated macrophages activates an inflammatory signature that promotes breast cancer metastasis. J. Exp. Med. 2015, 212, 1433–1448. [Google Scholar] [CrossRef] [PubMed]
- Butti, R.; Das, S.; Gunasekaran, V.P.; Yadav, A.S.; Kumar, D.; Kundu, G.C. Receptor tyrosine kinases (RTKs) in breast cancer: Signaling, therapeutic implications and challenges. Mol. Cancer 2018, 17, 34. [Google Scholar] [CrossRef]
- Ibrahim, S.A.; Gadalla, R.; El-Ghonaimy, E.A.; Samir, O.; Mohamed, H.T.; Hassan, H.; Greve, B.; El-Shinawi, M.; Mohamed, M.M.; Götte, M. Syndecan-1 is a novel molecular marker for triple negative inflammatory breast cancer and modulates the cancer stem cell phenotype via the IL-6/STAT3, Notch and EGFR signaling pathways. Mol. Cancer 2017, 16, 57. [Google Scholar] [CrossRef]
- Ring, A.; Dowsett, M. Mechanisms of tamoxifen resistance. Endocr.-Relat. Cancer 2004, 11, 643–658. [Google Scholar] [CrossRef]
- Schiff, R.; Massarweh, S.A.; Shou, J.; Bharwani, L.; Mohsin, S.K.; Osborne, C.K. Cross-talk between estrogen receptor and growth factor pathways as a molecular target for overcoming endocrine resistance. Clin. Cancer Res. 2004, 10, 331s–336s. [Google Scholar] [CrossRef]
- García-Becerra, R.; Santos, N.; Díaz, L.; Camacho, J. Mechanisms of resistance to endocrine therapy in breast cancer: Focus on signaling pathways, miRNAs and genetically based resistance. Int. J. Mol. Sci. 2013, 14, 108–145. [Google Scholar] [CrossRef]
- O’Brien, C.S.; Howell, S.J.; Farnie, G.; Clarke, R.B. Resistance to endocrine therapy: Are breast cancer stem cells the culprits? J. Mammary Gland. Biol. Neoplasia 2009, 14, 45–54. [Google Scholar] [CrossRef]
- Hiscox, S.; Baruah, B.; Smith, C.; Bellerby, R.; Goddard, L.; Jordan, N.; Poghosyan, Z.; Nicholson, R.I.; Barrett-Lee, P.; Gee, J. Overexpression of CD44 accompanies acquired tamoxifen resistance in MCF7 cells and augments their sensitivity to the stromal factors, heregulin and hyaluronan. BMC Cancer 2012, 12, 458. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.; Jordan, V.C. New insights into acquired endocrine resistance of breast cancer. Cancer Drug Resist. 2019, 2, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Campbell, R.A.; Bhat-Nakshatri, P.; Patel, N.M.; Constantinidou, D.; Ali, S.; Nakshatri, H. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor α: A new model for anti-estrogen resistance. J. Biol. Chem. 2001, 276, 9817–9824. [Google Scholar] [CrossRef] [PubMed]
- Yamnik, R.L.; Digilova, A.; Davis, D.C.; Brodt, Z.N.; Murphy, C.J.; Holz, M.K. S6 kinase 1 regulates estrogen receptor α in control of breast cancer cell proliferation. J. Biol. Chem. 2009, 284, 6361–6369. [Google Scholar] [CrossRef]
- Logan, S.K.; Falasca, M.; Hu, P.; Schlessinger, J. Phosphatidylinositol 3-kinase mediates epidermal growth factor-induced activation of the c-Jun N-terminal kinase signaling pathway. Mol. Cell. Biol. 1997, 17, 5784–5790. [Google Scholar] [CrossRef][Green Version]
- Dérijard, B.; Hibi, M.; Wu, I.H.; Barrett, T.; Su, B.; Deng, T.; Karin, M.; Davis, R.J. JNK1: A protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 1994, 76, 1025–1037. [Google Scholar] [CrossRef]
- Rauscher, F.J.; Cohen, D.R.; Curran, T.; Bos, T.J.; Vogt, P.K.; Bohmann, D.; Tjian, R.; Franza, B.R., Jr. Fos-associated protein p39 is the product of the jun proto-oncogene. Science 1988, 240, 1010–1016. [Google Scholar] [CrossRef]
- Miller, T.W.; Balko, J.M.; Arteaga, C.L. Phosphatidylinositol 3-kinase and antiestrogen resistance in breast cancer. J. Clin. Oncol. 2011, 29, 4452–4461. [Google Scholar] [CrossRef]
- Tomlinson, D.; Knowles, M.; Speirs, V. Mechanisms of FGFR3 actions in endocrine resistant breast cancer. Int. J. Cancer 2012, 130, 2857–2866. [Google Scholar] [CrossRef]
- Zhou, Y.; Wu, C.; Lu, G.; Hu, Z.; Chen, Q.; Du, X. FGF/FGFR signaling pathway involved resistance in various cancer types. J. Cancer 2020, 11, 2000–2007. [Google Scholar] [CrossRef]
- Wang, X.; Wang, G.; Zhao, Y.; Liu, X.; Ding, Q.; Shi, J.; Ding, Y.; Wang, S. STAT3 mediates resistance of CD44+ CD24−/low breast cancer stem cells to tamoxifen in vitro. J. Biomed. Res. 2012, 26, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Dubrovska, A.; Hartung, A.; Bouchez, L.; Walker, J.; Reddy, V.; Cho, C.; Schultz, P. CXCR4 activation maintains a stem cell population in tamoxifen-resistant breast cancer cells through AhR signalling. Br. J. Cancer 2012, 107, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Kordon, E.C.; Smith, G.H. An entire functional mammary gland may comprise the progeny from a single cell. Development 1998, 125, 1921–1930. [Google Scholar] [CrossRef] [PubMed]
- Van Keymeulen, A.; Rocha, A.S.; Ousset, M.; Beck, B.; Bouvencourt, G.; Rock, J.; Sharma, N.; Dekoninck, S.; Blanpain, C. Distinct stem cells contribute to mammary gland development and maintenance. Nature 2011, 479, 189–193. [Google Scholar] [CrossRef]
- Shackleton, M.; Vaillant, F.; Simpson, K.J.; Stingl, J.; Smyth, G.K.; Asselin-Labat, M.-L.; Wu, L.; Lindeman, G.J.; Visvader, J.E. Generation of a functional mammary gland from a single stem cell. Nature 2006, 439, 84–88. [Google Scholar] [CrossRef]
- Fu, N.Y.; Rios, A.C.; Pal, B.; Law, C.W.; Jamieson, P.; Liu, R.; Vaillant, F.; Jackling, F.; Liu, K.H.; Smyth, G.K. Identification of quiescent and spatially restricted mammary stem cells that are hormone responsive. Nat. Cell Biol. 2017, 19, 164–176. [Google Scholar] [CrossRef]
- Cai, S.; Kalisky, T.; Sahoo, D.; Dalerba, P.; Feng, W.; Lin, Y.; Qian, D.; Kong, A.; Yu, J.; Wang, F.; et al. A quiescent Bcl11b high stem cell population is required for maintenance of the mammary gland. Cell Stem Cell 2017, 20, 247.e5–260.e5. [Google Scholar] [CrossRef]
- Eirew, P.; Stingl, J.; Raouf, A.; Turashvili, G.; Aparicio, S.; Emerman, J.T.; Eaves, C.J. A method for quantifying normal human mammary epithelial stem cells with in vivo regenerative ability. Nat. Med. 2008, 14, 1384–1389. [Google Scholar] [CrossRef]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Gomez-Miragaya, J.; González-Suárez, E. Tumor-initiating CD49f cells are a hallmark of chemoresistant triple negative breast cancer. Mol. Cell. Oncol. 2017, 4, e1338208. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sansone, P.; Ceccarelli, C.; Berishaj, M.; Chang, Q.; Rajasekhar, V.K.; Perna, F.; Bowman, R.L.; Vidone, M.; Daly, L.; Nnoli, J.; et al. Self-renewal of CD133hi cells by IL6/Notch3 signalling regulates endocrine resistance in metastatic breast cancer. Nat. Commun. 2016, 7, 10442. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Sun, B.; Zhao, X.; Zhao, X.; Sun, T.; Gu, Q.; Yao, Z.; Dong, X.; Zhao, N.; Liu, N. CD133+ cells with cancer stem cell characteristics associates with vasculogenic mimicry in triple-negative breast cancer. Oncogene 2013, 32, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Ricardo, S.; Vieira, A.F.; Gerhard, R.; Leitão, D.; Pinto, R.; Cameselle-Teijeiro, J.F.; Milanezi, F.; Schmitt, F.; Paredes, J. Breast cancer stem cell markers CD44, CD24 and ALDH1: Expression distribution within intrinsic molecular subtype. J. Clin. Pathol. 2011, 64, 937–946. [Google Scholar] [CrossRef]
- Miyoshi, Y.; Shien, T.; Ogiya, A.; Ishida, N.; Yamazaki, K.; Horii, R.; Horimoto, Y.; Masuda, N.; Yasojima, H.; Inao, T.; et al. Differences in expression of the cancer stem cell marker aldehyde dehydrogenase 1 among estrogen receptor-positive/human epidermal growth factor receptor type 2-negative breast cancer cases with early, late, and no recurrence. Breast Cancer Res. 2016, 18, 73. [Google Scholar] [CrossRef]
- Piva, M.; Domenici, G.; Iriondo, O.; Rábano, M.; Simoes, B.M.; Comaills, V.; Barredo, I.; López-Ruiz, J.A.; Zabalza, I.; Kypta, R.; et al. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol. Med. 2014, 6, 66–79. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, H.-W.; Sun, X.-F.; Guo, X.-H.; He, Y.-N.; Cui, S.-D.; Fan, Q.-X. Tamoxifen-resistant breast cancer cells possess cancer stem-like cell properties. Chin. Med. J. 2013, 126, 3030–3034. [Google Scholar]
