
Proliferation and Immune Response Gene Signatures Associated with Clinical Outcome to Immunotherapy and Targeted Therapy in Metastatic Cutaneous Malignant Melanoma

 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
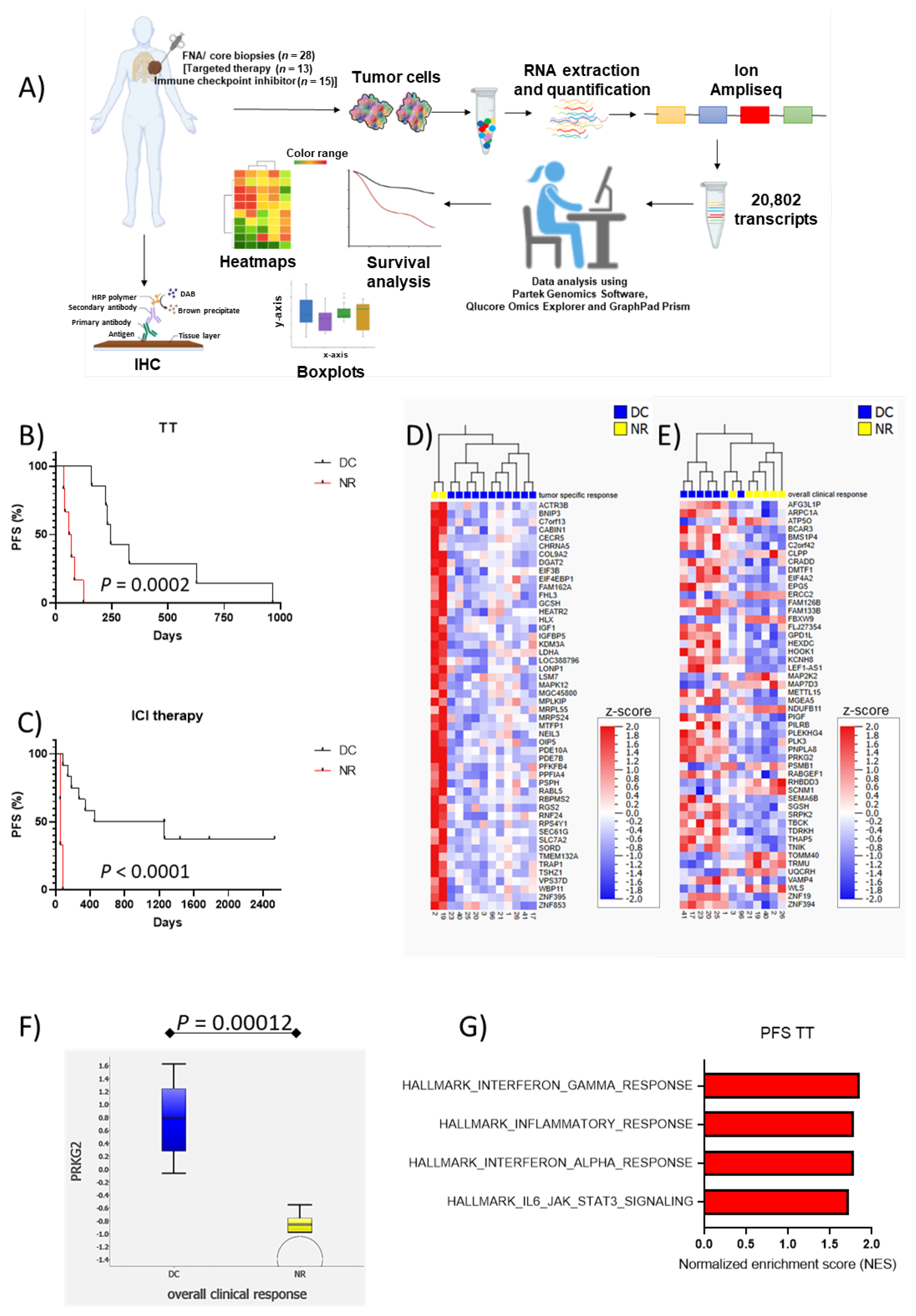
2. Material and Methods
2.1. Patients
2.2. RNA Extraction
2.3. Targeted Sequencing Using Ion AmpliSeq™
2.4. Immunohistochemistry
2.5. Statistical Analysis
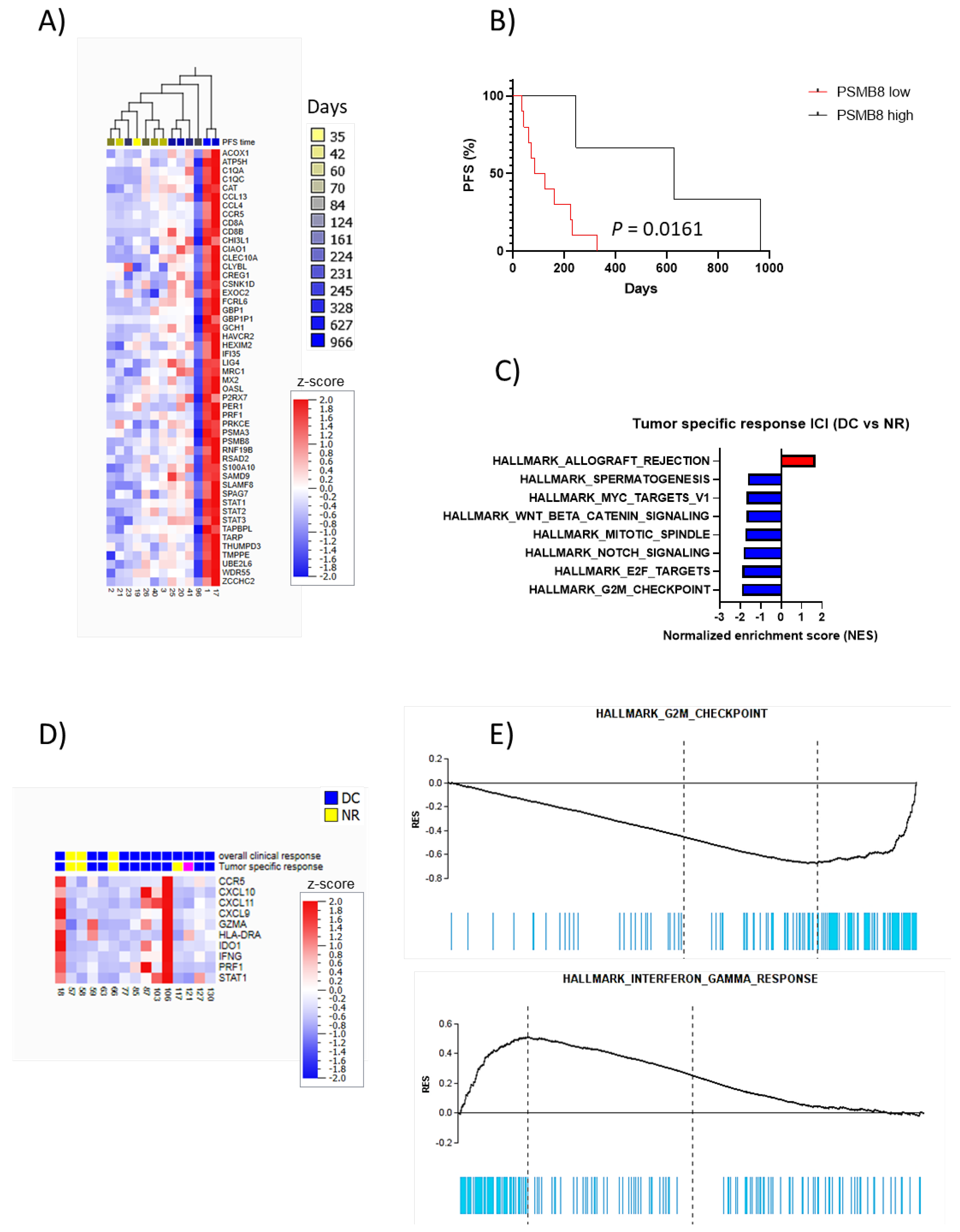
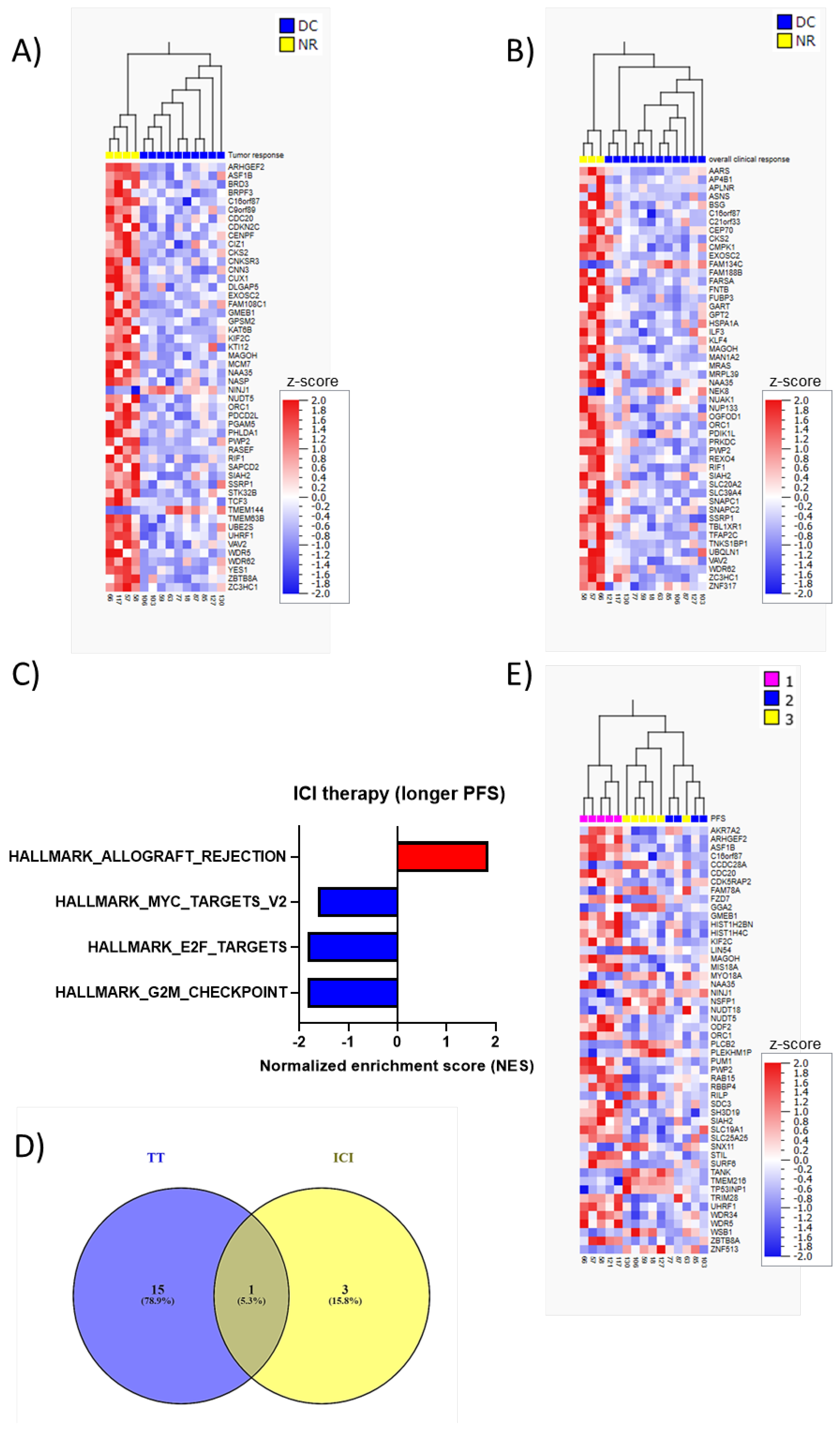
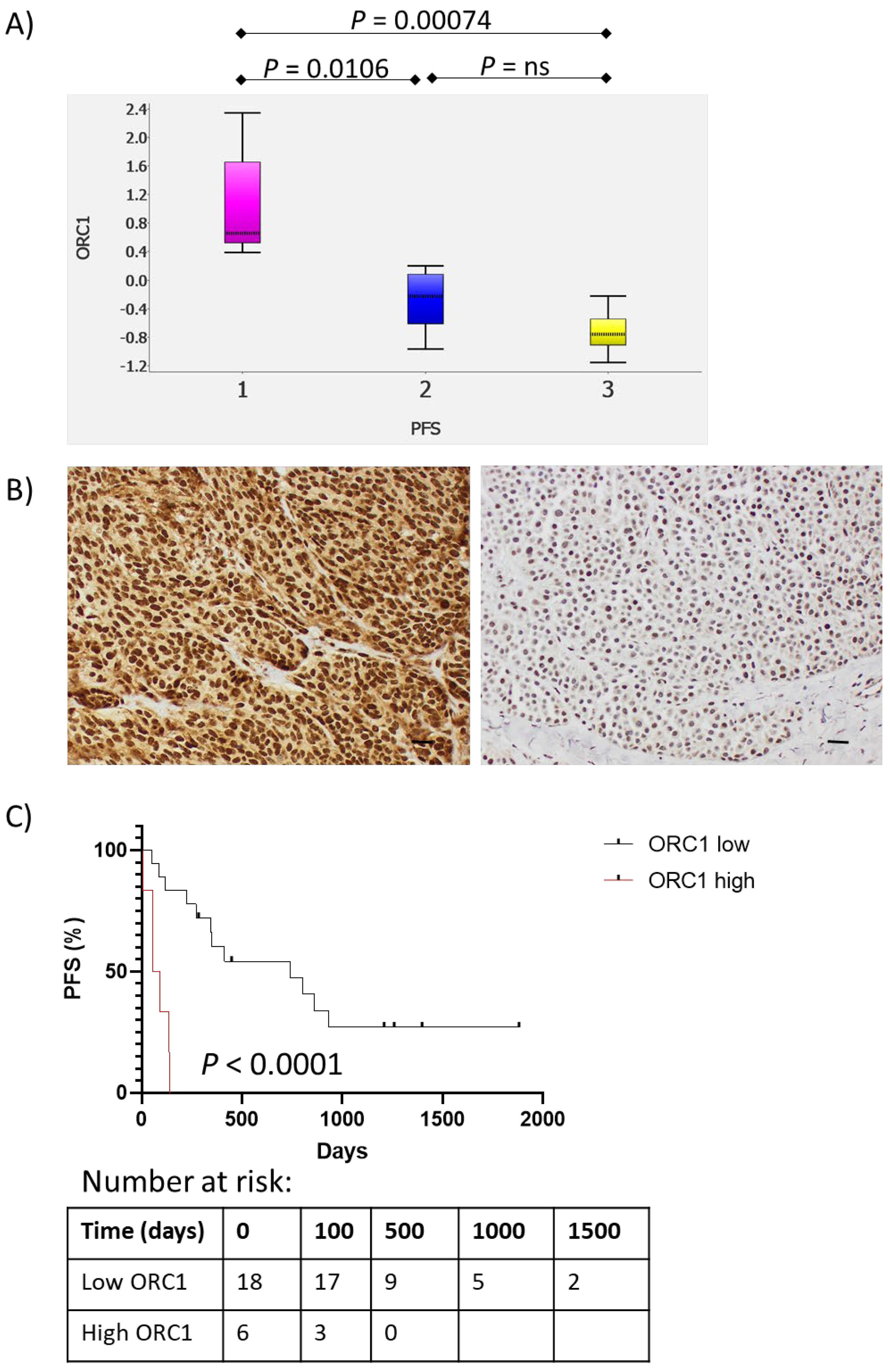
3. Results
Elevated Baseline LDH Levels Correlates with Shorter PFS in Patients Treated with Targeted Therapy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chapman, P.B.; Robert, C.; Larkin, J.; Haanen, J.B.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.; Testori, A.; Lorigan, P.C.; et al. Vemurafenib in patients with BRAFV600 mutation-positive metastatic melanoma: Final overall survival results of the randomized BRIM-3 study. Ann. Oncol. 2017, 28, 2581–2587. [Google Scholar] [CrossRef] [PubMed]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H., Jr.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; McArthur, G.A.; Dreno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandala, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.F.; Testori, A.; Grob, J.J.; et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1315–1327. [Google Scholar] [CrossRef]
