
Complement Factor D Is a Novel Biomarker and Putative Therapeutic Target in Cutaneous Squamous Cell Carcinoma

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
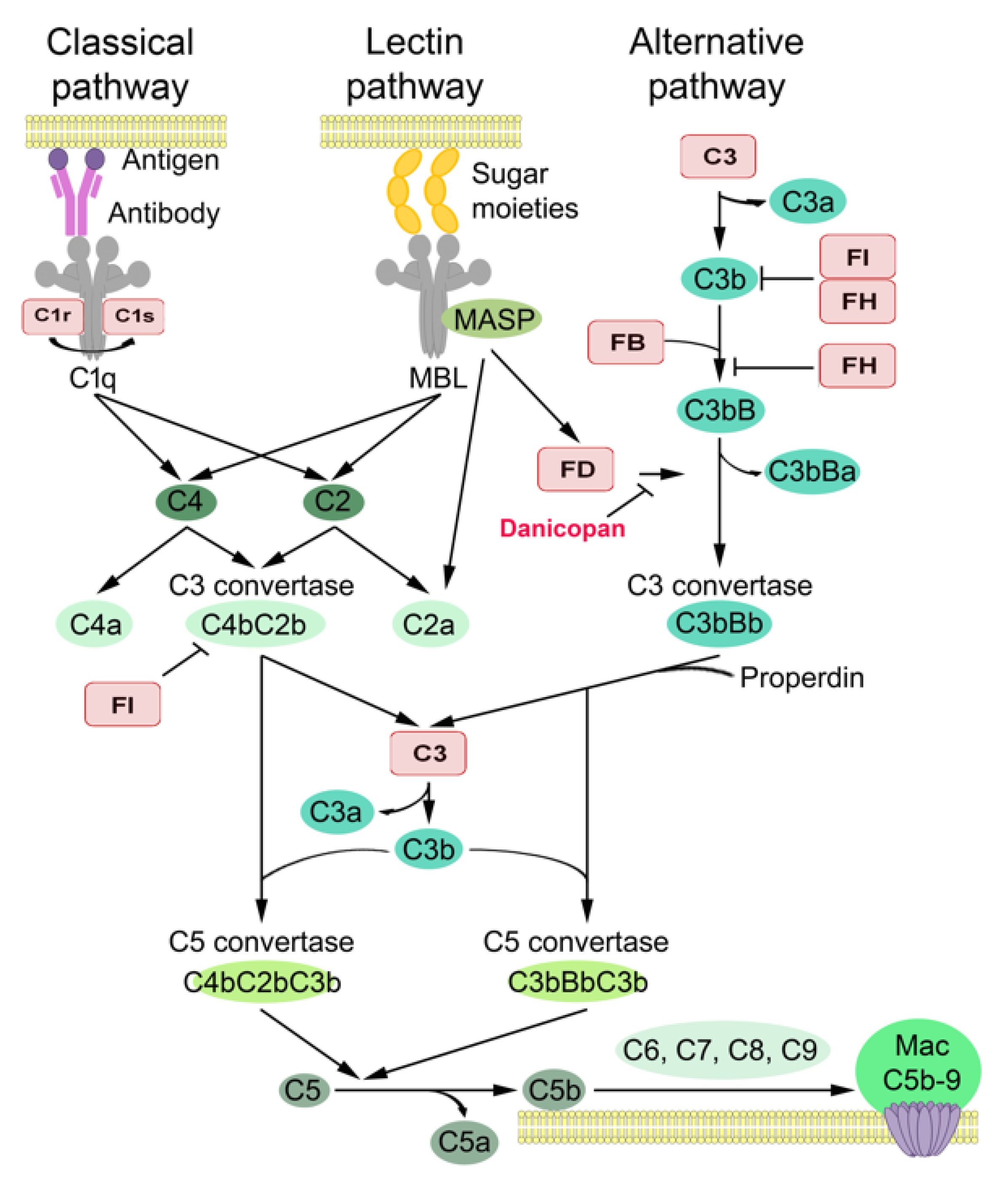{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Tissue RNA
2.3. Real-Time Quantitative PCR (RT-qPCR)
2.4. Western Blot Analysis
2.5. Tissue Samples and Immunohistochemistry
2.6. Human cSCC Xenografts
2.7. Targeted FD Inhibition and Cell Proliferation Assay
2.8. Statistical Analysis
3. Results
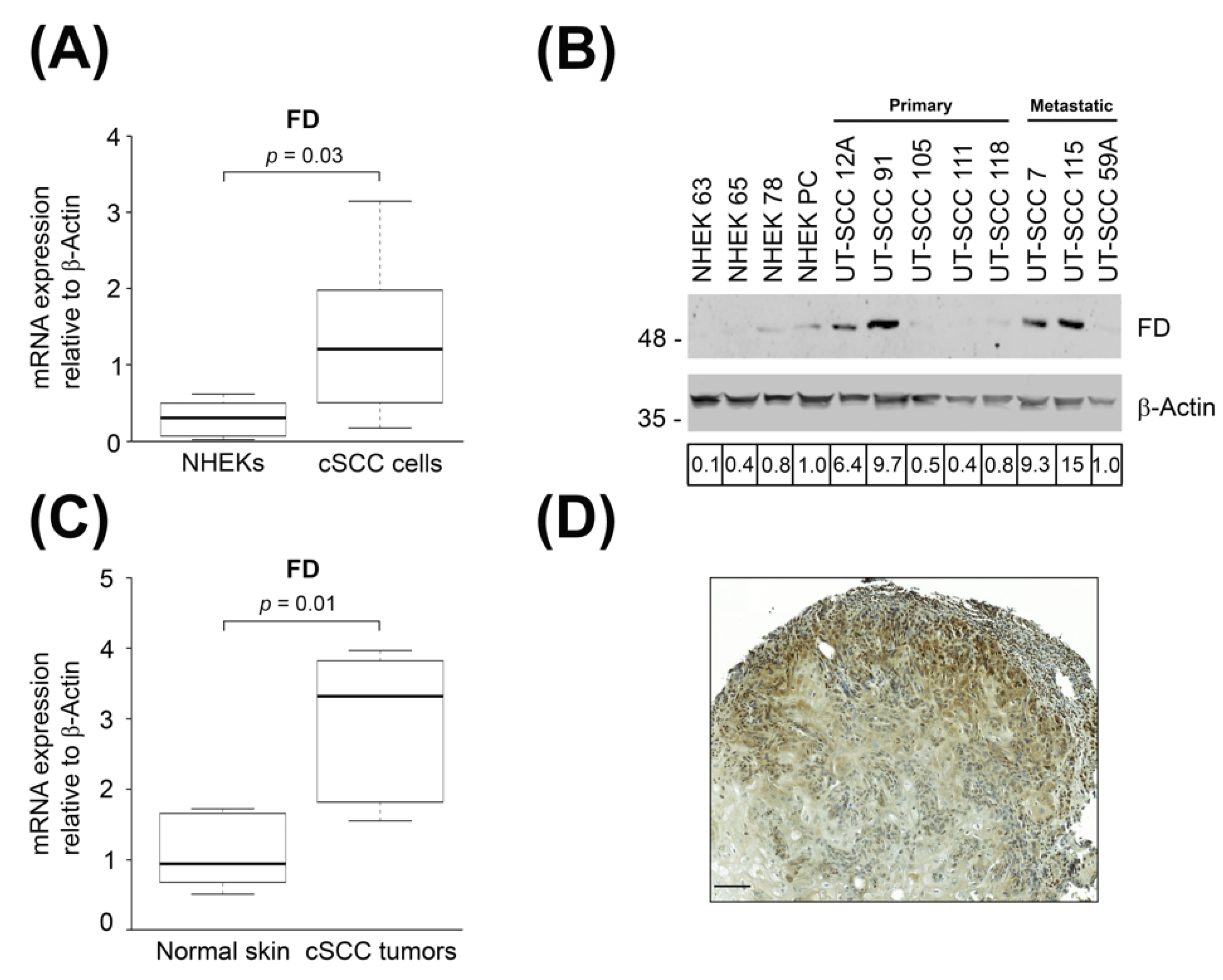
3.1. Expression of FD Is Up-Regulated in cSCC Cells and Tumors
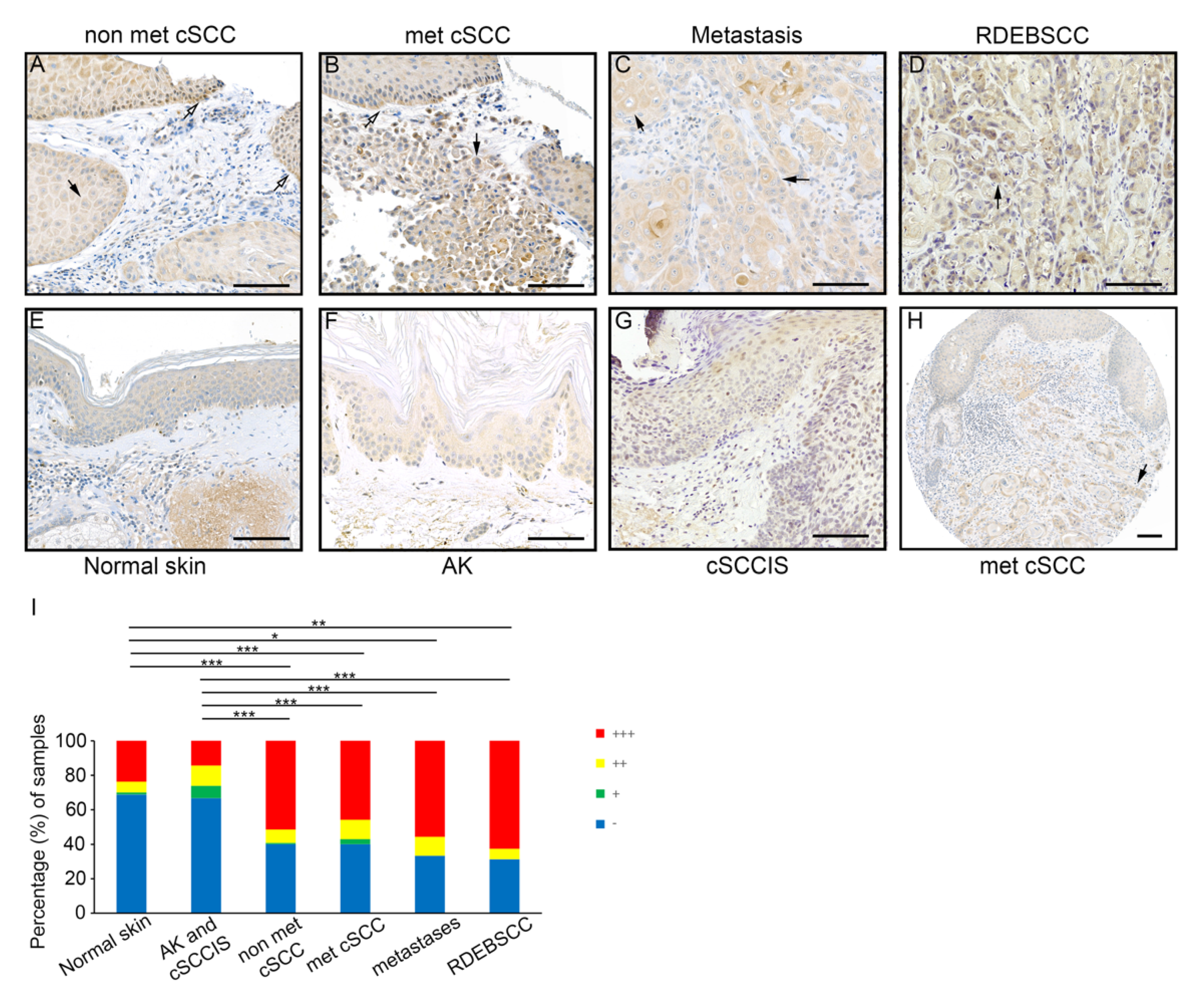
3.2. Expression of FD by Tumor Cells in cSCC In Vivo
3.3. Expression of FD in cSCC Cells Is Up-Regulated by IFN-γ and IL-1β
3.4. FD Expression by cSCC Cells Is Regulated by p38 MAPK Pathway
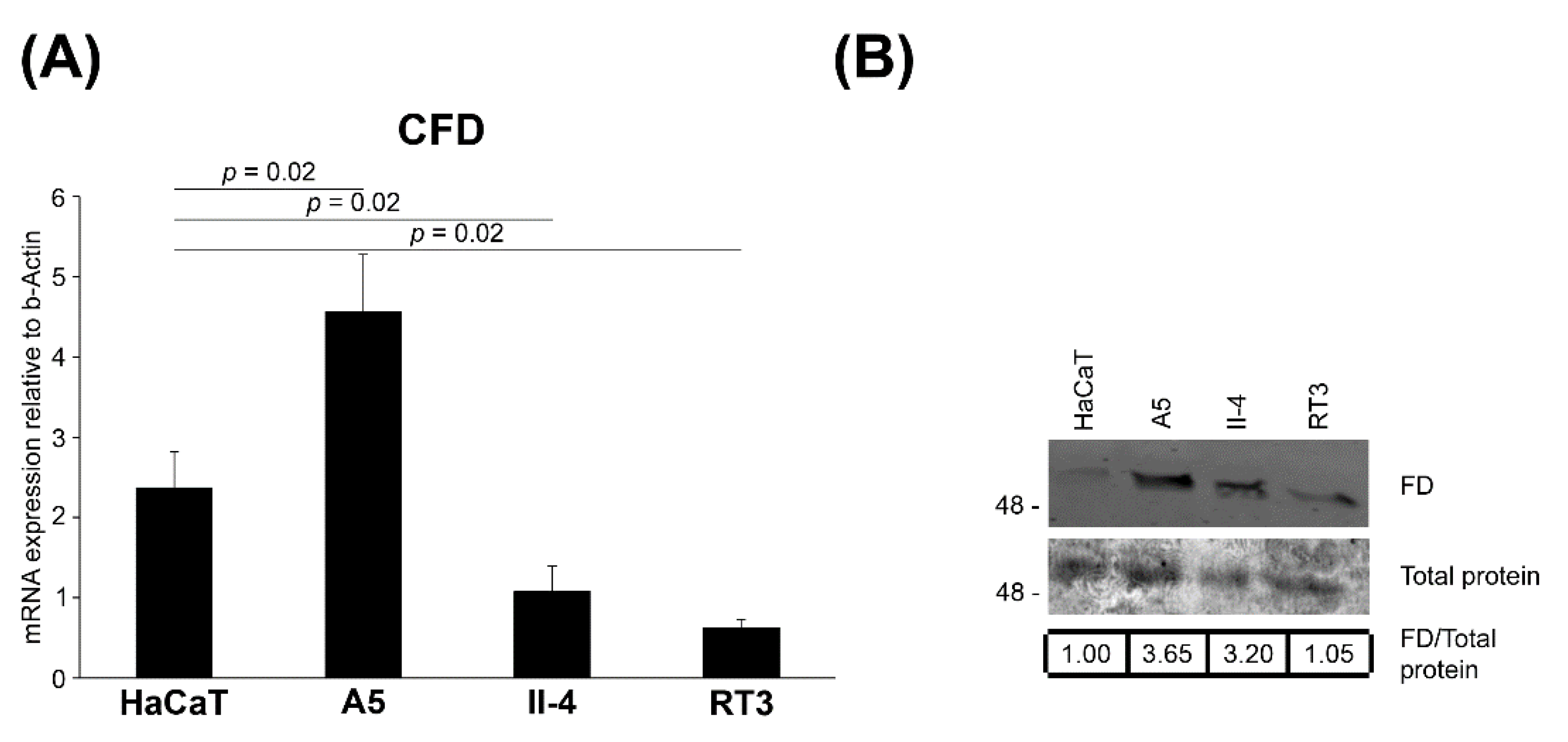
3.5. Expression of FD in Tumorigenic Ha-ras-Transformed HaCaT cells
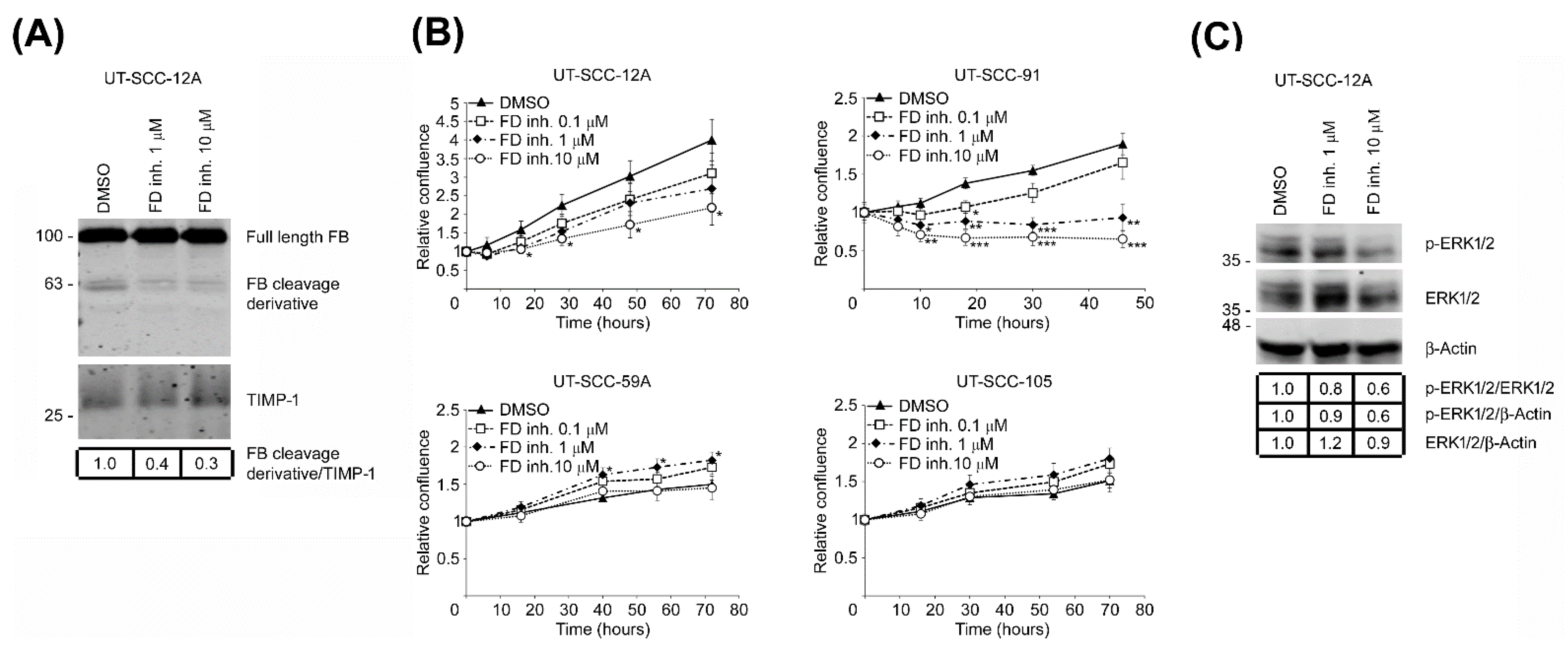
3.6. Targeted Inhibition of FD Inhibits Proliferation of cSCC Cells via Blockade of ERK1/2 Activation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nehal, K.S.; Bichakjian, C.K. Update on keratinocyte carcinomas. N. Engl. J. Med. 2018, 379, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, P.; Asgari, M.M.; Green, A.C.; Guhan, S.M.; Arron, S.T.; Proby, C.M.; Rollison, D.E.; Harwood, C.A.; Toland, A.E. Keratinocyte carcinomas: Current concepts and future research priorities. Clin. Cancer Res. 2019, 25, 2379–2391. [Google Scholar] [CrossRef]
- Green, A.C.; Olsen, C.M. Cutaneous squamous cell carcinoma: An epidemiological review. Br. J. Dermatol. 2017, 177, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Rogers, H.W.; Weinstock, M.A.; Feldman, S.R.; Coldiron, B.M. Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the U.S. population, 2012. JAMA Dermatol. 2015, 151, 1081–1086. [Google Scholar] [CrossRef]
