
Roles of Chromatin Remodelling and Molecular Heterogeneity in Therapy Resistance in Glioblastoma

Abstract
Simple Summary
Abstract
1. Introduction
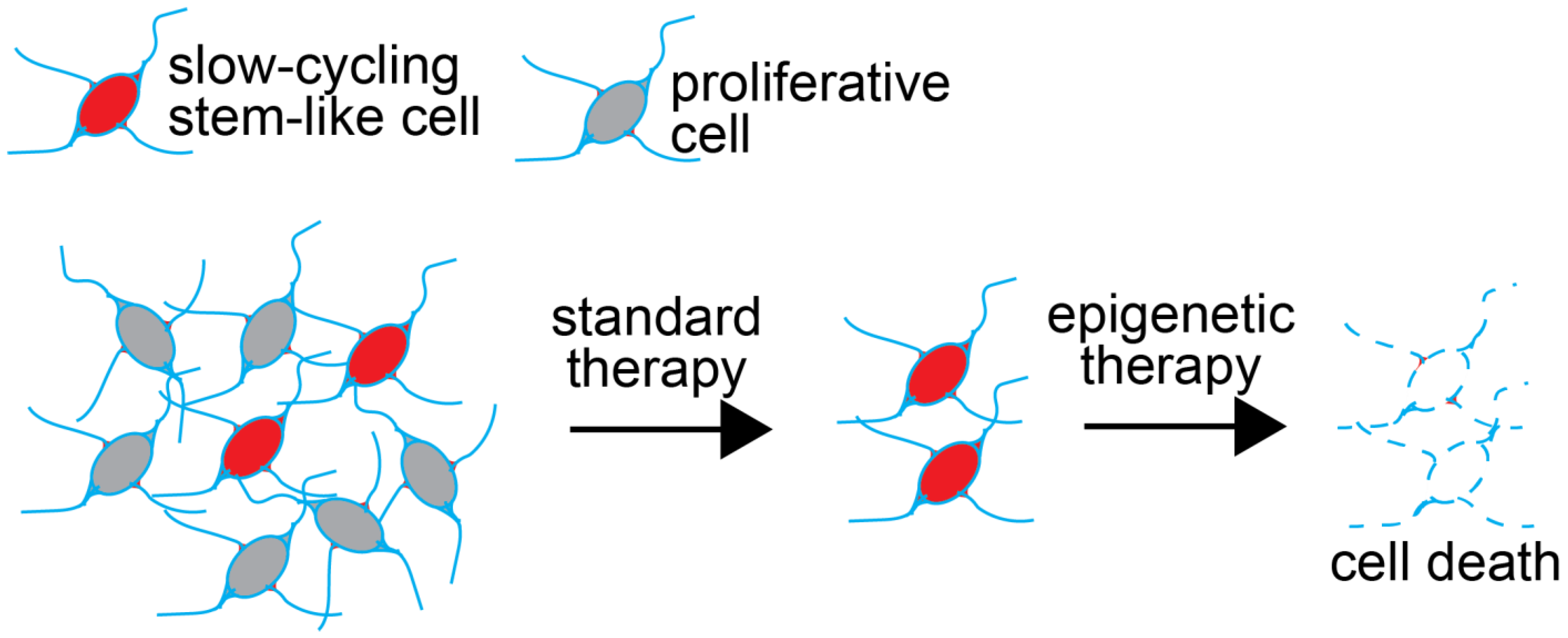
2. Cancer Stem Cells Represent Reservoirs of Therapy Resistance in GBM
3. Chromatin Dynamics Contribute to the Therapy-Resistance Properties of GSCs
4. DNA and Histone Methylation Regulate Self-Renewal Pathways
5. Epigenetic Roles of Long Noncoding RNAs in GBM
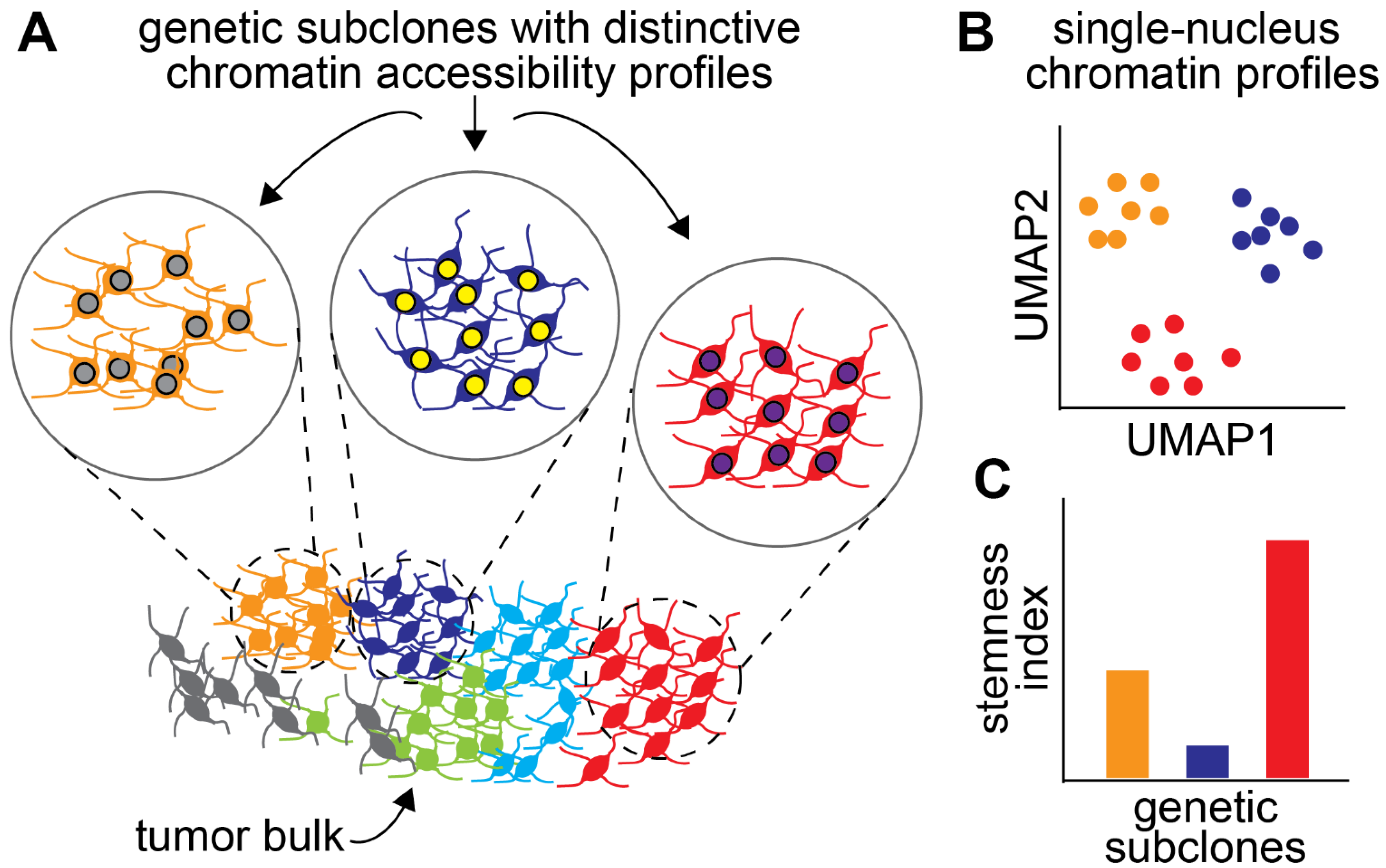
6. Intratumoural Epigenetic Heterogeneity and Its Role in Drug Resistance
7. Epigenetics, Plasticity, and the Tumour Microenvironment
8. Targeting the Epigenetic and Chromatin Factors in GBM
9. Epidrugs with Promising Action in Preclinical Models
10. Mechanisms of Resistance to Epigenetic Therapies
11. Epigenetic Therapy for GBM in the Clinic
12. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Johnstone, C.P.; Wang, N.B.; Sevier, S.A.; Galloway, K.E. Understanding and Engineering Chromatin as a Dynamical System across Length and Timescales. Cell Syst. 2020, 11, 424–448. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of Chromatin by Histone Modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Statello, L.; Guo, C.-J.; Chen, L.-L.; Huarte, M. Gene Regulation by Long Non-Coding RNAs and Its Biological Functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic Histone H3 Alterations in Pediatric Diffuse Intrinsic Pontine Gliomas and Non-Brainstem Glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.W.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.K.; Tönjes, M.; et al. Driver Mutations in Histone H3.3 and Chromatin Remodelling Genes in Paediatric Glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Pardal, R.; Clarke, M.F.; Morrison, S.J. Applying the Principles of Stem-Cell Biology to Cancer. Nat. Rev. Cancer 2003, 3, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B.; Terasaki, M.; et al. Identification of a Cancer Stem Cell in Human Brain Tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of Human Brain Tumour Initiating Cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.; van den Bent, M.J.; Weller, M.; Fisher, B.M.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Poon, M.T.C.; Sudlow, C.L.M.; Figueroa, J.D.; Brennan, P.M. Longer-Term (≥2 Years) Survival in Patients with Glioblastoma in Population-Based Studies Pre- and Post-2005: A Systematic Review and Meta-Analysis. Sci. Rep. 2020, 10, 11622. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; Hjelmeland, A.B.; Rich, J.N.; Dewhirst, M.W.; Shi, Q.; Hao, Y.; McLendon, R.E.; Bigner, D.D. Glioma Stem Cells Promote Radioresistance by Preferential Activation of the DNA Damage Response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic State of Glioma Stem Cells and Nontumorigenic Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar] [CrossRef] [PubMed]
- Soeda, A.; Park, M.; Lee, D.; Mintz, A.; Androutsellis-Theotokis, A.; McKay, R.D.; Engh, J.; Iwama, T.; Kunisada, T.; Kassam, A.B.; et al. Hypoxia Promotes Expansion of the CD133-Positive Glioma Stem Cells through Activation of HIF-1alpha. Oncogene 2009, 28, 3949–3959. [Google Scholar] [CrossRef]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; McLendon, R.E.; et al. Hypoxia-Inducible Factors Regulate Tumorigenic Capacity of Glioma Stem Cells. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A Restricted Cell Population Propagates Glioblastoma Growth after Chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef]
- Xie, X.P.; Laks, D.R.; Sun, D.; Ganbold, M.; Wang, Z.; Pedraza, A.M.; Bale, T.; Tabar, V.; Brennan, C.; Zhou, X.; et al. Quiescent Human Glioblastoma Cancer Stem Cells Drive Tumor Initiation, Expansion, and Recurrence Following Chemotherapy. Dev. Cell 2022, 57, 32–46.e8. [Google Scholar] [CrossRef]
- Antonica, F.; Santomaso, L.; Pernici, D.; Petrucci, L.; Aiello, G.; Cutarelli, A.; Conti, L.; Romanel, A.; Miele, E.; Tebaldi, T.; et al. A Slow-Cycling/Quiescent Cells Subpopulation Is Involved in Glioma Invasiveness. Nat. Commun. 2022, 13, 4767. [Google Scholar] [CrossRef] [PubMed]
- van Velthoven, C.T.J.; Rando, T.A. Stem Cell Quiescence: Dynamism, Restraint, and Cellular Idling. Cell Stem Cell 2019, 24, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Codega, P.; Silva-Vargas, V.; Paul, A.; Maldonado-Soto, A.R.; Deleo, A.M.; Pastrana, E.; Doetsch, F. Prospective Identification and Purification of Quiescent Adult Neural Stem Cells from Their in Vivo Niche. Neuron 2014, 82, 545–559. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.M.; Sarić, N.; Russell, J.P.; Andoniadou, C.L.; Scambler, P.J.; Basson, M.A. CHD7 Maintains Neural Stem Cell Quiescence and Prevents Premature Stem Cell Depletion in the Adult Hippocampus. Stem Cells 2015, 33, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Llorens-Bobadilla, E.; Zhao, S.; Baser, A.; Saiz-Castro, G.; Zwadlo, K.; Martin-Villalba, A. Single-Cell Transcriptomics Reveals a Population of Dormant Neural Stem Cells That Become Activated upon Brain Injury. Cell Stem Cell 2015, 17, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Ziebell, F.; Dehler, S.; Martin-Villalba, A.; Marciniak-Czochra, A. Revealing Age-Related Changes of Adult Hippocampal Neurogenesis Using Mathematical Models. Development 2018, 145, dev153544. [Google Scholar] [CrossRef] [PubMed]