- Maio, M.; Grob, J.J.; Aamdal, S.; Bondarenko, I.; Robert, C.; Thomas, L.; Garbe, C.; Chiarion-Sileni, V.; Testori, A.; Chen, T.T.; et al. Five-year survival rates for treatment-naive patients with advanced melanoma who received ipilimumab plus dacarbazine in a phase III trial. J. Clin. Oncol. 2015, 33, 1191–1196. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Long, G.V.; Robert, C.; Brady, B.; Dutriaux, C.; Di Giacomo, A.M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; et al. Survival Outcomes in Patients with Previously Untreated BRAF Wild-Type Advanced Melanoma Treated with Nivolumab Therapy: Three-Year Follow-up of a Randomized Phase 3 Trial. JAMA Oncol. 2019, 5, 187–194. [Google Scholar] [CrossRef]
- Schachter, J.; Ribas, A.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab for advanced melanoma: Final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 2017, 390, 1853–1862. [Google Scholar] [CrossRef]
- Robert, C.; Ribas, A.; Schachter, J.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): Post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 2019, 20, 1239–1251. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef]
- Buder-Bakhaya, K.; Hassel, J.C. Biomarkers for Clinical Benefit of Immune Checkpoint Inhibitor Treatment—A Review from the Melanoma Perspective and Beyond. Front. Immunol. 2018, 9, 1474. [Google Scholar] [CrossRef]
- Schadendorf, D.; Long, G.V.; Stroiakovski, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion-Sileni, V.; Schachter, J.; Garbe, C.; Dutriaux, C.; et al. Three-year pooled analysis of factors associated with clinical outcomes across dabrafenib and trametinib combination therapy phase 3 randomised trials. Eur. J. Cancer 2017, 82, 45–55. [Google Scholar] [CrossRef]