- Raffo, D.; Berardi, D.E.; Pontiggia, O.; Todaro, L.; de Kier Joffé, E.B.; Simian, M. Tamoxifen selects for breast cancer cells with mammosphere forming capacity and increased growth rate. Breast Cancer Res. Treat. 2013, 142, 537–548. [Google Scholar] [CrossRef]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef]
- Simões, B.M.; O’Brien, C.S.; Eyre, R.; Silva, A.; Yu, L.; Sarmiento-Castro, A.; Alférez, D.G.; Spence, K.; Santiago-Gómez, A.; Chemi, F.; et al. Anti-estrogen Resistance in Human Breast Tumors Is Driven by JAG1-NOTCH4-Dependent Cancer Stem Cell Activity. Cell Rep. 2015, 12, 1968–1977. [Google Scholar] [CrossRef]
- Ojo, D.; Lin, X.; Wu, Y.; Cockburn, J.; Bane, A.; Tang, D. Polycomb complex protein BMI1 confers resistance to tamoxifen in estrogen receptor positive breast cancer. Cancer Lett. 2018, 426, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Haughian, J.M.; Pinto, M.P.; Harrell, J.C.; Bliesner, B.S.; Joensuu, K.M.; Dye, W.W.; Sartorius, C.A.; Tan, A.C.; Heikkilä, P.; Perou, C.M.; et al. Maintenance of hormone responsiveness in luminal breast cancers by suppression of Notch. Proc. Natl. Acad. Sci. USA 2012, 109, 2742–2747. [Google Scholar] [CrossRef] [PubMed]
- Peiffer, D.S.; Wyatt, D.; Zlobin, A.; Piracha, A.; Ng, J.; Dingwall, A.K.; Albain, K.S.; Osipo, C. DAXX suppresses tumor-initiating cells in estrogen receptor–positive breast cancer following endocrine therapy. Cancer Res. 2019, 79, 4965–4977. [Google Scholar] [CrossRef]
- Smith, D.C.; Chugh, R.; Patnaik, A.; Papadopoulos, K.P.; Wang, M.; Kapoun, A.M.; Xu, L.; Dupont, J.; Stagg, R.J.; Tolcher, A. A phase 1 dose escalation and expansion study of Tarextumab (OMP-59R5) in patients with solid tumors. Investig. New Drugs 2019, 37, 722–730. [Google Scholar] [CrossRef]
- Gelsomino, L.; Panza, S.; Giordano, C.; Barone, I.; Gu, G.; Spina, E.; Catalano, S.; Fuqua, S.; Andò, S. Mutations in the estrogen receptor α hormone binding domain promote stem cell phenotype through notch activation in breast cancer cell lines. Cancer Lett. 2018, 428, 12–20. [Google Scholar] [CrossRef]
- Truong, T.H.; Hu, H.; Temiz, N.A.; Hagen, K.M.; Girard, B.J.; Brady, N.J.; Schwertfeger, K.L.; Lange, C.A.; Ostrander, J.H. Cancer Stem Cell Phenotypes in ER+ Breast Cancer Models Are Promoted by PELP1/AIB1 ComplexesPELP1/AIB1 Complexes Promote Cancer Stem Cell Phenotypes. Mol. Cancer Res. 2018, 16, 707–719. [Google Scholar] [CrossRef]
- Percharde, M.; Lavial, F.; Ng, J.-H.; Kumar, V.; Tomaz, R.A.; Martin, N.; Yeo, J.-C.; Gil, J.; Prabhakar, S.; Ng, H.-H. Ncoa3 functions as an essential Esrrb coactivator to sustain embryonic stem cell self-renewal and reprogramming. Genes Dev. 2012, 26, 2286–2298. [Google Scholar] [CrossRef]
- Hardt, O.; Wild, S.; Oerlecke, I.; Hofmann, K.; Luo, S.; Wiencek, Y.; Kantelhardt, E.; Vess, C.; Smith, G.P.; Schroth, G.P. Highly sensitive profiling of CD44+/CD24− breast cancer stem cells by combining global mRNA amplification and next generation sequencing: Evidence for a hyperactive PI3K pathway. Cancer Lett. 2012, 325, 165–174. [Google Scholar] [CrossRef]
- Pommier, S.J.; Hernandez, A.; Han, E.; Massimino, K.; Muller, P.; Diggs, B.; Chamberlain, E.; Murphy, J.; Hansen, J.; Naik, A. Fresh surgical specimens yield breast stem/progenitor cells and reveal their oncogenic abnormalities. Ann. Surg. Oncol. 2012, 19, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Karthik, G.-M.; Ma, R.; Lövrot, J.; Kis, L.L.; Lindh, C.; Blomquist, L.; Fredriksson, I.; Bergh, J.; Hartman, J. mTOR inhibitors counteract tamoxifen-induced activation of breast cancer stem cells. Cancer Lett. 2015, 367, 76–87. [Google Scholar] [CrossRef]
- Leung, E.Y.; Askarian-Amiri, M.E.; Sarkar, D.; Ferraro-Peyret, C.; Joseph, W.R.; Finlay, G.J.; Baguley, B.C. Endocrine therapy of estrogen receptor-positive breast cancer cells: Early differential effects on stem cell markers. Front. Oncol. 2017, 7, 184. [Google Scholar] [CrossRef] [PubMed]
- Domenici, G.; Aurrekoetxea-Rodríguez, I.; Simões, B.M.; Rábano, M.; Lee, S.Y.; Millán, J.S.; Comaills, V.; Oliemuller, E.; López-Ruiz, J.A.; Zabalza, I.; et al. A Sox2–Sox9 signalling axis maintains human breast luminal progenitor and breast cancer stem cells. Oncogene 2019, 38, 3151–3169. [Google Scholar] [CrossRef] [PubMed]
- Cittelly, D.M.; Das, P.M.; Spoelstra, N.S.; Edgerton, S.M.; Richer, J.K.; Thor, A.D.; Jones, F.E. Downregulation of miR-342 is associated with tamoxifen resistant breast tumors. Mol. Cancer 2010, 9, 317. [Google Scholar] [CrossRef] [PubMed]
- Cordenonsi, M.; Zanconato, F.; Azzolin, L.; Forcato, M.; Rosato, A.; Frasson, C.; Inui, M.; Montagner, M.; Parenti, A.R.; Poletti, A.; et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 2011, 147, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Guan, K.-L. Hippo signaling at a glance. J. Cell Sci. 2010, 123, 4001–4006. [Google Scholar] [CrossRef]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef]
- Callari, M.; Guffanti, A.; Soldà, G.; Merlino, G.; Fina, E.; Brini, E.; Moles, A.; Cappelletti, V.; Daidone, M.G. In-depth characterization of breast cancer tumor-promoting cell transcriptome by RNA sequencing and microarrays. Oncotarget 2016, 7, 976–994. [Google Scholar] [CrossRef]
- Fu, X.; Jeselsohn, R.; Pereira, R.; Hollingsworth, E.F.; Creighton, C.J.; Li, F.; Shea, M.; Nardone, A.; De Angelis, C.; Heiser, L.M.; et al. FOXA1 overexpression mediates endocrine resistance by altering the ER transcriptome and IL-8 expression in ER-positive breast cancer. Proc. Natl. Acad. Sci. USA 2016, 113, E6600–E6609. [Google Scholar] [CrossRef]
- Kitajima, S.; Yoshida, A.; Kohno, S.; Li, F.; Suzuki, S.; Nagatani, N.; Nishimoto, Y.; Sasaki, N.; Muranaka, H.; Wan, Y.; et al. The RB–IL-6 axis controls self-renewal and endocrine therapy resistance by fine-tuning mitochondrial activity. Oncogene 2017, 36, 5145–5157. [Google Scholar] [CrossRef]
- Hu, H.; Sun, J.; Wang, C.; Bu, X.; Liu, X.; Mao, Y.; Wang, H. IL-33 facilitates endocrine resistance of breast cancer by inducing cancer stem cell properties. Biochem. Biophys. Res. Commun. 2017, 485, 643–650. [Google Scholar] [CrossRef]
- Iorio, M.V.; Croce, C.M. microRNA involvement in human cancer. Carcinogenesis 2012, 33, 1126–1133. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, L.; Ravo, M.; Nassa, G.; Tarallo, R.; De Filippo, M.R.; Giurato, G.; Cirillo, F.; Stellato, C.; Silvestro, S.; Cantarella, C.; et al. Effects of oestrogen on microRNA expression in hormone-responsive breast cancer cells. Horm. Cancer 2012, 3, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Klinge, C.M. miRNAs regulated by estrogens, tamoxifen, and endocrine disruptors and their downstream gene targets. Mol. Cell Endocrinol. 2015, 418 Pt 3, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Rao, X.; Di Leva, G.; Li, M.; Fang, F.; Devlin, C.; Hartman-Frey, C.; Burow, M.; Ivan, M.; Croce, C.M.; Nephew, K.P. MicroRNA-221/222 confers breast cancer fulvestrant resistance by regulating multiple signaling pathways. Oncogene 2011, 30, 1082–1097. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Yang, Y.; Li, H.; Leng, Y.; Qian, K.; Huang, Q.; Zhang, C.; Lu, Z.; Chen, J.; Sun, T.; et al. MiR-873 regulates ERα transcriptional activity and tamoxifen resistance via targeting CDK3 in breast cancer cells. Oncogene 2015, 34, 3895–3907. [Google Scholar] [CrossRef]
- Zhao, J.-J.; Lin, J.; Yang, H.; Kong, W.; He, L.; Ma, X.; Coppola, D.; Cheng, J.Q. MicroRNA-221/222 negatively regulates estrogen receptorα and is associated with tamoxifen resistance in breast cancer. J. Biol. Chem. 2008, 283, 31079–31087. [Google Scholar] [CrossRef]
- Miller, T.E.; Ghoshal, K.; Ramaswamy, B.; Roy, S.; Datta, J.; Shapiro, C.L.; Jacob, S.; Majumder, S. MicroRNA-221/222 Confers Tamoxifen Resistance in Breast Cancer by Targeting p27Kip1. J. Biol. Chem. 2008, 283, 29897–29903. [Google Scholar] [CrossRef]