- Bonitto, K.; Sarathy, K.; Atai, K.; Mitra, M.; Coller, H.A. Is There a Histone Code for Cellular Quiescence? Front. Cell Dev. Biol. 2021, 9, 739780. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A Bivalent Chromatin Structure Marks Key Developmental Genes in Embryonic Stem Cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.-K.; Koche, R.P.; et al. Genome-Wide Maps of Chromatin State in Pluripotent and Lineage-Committed Cells. Nature 2007, 448, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Cui, K.; Zang, C.; Roh, T.-Y.; Schones, D.E.; Childs, R.W.; Peng, W.; Zhao, K. Chromatin Signatures in Multipotent Human Hematopoietic Stem Cells Indicate the Fate of Bivalent Genes during Differentiation. Cell Stem Cell 2009, 4, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-Cell RNA-Seq Highlights Intratumoral Heterogeneity in Primary Glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Liau, B.B.; Sievers, C.; Donohue, L.K.; Gillespie, S.M.; Flavahan, W.A.; Miller, T.E.; Venteicher, A.S.; Hebert, C.H.; Carey, C.D.; Rodig, S.J.; et al. Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem Cell 2017, 20, 233–246.e7. [Google Scholar] [CrossRef] [PubMed]
- Burgold, T.; Spreafico, F.; De Santa, F.; Totaro, M.G.; Prosperini, E.; Natoli, G.; Testa, G. The Histone H3 Lysine 27-Specific Demethylase Jmjd3 Is Required for Neural Commitment. PLoS ONE 2008, 3, e3034. [Google Scholar] [CrossRef] [PubMed]
- Kidder, B.L.; Hu, G.; Zhao, K. KDM5B Focuses H3K4 Methylation near Promoters and Enhancers during Embryonic Stem Cell Self-Renewal and Differentiation. Genome Biol. 2014, 15, R32. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.-Y.; Zhang, S.-F.; Dai, S.-K.; Liu, C.; Wang, Y.-Y.; Du, H.-Z.; Teng, Z.-Q.; Liu, C.-M. UTX Regulates Human Neural Differentiation and Dendritic Morphology by Resolving Bivalent Promoters. Stem Cell Rep. 2020, 15, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Hamed, A.A.; Kunz, D.J.; El-Hamamy, I.; Trinh, Q.M.; Subedar, O.D.; Richards, L.M.; Foltz, W.; Bullivant, G.; Ware, M.; Vladoiu, M.C.; et al. A Brain Precursor Atlas Reveals the Acquisition of Developmental-like States in Adult Cerebral Tumours. Nat. Commun. 2022, 13, 4178. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Cai, L.-D.; Liu, Y.-H.; Li, S.; Gan, W.-J.; Li, X.-M.; Wang, J.-R.; Guo, P.-D.; Zhou, Q.; Lu, X.-X.; et al. Ube2v1-Mediated Ubiquitination and Degradation of Sirt1 Promotes Metastasis of Colorectal Cancer by Epigenetically Suppressing Autophagy. J. Hematol. Oncol. 2018, 11, 95. [Google Scholar] [CrossRef]
- Pessoa Rodrigues, C.; Akhtar, A. Differential H4K16ac Levels Ensure a Balance between Quiescence and Activation in Hematopoietic Stem Cells. Sci. Adv. 2021, 7, eabi5987. [Google Scholar] [CrossRef]
- Ruthenburg, A.J.; Li, H.; Milne, T.A.; Dewell, S.; McGinty, R.K.; Yuen, M.; Ueberheide, B.; Dou, Y.; Muir, T.W.; Patel, D.J.; et al. Recognition of a Mononucleosomal Histone Modification Pattern by BPTF via Multivalent Interactions. Cell 2011, 145, 692–706. [Google Scholar] [CrossRef]
- Xu, B.; Cai, L.; Butler, J.M.; Chen, D.; Lu, X.; Allison, D.F.; Lu, R.; Rafii, S.; Parker, J.S.; Zheng, D.; et al. The Chromatin Remodeler BPTF Activates a Stemness Gene-Expression Program Essential for the Maintenance of Adult Hematopoietic Stem Cells. Stem Cell Rep. 2018, 10, 675–683. [Google Scholar] [CrossRef]
- Koludrovic, D.; Laurette, P.; Strub, T.; Keime, C.; Le Coz, M.; Coassolo, S.; Mengus, G.; Larue, L.; Davidson, I. Chromatin-Remodelling Complex NURF Is Essential for Differentiation of Adult Melanocyte Stem Cells. PLoS Genet. 2015, 11, e1005555. [Google Scholar] [CrossRef]
- Green, A.L.; DeSisto, J.; Flannery, P.; Lemma, R.; Knox, A.; Lemieux, M.; Sanford, B.; O’Rourke, R.; Ramkissoon, S.; Jones, K.; et al. BPTF Regulates Growth of Adult and Pediatric High-Grade Glioma through the MYC Pathway. Oncogene 2020, 39, 2305–2327. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Kim, G.W.; Yoo, J.; Lee, S.W.; Jeon, Y.H.; Kim, S.Y.; Kang, H.G.; Kim, D.-H.; Chun, K.-H.; Choi, J.; et al. Histone Demethylase KDM4C Controls Tumorigenesis of Glioblastoma by Epigenetically Regulating P53 and C-Myc. Cell Death Dis. 2021, 12, 89. [Google Scholar] [CrossRef] [PubMed]
