

A Potential Fatty Acid Metabolism-Related Gene Signature for Prognosis in Clear Cell Renal Cell Carcinoma

Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data source and Processing
2.2. Candidate Selection and Signature Establishment
2.3. Quantitative RT-qPCR and Risk Score Calculations of Clinical Cohort
2.4. Bioinformatics and Statistical Analyses
3. Results
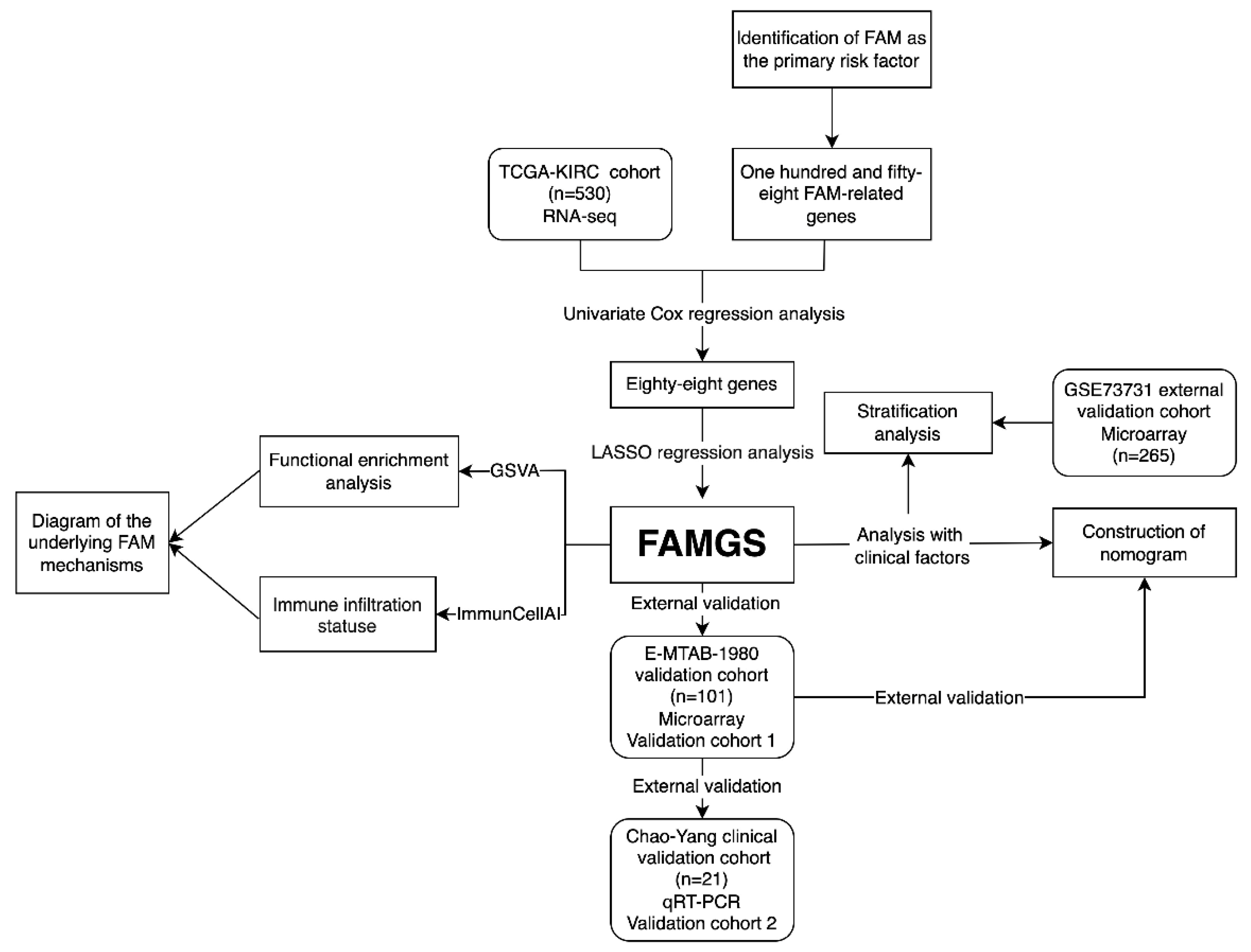
3.1. Study Design and Cohort Characteristics
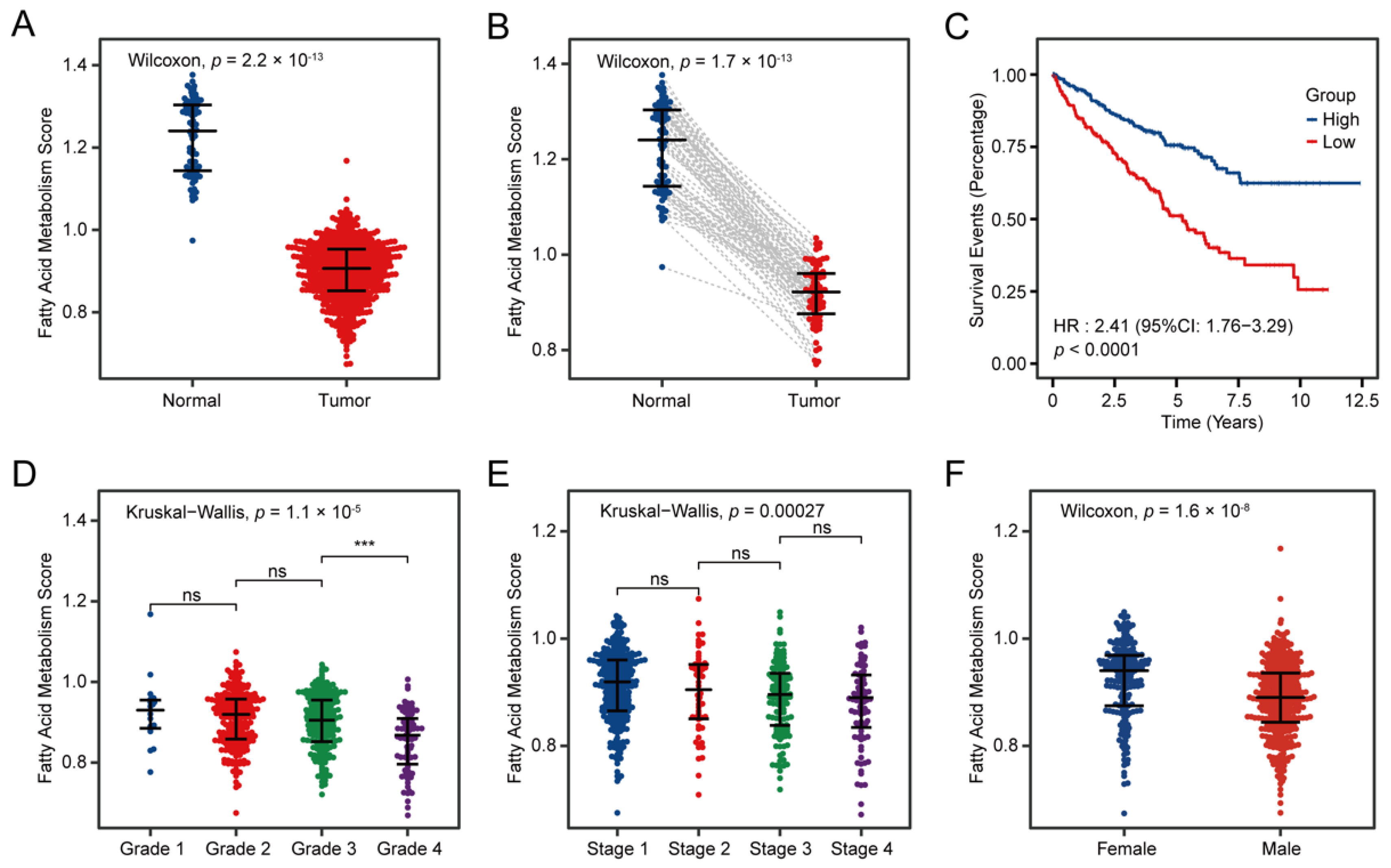
3.2. Fatty Acid Metabolism Confirmed as a Crucial Factor in ccRCC
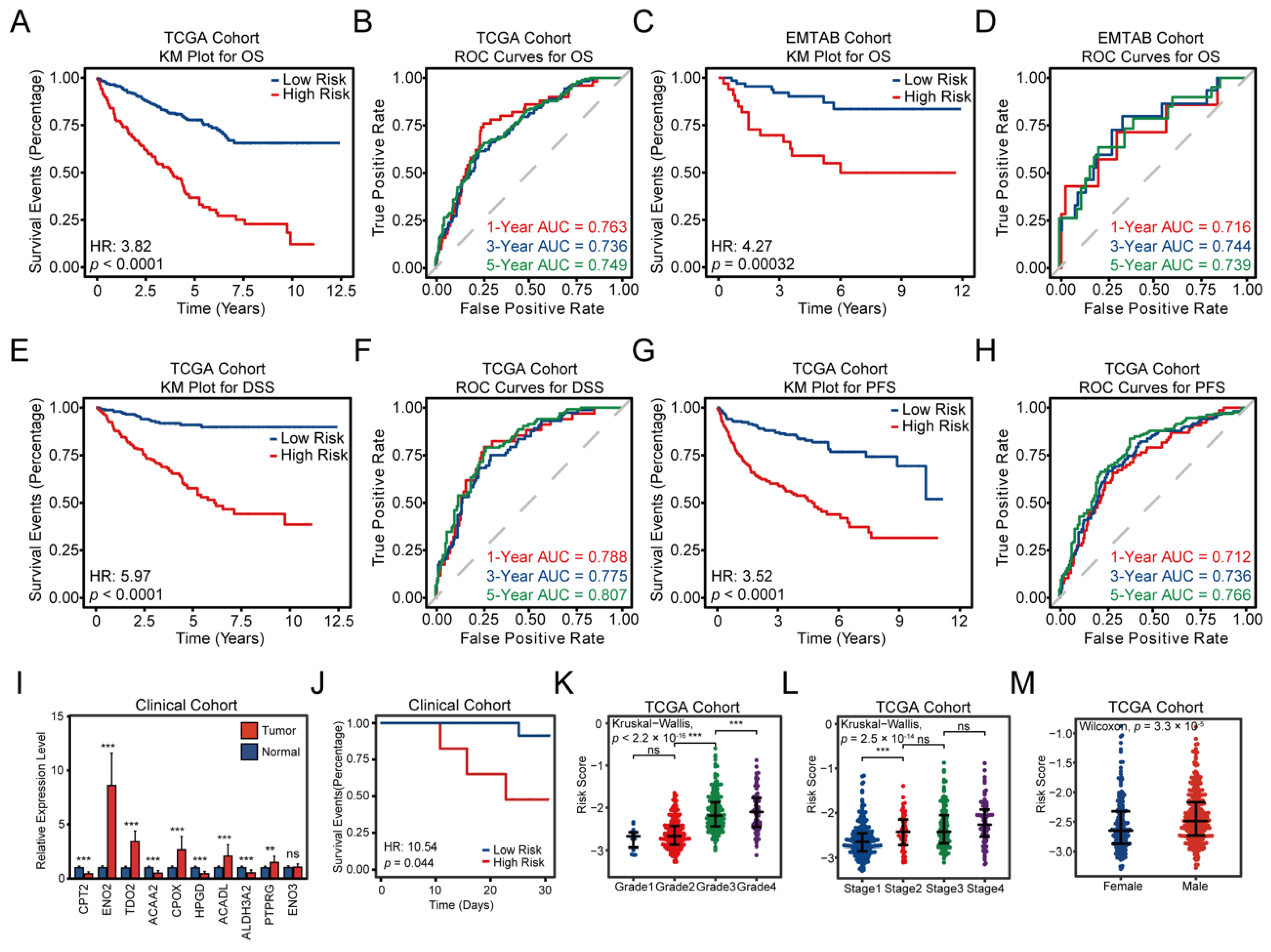
3.3. Construction and Validation of the FAMGS for Prognosis
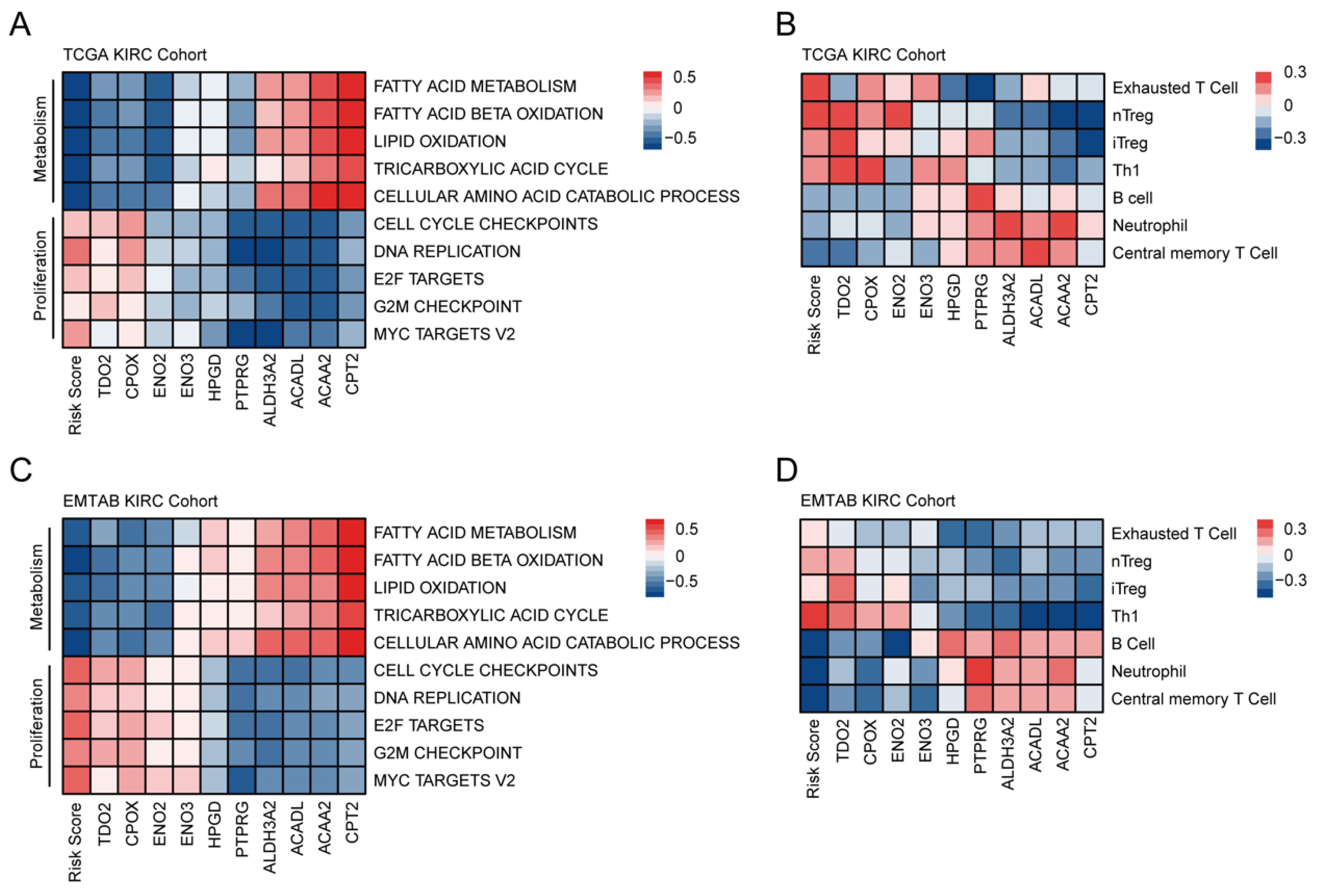
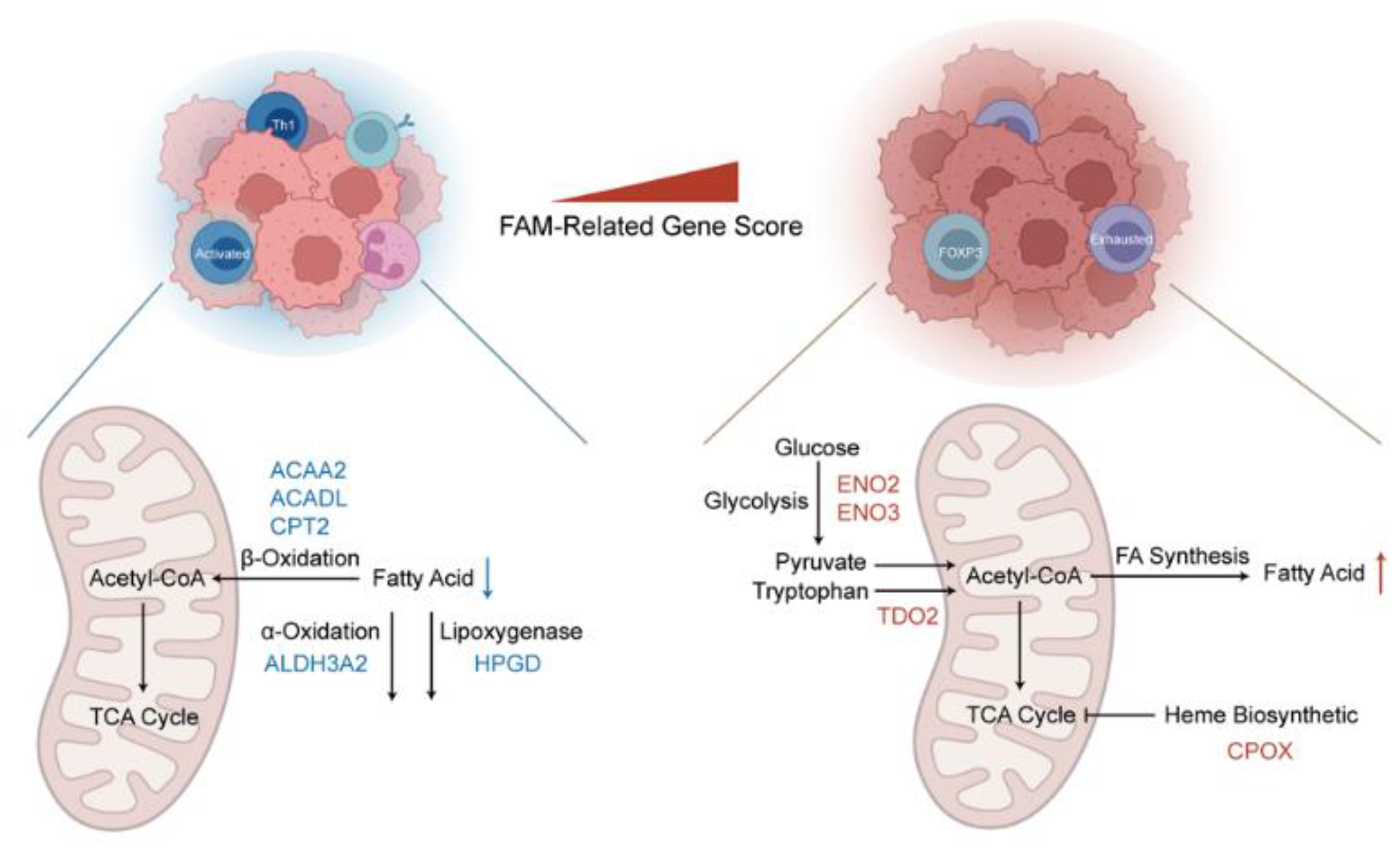