- Xin, F.; Li, M.; Balch, C.; Thomson, M.; Fan, M.; Liu, Y.; Hammond, S.M.; Kim, S.; Nephew, K.P. Computational analysis of microRNA profiles and their target genes suggests significant involvement in breast cancer antiestrogen resistance. Bioinformatics 2009, 25, 430–434. [Google Scholar] [CrossRef]
- Rodríguez-González, F.G.; Sieuwerts, A.M.; Smid, M.; Look, M.P.; Meijer-van Gelder, M.E.; de Weerd, V.; Sleijfer, S.; Martens, J.W.M.; Foekens, J.A. MicroRNA-30c expression level is an independent predictor of clinical benefit of endocrine therapy in advanced estrogen receptor positive breast cancer. Breast Cancer Res. Treat. 2011, 127, 43–51. [Google Scholar] [CrossRef]
- Huo, D.; Clayton, W.M.; Yoshimatsu, T.F.; Chen, J.; Olopade, O.I. Identification of a circulating microRNA signature to distinguish recurrence in breast cancer patients. Oncotarget 2016, 7, 55231–55248. [Google Scholar] [CrossRef]
- He, Y.J.; Wu, J.Z.; Ji, M.H.; Ma, T.; Qiao, E.Q.; Ma, R.; Tang, J.H. miR-342 is associated with estrogen receptor-α expression and response to tamoxifen in breast cancer. Exp. Ther. Med. 2013, 5, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Liang, G.; Zhang, Q.; Zhao, W. The Role of Long Noncoding RNAs in Antiestrogen Resistance in Breast Cancer: An Overview and Update. J. Breast Cancer 2020, 23, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Dinglin, X.; Cao, S.; Zheng, S.; Wu, C.; Chen, W.; Li, Q.; Hu, Q.; Zheng, F.; Wu, Z.; et al. Enhancer-Driven lncRNA BDNF-AS Induces Endocrine Resistance and Malignant Progression of Breast Cancer through the RNH1/TRIM21/mTOR Cascade. Cell Rep. 2020, 31, 107753. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Yang, Y.A.; Zhang, A.; Fong, K.W.; Kim, J.; Song, B.; Li, S.; Zhao, J.C.; Yu, J. LncRNA HOTAIR enhances ER signaling and confers tamoxifen resistance in breast cancer. Oncogene 2016, 35, 2746–2755. [Google Scholar] [CrossRef] [PubMed]
- Mitobe, Y.; Ikeda, K.; Suzuki, T.; Takagi, K.; Kawabata, H.; Horie-Inoue, K.; Inoue, S. ESR1-Stabilizing Long Noncoding RNA TMPO-AS1 Promotes Hormone-Refractory Breast Cancer Progression. Mol. Cell. Biol. 2019, 39, e00261-19. [Google Scholar] [CrossRef]
- Shi, Y.F.; Lu, H.; Wang, H.B. Downregulated lncRNA ADAMTS9-AS2 in breast cancer enhances tamoxifen resistance by activating microRNA-130a-5p. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 1563–1573. [Google Scholar] [CrossRef]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef]
- Chisholm, K.M.; Wan, Y.; Li, R.; Montgomery, K.D.; Chang, H.Y.; West, R.B. Detection of long non-coding RNA in archival tissue: Correlation with polycomb protein expression in primary and metastatic breast carcinoma. PLoS ONE 2012, 7, e47998. [Google Scholar] [CrossRef]
- Sørensen, K.P.; Thomassen, M.; Tan, Q.; Bak, M.; Cold, S.; Burton, M.; Larsen, M.J.; Kruse, T.A. Long non-coding RNA HOTAIR is an independent prognostic marker of metastasis in estrogen receptor-positive primary breast cancer. Breast Cancer Res. Treat. 2013, 142, 529–536. [Google Scholar] [CrossRef]
- Zhang, M.; Wu, K.; Zhang, P.; Qiu, Y.; Bai, F.; Chen, H. HOTAIR Facilitates Endocrine Resistance in Breast Cancer Through ESR1/miR-130b-3p Axis: Comprehensive Analysis of mRNA-miRNA-lncRNA Network. Int. J. Gen. Med. 2021, 14, 4653–4663. [Google Scholar] [CrossRef]
- Lu, R.; Zhang, J.; Zhang, W.; Huang, Y.; Wang, N.; Zhang, Q.; Qu, S. Circulating HOTAIR expression predicts the clinical response to neoadjuvant chemotherapy in patients with breast cancer. Cancer Biomark. 2018, 22, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhou, N.; Watabe, K.; Lu, Z.; Wu, F.; Xu, M.; Mo, Y.Y. Long non-coding RNA UCA1 promotes breast tumor growth by suppression of p27 (Kip1). Cell Death Dis. 2014, 5, e1008. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Shao, C.; Xu, M.; Ji, J.; Xie, Y.; Lei, Y.; Wang, X. Macrophage infiltration promotes invasiveness of breast cancer cells via activating long non-coding RNA UCA1. Int. J. Clin. Exp. Pathol. 2015, 8, 9052–9061. [Google Scholar] [PubMed]
- Wu, C.; Luo, J. Long Non-Coding RNA (lncRNA) Urothelial Carcinoma-Associated 1 (UCA1) Enhances Tamoxifen Resistance in Breast Cancer Cells via Inhibiting mTOR Signaling Pathway. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2016, 22, 3860–3867. [Google Scholar] [CrossRef]
- Li, X.; Wu, Y.; Liu, A.; Tang, X. Long non-coding RNA UCA1 enhances tamoxifen resistance in breast cancer cells through a miR-18a-HIF1α feedback regulatory loop. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2016, 37, 14733–14743. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yu, D.; Li, H.; Lv, Y.; Li, S. Long non-coding RNA UCA1 confers tamoxifen resistance in breast cancer endocrinotherapy through regulation of the EZH2/p21 axis and the PI3K/AKT signaling pathway. Int. J. Oncol. 2019, 54, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Meijer, D.; van Agthoven, T.; Bosma, P.T.; Nooter, K.; Dorssers, L.C. Functional screen for genes responsible for tamoxifen resistance in human breast cancer cells. Mol. Cancer Res. MCR 2006, 4, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Godinho, M.F.; Sieuwerts, A.M.; Look, M.P.; Meijer, D.; Foekens, J.A.; Dorssers, L.C.; van Agthoven, T. Relevance of BCAR4 in tamoxifen resistance and tumour aggressiveness of human breast cancer. Br. J. Cancer 2010, 103, 1284–1291. [Google Scholar] [CrossRef]
- Godinho, M.; Meijer, D.; Setyono-Han, B.; Dorssers, L.C.J.; Van Agthoven, T. Characterization of BCAR4, a novel oncogene causing endocrine resistance in human breast cancer cells. J. Cell. Physiol. 2011, 226, 1741–1749. [Google Scholar] [CrossRef]
- Godinho, M.F.; Wulfkuhle, J.D.; Look, M.P.; Sieuwerts, A.M.; Sleijfer, S.; Foekens, J.A.; Petricoin, E.F.; Dorssers, L.C.; van Agthoven, T. BCAR4 induces antioestrogen resistance but sensitises breast cancer to lapatinib. Br. J. Cancer 2012, 107, 947–955. [Google Scholar] [CrossRef]
- Huang, N.-s.; Chi, Y.-Y.; Xue, J.-Y.; Liu, M.-Y.; Huang, S.; Mo, M.; Zhou, S.-L.; Wu, J. Long non-coding RNA metastasis associated in lung adenocarcinoma transcript 1 (MALAT1) interacts with estrogen receptor and predicted poor survival in breast cancer. Oncotarget 2016, 7, 37957–37965. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Shao, F.; Wu, Q.; Zhang, X.; Xu, D.; Qian, K.; Xie, Y.; Wang, S.; Xu, N.; Wang, Y.; et al. miR-124 downregulation leads to breast cancer progression via LncRNA-MALAT1 regulation and CDK4/E2F1 signal activation. Oncotarget 2016, 7, 16205–16216. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Lu, Q.; Lin, Y.; Ma, L. Reciprocal regulation of Hsa-miR-1 and long noncoding RNA MALAT1 promotes triple-negative breast cancer development. Tumour Biol. 2016, 37, 7383–7394. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, Y.; Yang, Z.; Chen, B.; Huang, W.; Liu, Y.; Zhang, Y.J. MiR-204/ZEB2 axis functions as key mediator for MALAT1-induced epithelial–mesenchymal transition in breast cancer. Tumour Biol. 2017, 39, 1010428317690998. [Google Scholar] [CrossRef]
- Zhao, L.; Zhao, Y.; He, Y.; Li, Q.; Mao, Y. The functional pathway analysis and clinical significance of miR-20a and its related lncRNAs in breast cancer. Cell. Signal. 2018, 51, 152–165. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, B.; Zhu, H.; Qu, X.; Zhao, L.; Tan, Y.; Jiang, Y.; Liao, M.; Wu, X. Inhibition of long non-coding RNA ROR reverses resistance to Tamoxifen by inducing autophagy in breast cancer. Tumour Biol. 2017, 39, 1010428317705790. [Google Scholar] [CrossRef]
- Peng, W.-x.; Huang, J.-G.; Yang, L.; Gong, A.-H.; Mo, Y.-Y. Linc-RoR promotes MAPK/ERK signaling and confers estrogen-independent growth of breast cancer. Mol. Cancer 2017, 16, 161. [Google Scholar] [CrossRef]
- Ma, T.; Liang, Y.; Li, Y.; Song, X.; Zhang, N.; Li, X.; Chen, B.; Zhao, W.; Wang, L.; Yang, Q. LncRNA LINP1 confers tamoxifen resistance and negatively regulated by ER signaling in breast cancer. Cell. Signal. 2020, 68, 109536. [Google Scholar] [CrossRef]