- Mallm, J.-P.; Windisch, P.; Biran, A.; Gal, Z.; Schumacher, S.; Glass, R.; Herold-Mende, C.; Meshorer, E.; Barbus, M.; Rippe, K. Glioblastoma Initiating Cells Are Sensitive to Histone Demethylase Inhibition Due to Epigenetic Deregulation. Int. J. Cancer 2020, 146, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 Mutations in Gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-Derived Mutations in IDH1 Dominantly Inhibit IDH1 Catalytic Activity and Induce HIF-1alpha. Science 2009, 324, 261–265. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-Associated IDH1 Mutations Produce 2-Hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef]
- Tsukada, Y.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone Demethylation by a Family of JmjC Domain-Containing Proteins. Nature 2006, 439, 811–816. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef]
- Carey, B.W.; Finley, L.W.S.; Cross, J.R.; Allis, C.D.; Thompson, C.B. Intracellular α-Ketoglutarate Maintains the Pluripotency of Embryonic Stem Cells. Nature 2015, 518, 413–416. [Google Scholar] [CrossRef]
- TeSlaa, T.; Chaikovsky, A.C.; Lipchina, I.; Escobar, S.L.; Hochedlinger, K.; Huang, J.; Graeber, T.G.; Braas, D.; Teitell, M.A. α-Ketoglutarate Accelerates the Initial Differentiation of Primed Human Pluripotent Stem Cells. Cell Metab. 2016, 24, 485–493. [Google Scholar] [CrossRef]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.-M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.H.; Li, X.S.; Woon, E.C.Y.; Yang, M.; et al. The Oncometabolite 2-Hydroxyglutarate Inhibits Histone Lysine Demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH Mutation Impairs Histone Demethylation and Results in a Block to Cell Differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Andrade, J.; Shi, C.; Costa, A.S.H.; Choi, J.; Kim, J.; Doddaballapur, A.; Sugino, T.; Ong, Y.T.; Castro, M.; Zimmermann, B.; et al. Control of Endothelial Quiescence by FOXO-Regulated Metabolites. Nat. Cell Biol. 2021, 23, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Paik, J.; Ding, Z.; Narurkar, R.; Ramkissoon, S.; Muller, F.; Kamoun, W.S.; Chae, S.-S.; Zheng, H.; Ying, H.; Mahoney, J.; et al. FoxOs Cooperatively Regulate Diverse Pathways Governing Neural Stem Cell Homeostasis. Cell Stem Cell 2009, 5, 540–553. [Google Scholar] [CrossRef] [PubMed]
- Bulstrode, H.; Johnstone, E.; Marques-Torrejon, M.A.; Ferguson, K.M.; Bressan, R.B.; Blin, C.; Grant, V.; Gogolok, S.; Gangoso, E.; Gagrica, S.; et al. Elevated FOXG1 and SOX2 in Glioblastoma Enforces Neural Stem Cell Identity through Transcriptional Control of Cell Cycle and Epigenetic Regulators. Genes Dev. 2017, 31, 757–773. [Google Scholar] [CrossRef] [PubMed]
- Kusi, M.; Zand, M.; Lin, L.-L.; Chen, M.; Lopez, A.; Lin, C.-L.; Wang, C.-M.; Lucio, N.D.; Kirma, N.B.; Ruan, J.; et al. 2-Hydroxyglutarate Destabilizes Chromatin Regulatory Landscape and Lineage Fidelity to Promote Cellular Heterogeneity. Cell Rep. 2022, 38, 110220. [Google Scholar] [CrossRef]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many Human Large Intergenic Noncoding RNAs Associate with Chromatin-Modifying Complexes and Affect Gene Expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef]
- Zhang, G.; Lan, Y.; Xie, A.; Shi, J.; Zhao, H.; Xu, L.; Zhu, S.; Luo, T.; Zhao, T.; Xiao, Y.; et al. Comprehensive Analysis of Long Noncoding RNA (LncRNA)-Chromatin Interactions Reveals LncRNA Functions Dependent on Binding Diverse Regulatory Elements. J. Biol. Chem. 2019, 294, 15613–15622. [Google Scholar] [CrossRef]
- Zhang, P.; Wu, W.; Chen, Q.; Chen, M. Non-Coding RNAs and Their Integrated Networks. J. Integr. Bioinform. 2019, 16, 20190027. [Google Scholar] [CrossRef]
- Zhao, J.; Sun, B.K.; Erwin, J.A.; Song, J.-J.; Lee, J.T. Polycomb Proteins Targeted by a Short Repeat RNA to the Mouse X Chromosome. Science 2008, 322, 750–756. [Google Scholar] [CrossRef]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional Demarcation of Active and Silent Chromatin Domains in Human HOX Loci by Noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, A.; He, X.C.; Thorvaldsen, J.L.; Sugimura, R.; Perry, J.M.; Tao, F.; Zhao, M.; Christenson, M.K.; Sanchez, R.; Yu, J.Y.; et al. Maternal Imprinting at the H19-Igf2 Locus Maintains Adult Haematopoietic Stem Cell Quiescence. Nature 2013, 500, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Monnier, P.; Martinet, C.; Pontis, J.; Stancheva, I.; Ait-Si-Ali, S.; Dandolo, L. H19 LncRNA Controls Gene Expression of the Imprinted Gene Network by Recruiting MBD1. Proc. Natl. Acad. Sci. USA 2013, 110, 20693–20698. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Li, Z.; Ma, K.; Li, X.; Tian, N.; Duan, J.; Xiao, X.; Wang, Y. Long Non-Coding RNA XIST Promotes Glioma Tumorigenicity and Angiogenesis by Acting as a Molecular Sponge of MiR-429. J. Cancer 2017, 8, 4106–4116. [Google Scholar] [CrossRef] [PubMed]