3.4. Comprehensive Enrichment Analyses and Immune Infiltration
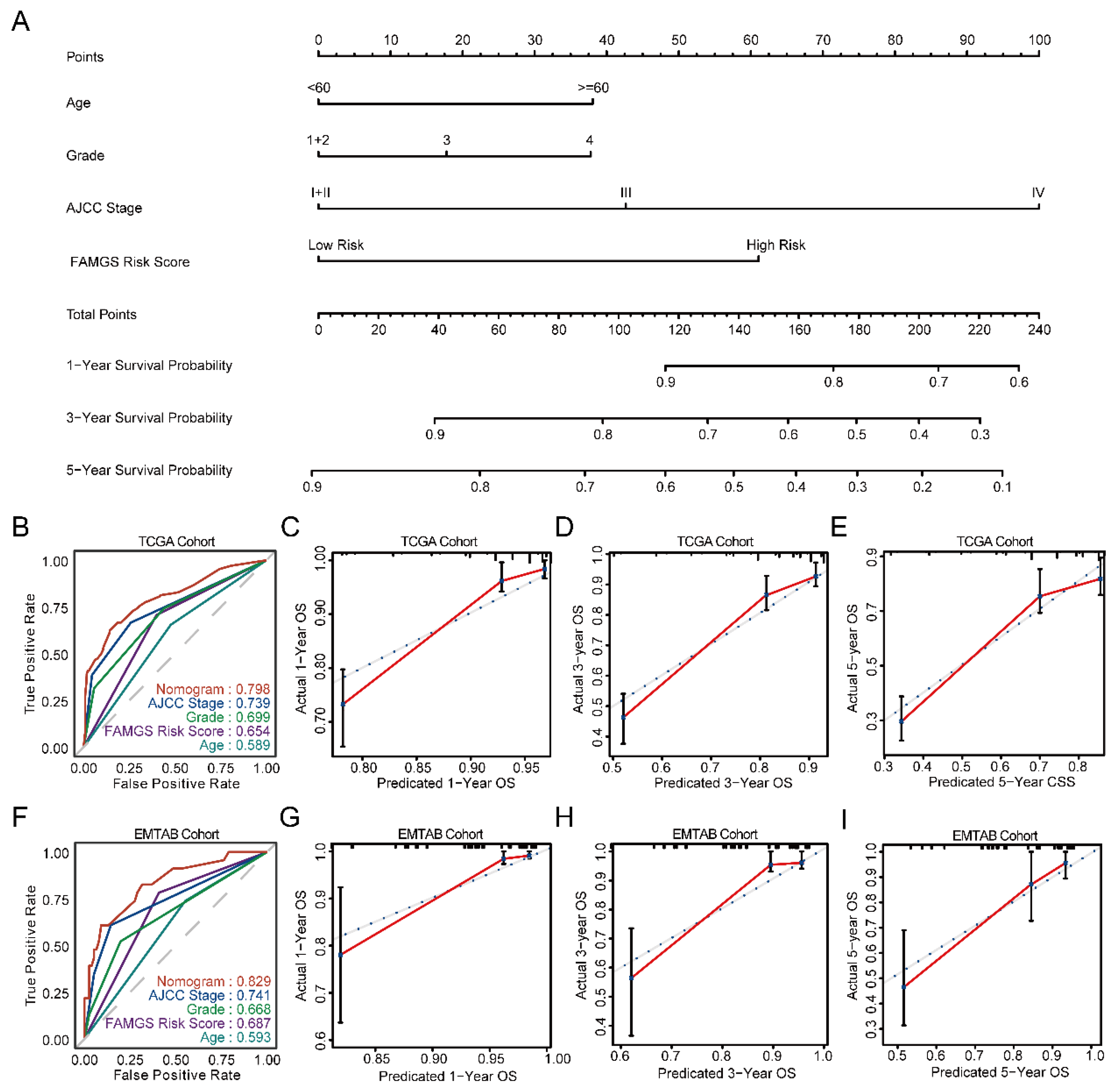
3.5. Establishment and Verification of a Nomogram Model According to the FAMGS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal Cell Carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009. [Google Scholar] [CrossRef]
- Corn, K.C.; Windham, M.A.; Rafat, M. Lipids in the Tumor Microenvironment: From Cancer Progression to Treatment. Prog. Lipid Res. 2020, 80, 101055. [Google Scholar] [CrossRef] [PubMed]
- Koundouros, N.; Poulogiannis, G. Reprogramming of Fatty Acid Metabolism in Cancer. Brit. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s Contributions to Current Concepts of Cancer Metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, A.; Asano, T.; Asano, T.; Ito, K.; Sumitomo, M.; Hayakawa, M. Fatty Acid Synthase over Expression Is an Indicator of Tumor Aggressiveness and Poor Prognosis in Renal Cell Carcinoma. J. Urol. 2008, 180, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Wettersten, H.I.; Aboud, O.A.; Lara, P.N.; Weiss, R.H. Metabolic Reprogramming in Clear Cell Renal Cell Carcinoma. Nat. Rev. Nephrol. 2017, 13, 410–419. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Program-NCI. Available online: https://portal.gdc.cancer.gov (accessed on 24 July 2022).
- E-MTAB-1980 <Browse <ArrayExpress <EMBL-EBI. Available online: https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-1980/ (accessed on 24 July 2022).
- GEO Accession Viewer. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE73731 (accessed on 24 July 2022).