- Liang, Y.; Li, Y.; Song, X.; Zhang, N.; Sang, Y.; Zhang, H.; Liu, Y.; Chen, B.; Zhao, W.; Wang, L.; et al. Long noncoding RNA LINP1 acts as an oncogene and promotes chemoresistance in breast cancer. Cancer Biol. Ther. 2018, 19, 120–131. [Google Scholar] [CrossRef]
- Lim, E.; Jhaveri, K.L.; Perez-Fidalgo, J.A.; Bellet, M.; Boni, V.; Garcia, J.M.P.; Estevez, L.; Bardia, A.; Turner, N.C.; Villanueva, R.; et al. A phase Ib study to evaluate the oral selective estrogen receptor degrader GDC-9545 alone or combined with palbociclib in metastatic ER-positive HER2-negative breast cancer. J. Clin. Oncol. 2020, 38, 1023. [Google Scholar] [CrossRef]
- Liu, Y.; Li, M.; Yu, H.; Piao, H. lncRNA CYTOR promotes tamoxifen resistance in breast cancer cells via sponging miR-125a-5p. Int. J. Mol. Med. 2020, 45, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Di Leo, A.; Jerusalem, G.; Petruzelka, L.; Torres, R.; Bondarenko, I.N.; Khasanov, R.; Verhoeven, D.; Pedrini, J.L.; Smirnova, I.; Lichinitser, M.R.; et al. Results of the CONFIRM phase III trial comparing fulvestrant 250 mg with fulvestrant 500 mg in postmenopausal women with estrogen receptor-positive advanced breast cancer. J. Clin. Oncol. 2010, 28, 4594–4600. [Google Scholar] [CrossRef] [PubMed]
- Fawell, S.E.; White, R.; Hoare, S.; Sydenham, M.; Page, M.; Parker, M.G. Inhibition of estrogen receptor-DNA binding by the “pure” antiestrogen ICI 164,384 appears to be mediated by impaired receptor dimerization. Proc. Natl. Acad. Sci. USA 1990, 87, 6883–6887. [Google Scholar] [CrossRef]
- Dauvois, S.; White, R.; Parker, M.G. The antiestrogen ICI 182780 disrupts estrogen receptor nucleocytoplasmic shuttling. J. Cell Sci. 1993, 106 Pt 4, 1377–1388. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K.; Wakeling, A.; Nicholson, R.I. Fulvestrant: An oestrogen receptor antagonist with a novel mechanism of action. Br. J. Cancer 2004, 90 (Suppl. S1), S2–S6. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Nephew, K.P. Fulvestrant (ICI 182,780)-dependent interacting proteins mediate immobilization and degradation of estrogen receptor-α. J. Biol. Chem. 2006, 281, 9607–9615. [Google Scholar] [CrossRef] [PubMed]
- Shomali, M.; Cheng, J.; Sun, F.; Koundinya, M.; Guo, Z.; Hebert, A.T.; McManus, J.; Levit, M.N.; Hoffmann, D.; Courjaud, A.; et al. SAR439859, a Novel Selective Estrogen Receptor Degrader (SERD), Demonstrates Effective and Broad Antitumor Activity in Wild-Type and Mutant ER-Positive Breast Cancer Models. Mol. Cancer Ther. 2021, 20, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Hernando, C.; Ortega-Morillo, B.; Tapia, M.; Moragón, S.; Martínez, M.T.; Eroles, P.; Garrido-Cano, I.; Adam-Artigues, A.; Lluch, A.; Bermejo, B.; et al. Oral Selective Estrogen Receptor Degraders (SERDs) as a Novel Breast Cancer Therapy: Present and Future from a Clinical Perspective. Int. J. Mol. Sci. 2021, 22, 57812. [Google Scholar] [CrossRef]
- Andreano, K.J.; Wardell, S.E.; Baker, J.G.; Desautels, T.K.; Baldi, R.; Chao, C.A.; Heetderks, K.A.; Bae, Y.; Xiong, R.; Tonetti, D.A.; et al. G1T48, an oral selective estrogen receptor degrader, and the CDK4/6 inhibitor lerociclib inhibit tumor growth in animal models of endocrine-resistant breast cancer. Breast Cancer Res. Treat. 2020, 180, 635–646. [Google Scholar] [CrossRef]
- Scott, J.S.; Moss, T.A.; Balazs, A.; Barlaam, B.; Breed, J.; Carbajo, R.J.; Chiarparin, E.; Davey, P.R.J.; Delpuech, O.; Fawell, S.; et al. Discovery of AZD9833, a Potent and Orally Bioavailable Selective Estrogen Receptor Degrader and Antagonist. J. Med. Chem. 2020, 63, 14530–14559. [Google Scholar] [CrossRef]
- Tria, G.S.; Abrams, T.; Baird, J.; Burks, H.E.; Firestone, B.; Gaither, L.A.; Hamann, L.G.; He, G.; Kirby, C.A.; Kim, S.; et al. Discovery of LSZ102, a Potent, Orally Bioavailable Selective Estrogen Receptor Degrader (SERD) for the Treatment of Estrogen Receptor Positive Breast Cancer. J. Med. Chem. 2018, 61, 2837–2864. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, K.; Juric, D.; Yap, Y.S.; Cresta, S.; Layman, R.M.; Duhoux, F.P.; Terret, C.; Takahashi, S.; Huober, J.; Kundamal, N.; et al. A Phase I Study of LSZ102, an Oral Selective Estrogen Receptor Degrader, with or without Ribociclib or Alpelisib, in Patients with Estrogen Receptor-Positive Breast Cancer. Clin. Cancer Res. 2021, 27, 5760–5770. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A.; Kaklamani, V.; Wilks, S.; Weise, A.; Richards, D.; Harb, W.; Osborne, C.; Wesolowski, R.; Karuturi, M.; Conkling, P.; et al. Phase I Study of Elacestrant (RAD1901), a Novel Selective Estrogen Receptor Degrader, in ER-Positive, HER2-Negative Advanced Breast Cancer. J. Clin. Oncol. 2021, 39, 1360–1370. [Google Scholar] [CrossRef]
- Moore, H.M.; Boni, V.; Bellet, M.; Heras, B.B.D.L.; Cortés, M.G.; Oakman, C.; Schmid, P.; Trinh, X.B.; Wheatley, D.; Jhaveri, K.L.; et al. Evaluation of pharmacodynamic (PD) and biologic activity in a preoperative window-of-opportunity (WOO) study of giredestrant (GDC-9545) in postmenopausal patients (pts) with estrogen receptor-positive, HER2-negative (ER+/HER2−) operable breast cancer (BC). J. Clin. Oncol. 2021, 39, 577. [Google Scholar] [CrossRef]
- Bardia, A.; Chandarlapaty, S.; Linden, H.M.; Ulaner, G.A.; Gosselin, A.; Cartot-Cotton, S.; Cohen, P.; Doroumian, S.; Paux, G.; Celanovic, M.; et al. AMEERA-1 phase 1/2 study of amcenestrant, SAR439859, in postmenopausal women with ER-positive/HER2-negative advanced breast cancer. Nat. Commun. 2022, 13, 4116. [Google Scholar] [CrossRef]
- Oliveira, M.; Hamilton, E.P.; Incorvati, J.; Heras, B.B.d.l.; Calvo, E.; García-Corbacho, J.; Ruiz-Borrego, M.; Vaklavas, C.; Turner, N.C.; Ciruelos, E.M.; et al. Serena-1: Updated analyses from a phase 1 study (parts C/D) of the next-generation oral SERD camizestrant (AZD9833) in combination with palbociclib, in women with ER-positive, HER2-negative advanced breast cancer. J. Clin. Oncol. 2022, 40, 1032. [Google Scholar] [CrossRef]
- Baird, R.; Oliveira, M.; Gil, E.M.C.; Patel, M.R.; de las Heras, B.B.; Ruiz-Borrego, M.; Garcia-Corbacho, J.; Armstrong, A.C.; Banerji, U.; Twelves, C.; et al. Updated data from SERENA-1: A Phase 1 dose escalation and expansion study of the next generation oral SERD AZD9833 as a monotherapy and in combination with palbociclib, in women with ER-positive, HER2-negative advanced breast cancer. Cancer Res. 2021, 81, PS11-05. [Google Scholar] [CrossRef]
- Du Rusquec, P.; Blonz, C.; Frenel, J.S.; Campone, M. Targeting the PI3K/Akt/mTOR pathway in estrogen-receptor positive HER2 negative advanced breast cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835920940939. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Abrams, S.L.; Lee, J.T.; Chang, F.; Bertrand, F.E.; Navolanic, P.M.; Terrian, D.M.; Franklin, R.A.; D’Assoro, A.B.; et al. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv. Enzym. Regul. 2006, 46, 249–279. [Google Scholar] [CrossRef]
- Ciruelos Gil, E.M. Targeting the PI3K/AKT/mTOR pathway in estrogen receptor-positive breast cancer. Cancer Treat. Rev. 2014, 40, 862–871. [Google Scholar] [CrossRef]
- Paplomata, E.; Zelnak, A.; O’Regan, R. Everolimus: Side effect profile and management of toxicities in breast cancer. Breast Cancer Res. Treat. 2013, 140, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.W.; Rexer, B.N.; Garrett, J.T.; Arteaga, C.L. Mutations in the phosphatidylinositol 3-kinase pathway: Role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011, 13, 224. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Sáez, O.; Chic, N.; Pascual, T.; Adamo, B.; Vidal, M.; González-Farré, B.; Sanfeliu, E.; Schettini, F.; Conte, B.; Brasó-Maristany, F.; et al. Frequency and spectrum of PIK3CA somatic mutations in breast cancer. Breast Cancer Res. 2020, 22, 45. [Google Scholar] [CrossRef] [PubMed]