- Jia, P.; Cai, H.; Liu, X.; Chen, J.; Ma, J.; Wang, P.; Liu, Y.; Zheng, J.; Xue, Y. Long Non-Coding RNA H19 Regulates Glioma Angiogenesis and the Biological Behavior of Glioma-Associated Endothelial Cells by Inhibiting MicroRNA-29a. Cancer Lett. 2016, 381, 359–369. [Google Scholar] [CrossRef]
- Zhang, K.; Sun, X.; Zhou, X.; Han, L.; Chen, L.; Shi, Z.; Zhang, A.; Ye, M.; Wang, Q.; Liu, C.; et al. Long Non-Coding RNA HOTAIR Promotes Glioblastoma Cell Cycle Progression in an EZH2 Dependent Manner. Oncotarget 2015, 6, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Yadav, B.; Pal, S.; Rubstov, Y.; Goel, A.; Garg, M.; Pavlyukov, M.; Pandey, A.K. LncRNAs Associated with Glioblastoma: From Transcriptional Noise to Novel Regulators with a Promising Role in Therapeutics. Mol. Ther. Nucleic Acids 2021, 24, 728–742. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Diehn, M.; Watson, N.; Bollen, A.W.; Aldape, K.D.; Nicholas, M.K.; Lamborn, K.R.; Berger, M.S.; Botstein, D.; Brown, P.O.; et al. Gene Expression Profiling Reveals Molecularly and Clinically Distinct Subtypes of Glioblastoma Multiforme. Proc. Natl. Acad. Sci. USA 2005, 102, 5814–5819. [Google Scholar] [CrossRef]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor Heterogeneity in Human Glioblastoma Reflects Cancer Evolutionary Dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational Analysis Reveals the Origin and Therapy-Driven Evolution of Recurrent Glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Snuderl, M.; Fazlollahi, L.; Le, L.P.; Nitta, M.; Zhelyazkova, B.H.; Davidson, C.J.; Akhavanfard, S.; Cahill, D.P.; Aldape, K.D.; Betensky, R.A.; et al. Mosaic Amplification of Multiple Receptor Tyrosine Kinase Genes in Glioblastoma. Cancer Cell 2011, 20, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Little, S.E.; Popov, S.; Jury, A.; Bax, D.A.; Doey, L.; Al-Sarraj, S.; Jurgensmeier, J.M.; Jones, C. Receptor Tyrosine Kinase Genes Amplified in Glioblastoma Exhibit a Mutual Exclusivity in Variable Proportions Reflective of Individual Tumor Heterogeneity. Cancer Res. 2012, 72, 1614–1620. [Google Scholar] [CrossRef] [PubMed]
- Szerlip, N.J.; Pedraza, A.; Chakravarty, D.; Azim, M.; McGuire, J.; Fang, Y.; Ozawa, T.; Holland, E.C.; Huse, J.T.; Jhanwar, S.; et al. Intratumoral Heterogeneity of Receptor Tyrosine Kinases EGFR and PDGFRA Amplification in Glioblastoma Defines Subpopulations with Distinct Growth Factor Response. Proc. Natl. Acad. Sci. USA 2012, 109, 3041–3046. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Maley, C.C. Clonal Evolution in Cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Jordan, C.T.; Guzman, M.L.; Noble, M. Cancer Stem Cells. N. Engl. J. Med. 2006, 355, 1253–1261. [Google Scholar] [CrossRef]
- Salgia, R.; Kulkarni, P. The Genetic/Non-Genetic Duality of Drug “Resistance” in Cancer. Trends Cancer 2018, 4, 110–118. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Eyler, C.E.; Matsunaga, H.; Hovestadt, V.; Vantine, S.J.; van Galen, P.; Bernstein, B.E. Single-Cell Lineage Analysis Reveals Genetic and Epigenetic Interplay in Glioblastoma Drug Resistance. Genome Biol. 2020, 21, 174. [Google Scholar] [CrossRef] [PubMed]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, A.; Singhal, D.; Ellestad, K.; Johnston, M.; Shen, Y.; Gillmor, A.; Morrissy, S.; Cairncross, J.G.; Jones, S.; Lupien, M.; et al. Copy-ScAT: Deconvoluting Single-Cell Chromatin Accessibility of Genetic Subclones in Cancer. Sci. Adv. 2021, 7, eabg6045. [Google Scholar] [CrossRef] [PubMed]
- Mazor, T.; Pankov, A.; Johnson, B.E.; Hong, C.; Hamilton, E.G.; Bell, R.J.A.; Smirnov, I.V.; Reis, G.F.; Phillips, J.J.; Barnes, M.J.; et al. DNA Methylation and Somatic Mutations Converge on the Cell Cycle and Define Similar Evolutionary Histories in Brain Tumors. Cancer Cell 2015, 28, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.S.; Costello, M.A.; Talsma, C.E.; Flack, C.G.; Crowley, J.G.; Hamm, L.L.; He, X.; Hervey-Jumper, S.L.; Heth, J.A.; Muraszko, K.M.; et al. Endothelial Cells Create a Stem Cell Niche in Glioblastoma by Providing NOTCH Ligands That Nurture Self-Renewal of Cancer Stem-like Cells. Cancer Res. 2011, 71, 6061–6072. [Google Scholar] [CrossRef]
- Heddleston, J.M.; Wu, Q.; Rivera, M.; Minhas, S.; Lathia, J.D.; Sloan, A.E.; Iliopoulos, O.; Hjelmeland, A.B.; Rich, J.N. Hypoxia-Induced Mixed-Lineage Leukemia 1 Regulates Glioma Stem Cell Tumorigenic Potential. Cell Death Differ. 2012, 19, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Heddleston, J.M.; Li, Z.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. The Hypoxic Microenvironment Maintains Glioblastoma Stem Cells and Promotes Reprogramming towards a Cancer Stem Cell Phenotype. Cell Cycle 2009, 8, 3274–3284. [Google Scholar] [CrossRef]