- Azimi, A.; Tuominen, R.; Costa Svedman, F.; Caramuta, S.; Pernemalm, M.; Frostvik Stolt, M.; Kanter, L.; Kharaziha, P.; Lehtio, J.; Hertzman Johansson, C.; et al. Silencing FLI or targeting CD13/ANPEP lead to dephosphorylation of EPHA2, a mediator of BRAF inhibitor resistance, and induce growth arrest or apoptosis in melanoma cells. Cell Death Dis. 2017, 8, e3029. [Google Scholar] [CrossRef]
- Kakadia, S.; Yarlagadda, N.; Awad, R.; Kundranda, M.; Niu, J.; Naraev, B.; Mina, L.; Dragovich, T.; Gimbel, M.; Mahmoud, F. Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of US Food and Drug Administration—Approved targeted therapy in advanced melanoma. OncoTargets Ther. 2018, 11, 7095–7107. [Google Scholar] [CrossRef]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martin-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949.e16. [Google Scholar] [CrossRef]
- Grasso, C.S.; Tsoi, J.; Onyshchenko, M.; Abril-Rodriguez, G.; Ross-Macdonald, P.; Wind-Rotolo, M.; Champhekar, A.; Medina, E.; Torrejon, D.Y.; Shin, D.S.; et al. Conserved Interferon-gamma Signaling Drives Clinical Response to Immune Checkpoint Blockade Therapy in Melanoma. Cancer Cell 2020, 38, 500–515.e3. [Google Scholar] [CrossRef]
- Balch, C.M.; Gershenwald, J.E.; Soong, S.J.; Thompson, J.F.; Atkins, M.B.; Byrd, D.R.; Buzaid, A.C.; Cochran, A.J.; Coit, D.G.; Ding, S.; et al. Final version of 2009 AJCC melanoma staging and classification. J. Clin. Oncol. 2009, 27, 6199–6206. [Google Scholar] [CrossRef]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Jiang, L.; Chen, Y.; Li, Y.; Lan, T.; Wu, M.; Wang, Y.; Qian, H. Type II cGMP-dependent protein kinase inhibits ligand-induced activation of EGFR in gastric cancer cells. Mol. Med. Rep. 2014, 9, 1405–1409. [Google Scholar] [CrossRef][Green Version]