- Miricescu, D.; Totan, A.; Stanescu, S., II; Badoiu, S.C.; Stefani, C.; Greabu, M. PI3K/AKT/mTOR Signaling Pathway in Breast Cancer: From Molecular Landscape to Clinical Aspects. Int. J. Mol. Sci. 2020, 22, 173. [Google Scholar] [CrossRef] [PubMed]
- Paplomata, E.; O’Regan, R. The PI3K/AKT/mTOR pathway in breast cancer: Targets, trials and biomarkers. Ther. Adv. Med. Oncol. 2014, 6, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Houghton, P.J. Everolimus. Clin. Cancer Res. 2010, 16, 1368–1372. [Google Scholar] [CrossRef]
- Lee, J.J.; Loh, K.; Yap, Y.S. PI3K/Akt/mTOR inhibitors in breast cancer. Cancer Biol. Med. 2015, 12, 342–354. [Google Scholar] [CrossRef]
- Baselga, J.; Campone, M.; Piccart, M.; Burris, H.A., 3rd; Rugo, H.S.; Sahmoud, T.; Noguchi, S.; Gnant, M.; Pritchard, K.I.; Lebrun, F.; et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 2012, 366, 520–529. [Google Scholar] [CrossRef]
- Schmid, P.; Zaiss, M.; Harper-Wynne, C.; Ferreira, M.; Dubey, S.; Chan, S.; Makris, A.; Nemsadze, G.; Brunt, A.M.; Kuemmel, S.; et al. Fulvestrant Plus Vistusertib vs Fulvestrant Plus Everolimus vs Fulvestrant Alone for Women with Hormone Receptor-Positive Metastatic Breast Cancer: The MANTA Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1556–1564. [Google Scholar] [CrossRef]
- Bachelot, T.; Bourgier, C.; Cropet, C.; Ray-Coquard, I.; Ferrero, J.M.; Freyer, G.; Abadie-Lacourtoisie, S.; Eymard, J.C.; Debled, M.; Spaëth, D.; et al. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: A GINECO study. J. Clin. Oncol. 2012, 30, 2718–2724. [Google Scholar] [CrossRef]
- Boulay, A.; Rudloff, J.; Ye, J.; Zumstein-Mecker, S.; O’Reilly, T.; Evans, D.B.; Chen, S.; Lane, H.A. Dual inhibition of mTOR and estrogen receptor signaling in vitro induces cell death in models of breast cancer. Clin. Cancer Res. 2005, 11, 5319–5328. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Semiglazov, V.; van Dam, P.; Manikhas, A.; Bellet, M.; Mayordomo, J.; Campone, M.; Kubista, E.; Greil, R.; Bianchi, G.; et al. Phase II randomized study of neoadjuvant everolimus plus letrozole compared with placebo plus letrozole in patients with estrogen receptor-positive breast cancer. J. Clin. Oncol. 2009, 27, 2630–2637. [Google Scholar] [CrossRef] [PubMed]
- Campone, M.; Im, S.A.; Iwata, H.; Clemons, M.; Ito, Y.; Awada, A.; Chia, S.; Jagiełło-Gruszfeld, A.; Pistilli, B.; Tseng, L.M.; et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant for postmenopausal, hormone receptor-positive, human epidermal growth factor receptor 2-negative, advanced breast cancer: Overall survival results from BELLE-2. Eur. J. Cancer 2018, 103, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Campone, M.; Ciruelos, E.M.; Iwata, H.; Loibl, S.; Rugo, H.S.; Wilke, C.; Mills, D.; Chol, M.; Longin, A.-S.; et al. SOLAR-1: A phase III study of alpelisib + fulvestrant in men and postmenopausal women with HR+/HER2– advanced breast cancer (BC) progressing on or after prior aromatase inhibitor therapy. J. Clin. Oncol. 2016, 34, TPS618. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Lapenna, S.; Giordano, A. Cell cycle kinases as therapeutic targets for cancer. Nat. Rev. Drug Discov. 2009, 8, 547–566. [Google Scholar] [CrossRef]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef]
- Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [CrossRef]
- Thangavel, C.; Dean, J.L.; Ertel, A.; Knudsen, K.E.; Aldaz, C.M.; Witkiewicz, A.K.; Clarke, R.; Knudsen, E.S. Therapeutically activating RB: Reestablishing cell cycle control in endocrine therapy-resistant breast cancer. Endocr.-Relat. Cancer 2011, 18, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet. Oncol. 2015, 16, 25–35. [Google Scholar] [CrossRef]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, N.; D’Angelo, A.; Pittacolo, M.; Roviello, G.; Miccoli, A.; Corona, S.P.; Bernocchi, O.; Generali, D.; Otto, T. Updates on the CDK4/6 Inhibitory Strategy and Combinations in Breast Cancer. Cells 2019, 8, 321. [Google Scholar] [CrossRef] [PubMed]
- Dickler, M.N.; Tolaney, S.M.; Rugo, H.S.; Cortés, J.; Diéras, V.; Patt, D.; Wildiers, H.; Hudis, C.A.; O’Shaughnessy, J.; Zamora, E.; et al. MONARCH 1, A Phase II Study of Abemaciclib, a CDK4 and CDK6 Inhibitor, as a Single Agent, in Patients with Refractory HR(+)/HER2(−) Metastatic Breast Cancer. Clin. Cancer Res. 2017, 23, 5218–5224. [Google Scholar] [CrossRef]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Blackwell, K.L.; André, F.; Winer, E.P.; et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1738–1748. [Google Scholar] [CrossRef]
- Nichols, M. New directions for drug-resistant breast cancer: The CDK4/6 inhibitors. Future Med. Chem. 2015, 7, 1473–1481. [Google Scholar] [CrossRef]
- Turner, N.C.; Ro, J.; André, F.; Loi, S.; Verma, S.; Iwata, H.; Harbeck, N.; Loibl, S.; Huang Bartlett, C.; Zhang, K.; et al. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2015, 373, 209–219. [Google Scholar] [CrossRef]
- Sledge, G.W., Jr.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. MONARCH 2: Abemaciclib in Combination with Fulvestrant in Women with HR+/HER2− Advanced Breast Cancer Who Had Progressed While Receiving Endocrine Therapy. J. Clin. Oncol. 2017, 35, 2875–2884. [Google Scholar] [CrossRef]
- Im, S.A.; Lu, Y.S.; Bardia, A.; Harbeck, N.; Colleoni, M.; Franke, F.; Chow, L.; Sohn, J.; Lee, K.S.; Campos-Gomez, S.; et al. Overall Survival with Ribociclib plus Endocrine Therapy in Breast Cancer. N. Engl. J. Med. 2019, 381, 307–316. [Google Scholar] [CrossRef]
- Sledge, G.W., Jr.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. The Effect of Abemaciclib Plus Fulvestrant on Overall Survival in Hormone Receptor-Positive, ERBB2-Negative Breast Cancer That Progressed on Endocrine Therapy-MONARCH 2: A Randomized Clinical Trial. JAMA Oncol. 2020, 6, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Goetz, M.P.; Toi, M.; Campone, M.; Sohn, J.; Paluch-Shimon, S.; Huober, J.; Park, I.H.; Trédan, O.; Chen, S.C.; Manso, L.; et al. MONARCH 3: Abemaciclib As Initial Therapy for Advanced Breast Cancer. J. Clin. Oncol. 2017, 35, 3638–3646. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Gray, R.; Braybrooke, J.; Davies, C.; Taylor, C.; McGale, P.; Peto, R.; Pritchard, K.I.; Bergh, J.; Dowsett, M.; et al. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. N. Engl. J. Med. 2017, 377, 1836–1846. [Google Scholar] [CrossRef]
- Xu, H.; Wang, Y.; Han, Y.; Wu, Y.; Wang, J.; Xu, B. CDK4/6 inhibitors versus PI3K/AKT/mTOR inhibitors in women with hormone receptor-positive, HER2-negative metastatic breast cancer: An updated systematic review and network meta-analysis of 28 randomized controlled trials. Front. Oncol. 2022, 12, 956464. [Google Scholar] [CrossRef] [PubMed]
- Juric, D.; Rodon, J.; Tabernero, J.; Janku, F.; Burris, H.A.; Schellens, J.H.M.; Middleton, M.R.; Berlin, J.; Schuler, M.; Gil-Martin, M.; et al. Phosphatidylinositol 3-Kinase α-Selective Inhibition with Alpelisib (BYL719) in PIK3CA-Altered Solid Tumors: Results From the First-in-Human Study. J. Clin. Oncol. 2018, 36, 1291–1299. [Google Scholar] [CrossRef]
- Vora, S.R.; Juric, D.; Kim, N.; Mino-Kenudson, M.; Huynh, T.; Costa, C.; Lockerman, E.L.; Pollack, S.F.; Liu, M.; Li, X.; et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell 2014, 26, 136–149. [Google Scholar] [CrossRef]
- O’Brien, N.A.; McDermott, M.S.J.; Conklin, D.; Luo, T.; Ayala, R.; Salgar, S.; Chau, K.; DiTomaso, E.; Babbar, N.; Su, F.; et al. Targeting activated PI3K/mTOR signaling overcomes acquired resistance to CDK4/6-based therapies in preclinical models of hormone receptor-positive breast cancer. Breast Cancer Res. 2020, 22, 89. [Google Scholar] [CrossRef]
- Herrera-Abreu, M.T.; Palafox, M.; Asghar, U.; Rivas, M.A.; Cutts, R.J.; Garcia-Murillas, I.; Pearson, A.; Guzman, M.; Rodriguez, O.; Grueso, J.; et al. Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Res. 2016, 76, 2301–2313. [Google Scholar] [CrossRef]