- Brooks, L.J.; Clements, M.P.; Burden, J.J.; Kocher, D.; Richards, L.; Devesa, S.C.; Zakka, L.; Woodberry, M.; Ellis, M.; Jaunmuktane, Z.; et al. The White Matter Is a Pro-Differentiative Niche for Glioblastoma. Nat. Commun. 2021, 12, 2184. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.C.; Anderson, K.J.; Courtois, E.T.; Gujar, A.D.; Barthel, F.P.; Varn, F.S.; Luo, D.; Seignon, M.; Yi, E.; Kim, H.; et al. Single-Cell Multimodal Glioma Analyses Identify Epigenetic Regulators of Cellular Plasticity and Environmental Stress Response. Nat. Genet. 2021, 53, 1456–1468. [Google Scholar] [CrossRef] [PubMed]
- Ravi, V.M.; Will, P.; Kueckelhaus, J.; Sun, N.; Joseph, K.; Salié, H.; Vollmer, L.; Kuliesiute, U.; von Ehr, J.; Benotmane, J.K.; et al. Spatially Resolved Multi-Omics Deciphers Bidirectional Tumor-Host Interdependence in Glioblastoma. Cancer Cell 2022, 40, 639–655.e13. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; DeCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef] [PubMed]
- Varn, F.S.; Johnson, K.C.; Martinek, J.; Huse, J.T.; Nasrallah, M.P.; Wesseling, P.; Cooper, L.A.D.; Malta, T.M.; Wade, T.E.; Sabedot, T.S.; et al. Glioma Progression Is Shaped by Genetic Evolution and Microenvironment Interactions. Cell 2022, 185, 2184–2199.e16. [Google Scholar] [CrossRef] [PubMed]
- Pombo Antunes, A.R.; Scheyltjens, I.; Duerinck, J.; Neyns, B.; Movahedi, K.; Van Ginderachter, J.A. Understanding the Glioblastoma Immune Microenvironment as Basis for the Development of New Immunotherapeutic Strategies. Elife 2020, 9, e52176. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin Secreted by Glioblastoma Stem Cells Recruits M2 Tumour-Associated Macrophages and Promotes Malignant Growth. Nat. Cell Biol. 2015, 17, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Chanoch-Myers, R.; Mathewson, N.D.; Myskiw, C.; Atta, L.; Bussema, L.; Eichhorn, S.W.; Greenwald, A.C.; Kinker, G.S.; Rodman, C.; et al. Interactions between Cancer Cells and Immune Cells Drive Transitions to Mesenchymal-like States in Glioblastoma. Cancer Cell 2021, 39, 779–792.e11. [Google Scholar] [CrossRef] [PubMed]
- Mathewson, N.D.; Ashenberg, O.; Tirosh, I.; Gritsch, S.; Perez, E.M.; Marx, S.; Jerby-Arnon, L.; Chanoch-Myers, R.; Hara, T.; Richman, A.R.; et al. Inhibitory CD161 Receptor Identified in Glioma-Infiltrating T Cells by Single-Cell Analysis. Cell 2021, 184, 1281–1298.e26. [Google Scholar] [CrossRef] [PubMed]
- Babikir, H.; Wang, L.; Shamardani, K.; Catalan, F.; Sudhir, S.; Aghi, M.K.; Raleigh, D.R.; Phillips, J.J.; Diaz, A.A. ATRX Regulates Glial Identity and the Tumor Microenvironment in IDH-Mutant Glioma. Genome Biol. 2021, 22, 311. [Google Scholar] [CrossRef]
- Gangoso, E.; Southgate, B.; Bradley, L.; Rus, S.; Galvez-Cancino, F.; McGivern, N.; Güç, E.; Kapourani, C.-A.; Byron, A.; Ferguson, K.M.; et al. Glioblastomas Acquire Myeloid-Affiliated Transcriptional Programs via Epigenetic Immunoediting to Elicit Immune Evasion. Cell 2021, 184, 2454–2470.e26. [Google Scholar] [CrossRef] [PubMed]
- Parmigiani, E.; Ivanek, R.; Rolando, C.; Hafen, K.; Turchinovich, G.; Lehmann, F.M.; Gerber, A.; Brkic, S.; Frank, S.; Meyer, S.C.; et al. Interferon-γ Resistance and Immune Evasion in Glioma Develop via Notch-Regulated Co-Evolution of Malignant and Immune Cells. Dev. Cell 2022, 57, 1847–1865.e9. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A.; Tang, Y.; Nagaraja, S.; Gibson, E.M.; Mount, C.W.; Polepalli, J.; Mitra, S.S.; et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell 2015, 161, 803–816. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Tam, L.T.; Woo, P.J.; Lennon, J.; Nagaraja, S.; Gillespie, S.M.; Ni, J.; Duveau, D.Y.; Morris, P.J.; Zhao, J.J.; et al. Targeting Neuronal Activity-Regulated Neuroligin-3 Dependency in High-Grade Glioma. Nature 2017, 549, 533–537. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and Synaptic Integration of Glioma into Neural Circuits. Nature 2019, 573, 539–545. [Google Scholar] [CrossRef]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Körber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic Synaptic Input to Glioma Cells Drives Brain Tumour Progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef]
- Larson, J.D.; Kasper, L.H.; Paugh, B.S.; Jin, H.; Wu, G.; Kwon, C.H.; Fan, Y.; Shaw, T.I.; Silveira, A.B.; Qu, C.; et al. Histone H3.3 K27M Accelerates Spontaneous Brainstem Glioma and Drives Restricted Changes in Bivalent Gene Expression. Cancer Cell 2019, 35, 140–155. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic Protein Families: A New Frontier for Drug Discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [CrossRef] [PubMed]