- Karia, P.S.; Han, J.; Schmults, C.D. Cutaneous squamous cell carcinoma: Estimated incidence of disease, nodal metastasis, and deaths from disease in the United States, 2012. J. Am. Acad Derm. 2013, 68, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Que, S.K.T.; Zwald, F.O.; Schmults, C.D. Cutaneous squamous cell carcinoma: Incidence, risk factors, diagnosis, and staging. J. Am. Acad. Dermatol. 2018, 78, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Schmults, C.D.; Karia, P.S.; Carter, J.B.; Han, J.; Qureshi, A.A. Factors predictive of recurrence and death from cutaneous squamous cell carcinoma: A 10-year, single-institution cohort study. JAMA Dermatol. 2013, 149, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Knuutila, J.S.; Riihilä, P.; Kurki, S.; Nissinen, L.; Kähäri, V.M. Risk factors and prognosis for metastatic cutaneous squamous cell carcinoma: A cohort study. Acta Derm. Venereol. 2020, 100, adv00266. [Google Scholar] [CrossRef] [PubMed]
- Ratushny, V.; Gober, M.D.; Hick, R.; Ridky, T.W.; Seykora, J.T. From keratinocyte to cancer: The pathogenesis and modeling of cutaneous squamous cell carcinoma. J. Clin. Invest. 2012, 122, 464–472. [Google Scholar] [CrossRef]
- Riihilä, P.; Nissinen, L.; Kähäri, V.-M. Matrix metalloproteinases in keratinocyte carcinomas. Exp. Dermatol. 2021, 30, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Riihilä, P.; Nissinen, L.; Knuutila, J.; Rahmati Nezhad, P.; Viiklepp, K.; Kähäri, V.-M. Complement system in cutaneous squamous cell carcinoma. Int. J. Mol. Sci. 2019, 20, 3550. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef]
- Serna, M.; Giles, J.L.; Morgan, B.P.; Bubeck, D. Structural basis of complement membrane attack complex formation. Nat. Commun. 2016, 7, 10587. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, M.J.; Sughrue, M.E.; Kane, A.J.; Ahn, B.J.; Fang, S.; Parsa, A.T. The complement cascade as a mediator of tissue growth and regeneration. Inflamm. Res. 2010, 59, 897–905. [Google Scholar] [CrossRef][Green Version]
- Bohlson, S.S.; Garred, P.; Kemper, C.; Tenner, A.J. Complement nomenclature deconvoluted. Front. Immunol. 2019, 10, 1308. [Google Scholar] [CrossRef]
- Dobó, J.; Szakács, D.; Oroszlán, G.; Kortvely, E.; Kiss, B.; Boros, E.; Szász, R.; Závodszky, P.; Gál, P.; Pál, G. MASP-3 is the exclusive pro-factor D activator in resting blood: The lectin and the alternative complement pathways are fundamentally linked. Sci. Rep. 2016, 6, 31877. [Google Scholar] [CrossRef]