- Rugo, H.S.; Lerebours, F.; Ciruelos, E.; Drullinsky, P.; Ruiz-Borrego, M.; Neven, P.; Park, Y.H.; Prat, A.; Bachelot, T.; Juric, D.; et al. Alpelisib plus fulvestrant in PIK3CA-mutated, hormone receptor-positive advanced breast cancer after a CDK4/6 inhibitor (BYLieve): One cohort of a phase 2, multicentre, open-label, non-comparative study. Lancet. Oncol. 2021, 22, 489–498. [Google Scholar] [CrossRef]
- Forero-Torres, A.; Han, H.; Dees, E.C.; Wesolowski, R.; Bardia, A.; Kabos, P.; Layman, R.M.; Lu, J.M.; Kern, K.A.; Perea, R.; et al. Phase Ib study of gedatolisib in combination with palbociclib and endocrine therapy (ET) in women with estrogen receptor (ER) positive (+) metastatic breast cancer (MBC) (B2151009). J. Clin. Oncol. 2018, 36, 1040. [Google Scholar] [CrossRef]
- Bardia, A.; Hurvitz, S.A.; DeMichele, A.; Clark, A.S.; Zelnak, A.B.; Yardley, D.A.; Karuturi, M.S.; Sanft, T.B.; Blau, S.; Hart, L.L.; et al. Triplet therapy (continuous ribociclib, everolimus, exemestane) in HR+/HER2− advanced breast cancer postprogression on a CDK4/6 inhibitor (TRINITI-1): Efficacy, safety, and biomarker results. J. Clin. Oncol. 2019, 37, 1016. [Google Scholar] [CrossRef]
- Ahmadzada, T.; Reid, G.; McKenzie, D.R. Fundamentals of siRNA and miRNA therapeutics and a review of targeted nanoparticle delivery systems in breast cancer. Biophys. Rev. 2018, 10, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Chen, W.; Yu, W.; Huang, W.; Deng, W. Small interfering RNA-based molecular therapy of cancers. Chin. J. Cancer 2013, 32, 488–493. [Google Scholar] [CrossRef]
- Kim, D.H.; Rossi, J.J. Strategies for silencing human disease using RNA interference. Nat. Rev. Genet. 2007, 8, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, T.C.; El-Osta, A. RNA interference and potential therapeutic applications of short interfering RNAs. Cancer Gene Ther. 2005, 12, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Cui, J.; Leonard, M.; Nephew, K.; Li, Y.; Zhang, X. Silencing MED1 sensitizes breast cancer cells to pure anti-estrogen fulvestrant in vitro and in vivo. PLoS ONE 2013, 8, e70641. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Zhang, K.; Mu, K.; Xu, J.; Yang, H.; Liu, Y.; Wang, B.; Wang, Z.; Li, Z.; Kong, Q.; et al. Regulation of estrogen signaling and breast cancer proliferation by an ubiquitin ligase TRIM56. Oncogenesis 2019, 8, 30. [Google Scholar] [CrossRef]
- Martin, L.A.; Pancholi, S.; Ribas, R.; Gao, Q.; Simigdala, N.; Nikitorowicz, J.; Johnston, S.R.; Dowsett, M. Abstract P3-03-09: Resistance to palbociclib depends on multiple targetable mechanisms highlighting the potential of drug holidays and drug switching to improve therapeutic outcome. Cancer Res. 2017, 77, P3-03. [Google Scholar] [CrossRef]
- Park, J.; Kim, G.H.; Lee, J.; Phuong, B.T.C.; Kong, B.; Won, J.E.; Won, G.W.; Lee, Y.H.; Han, H.D.; Lee, Y. MST2 silencing induces apoptosis and inhibits tumor growth for estrogen receptor α-positive MCF-7 breast cancer. Toxicol. Appl. Pharmacol. 2020, 408, 115257. [Google Scholar] [CrossRef]
- Bouclier, C.; Marsaud, V.; Bawa, O.; Nicolas, V.; Moine, L.; Opolon, P.; Renoir, J.-M. Coadministration of nanosystems of short silencing RNAs targeting oestrogen receptor α and anti-oestrogen synergistically induces tumour growth inhibition in human breast cancer xenografts. Breast Cancer Res. Treat. 2010, 122, 145–158. [Google Scholar] [CrossRef]
- Pandey, K.; Park, N.; Park, K.-S.; Hur, J.; Cho, Y.B.; Kang, M.; An, H.-J.; Kim, S.; Hwang, S.; Moon, Y.W. Combined CDK2 and CDK4/6 Inhibition Overcomes Palbociclib Resistance in Breast Cancer by Enhancing Senescence. Cancers 2020, 12, 3566. [Google Scholar] [CrossRef] [PubMed]
- Tekedereli, I.; Alpay, S.N.; Akar, U.; Yuca, E.; Ayugo-Rodriguez, C.; Han, H.-D.; Sood, A.K.; Lopez-Berestein, G.; Ozpolat, B. Therapeutic silencing of Bcl-2 by systemically administered siRNA nanotherapeutics inhibits tumor growth by autophagy and apoptosis and enhances the efficacy of chemotherapy in orthotopic xenograft models of ER (−) and ER (+) breast cancer. Mol. Ther. Nucleic Acids 2013, 2, e121. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.; Sun, T.; Ma, G.; Cao, J.; Hu, Q.; Chen, L.; Wang, Y.; Wang, Q.; Sun, J.; Wu, R.; et al. LEM4 confers tamoxifen resistance to breast cancer cells by activating cyclin D-CDK4/6-Rb and ERα pathway. Nat. Commun. 2018, 9, 4180. [Google Scholar] [CrossRef] [PubMed]
- Jansen, V.M.; Bhola, N.E.; Bauer, J.A.; Formisano, L.; Lee, K.-M.; Hutchinson, K.E.; Witkiewicz, A.K.; Moore, P.D.; Estrada, M.V.; Sánchez, V.; et al. Kinome-Wide RNA Interference Screen Reveals a Role for PDK1 in Acquired Resistance to CDK4/6 Inhibition in ER-Positive Breast Cancer. Cancer Res. 2017, 77, 2488–2499. [Google Scholar] [CrossRef]
- Yang, W.; Hosford, S.R.; Traphagen, N.A.; Shee, K.; Demidenko, E.; Liu, S.; Miller, T.W. Autophagy promotes escape from phosphatidylinositol 3-kinase inhibition in estrogen receptor-positive breast cancer. FASEB J. 2018, 32, 1222–1235. [Google Scholar] [CrossRef]
- Yang, H.; Lv, X.; Li, X.; Mao, L.; Niu, Z.; Wang, T.; Zhuang, T.; Huang, Q. ZNF213 Facilitates ER Alpha Signaling in Breast Cancer Cells. Front. Oncol. 2021, 11, 638751. [Google Scholar] [CrossRef]
- Gustafsson, N.; Zhao, C.; Gustafsson, J.-Å.; Dahlman-Wright, K. RBCK1 drives breast cancer cell proliferation by promoting transcription of estrogen receptor α and cyclin B1. Cancer Res. 2010, 70, 1265–1274. [Google Scholar] [CrossRef]
- Choi, H.-N.; Jin, H.-O.; Kim, J.-H.; Hong, S.-E.; Kim, H.-A.; Kim, E.-K.; Lee, J.K.; Park, I.-C.; Noh, W.C. Inhibition of S6K1 enhances glucose deprivation-induced cell death via downregulation of anti-apoptotic proteins in MCF-7 breast cancer cells. Biochem. Biophys. Res. Commun. 2013, 432, 123–128. [Google Scholar] [CrossRef]
- Xia, X.; Huang, C.; Liao, Y.; Liu, Y.; He, J.; Shao, Z.; Hu, T.; Yu, C.; Jiang, L.; Liu, J.; et al. The deubiquitinating enzyme USP15 stabilizes ERα and promotes breast cancer progression. Cell Death Dis. 2021, 12, 329. [Google Scholar] [CrossRef]
- Hampsch, R.A.; Wells, J.D.; Traphagen, N.A.; McCleery, C.F.; Fields, J.L.; Shee, K.; Dillon, L.M.; Pooler, D.B.; Lewis, L.D.; Demidenko, E. AMPK activation by metformin promotes survival of dormant ER+ breast cancer cells. Clin. Cancer Res. 2020, 26, 3707–3719. [Google Scholar] [CrossRef]
- Attia, Y.M.; Shouman, S.A.; Salama, S.A.; Ivan, C.; Elsayed, A.M.; Amero, P.; Rodriguez-Aguayo, C.; Lopez-Berestein, G. Blockade of CDK7 reverses endocrine therapy resistance in breast cancer. Int. J. Mol. Sci. 2020, 21, 2974. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.D.; Ratnam, M.; Patki, M.; Kisovic, I.; Trumbly, R.; Iman, M.; Ratnam, M. During hormone depletion or tamoxifen treatment of breast cancer cells the estrogen receptor apoprotein supports cell cycling through the retinoic acid receptor α1 apoprotein. Breast Cancer Res. 2011, 13, R18. [Google Scholar] [CrossRef]
- Iida, M.; Nakamura, M.; Tokuda, E.; Toyosawa, D.; Niwa, T.; Ohuchi, N.; Ishida, T.; Hayashi, S.I. The p21 levels have the potential to be a monitoring marker for ribociclib in breast cancer. Oncotarget 2019, 10, 4907–4918. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.V.; Croce, C.M. MicroRNA dysregulation in cancer: Diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol. Med. 2012, 4, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Horie-Inoue, K.; Ueno, T.; Suzuki, T.; Sato, W.; Shigekawa, T.; Osaki, A.; Saeki, T.; Berezikov, E.; Mano, H.; et al. miR-378a-3p modulates tamoxifen sensitivity in breast cancer MCF-7 cells through targeting GOLT1A. Sci. Rep. 2015, 5, 13170. [Google Scholar] [CrossRef]
- Iorio, M.; Ferracin, M.; Liu, C.; Veronese, A.; Spizzo, R.; Sabbioni, S.; Magri, E.; Pedriali, M.; Fabbri, M.; Campiglio, M. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005, 65, 7065–7070. [Google Scholar] [CrossRef]
- Hydbring, P.; Badalian-Very, G. Clinical applications of microRNAs. F1000Research 2013, 2, 136. [Google Scholar] [CrossRef]