- Scheer, S.; Ackloo, S.; Medina, T.S.; Schapira, M.; Li, F.; Ward, J.A.; Lewis, A.M.; Northrop, J.P.; Richardson, P.L.; Kaniskan, H.Ü.; et al. A Chemical Biology Toolbox to Study Protein Methyltransferases and Epigenetic Signaling. Nat. Commun. 2019, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Perumal, N.B.; Oldfield, C.J.; Su, E.W.; Uversky, V.N.; Dunker, A.K. Intrinsic Disorder in Transcription Factors. Biochemistry 2006, 45, 6873–6888. [Google Scholar] [CrossRef]
- Wheeler, R.J. Therapeutics-How to Treat Phase Separation-Associated Diseases. Emerg. Top. Life Sci. 2020, 4, 307–318. [Google Scholar] [CrossRef]
- Han, X.; Yu, D.; Gu, R.; Jia, Y.; Wang, Q.; Jaganathan, A.; Yang, X.; Yu, M.; Babault, N.; Zhao, C.; et al. Roles of the BRD4 Short Isoform in Phase Separation and Active Gene Transcription. Nat. Struct. Mol. Biol. 2020, 27, 333–341. [Google Scholar] [CrossRef]
- Sabari, B.R.; Dall’Agnese, A.; Boija, A.; Klein, I.A.; Coffey, E.L.; Shrinivas, K.; Abraham, B.J.; Hannett, N.M.; Zamudio, A.V.; Manteiga, J.C.; et al. Coactivator Condensation at Super-Enhancers Links Phase Separation and Gene Control. Science 2018, 361, eaar3958. [Google Scholar] [CrossRef]
- Klein, I.A.; Boija, A.; Afeyan, L.K.; Hawken, S.W.; Fan, M.; Dall’Agnese, A.; Oksuz, O.; Henninger, J.E.; Shrinivas, K.; Sabari, B.R.; et al. Partitioning of Cancer Therapeutics in Nuclear Condensates. Science 2020, 368, 1386–1392. [Google Scholar] [CrossRef]
- Triarico, S.; Maurizi, P.; Mastrangelo, S.; Attinà, G.; Capozza, M.A.; Ruggiero, A. Improving the Brain Delivery of Chemotherapeutic Drugs in Childhood Brain Tumors. Cancers 2019, 11, 824. [Google Scholar] [CrossRef]
- Stazi, G.; Taglieri, L.; Nicolai, A.; Romanelli, A.; Fioravanti, R.; Morrone, S.; Sabatino, M.; Ragno, R.; Taurone, S.; Nebbioso, M.; et al. Dissecting the Role of Novel EZH2 Inhibitors in Primary Glioblastoma Cell Cultures: Effects on Proliferation, Epithelial-Mesenchymal Transition, Migration, and on the pro-Inflammatory Phenotype. Clin. Epigenet. 2019, 11, 173. [Google Scholar] [CrossRef]
- Qi, L.; Lindsay, H.; Kogiso, M.; Du, Y.; Braun, F.K.; Zhang, H.; Guo, L.; Zhao, S.; Injac, S.G.; Baxter, P.A.; et al. Evaluation of an EZH2 Inhibitor in Patient-Derived Orthotopic Xenograft Models of Pediatric Brain Tumors Alone and in Combination with Chemo- and Radiation Therapies. Lab. Investig. 2022, 102, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Wang, Y.; Hu, Q.; Wu, W.; Wu, Y.; Wei, W.; Han, D.; You, Y.; Lin, N.; Liu, N. The EZH2 Inhibitor GSK343 Suppresses Cancer Stem-like Phenotypes and Reverses Mesenchymal Transition in Glioma Cells. Oncotarget 2017, 8, 98348–98359. [Google Scholar] [CrossRef] [PubMed]
- Del Moral-Morales, A.; González-Orozco, J.C.; Hernández-Vega, A.M.; Hernández-Ortega, K.; Peña-Gutiérrez, K.M.; Camacho-Arroyo, I. EZH2 Mediates Proliferation, Migration, and Invasion Promoted by Estradiol in Human Glioblastoma Cells. Front. Endocrinol. 2022, 13, 703733. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Qiu, S.; Li, X.; Huang, B.; Xu, Y.; Peng, Y. EZH2 Suppression in Glioblastoma Shifts Microglia toward M1 Phenotype in Tumor Microenvironment. J. Neuroinflamm. 2017, 14, 220. [Google Scholar] [CrossRef] [PubMed]
- De La Rosa, J.; Urdiciain, A.; Zazpe, I.; Zelaya, M.V.; Meléndez, B.; Rey, J.A.; Idoate, M.A.; Castresana, J.S. The Synergistic Effect of DZ-NEP, Panobinostat and Temozolomide Reduces Clonogenicity and Induces Apoptosis in Glioblastoma Cells. Int. J. Oncol. 2020, 56, 283–300. [Google Scholar] [CrossRef]
- Leone, G.; Teofili, L.; Voso, M.T.; Lübbert, M. DNA Methylation and Demethylating Drugs in Myelodysplastic Syndromes and Secondary Leukemias. Haematologica 2002, 87, 1324–1341. [Google Scholar]
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via DsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef]
- Mehdipour, P.; Murphy, T.; De Carvalho, D.D. The Role of DNA-Demethylating Agents in Cancer Therapy. Pharmacol. Ther. 2020, 205, 107416. [Google Scholar] [CrossRef]
- Yamashita, A.S.; da Costa Rosa, M.; Borodovsky, A.; Festuccia, W.T.; Chan, T.; Riggins, G.J. Demethylation and Epigenetic Modification with 5-Azacytidine Reduces IDH1 Mutant Glioma Growth in Combination with Temozolomide. Neuro. Oncol. 2019, 21, 189–200. [Google Scholar] [CrossRef]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.M.; Lu, C.; Ward, P.S.; et al. IDH1 Mutation Is Sufficient to Establish the Glioma Hypermethylator Phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Purow, B. Repurposing Existing Agents as Adjunct Therapies for Glioblastoma. Neuro-Oncol. Pract. 2016, 3, 154–163. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gallo, M.; Ho, J.; Coutinho, F.; Vanner, R.; Lee, L.; Head, R.; Ling, E.; Clarke, I.; Dirks, P. A Tumorigenic MLL-Homeobox Network in Human Glioblastoma Stem Cells. Cancer Res. 2013, 73, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Song, J.-J.; Kingston, R.E. WDR5 Interacts with Mixed Lineage Leukemia (MLL) Protein via the Histone H3-Binding Pocket. J. Biol. Chem. 2008, 283, 35258–35264. [Google Scholar] [CrossRef]