- White, R.T.; Damm, D.; Hancock, N.; Rosen, B.S.; Lowell, B.B.; Usher, P.; Flier, J.S.; Spiegelman, B.M. Human adipsin is identical to complement factor D and is expressed at high levels in adipose tissue. J. Biol. Chem. 1992, 267, 9210–9213. [Google Scholar] [CrossRef]
- Maibaum, J.; Liao, S.-M.; Vulpetti, A.; Ostermann, N.; Randl, S.; Rüdisser, S.; Lorthiois, E.; Erbel, P.; Kinzel, B.; Kolb, F.A.; et al. Small-molecule factor D inhibitors targeting the alternative complement pathway. Nat. Chem. Biol. 2016, 12, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Jlstra, A.; Webers, C.A.B.; Berendschot, T.T.J.M. Lutein and factor D: Two intriguing players in the field of age-related macular degeneration. Arch Biochem. Biophys. 2015, 572, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Mamidi, S.; Höne, S.; Kirschfink, M. The complement system in cancer: Ambivalence between tumour destruction and promotion. Immunobiology 2017, 222, 45–54. [Google Scholar] [CrossRef]
- Afshar-Kharghan, V. The role of the complement system in cancer. J. Clin. Investig. 2017, 127, 780–789. [Google Scholar] [CrossRef]
- Reis, E.S.; Mastellos, D.C.; Ricklin, D.; Mantovani, A.; Lambris, J.D. Complement in cancer: Untangling an intricate relationship. Nat. Rev. Immunol. 2018, 18, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Roumenina, L.T.; Daugan, M.V.; Petitprez, F.; Sautés-Fridman, C.; Fridman, W.H. Context-dependent roles of complement in cancer. Nat. Rev. Cancer 2019, 19, 698–715. [Google Scholar] [CrossRef] [PubMed]
- Netti, G.S.; Franzin, R.; Stasi, A.; Spadaccino, F.; Dello Strologo, A.; Infante, B.; Gesualdo, L.; Castellano, G.; Ranieri, E.; Stallone, G.; et al. Role of complement in regulating inflammation processes in renal and prostate cancers. Cells. 2021, 10, 2426. [Google Scholar] [CrossRef]