- Ebert, M.S.; Neilson, J.R.; Sharp, P.A. MicroRNA sponges: Competitive inhibitors of small RNAs in mammalian cells. Nat. Methods 2007, 4, 721–726. [Google Scholar] [CrossRef]
- Ouyang, Y.X.; Feng, J.; Wang, Z.; Zhang, G.J.; Chen, M. miR-221/222 sponge abrogates tamoxifen resistance in ER-positive breast cancer cells through restoring the expression of ERα. Mol. Biomed. 2021, 2, 20. [Google Scholar] [CrossRef]
- Di Leva, G.; Gasparini, P.; Piovan, C.; Ngankeu, A.; Garofalo, M.; Taccioli, C.; Iorio, M.V.; Li, M.; Volinia, S.; Alder, H.; et al. MicroRNA cluster 221-222 and estrogen receptor α interactions in breast cancer. J. Natl. Cancer Inst. 2010, 102, 706–721. [Google Scholar] [CrossRef]
- Martin, E.C.; Conger, A.K.; Yan, T.J.; Hoang, V.T.; Miller, D.F.; Buechlein, A.; Rusch, D.B.; Nephew, K.P.; Collins-Burow, B.M.; Burow, M.E. MicroRNA-335-5p and -3p synergize to inhibit estrogen receptor α expression and promote tamoxifen resistance. FEBS Lett. 2017, 591, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.-F.; Li, D.-M.; Shi, Z.-M.; Wang, L.; Liu, M.-M.; Ge, X.; Liu, X.; Qian, Y.-C.; Wen, Y.-Y.; Zhen, L.-L.; et al. Estrogen regulates miRNA expression: Implication of estrogen receptor and miR-124/AKT2 in tumor growth and angiogenesis. Oncotarget 2016, 7, 36940–36955. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Li, H.; Qu, Q. miR-484 suppresses endocrine therapy-resistant cells by inhibiting KLF4-induced cancer stem cells in estrogen receptor-positive cancers. Breast Cancer 2021, 28, 175–186. [Google Scholar] [CrossRef]
- Liu, L.; Shen, W.; Zhu, Z.; Lin, J.; Fang, Q.; Ruan, Y.; Zhao, H. Combined inhibition of EGFR and c-ABL suppresses the growth of fulvestrant-resistant breast cancer cells through miR-375-autophagy axis. Biochem. Biophys. Res. Commun. 2018, 498, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Luo, A.; Liu, Y.; Wang, S.; Li, Y.; Shi, W.; Liu, Z.; Qu, X. MiR-214 increases the sensitivity of breast cancer cells to tamoxifen and fulvestrant through inhibition of autophagy. Mol. Cancer 2015, 14, 208. [Google Scholar] [CrossRef]
- Yu, X.; Li, R.; Shi, W.; Jiang, T.; Wang, Y.; Li, C.; Qu, X. Silencing of MicroRNA-21 confers the sensitivity to tamoxifen and fulvestrant by enhancing autophagic cell death through inhibition of the PI3K-AKT-mTOR pathway in breast cancer cells. Biomed. Pharmacother. 2016, 77, 37–44. [Google Scholar] [CrossRef]
- Yao, X.; Tu, Y.; Xu, Y.; Guo, Y.; Yao, F.; Zhang, X. Endoplasmic reticulum stress-induced exosomal miR-27a-3p promotes immune escape in breast cancer via regulating PD-L1 expression in macrophages. J. Cell. Mol. Med. 2020, 24, 9560–9573. [Google Scholar] [CrossRef]
- Liu, S.S.; Li, Y.; Zhang, H.; Zhang, D.; Zhang, X.B.; Wang, X.; Yu, Y. The ERα-miR-575-p27 feedback loop regulates tamoxifen sensitivity in ER-positive Breast Cancer. Theranostics 2020, 10, 10729–10742. [Google Scholar] [CrossRef]
- Tokumaru, Y.; Asaoka, M.; Oshi, M.; Katsuta, E.; Yan, L.; Narayanan, S.; Sugito, N.; Matsuhashi, N.; Futamura, M.; Akao, Y.; et al. High Expression of microRNA-143 is Associated with Favorable Tumor Immune Microenvironment and Better Survival in Estrogen Receptor Positive Breast Cancer. Int. J. Mol. Sci. 2020, 21, 3213. [Google Scholar] [CrossRef]
- Nair, M.G.; Prabhu, J.S.; Korlimarla, A.; Rajarajan, S.; Hari, P.S.; Kaul, R.; Alexander, A.; Raghavan, R.; Srinath, B.S.; Sridhar, T.S. miR-18a activates Wnt pathway in ER-positive breast cancer and is associated with poor prognosis. Cancer Med. 2020, 9, 5587–5597. [Google Scholar] [CrossRef]
- Liu, J.; Li, X.; Wang, M.; Xiao, G.; Yang, G.; Wang, H.; Li, Y.; Sun, X.; Qin, S.; Du, N.; et al. A miR-26a/E2F7 feedback loop contributes to tamoxifen resistance in ER-positive breast cancer. Int. J. Oncol. 2018, 53, 1601–1612. [Google Scholar] [CrossRef] [PubMed]
- Gan, R.; Yang, Y.; Yang, X.; Zhao, L.; Lu, J.; Meng, Q.H. Downregulation of miR-221/222 enhances sensitivity of breast cancer cells to tamoxifen through upregulation of TIMP3. Cancer Gene Ther. 2014, 21, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhu, S.; Tang, W.; Huang, Q.; Mei, Y.; Yang, H. Exosomes from tamoxifen-resistant breast cancer cells transmit drug resistance partly by delivering miR-9-5p. Cancer Cell Int. 2021, 21, 55. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Park, S.J.; Lee, Y.S.; Kong, H.K.; Park, J.H. miRNAs involved in LY6K and estrogen receptor α contribute to tamoxifen-susceptibility in breast cancer. Oncotarget 2016, 7, 42261–42273. [Google Scholar] [CrossRef]
- Wang, B.; Li, D.; Filkowski, J.; Rodriguez-Juarez, R.; Storozynsky, Q.; Malach, M.; Carpenter, E.; Kovalchuk, O. A dual role of miR-22 modulated by RelA/p65 in resensitizing fulvestrant-resistant breast cancer cells to fulvestrant by targeting FOXP1 and HDAC4 and constitutive acetylation of p53 at Lys382. Oncogenesis 2018, 7, 54. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Q.; Li, X.; Luo, G.; Shen, M.; Shi, J.; Wang, X.; Tang, L. Paeoniflorin Sensitizes Breast Cancer Cells to Tamoxifen by Downregulating microRNA-15b via the FOXO1/CCND1/β-Catenin Axis. Drug Des. Dev. Ther. 2021, 15, 245–257. [Google Scholar] [CrossRef]
- Zhang, H.; Fan, Q. MicroRNA-205 inhibits the proliferation and invasion of breast cancer by regulating AMOT expression. Oncol. Rep. 2015, 34, 2163–2170. [Google Scholar] [CrossRef][Green Version]
- Wysokinski, D.; Blasiak, J.; Pawlowska, E. Role of RUNX2 in Breast Carcinogenesis. Int. J. Mol. Sci. 2015, 16, 20969–20993. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, W.; Liu, C.; Li, G. miR-200 affects tamoxifen resistance in breast cancer cells through regulation of MYB. Sci. Rep. 2019, 9, 18844. [Google Scholar] [CrossRef]
- Xu, E.; Hu, M.; Ge, R.; Tong, D.; Fan, Y.; Ren, X.; Liu, Y. LncRNA-42060 Regulates Tamoxifen Sensitivity and Tumor Development via Regulating the miR-204-5p/SOX4 Axis in Canine Mammary Gland Tumor Cells. Front. Vet. Sci. 2021, 8, 654694. [Google Scholar] [CrossRef]
- Hui, Z.; Yiling, C.; Wenting, Y.; XuQun, H.; ChuanYi, Z.; Hui, L. miR-491-5p functions as a tumor suppressor by targeting JMJD2B in ERα-positive breast cancer. FEBS Lett. 2015, 589, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Kondo, N.; Toyama, T.; Sugiura, H.; Fujii, Y.; Yamashita, H. miR-206 Expression is down-regulated in estrogen receptor α-positive human breast cancer. Cancer Res. 2008, 68, 5004–5008. [Google Scholar] [CrossRef] [PubMed]
- Cornell, L.; Wander, S.A.; Visal, T.; Wagle, N.; Shapiro, G.I. MicroRNA-Mediated Suppression of the TGF-β Pathway Confers Transmissible and Reversible CDK4/6 Inhibitor Resistance. Cell Rep. 2019, 26, 2667.e2667–2680.e2667. [Google Scholar] [CrossRef] [PubMed]
- Citron, F.; Segatto, I.; Vinciguerra, G.L.R.; Musco, L.; Russo, F.; Mungo, G.; D’Andrea, S.; Mattevi, M.C.; Perin, T.; Schiappacassi, M.; et al. Downregulation of miR-223 expression is an early event during mammary transformation and confers resistance to CDK4/6 inhibitors in luminal breast cancer. Cancer Res. 2020, 80, 1064–1077. [Google Scholar] [CrossRef]
- Feng, J.; Wen, T.; Li, Z.; Feng, L.; Zhou, L.; Yang, Z.; Xu, L.; Shi, S.; Hou, K.; Shen, J.; et al. Cross-talk between the ER pathway and the lncRNA MAFG-AS1/miR-339-5p/CDK2 axis promotes progression of ER+ breast cancer and confers tamoxifen resistance. Aging 2020, 12, 20658–20683. [Google Scholar] [CrossRef]
- Thangavel, C.; Boopathi, E.; Ertel, A.; Lim, M.; Addya, S.; Fortina, P.; Witkiewicz, A.K.; Knudsen, E.S. Regulation of miR106b cluster through the RB pathway: Mechanism and functional targets. Cell Cycle 2013, 12, 98–111. [Google Scholar] [CrossRef]
- Ward, A.; Shukla, K.; Balwierz, A.; Soons, Z.; König, R.; Sahin, O.; Wiemann, S. MicroRNA-519a is a novel oncomir conferring tamoxifen resistance by targeting a network of tumour-suppressor genes in ER+ breast cancer. J. Pathol. 2014, 233, 368–379. [Google Scholar] [CrossRef]