- Wu, Y.; Yao, X.; Zhu, M.; Qian, H.; Jiang, L.; Lan, T.; Wu, M.; Pang, J.; Chen, Y. PKG II reverses HGF-triggered cellular activities by phosphorylating serine 985 of c-Met in gastric cancer cells. Oncotarget 2016, 7, 34190–34200. [Google Scholar] [CrossRef]
- Wongchenko, M.J.; McArthur, G.A.; Dreno, B.; Larkin, J.; Ascierto, P.A.; Sosman, J.; Andries, L.; Kockx, M.; Hurst, S.D.; Caro, I.; et al. Gene Expression Profiling in BRAF-Mutated Melanoma Reveals Patient Subgroups with Poor Outcomes to Vemurafenib That May Be Overcome by Cobimetinib Plus Vemurafenib. Clin. Cancer Res. 2017, 23, 5238–5245. [Google Scholar] [CrossRef]
- Hauschild, A.; Larkin, J.; Ribas, A.; Dreno, B.; Flaherty, K.T.; Ascierto, P.A.; Lewis, K.D.; McKenna, E.; Zhu, Q.; Mun, Y.; et al. Modeled Prognostic Subgroups for Survival and Treatment Outcomes in BRAF V600-Mutated Metastatic Melanoma: Pooled Analysis of 4 Randomized Clinical Trials. JAMA Oncol. 2018, 4, 1382–1388. [Google Scholar] [CrossRef]
- Dummer, R.; Brase, J.C.; Garrett, J.; Campbell, C.D.; Gasal, E.; Squires, M.; Gusenleitner, D.; Santinami, M.; Atkinson, V.; Mandala, M.; et al. Adjuvant dabrafenib plus trametinib versus placebo in patients with resected, BRAF(V600)-mutant, stage III melanoma (COMBI-AD): Exploratory biomarker analyses from a randomised, phase 3 trial. Lancet Oncol. 2020, 21, 358–372. [Google Scholar] [CrossRef]
- Hugo, W.; Shi, H.; Sun, L.; Piva, M.; Song, C.; Kong, X.; Moriceau, G.; Hong, A.; Dahlman, K.B.; Johnson, D.B.; et al. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell 2015, 162, 1271–1285. [Google Scholar] [CrossRef] [PubMed]