- Riihilä, P.M.; Nissinen, L.M.; Ala-aho, R.; Kallajoki, M.; Grénman, R.; Meri, S.; Peltonen, S.; Peltonen, J.; Kähäri, V.M. Complement factor H: A biomarker for progression of cutaneous squamous cell carcinoma. J. Investig. Dermatol. 2014, 134, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Riihilä, P.; Nissinen, L.; Farshchian, M.; Kivisaari, A.; Ala-aho, R.; Kallajoki, M.; Grénman, R.; Meri, S.; Peltonen, S.; Peltonen, J.; et al. Complement factor I promotes progression of cutaneous squamous cell carcinoma. J. Investig. Dermatol. 2015, 135, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Riihilä, P.; Nissinen, L.; Farshchian, M.; Kallajoki, M.; Kivisaari, A.; Meri, S.; Grénman, R.; Peltonen, S.; Peltonen, J.; Pihlajaniemi, T.; et al. Complement component C3 and complement factor B promote growth of cutaneous squamous cell carcinoma. Am. J. Pathol. 2017, 187, 1186–1197. [Google Scholar] [CrossRef] [PubMed]
- Riihilä, P.; Viiklepp, K.; Nissinen, L.; Farshchian, M.; Kallajoki, M.; Kivisaari, A.; Meri, S.; Peltonen, J.; Peltonen, S.; Kähäri, V.M. Tumour-cell-derived complement components C1r and C1s promote growth of cutaneous squamous cell carcinoma. Br. J. Dermatol. 2020, 182, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Viiklepp, K.; Nissinen, L.; Ojalill, M.; Riihilä, P.; Kallajoki, M.; Meri, S.; Heino, J.; Kähäri, V.M. C1r upregulates production of matrix metalloproteinase-13 and promotes invasion of cutaneous squamous cell carcinoma. J Investig. Dermatol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Rahmati Nezhad, P.; Riihilä, P.; Piipponen, M.; Kallajoki, M.; Meri, S.; Nissinen, L.; Kähäri, V.M. Complement factor I upregulates expression of matrix metalloproteinase-13 and -2 and promotes invasion of cutaneous squamous carcinoma cells. Exp. Dermatol. 2021, 30, 1631–1641. [Google Scholar] [CrossRef]
- Farshchian, M.; Kivisaari, A.; Ala-aho, R.; Riihilä, P.; Kallajoki, M.; Grénman, R.; Peltonen, J.; Pihlajaniemi, T.; Heljasvaara, R.; Kähäri, V.M. Serpin peptidase inhibitor clade A member 1 (SerpinA1) is a novel biomarker for progression of cutaneous squamous cell carcinoma. Am. J. Pathol. 2011, 179, 1110–1119. [Google Scholar] [CrossRef] [PubMed]