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular Signatures Database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Tibshirani, R. The Lasso Method for Variable Selection in the Cox Model. Stat. Med. 1997, 16, 385–395. [Google Scholar] [CrossRef]
- Dyrskjøt, L.; Reinert, T.; Novoradovsky, A.; Zuiverloon, T.C.M.; Beukers, W.; Zwarthoff, E.; Malats, N.; Real, F.X.; Segersten, U.; Malmström, P.-U.; et al. Analysis of Molecular Intra-Patient Variation and Delineation of a Prognostic 12-Gene Signature in Non-Muscle Invasive Bladder Cancer; Technology Transfer from Microarrays to PCR. Br. J. Cancer 2012, 107, 1392–1398. [Google Scholar] [CrossRef]
- Dyrskjøt, L.; Reinert, T.; Algaba, F.; Christensen, E.; Nieboer, D.; Hermann, G.G.; Mogensen, K.; Beukers, W.; Marquez, M.; Segersten, U.; et al. Prognostic Impact of a 12-Gene Progression Score in Non–Muscle-Invasive Bladder Cancer: A Prospective Multicentre Validation Study. Eur. Urol. 2017, 72, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Therneau, T.M.; Lumley, T.; Elizabeth, A.; Cynthia, C. Survival: Survival Analysis. 2022. Available online: https://github.com/therneau/survival (accessed on 24 July 2022).
- Heagerty, P.J.; Lumley, T.; Pepe, M.S. Time-Dependent ROC Curves for Censored Survival Data and a Diagnostic Marker. Biometrics 2000, 56, 337–344. [Google Scholar] [CrossRef]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene Set Variation Analysis for Microarray and RNA-Seq Data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.-R.; Zhang, Q.; Lei, Q.; Luo, M.; Xie, G.-Y.; Wang, H.; Guo, A.-Y. ImmuCellAI: A Unique Method for Comprehensive T-Cell Subsets Abundance Prediction and Its Application in Cancer Immunotherapy. Adv. Sci. (Weinh) 2020, 7, 1902880. [Google Scholar] [CrossRef] [PubMed]
- Kolde, R. Pheatmap: Pretty Heatmaps. 2019. Available online: https://github.com/raivokolde/pheatmap (accessed on 24 July 2022).
- Zhang, Z.; Kattan, M.W. Drawing Nomograms with R: Applications to Categorical Outcome and Survival Data. Ann. Transl. Med. 2017, 5, 211. [Google Scholar] [CrossRef] [PubMed]
- Ljungberg, B.; Bensalah, K.; Canfield, S.; Dabestani, S.; Hofmann, F.; Hora, M.; Kuczyk, M.A.; Lam, T.; Marconi, L.; Merseburger, A.S.; et al. EAU Guidelines on Renal Cell Carcinoma: 2014 Update. Eur. Urol. 2015, 67, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic Reprogramming and Cancer Progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef] [PubMed]
- Röhrig, F.; Schulze, A. The Multifaceted Roles of Fatty Acid Synthesis in Cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer Metabolism: Fatty Acid Oxidation in the Limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef]
- Yuan, Y.; Yang, X.; Li, Y.; Liu, Q.; Wu, F.; Qu, H.; Gao, H.; Ge, J.; Xu, Y.; Wang, H.; et al. Expression and Prognostic Significance of Fatty Acid Synthase in Clear Cell Renal Cell Carcinoma. Pathol.-Res. Pract. 2020, 216, 153227. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Cai, L.; Huang, W.; Weng, Q.; Lin, X.; You, M.; Liao, S. Prognostic Value of Fatty Acid Metabolism-Related Genes in Patients with Hepatocellular Carcinoma. Aging 2021, 13, 17847–17863. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Xu, C.; Wen, J.; Zhang, Y.; Wang, M.; Liu, X.; Zhao, K.; Wang, Z.; Liu, Y.; Zhang, T. Fatty Acid Metabolism-Related LncRNAs Are Potential Biomarkers for Predicting the Overall Survival of Patients With Colorectal Cancer. Front. Oncol. 2021, 11, 704038. [Google Scholar] [CrossRef]