- Kalaora, S.; Lee, J.S.; Barnea, E.; Levy, R.; Greenberg, P.; Alon, M.; Yagel, G.; Bar Eli, G.; Oren, R.; Peri, A.; et al. Immunoproteasome expression is associated with better prognosis and response to checkpoint therapies in melanoma. Nat. Commun. 2020, 11, 896. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, A.; Klein, O.; McDermott, D.F.; Wang, W.; Ibrahim, N.; Lawrence, D.P.; Gunturi, A.; Flaherty, K.T.; Hodi, F.S.; Kefford, R.; et al. Outcomes of patients with metastatic melanoma treated with immunotherapy prior to or after BRAF inhibitors. Cancer 2014, 120, 1695–1701. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Lee, E.K.; Diehl, J.A. Induction of Therapeutic Senescence in Vemurafenib-Resistant Melanoma by Extended Inhibition of CDK4/6. Cancer Res. 2016, 76, 2990–3002. [Google Scholar] [CrossRef]
- Lau, E.; Tsuji, T.; Guo, L.; Lu, S.H.; Jiang, W. The role of pre-replicative complex (pre-RC) components in oncogenesis. FASEB J. 2007, 21, 3786–3794. [Google Scholar] [CrossRef]
- Tsaniras, S.C.; Kanellakis, N.; Symeonidou, I.E.; Nikolopoulou, P.; Lygerou, Z.; Taraviras, S. Licensing of DNA replication, cancer, pluripotency and differentiation: An interlinked world? Semin. Cell Dev. Biol. 2014, 30, 174–180. [Google Scholar] [CrossRef]
- Liu, Y.Z.; Jiang, Y.Y.; Hao, J.J.; Lu, S.S.; Zhang, T.T.; Shang, L.; Cao, J.; Song, X.; Wang, B.S.; Cai, Y.; et al. Prognostic significance of MCM7 expression in the bronchial brushings of patients with non-small cell lung cancer (NSCLC). Lung Cancer 2012, 77, 176–182. [Google Scholar] [CrossRef]