- Farshchian, M.; Nissinen, L.; Grénman, R.; Kähäri, V.-M. Dasatinib promotes apoptosis of cutaneous squamous carcinoma cells by regulating activation of ERK1/2. Exp. Dermatol. 2017, 26, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Farshchian, M.; Nissinen, L.; Siljamäki, E.; Riihilä, P.; Toriseva, M.; Kivisaari, A.; Ala-aho, R.; Kallajoki, M.; Veräjänkorva, E.; Honkanen, H.K.; et al. EphB2 promotes progression of cutaneous squamous cell carcinoma. J. Investig. Dermatol. 2015, 135, 1882–1892. [Google Scholar] [CrossRef] [PubMed]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef]
- Boukamp, P.; Stanbridge, E.J.; Foo, D.Y.; Cerutti, P.A.; Fusenig, N.E. c-Ha-ras oncogene expression in immortalized human keratinocytes (HaCaT) alters growth potential in vivo but lacks correlation with malignancy. Cancer Res. 1990, 50, 2840–2847. [Google Scholar] [PubMed]
- Mueller, M.M.; Peter, W.; Mappes, M.; Huelsen, A.; Steinbauer, H.; Boukamp, P.; Vaccariello, M.; Garlick, J.; Fusenig, N.E. Tumor progression of skin carcinoma cells in vivo promoted by clonal selection, mutagenesis, and autocrine growth regulation by granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor. Am. J. Pathol. 2001, 159, 1567–1579. [Google Scholar] [CrossRef][Green Version]
- Stokes, A.; Joutsa, J.; Ala-aho, R.; Pitchers, M.; Pennington, C.J.; Martin, C.; Premachandra, D.J.; Okada, Y.; Peltonen, J.; Grénman, R.; et al. Expression profiles and clinical correlations of degradome components in the tumor microenvironment of head and neck squamous cell carcinoma. Clin. Cancer Res. 2010, 16, 2022–2035. [Google Scholar] [CrossRef]
- Kivisaari, A.K.; Kallajoki, M.; Mirtti, T.; McGrath, J.A.; Bauer, J.W.; Weber, F.; Königová, R.; Sawamura, D.; Sato-Matsumura, K.C.; Shimizu, H.; et al. Transformation-specific matrix metalloproteinases (MMP)-7 and MMP-13 are expressed by tumour cells in epidermolysis bullosa-associated squamous cell carcinomas. Br. J. Dermatol. 2008, 158, 778–785. [Google Scholar] [CrossRef]
- Kivisaari, A.K.; Kallajoki, M.; Ala-aho, R.; McGrath, J.A.; Bauer, J.W.; Königová, R.; Medvecz, M.; Beckert, W.; Grénman, R.; Kähäri, V.M. Matrix metalloproteinase-7 activates heparin-binding epidermal growth factor-like growth factor in cutaneous squamous cell carcinoma. Br. J. Dermatol. 2010, 163, 726–735. [Google Scholar] [CrossRef]