- Southall, S.M.; Wong, P.-S.; Odho, Z.; Roe, S.M.; Wilson, J.R. Structural Basis for the Requirement of Additional Factors for MLL1 SET Domain Activity and Recognition of Epigenetic Marks. Mol. Cell 2009, 33, 181–191. [Google Scholar] [CrossRef]
- Mitchell, K.; Shakya, S.; Silver, D.J.; Goins, C.M.; Wallace, L.; Roversi, G.; Schafer, R.; Kay, K.; Miller, T.E.; Lauko, A.; et al. The WRAD Complex Represents a Therapeutically Exploitable Target for Cancer Stem Cells in Glioblastoma. bioRxiv 2021. [Google Scholar] [CrossRef]
- Caslini, C.; Yang, Z.; El-Osta, M.; Milne, T.A.; Slany, R.K.; Hess, J.L. Interaction of MLL Amino Terminal Sequences with Menin Is Required for Transformation. Cancer Res. 2007, 67, 7275–7283. [Google Scholar] [CrossRef]
- Borkin, D.; He, S.; Miao, H.; Kempinska, K.; Pollock, J.; Chase, J.; Purohit, T.; Malik, B.; Zhao, T.; Wang, J.; et al. Pharmacologic Inhibition of the Menin-MLL Interaction Blocks Progression of MLL Leukemia in Vivo. Cancer Cell 2015, 27, 589–602. [Google Scholar] [CrossRef]
- Grembecka, J.; He, S.; Shi, A.; Purohit, T.; Muntean, A.G.; Sorenson, R.J.; Showalter, H.D.; Murai, M.J.; Belcher, A.M.; Hartley, T.; et al. Menin-MLL Inhibitors Reverse Oncogenic Activity of MLL Fusion Proteins in Leukemia. Nat. Chem. Biol. 2012, 8, 277–284. [Google Scholar] [CrossRef]
- Krivtsov, A.V.; Evans, K.; Gadrey, J.Y.; Eschle, B.K.; Hatton, C.; Uckelmann, H.J.; Ross, K.N.; Perner, F.; Olsen, S.N.; Pritchard, T.; et al. A Menin-MLL Inhibitor Induces Specific Chromatin Changes and Eradicates Disease in Models of MLL-Rearranged Leukemia. Cancer Cell 2019, 36, 660–673. [Google Scholar] [CrossRef]
- Gallo, M.; Coutinho, F.J.; Vanner, R.J.; Gayden, T.; Mack, S.C.; Murison, A.; Remke, M.; Li, R.; Takayama, N.; Desai, K.; et al. MLL5 Orchestrates a Cancer Self-Renewal State by Repressing the Histone Variant H3.3 and Globally Reorganizing Chromatin. Cancer Cell 2015, 28, 715–729. [Google Scholar] [CrossRef]
- Lan, X.; Jörg, D.J.; Cavalli, F.M.G.; Richards, L.M.; Nguyen, L.V.; Vanner, R.J.; Guilhamon, P.; Lee, L.; Kushida, M.M.; Pellacani, D.; et al. Fate Mapping of Human Glioblastoma Reveals an Invariant Stem Cell Hierarchy. Nature 2017, 549, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Wood, K.; Tellier, M.; Murphy, S. DOT1L and H3K79 Methylation in Transcription and Genomic Stability. Biomolecules 2018, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.E.; Liau, B.B.; Wallace, L.C.; Morton, A.R.; Xie, Q.; Dixit, D.; Factor, D.C.; Kim, L.J.Y.; Morrow, J.J.; Wu, Q.; et al. Transcription Elongation Factors Represent in Vivo Cancer Dependencies in Glioblastoma. Nature 2017, 547, 355–359. [Google Scholar] [CrossRef]
- MacLeod, G.; Bozek, D.A.; Rajakulendran, N.; Monteiro, V.; Ahmadi, M.; Steinhart, Z.; Kushida, M.M.; Yu, H.; Coutinho, F.J.; Cavalli, F.M.G.; et al. Genome-Wide CRISPR-Cas9 Screens Expose Genetic Vulnerabilities and Mechanisms of Temozolomide Sensitivity in Glioblastoma Stem Cells. Cell Rep. 2019, 27, 971–986.e9. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of Radiotherapy with Concomitant and Adjuvant Temozolomide versus Radiotherapy Alone on Survival in Glioblastoma in a Randomised Phase III Study: 5-Year Analysis of the EORTC-NCIC Trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- de Vries, N.A.; Hulsman, D.; Akhtar, W.; de Jong, J.; Miles, D.C.; Blom, M.; van Tellingen, O.; Jonkers, J.; van Lohuizen, M. Prolonged Ezh2 Depletion in Glioblastoma Causes a Robust Switch in Cell Fate Resulting in Tumor Progression. Cell Rep. 2015, 10, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Babikir, H.; Müller, S.; Yagnik, G.; Shamardani, K.; Catalan, F.; Kohanbash, G.; Alvarado, B.; Di Lullo, E.; Kriegstein, A.; et al. The Phenotypes of Proliferating Glioblastoma Cells Reside on a Single Axis of Variation. Cancer Discov. 2019, 9, 1708–1719. [Google Scholar] [CrossRef] [PubMed]