- Choy, B.; LaLonde, A.; Que, J.; Wu, T.; Zhou, Z. MCM4 and MCM7, potential novel proliferation markers, significantly correlated with Ki-67, Bmi1, and cyclin E expression in esophageal adenocarcinoma, squamous cell carcinoma, and precancerous lesions. Hum. Pathol. 2016, 57, 126–135. [Google Scholar] [CrossRef]
- Gardner, N.J.; Gillespie, P.J.; Carrington, J.T.; Shanks, E.J.; McElroy, S.P.; Haagensen, E.J.; Frearson, J.A.; Woodland, A.; Blow, J.J. The High-Affinity Interaction between ORC and DNA that is Required for Replication Licensing is Inhibited by 2-Arylquinolin-4-Amines. Cell Chem. Biol. 2017, 24, 981–992.e4. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodriguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef]
- Powles, T.; Plimack, E.R.; Soulieres, D.; Waddell, T.; Stus, V.; Gafanov, R.; Nosov, D.; Pouliot, F.; Melichar, B.; Vynnychenko, I.; et al. Pembrolizumab plus axitinib versus sunitinib monotherapy as first-line treatment of advanced renal cell carcinoma (KEYNOTE-426): Extended follow-up from a randomised, open-label, phase 3 trial. Lancet Oncol. 2020, 21, 1563–1573. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Grzywa, T.M.; Paskal, W.; Wlodarski, P.K. Intratumor and Intertumor Heterogeneity in Melanoma. Transl. Oncol. 2017, 10, 956–975. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | TT (n = 13) | ICI (n = 15) |
|---|---|---|
| Gender | ||
| Male | 9 | 9 |
| Female | 4 | 6 |
| Age (years old) | ||
| Median (range) | 61 (42–86) | 73 (49–84) |
| M1 1 stage | ||
| M1a | 1 | 2 |
| M1b | 1 | 4 |
| M1c | 11 | 9 |
| LDH (microKat/L) 2 | ||
| Median (range) | 5.3 (2.6–30.1) | 4.1 (2.6–14.2) 3 |
| BRAF status | ||
| Mutation | 13 | 4 |
| Wild type | 0 | 11 |
| Therapy | ||
| BRAFi | 10 | 0 |
| BRAFi+MEKi | 3 | 0 |
| anti-PD-1 4 | 0 | 14 |
| anti-CTLA-4 | 0 | 1 |