- Kononen, J.; Bubendorf, L.; Kallioniemi, A.; Bärlund, M.; Schraml, P.; Leighton, S.; Torhorst, J.; Mihatsch, M.J.; Sauter, G.; Kallioniemi, O.P. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat. Med. 1998, 4, 844–847. [Google Scholar] [CrossRef]
- Yuan, X.; Gavriilaki, E.; Thanassi, J.A.; Yang, G.; Baines, A.C.; Podos, S.D.; Huang, Y.; Huang, M.; Brodsky, R.A. Small-molecule factor D inhibitors selectively block the alternative pathway of complement in paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. Haematologica 2017, 102, 466–475. [Google Scholar] [CrossRef]
- Junttila, M.R.; Ala-aho, R.; Jokilehto, T.; Peltonen, J.; Kallajoki, M.; Grenman, R.; Jaakkola, P.; Westermarck, J.; Kähäri, V.M. p38α and p38δ mitogen-activated protein kinase isoforms regulate invasion and growth of head and neck squamous carcinoma cells. Oncogene 2007, 26, 5267–5279. [Google Scholar] [CrossRef] [PubMed]
- Toriseva, M.; Ala-aho, R.; Peltonen, S.; Peltonen, J.; Grénman, R.; Kähäri, V.M. Keratinocyte growth factor induces gene expression signature associated with suppression of malignant phenotype of cutaneous squamous carcinoma cells. PLoS ONE 2012, 7, e33041. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Lear, J.T.; Szeimies, R.-M. Non-melanoma skin cancer. Lancet 2010, 375, 673–685. [Google Scholar] [CrossRef]
- Lee, S.H.; Jang, J.J.; Lee, J.Y.; Kim, S.Y.; Park, W.S.; Shin, M.S.; Dong, S.M.; Na, E.Y.; Kim, K.M.; Kim, C.S.; et al. Fas ligand is expressed in normal skin and in some cutaneous malignancies. Br. J. Dermatol. 1998, 139, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Modlin, R.L.; Moy, R.L.; Dubinett, S.M.; McHugh, T.; Nickoloff, B.J.; Uyemura, K. IL-10 production in cutaneous basal and squamous cell carcinomas. A mechanism for evading the local T cell immune response. J. Immunol. 1995, 155, 2240–2247. [Google Scholar] [PubMed]
- Johansson, N.; Ala-aho, R.; Uitto, V.; Grénman, R.; Fusenig, N.E.; López-Otín, C.; Kähäri, V.M. Expression of collagenase-3 (MMP-13) and collagenase-1 (MMP-1) by transformed keratinocytes is dependent on the activity of p38 mitogen-activated protein kinase. J. Cell Sci. 2000, 113, 227–235. [Google Scholar] [CrossRef]