- Richards, L.M.; Whitley, O.K.N.; MacLeod, G.; Cavalli, F.M.G.; Coutinho, F.J.; Jaramillo, J.E.; Svergun, N.; Riverin, M.; Croucher, D.C.; Kushida, M.; et al. Gradient of Developmental and Injury Response Transcriptional States Defines Functional Vulnerabilities Underpinning Glioblastoma Heterogeneity. Nat. Cancer 2021, 2, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Guilhamon, P.; Chesnelong, C.; Kushida, M.M.; Nikolic, A.; Singhal, D.; MacLeod, G.; Madani Tonekaboni, S.A.; Cavalli, F.M.; Arlidge, C.; Rajakulendran, N.; et al. Single-Cell Chromatin Accessibility Profiling of Glioblastoma Identifies an Invasive Cancer Stem Cell Population Associated with Lower Survival. Elife 2021, 10, e64090. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Kim, L.J.Y.; Wu, Q.; Wallace, L.C.; Prager, B.C.; Sanvoranart, T.; Gimple, R.C.; Wang, X.; Mack, S.C.; Miller, T.E.; et al. Targeting Glioma Stem Cells through Combined BMI1 and EZH2 Inhibition. Nat. Med. 2017, 23, 1352–1361. [Google Scholar] [CrossRef]
- Yi, Y.; Li, Y.; Meng, Q.; Li, Q.; Li, F.; Lu, B.; Shen, J.; Fazli, L.; Zhao, D.; Li, C.; et al. A PRC2-Independent Function for EZH2 in Regulating RRNA 2’-O Methylation and IRES-Dependent Translation. Nat. Cell Biol. 2021, 23, 341–354. [Google Scholar] [CrossRef]
- Ghodke-Puranik, Y.; Thorn, C.F.; Lamba, J.K.; Leeder, J.S.; Song, W.; Birnbaum, A.K.; Altman, R.B.; Klein, T.E. Valproic Acid Pathway: Pharmacokinetics and Pharmacodynamics. Pharmacogenet. Genom. 2013, 23, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Kim, M.; Woo, D.H.; Shin, Y.; Shin, J.; Chang, N.; Oh, Y.T.; Kim, H.; Rheey, J.; Nakano, I.; et al. Phosphorylation of EZH2 Activates STAT3 Signaling via STAT3 Methylation and Promotes Tumorigenicity of Glioblastoma Stem-like Cells. Cancer Cell 2013, 23, 839–852. [Google Scholar] [CrossRef]
- Carro, M.S.; Lim, W.K.; Alvarez, M.J.; Bollo, R.J.; Zhao, X.; Snyder, E.Y.; Sulman, E.P.; Anne, S.L.; Doetsch, F.; Colman, H.; et al. The Transcriptional Network for Mesenchymal Transformation of Brain Tumours. Nature 2010, 463, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Halliday, J.; Helmy, K.; Pattwell, S.S.; Pitter, K.L.; LaPlant, Q.; Ozawa, T.; Holland, E.C. In Vivo Radiation Response of Proneural Glioma Characterized by Protective P53 Transcriptional Program and Proneural-Mesenchymal Shift. Proc. Natl. Acad. Sci. USA 2014, 111, 5248–5253. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.; Li, X.; Wang, H.; Chen, G.; Feng, Z.; Wu, Y.; Yin, H.; Zhao, G.; Deng, Z.; Zhao, C.; et al. BRD4 Regulates Self-Renewal Ability and Tumorigenicity of Glioma-Initiating Cells by Enrichment in the Notch1 Promoter Region. Clin. Transl. Med. 2020, 10, e181. [Google Scholar] [CrossRef] [PubMed]
- Gusyatiner, O.; Bady, P.; Pham, M.D.T.; Lei, Y.; Park, J.; Daniel, R.T.; Delorenzi, M.; Hegi, M.E. BET Inhibitors Repress Expression of Interferon-Stimulated Genes and Synergize with HDAC Inhibitors in Glioblastoma. Neuro. Oncol. 2021, 23, 1680–1692. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Jia, S.; Wang, D.; Chen, X.; Kalvakolanu, D.V.; Zheng, H.; Wei, X.; Wen, N.; Liang, H.; Guo, B.; et al. A Combination of BRD4 and HDAC3 Inhibitors Synergistically Suppresses Glioma Stem Cell Growth by Blocking GLI1/IL6/STAT3 Signaling Axis. Mol. Cancer Ther. 2020, 19, 2542–2553. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Epigenetic Factors | Tool Compounds | Clinically Available Drugs | Reference |
|---|---|---|---|
| PRC2/EZH2 | UNC1999, GSK343 | Tazemetostat | [110,111] |
| DNA methylation | 5-Azacytidine, decitabine | [116] (review) | |
| WDR5 | C16 | [126] | |
| Menin/MLL1 | MI-2, MI-3 | SNDX-5613, KO-539 | [128,129,130] |
| DOT1L | Pinometostat | [134,135] |
| Compound | Mode of Action | Other Treatments | Phase | Clinical Trial Number |
|---|---|---|---|---|
| Azacitidine | DNA methylation inhibitor | FT-2102 | 1b/2 | NCT03684811 |
| CC-486 (oral azacitidine) | DNA methylation inhibitor | N/A | 1 | NCT02223052 |
| ST101 | CEBPB inhibitor | N/A | 1/2 | NCT04478279 |
| Borabresib (MK-8628) | BRD2/3/4 inhibitor | N/A | 2 | NCT02296476 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.-M.; Nikolic, A.; Singhal, D.; Gallo, M. Roles of Chromatin Remodelling and Molecular Heterogeneity in Therapy Resistance in Glioblastoma. Cancers 2022, 14, 4942. https://doi.org/10.3390/cancers14194942
Chen H-M, Nikolic A, Singhal D, Gallo M. Roles of Chromatin Remodelling and Molecular Heterogeneity in Therapy Resistance in Glioblastoma. Cancers. 2022; 14(19):4942. https://doi.org/10.3390/cancers14194942
Chicago/Turabian StyleChen, Huey-Miin, Ana Nikolic, Divya Singhal, and Marco Gallo. 2022. "Roles of Chromatin Remodelling and Molecular Heterogeneity in Therapy Resistance in Glioblastoma" Cancers 14, no. 19: 4942. https://doi.org/10.3390/cancers14194942
APA StyleChen, H.-M., Nikolic, A., Singhal, D., & Gallo, M. (2022). Roles of Chromatin Remodelling and Molecular Heterogeneity in Therapy Resistance in Glioblastoma. Cancers, 14(19), 4942. https://doi.org/10.3390/cancers14194942