| Line of treatment | ||
| 1st | 13 | 14 |
| 2nd | 0 | 0 |
| 3rd | 0 | 1 5 |
| Response (biopsied tumor) | ||
| Disease control | 11 | 10 |
| Non-responder | 2 | 4 |
| Not evaluable | 0 | 1 6 |
| PFS | ||
| Median (range), days | 161 (35–966) | 343 (52–2533) 7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa Svedman, F.; Das, I.; Tuominen, R.; Darai Ramqvist, E.; Höiom, V.; Egyhazi Brage, S. Proliferation and Immune Response Gene Signatures Associated with Clinical Outcome to Immunotherapy and Targeted Therapy in Metastatic Cutaneous Malignant Melanoma. Cancers 2022, 14, 3587. https://doi.org/10.3390/cancers14153587
Costa Svedman F, Das I, Tuominen R, Darai Ramqvist E, Höiom V, Egyhazi Brage S. Proliferation and Immune Response Gene Signatures Associated with Clinical Outcome to Immunotherapy and Targeted Therapy in Metastatic Cutaneous Malignant Melanoma. Cancers. 2022; 14(15):3587. https://doi.org/10.3390/cancers14153587
Chicago/Turabian StyleCosta Svedman, Fernanda, Ishani Das, Rainer Tuominen, Eva Darai Ramqvist, Veronica Höiom, and Suzanne Egyhazi Brage. 2022. "Proliferation and Immune Response Gene Signatures Associated with Clinical Outcome to Immunotherapy and Targeted Therapy in Metastatic Cutaneous Malignant Melanoma" Cancers 14, no. 15: 3587. https://doi.org/10.3390/cancers14153587
APA StyleCosta Svedman, F., Das, I., Tuominen, R., Darai Ramqvist, E., Höiom, V., & Egyhazi Brage, S. (2022). Proliferation and Immune Response Gene Signatures Associated with Clinical Outcome to Immunotherapy and Targeted Therapy in Metastatic Cutaneous Malignant Melanoma. Cancers, 14(15), 3587. https://doi.org/10.3390/cancers14153587