- Piipponen, M.; Nissinen, L.; Farshchian, M.; Riihilä, P.; Kivisaari, A.; Kallajoki, M.; Peltonen, J.; Peltonen, S.; Kähäri, V.M. Long noncoding RNA PICSAR promotes growth of cutaneous squamous cell carcinoma by regulating ERK1/2 activity. J. Investig. Dermatol. 2016, 136, 1701–1710. [Google Scholar] [CrossRef]
- Piipponen, M.; Riihilä, P.; Nissinen, L.; Kähäri, V.M. The Role of p53 in Progression of cutaneous squamous cell carcinoma. Cancers 2021, 13, 4507. [Google Scholar] [CrossRef]
- Kitano, E.; Kitamura, H. Synthesis of factor D by gastric cancer-derived cell lines. Int. Immunopharmacol. 2002, 2, 843–848. [Google Scholar] [CrossRef]
- Barnum, S.R.; Ishii, Y.; Agrawal, A.; Volanakis, J.E. Production and interferon-gamma-mediated regulation of complement component C2 and factors B and D by the astroglioma cell line U105-MG. Biochem. J. 1992, 287, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Goto, H.; Shimono, Y.; Funakoshi, Y.; Imamura, Y.; Toyoda, M.; Kiyota, N.; Kono, S.; Takao, S.; Mukohara, T.; Minami, H. Adipose-derived stem cells enhance human breast cancer growth and cancer stem cell-like properties through adipsin. Oncogene 2019, 38, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.D.; Gulino, A.; Fossati-Jimack, L.; Castro Seoane, R.; Tian, K.; Best, K.; Köhl, J.; Belmonte, B.; Strid, J.; Botto, M.J. C3 drives inflammatory skin carcinogenesis independently of C5. J. Investig. Dermatol. 2021, 141, 404–414.e6. [Google Scholar] [CrossRef] [PubMed]
- Bareke, H.; Akbuga, J. Complement system’s role in cancer and its therapeutic potential in ovarian cancer. Scand. J. Immunol. 2018, 88, e12672. [Google Scholar] [CrossRef] [PubMed]
- A Long-Term Treatment Study of ACH-0144471 in Participants with Paroxysmal Nocturnal Hemoglobinuria (PNH). Available online: https://clinicaltrials.gov/ct2/show/NCT03181633?term=ACH-0144471+in+Participants+With+Paroxysmal+Nocturnal+Hemoglobinuria+%28PNH%29&draw=2&rank=1 (accessed on 1 December 2021).