- Ujihira, T.; Ikeda, K.; Suzuki, T.; Yamaga, R.; Sato, W.; Horie-Inoue, K.; Shigekawa, T.; Osaki, A.; Saeki, T.; Okamoto, K.; et al. MicroRNA-574-3p, identified by microRNA library-based functional screening, modulates tamoxifen response in breast cancer. Sci. Rep. 2015, 5, 7641. [Google Scholar] [CrossRef]
- Li, J.; Lu, M.; Jin, J.; Lu, X.; Xu, T.; Jin, S. miR-449a Suppresses Tamoxifen Resistance in Human Breast Cancer Cells by Targeting ADAM22. Cell. Physiol. Biochem. 2018, 50, 136–149. [Google Scholar] [CrossRef]
- Ahmad, A.; Ginnebaugh, K.R.; Yin, S.; Bollig-Fischer, A.; Reddy, K.B.; Sarkar, F.H. Functional role of miR-10b in tamoxifen resistance of ER-positive breast cancer cells through down-regulation of HDAC4. BMC Cancer 2015, 15, 540. [Google Scholar] [CrossRef]





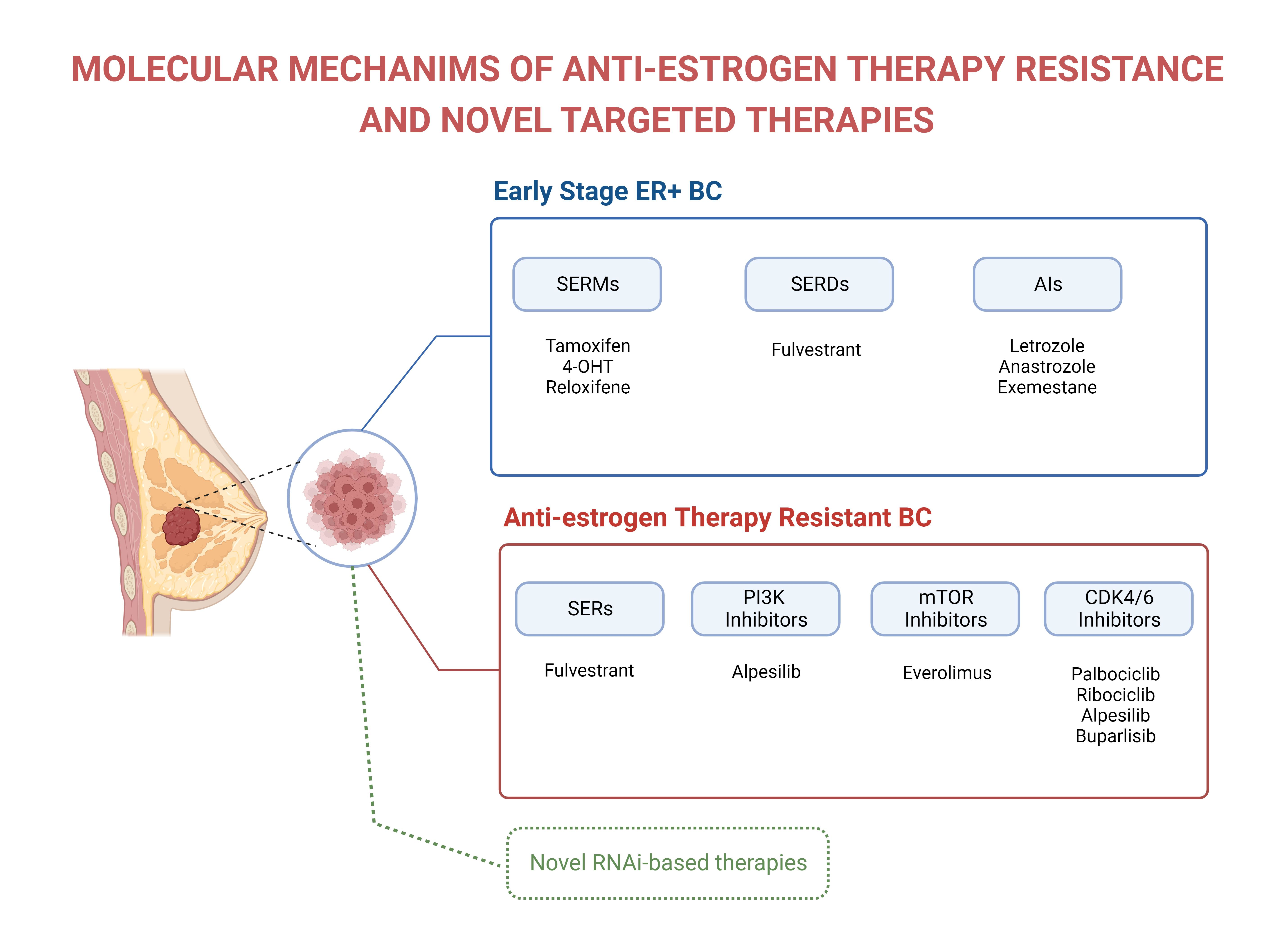{kind=link}
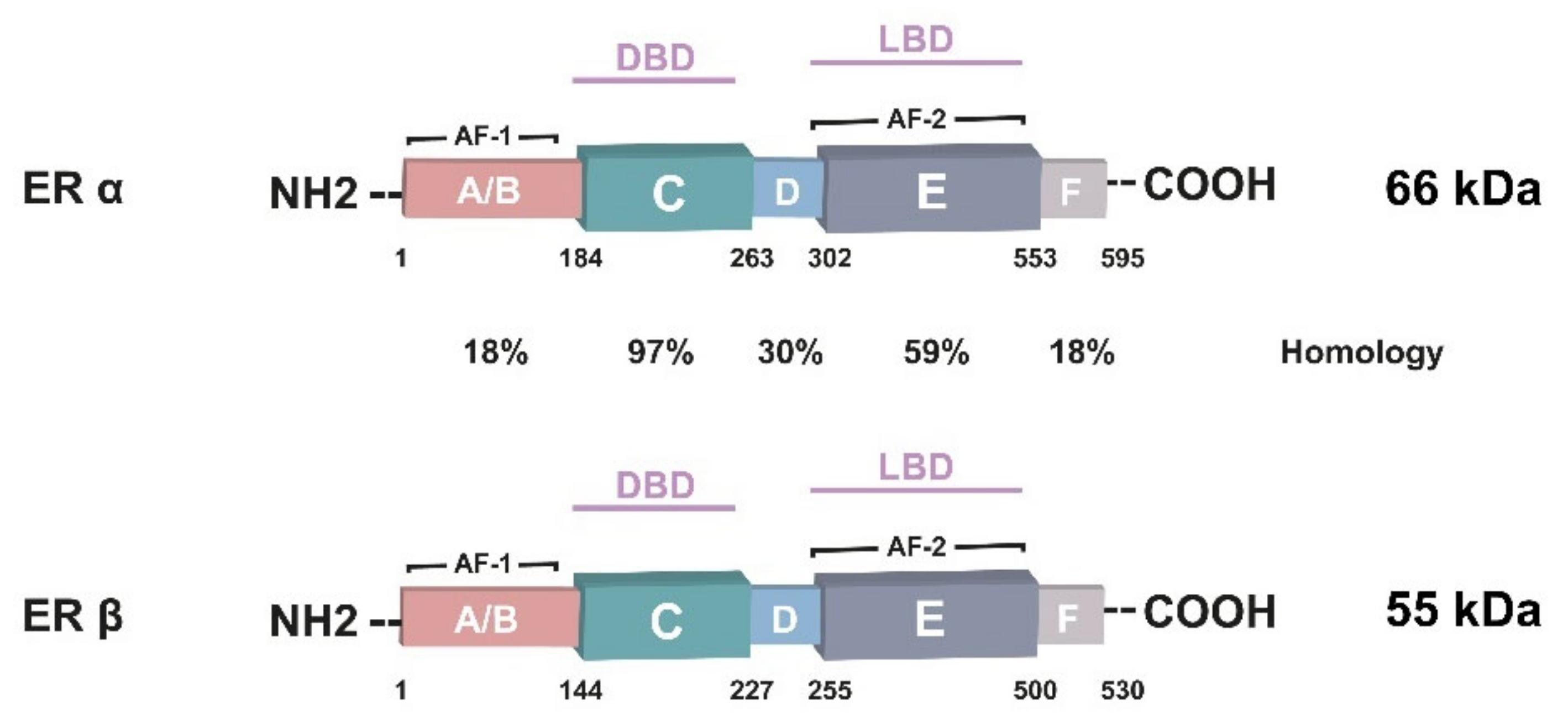{kind=link}
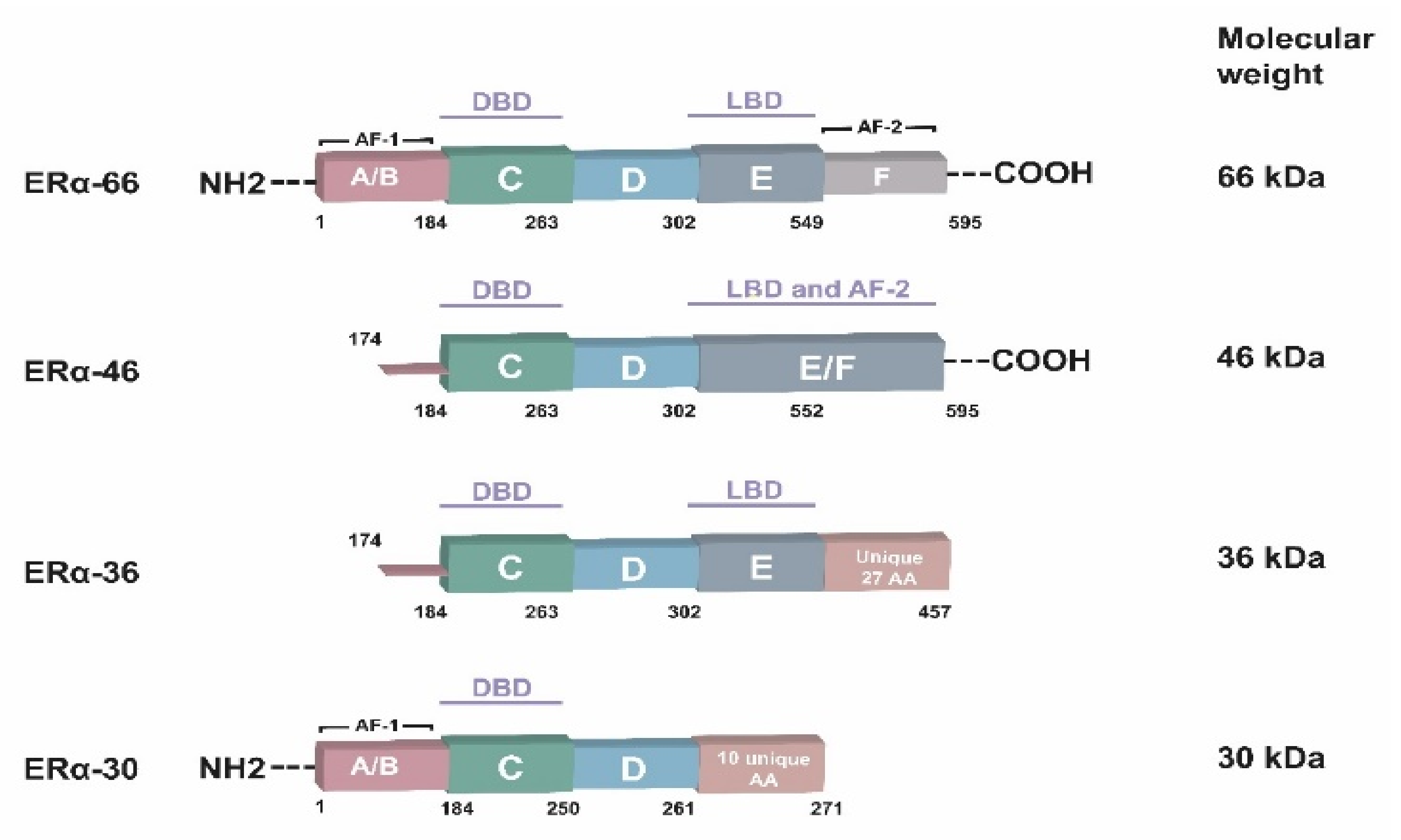{kind=link}
{kind=link}
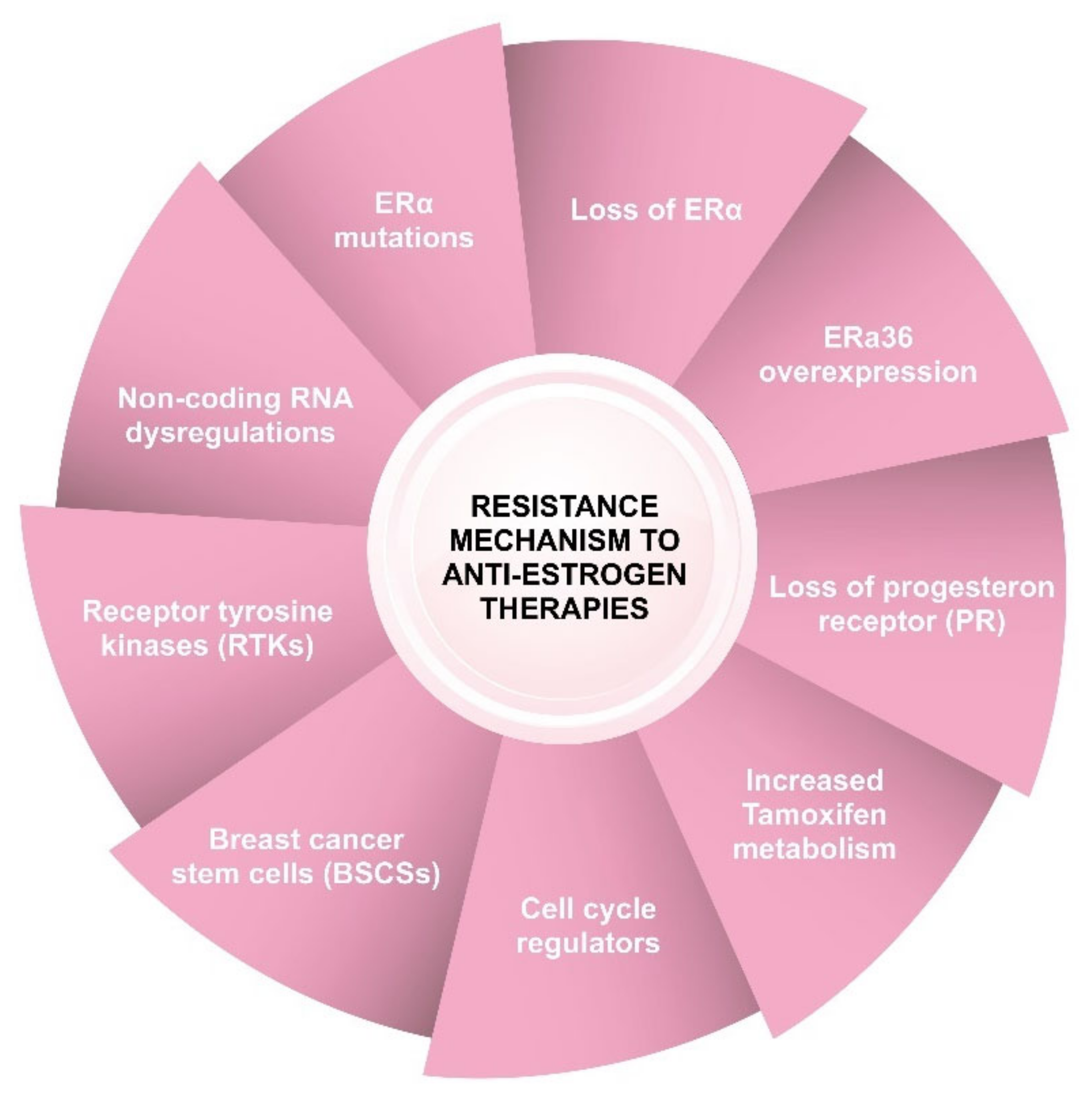{kind=link}
| Drug | Treatments | Clinical Trial Phase | Clinical Trial Registry Number |
|---|---|---|---|
| SERDs | |||
| Fulvestrant | Single agent | Phase III, active | NCT00099437 |
| LSZ102 | Single agent and combination with ribociclib or alpelisib | Phase I, terminated | NCT02734615 |
| RAD1901 (Elacestrant) | Single agent | Phase III, active | NCT03778931 |
| GDC-9545 (Giredestrant) | Single agent and combination with palbociclib and/or luteinizing hormone-releasing hormone (LHRH) agonist | Phase I, active | NCT03332797 |
| GDC-954 single agent compared with physician’s choice of endocrine monotherapy | Phase II, active | NCT04576455 | |
| GDC-9545 plus palbociclib compared with anastrozole plus Palbociclib | Phase II, completed | NCT04436744 | |
| GDC-9545 combined with palbociclib compared with letrozole combined with palbociclib | Phase III, recruiting | NCT04546009 | |
| Monotherapy | Phase I, completed | NCT03916744 | |
| SAR439859 (Amcenestrant) | Single agent and in combination with other anti-cancer therapies | Phase I/II, active | NCT03284957 |
| SAR439859 in comparison to endocrine monotherapy of the choice of the physician | Phase II, active | NCT04059484 | |
| G1T48 | Single agent and in combination with palbociclib | Phase I, active | NCT03455270 |
| AZD9833 | Single agent and combination with palbociclib or everolimus or abemaciclib or capivasertib | Phase I, recruiting | NCT03616587 |
| AZD9833 versus intramuscular (IM) fulvestrant | Phase II, active | NCT04214288 | |
| Single agent | Phase II, recruiting | NCT04588298 | |
| AZD9833 plus palbociclib versus anastrozole plus palbociclib | Phase III, recruiting | NCT04711252 | |
| PI3K/AKT/mTOR Inhibitors | |||
| Everolimus (RAD001) | Single agent | Phase III, active | NCT01805271 |
| Everolimus in combination with letrozole | Phase II, completed | NCT01231659 | |
| Everolimus combination with exemestane | Phase IV, completed | NCT01743560 | |
| Everolimus plus letrozole versus neoadjuvant fluorouracil, epirubicin plus cyclophosphamide (FEC) | Phase II, completed | NCT02742051 | |
| Everolimus combined with anti-estrogen therapy | Phase II, completed | NCT02291913 | |
| Everolimus in combination with exemestane | Phase III, completed | NCT00863655 | |
| Tamoxifen plus everolimus versus tamoxifen alone | Phase II, completed | NCT01298713 | |
| Buparlisib (BKM120) | Buparlisib plus fulvestrant versus fulvestrant plus placebo | Phase III, completed | NCT01610284 |
| Alpelisib | Alpelisib plus fulvestrant | Phase III, active | NCT02437318 |
| CDK4/CDK6 Inhibitors | |||
| Palbociclib | Palbociclib in combination with fulvestrant | Phase III, active | NCT01942135 |
| Palbociclib with everolimus plus exemestane | Phase I/II, completed | NCT02871791 | |
| Ribociclib (LEE011) | Ribociclib in combination with anti-estrogen therapy | Phase III, active | NCT02278120 |
| Ribociclib with everolimus plus exemestane | Phase I/II, completed | NCT02732119 | |
| Ribociclib with everolimus and exemestane | Phase I, completed | NCT01857193 | |
| Triplet combination of ribociclib, everolimus and exemastane | Phase I/II, completed | NCT02732119 | |
| Abemaciclib (LY2835219) | Abemaciclib plus fulvestrant or fulvestrant alone | Phase III, active | NCT02107703 |
| Abemaciclib in combination therapies | Phase I, active | NCT02057133 | |
| Cancer Hallmarks | In Vitro Study | In Vivo Study | Effect(s) | Reference |
|---|---|---|---|---|
| MED1 | + | + | Decreases cell viability, induces cell-cycle arrest, inhibits tumor growth | [266] |
| TRIM56 | + | + | Suppresses cell proliferation and tumor growth | [267] |
| Wee1, CDK4, CDK7 | + | + | Decreases tumor growth | [268] |
| MST2 | + | + | Induces apoptosis and decreases tumor growth | [269] |
| ERα | + | + | Decreases proliferation and vascularization of solid tumors | [270] |
| CDK2 | + | + | Inhibits cell proliferation and tumor growth | [271] |
| Bcl2 | + | + | Inhibits cell growth and tumor growth | [272] |
| LEM4 | + | + | Decreases cell proliferation and tumor growth | [273] |
| PDK1 | + | - | Decreases cell proliferation | [274] |
| ATG5, ATG7 | + | - | Inhibits autophagy | [275] |
| ZNF213 | + | - | Inhibits cell proliferation | [276] |
| RBCK1 | + | - | Reduces cell proliferation and G2-M arrest | [277] |
| S6 kinase 1 | + | - | Induces cell death via glucose deprivation | [278] |
| USP15 | + | + | Suppresses cell viability, triggers cell-cycle arrest, decreases tumor growth | [279] |
| AMPKα1/2 | + | - | Decreases cell survival | [280] |
| CDK7 | + | - | Decreases cell viability and increases apoptosis | [281] |
| MiRNA | Tumor Suppressor | Oncogenic | Target | In Vitro/in Vivo Study | Reference |
|---|---|---|---|---|---|
| miR-221 | - | + | ERα, TIMP3 | +/- | [166,302] |
| miR-222 | - | + | ERα | +/- | [166] |
| miR-192-5p | - | + | ERα, LY6K | +/- | [304] |
| miR-500-3p | + | - | ERα, LY6K | +/- | [304] |
| miR-335 | - | + | ERα | +/- | [291] |
| miR-378a-3p | + | - | GOLT1A | +/- | [285] |
| miR-484 | + | - | KLF4 | +/- | [293] |
| miR-22 | - | + | FOXP1, HDAC4 | +/- | [305] |
| miR-375 | + | - | ATG7, LC3-II | +/- | [294] |
| miR-214 | + | - | UCP2 | +/- | [295] |
| miR-21 | - | + | PTEN | +/- | [296] |
| miR-27a-3p | - | + | PDL1 | +/- | [297] |
| miR-575 | - | + | ERα, CDKN1B, BRCA1 | +/+ | [298] |
| miR-143 | + | - | KRAS | +/+ | [299] |
| miR-9-5p | - | + | ADIPOQ, ESR1 | +/+ | [303] |
| miR-18a | - | + | ERα, WNT | +/- | [300] |
| miR-26a | + | - | E2F7 | +/- | [301] |
| miR-15b | - | + | FOXO1 | +/- | [306] |
| miR-30c | + | - | ERα, HER4 | +/- | [180] |
| miR-205 | + | - | RUNX2, AMOT | +/- | [307,308] |
| miR-200c | + | - | CMYB | +/- | [309] |
| miR-204 | + | - | SOX4 | +/- | [310] |
| miR-491-5p | + | - | JMJD2B | +/- | [311] |
| miR-206 | + | - | ERα | +/- | [312] |
| miR-124 | + | - | AKT2 | +/+ | [292] |
| miR-432-5p | - | + | CDK6 | +/- | [313] |
| miR-223 | + | - | E2F1 | +/+ | [314] |
| miR-339-5p | + | - | CDK2 | +/- | [315] |
| miR-106b | - | + | p21 and PTEN | +/- | [316] |
| miR-519a | - | + | PTEN | +/- | [317] |
| miR-574-3p | + | - | CLTC | +/- | [318] |
| miR-449a | + | - | ADAM22 | +/- | [319] |
| miR-10b | - | + | HDAC4 | +/- | [320] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ozyurt, R.; Ozpolat, B. Molecular Mechanisms of Anti-Estrogen Therapy Resistance and Novel Targeted Therapies. Cancers 2022, 14, 5206. https://doi.org/10.3390/cancers14215206
Ozyurt R, Ozpolat B. Molecular Mechanisms of Anti-Estrogen Therapy Resistance and Novel Targeted Therapies. Cancers. 2022; 14(21):5206. https://doi.org/10.3390/cancers14215206
Chicago/Turabian StyleOzyurt, Rumeysa, and Bulent Ozpolat. 2022. "Molecular Mechanisms of Anti-Estrogen Therapy Resistance and Novel Targeted Therapies" Cancers 14, no. 21: 5206. https://doi.org/10.3390/cancers14215206
APA StyleOzyurt, R., & Ozpolat, B. (2022). Molecular Mechanisms of Anti-Estrogen Therapy Resistance and Novel Targeted Therapies. Cancers, 14(21), 5206. https://doi.org/10.3390/cancers14215206