- Study of Danicopan in Participants with Paroxysmal Nocturnal Hemoglobinuria with Inadequate Response to Eculizumab (pnh). Available online: https://clinicaltrials.gov/ct2/show/NCT03472885?term=Danicopan+in+Participants+With+Paroxysmal+Nocturnal+HemoglobinHemo&draw=2&rank=2 (accessed on 1 December 2021).
- Danicopan as Add-On Therapy to a c5 Inhibitor in Paroxysmal Nocturnal Hemoglobinuria (PNH) Participants Who Have Clinically Evident Extravascular Hemolysis (Evh)(Alpha). Available online: https://clinicaltrials.gov/ct2/show/NCT04469465?term=Danicopan+in+Participants+With+Paroxysmal+Nocturnal+Hemoglobinuria&draw=2&rank=3 (accessed on 1 December 2021).
- A Proof of Concept Study for a 12 Month Treatment in Patients with c3 Glomerulopathy (c3g) or Immune-Complex Membranoproliferative Glomerulonephritis (ic-mpgn). Available online: https://clinicaltrials.gov/ct2/show/NCT03459443?term=Danicopan&draw=3&rank=15 (accessed on 1 December 2021).
- A Study of Danicopan in Participants with Geographic Atrophy Secondary to Age-Related Macular Degeneration. Available online: https://clinicaltrials.gov/ct2/show/NCT05019521?term=Danicopan&draw=5&rank=5 (accessed on 1 December 2021).
- ACTIV-5/Big Effect Trial (bet-c) for the Treatment of COVID-19. Available online: https://clinicaltrials.gov/ct2/show/NCT04988035?term=Danicopan&draw=4&rank=22 (accessed on 1 December 2021).
- Proof of Concept Study of Ach-0145228 in Paroxysmal Nocturnal Hemoglobinuria (PNH). Available online: https://clinicaltrials.gov/ct2/show/NCT04170023?term=Danicopan&draw=4&rank=23 (accessed on 1 December 2021).
- A Long Term Safety Study of bcx9930 in subjects with Paroxysmal Nocturnal Hemoglobinuria (PNH). Available online: https://clinicaltrials.gov/ct2/show/NCT04702568?term=BCX9930&draw=2&rank=1 (accessed on 1 December 2021).
- IPH5401 (Anti-C5aR) in Combination with Durvalumab in Patients with Advanced Solid Tumors (STELLAR-001). Available online: https://clinicaltrials.gov/ct2/show/NCT03665129?term=5401+%28Anti-C5aR%29+in+Combination+With&draw=2&rank=1 (accessed on 1 December 2021).
- First-In-Human Trial with Single-Dose c5a-Neutralizing aon-d21 in Healthy Male Subjects. Available online: https://clinicaltrials.gov/ct2/show/NCT05018403?term=AON-D21&draw=2&rank=1 (accessed on 1 December 2021).






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahmati Nezhad, P.; Riihilä, P.; Knuutila, J.S.; Viiklepp, K.; Peltonen, S.; Kallajoki, M.; Meri, S.; Nissinen, L.; Kähäri, V.-M. Complement Factor D Is a Novel Biomarker and Putative Therapeutic Target in Cutaneous Squamous Cell Carcinoma. Cancers 2022, 14, 305. https://doi.org/10.3390/cancers14020305
Rahmati Nezhad P, Riihilä P, Knuutila JS, Viiklepp K, Peltonen S, Kallajoki M, Meri S, Nissinen L, Kähäri V-M. Complement Factor D Is a Novel Biomarker and Putative Therapeutic Target in Cutaneous Squamous Cell Carcinoma. Cancers. 2022; 14(2):305. https://doi.org/10.3390/cancers14020305
Chicago/Turabian StyleRahmati Nezhad, Pegah, Pilvi Riihilä, Jaakko S. Knuutila, Kristina Viiklepp, Sirkku Peltonen, Markku Kallajoki, Seppo Meri, Liisa Nissinen, and Veli-Matti Kähäri. 2022. "Complement Factor D Is a Novel Biomarker and Putative Therapeutic Target in Cutaneous Squamous Cell Carcinoma" Cancers 14, no. 2: 305. https://doi.org/10.3390/cancers14020305
APA StyleRahmati Nezhad, P., Riihilä, P., Knuutila, J. S., Viiklepp, K., Peltonen, S., Kallajoki, M., Meri, S., Nissinen, L., & Kähäri, V.-M. (2022). Complement Factor D Is a Novel Biomarker and Putative Therapeutic Target in Cutaneous Squamous Cell Carcinoma. Cancers, 14(2), 305. https://doi.org/10.3390/cancers14020305