- Qi, Y.; Chen, D.; Lu, Q.; Yao, Y.; Ji, C. Bioinformatic Profiling Identifies a Fatty Acid Metabolism-Related Gene Risk Signature for Malignancy, Prognosis, and Immune Phenotype of Glioma. Dis. Markers 2019, 2019, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Cheng, G.; Ye, Y.; Le, C.; Miao, Q.; Chen, J.; Yang, H.; Zhang, X. A Fatty Acid Metabolism Signature Associated With Clinical Therapy in Clear Cell Renal Cell Carcinoma. Front. Genet. 2022, 13, 18. [Google Scholar] [CrossRef]
- Jonasch, E.; Walker, C.L.; Rathmell, W.K. Clear Cell Renal Cell Carcinoma Ontogeny and Mechanisms of Lethality. Nat. Rev. Nephrol. 2021, 17, 245–261. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and Cellular Insights into T Cell Exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Chen, Z.; Bao, L.; Zhou, L.; Hou, Y.; Liu, L.; Xiong, M.; Zhang, Y.; Wang, B.; Tao, Z.; et al. Single-Cell Transcriptome Analysis Reveals Intratumoral Heterogeneity in CcRCC, Which Results in Different Clinical Outcomes. Mol. Ther. 2020, 28, 1658–1672. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.A. Lipid Signalling Enforces Functional Specialization of Treg Cells in Tumours. Nature 2021, 591, 306–311. [Google Scholar] [CrossRef]
- González-Romero, F.; Mestre, D.; Aurrekoetxea, I.; O’Rourke, C.J.; Andersen, J.B.; Woodhoo, A.; Tamayo-Caro, M.; Varela-Rey, M.; Palomo-Irigoyen, M.; Gómez-Santos, B.; et al. E2F1 and E2F2-Mediated Repression of CPT2 Establishes a Lipid-Rich Tumor-Promoting Environment. Cancer Res. 2021, 81, 2874–2887. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Guo, D.; Zhang, X.; Zhu, Y.; Wang, Z.; Jin, Y.; Guo, W.; Zhang, S. ENO3 Inhibits Growth and Metastasis of Hepatocellular Carcinoma via Wnt/β-Catenin Signaling Pathway. Front. Cell Dev. Biol. 2021, 9, 797102. [Google Scholar] [CrossRef] [PubMed]
- Cheong, J.E.; Sun, L. Targeting the IDO1/TDO2–KYN–AhR Pathway for Cancer Immunotherapy–Challenges and Opportunities. Trends Pharmacol. Sci. 2018, 39, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, M.; Liu, M.; Xu, Y.; Wu, G. Glycolysis-Related Genes Serve as Potential Prognostic Biomarkers in Clear Cell Renal Cell Carcinoma. Oxidative Med. Cell. Longev. 2021, 2021, 6699808. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Chu, W.; Yang, W.; Cheng, Y.; Chu, C.; Pan, X.; Ye, J.; Cao, J.; Gan, S.; Cui, X. Identification of RNA Transcript Makers Associated With Prognosis of Kidney Renal Clear Cell Carcinoma by a Competing Endogenous RNA Network Analysis. Front. Genet. 2020, 11, 540094. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Li, L.; Hu, Z.; Peng, D. Integrated Multi-Omics Analysis Identified PTPRG and CHL1 as Key Regulators of Immunophenotypes in Clear Cell Renal Cell Carcinoma(CcRCC). Front. Oncol. 2022, 12, 832027. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Tao, Z.; Chen, X. A Three-Metabolic-Genes Risk Score Model Predicts Overall Survival in Clear Cell Renal Cell Carcinoma Patients. Front. Oncol. 2020, 10, 570281. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | TCGA Training Cohort | EMTAB Validation Cohort | Chao-Yang Validation Cohort | |
|---|---|---|---|---|
| Number of Patients | 530 | 101 | 21 | |
| Overall Survival (IQR) | 1181.5 (520, 1912) | 1530 (1020, 2430) | 848 (712,916) | |
| Overall Survival Status (%) | Survival | 357 (67.36) | 78 (77.23) | 17 (80.95%) |
| Deceased | 173 (32.64) | 23 (22.77) | 4 (19.05%) | |
| Age (IQR) | 61 (52, 70) | 64 (56, 72) | 67 (62.5, 72.5) | |
| Gender (%) | Male | 344 (64.91) | 77 (76.24) | 16 (76.19%) |
| Female | 186 (35.09) | 24 (23.76) | 5 (23.81%) | |
| Grade (%) | G1 | 14 (2.64) | 13 (12.87) | 2 (9.52%) |
| G2 | 227 (42.83) | 59 (58.42) | 14 (57.14%) | |
| G3 | 207 (39.06) | 22 (21.78) | 5 (23.81%) | |
| G4 | 74 (13.96) | 5 (4.95) | 0 (0.00) | |
| Not Available | 8 (1.51) | 2 (1.98) | 0 (0.00) | |
| AJCC Stage (%) | Stage I | 265 (50.00) | 66 (65.35) | 13 (61.91%) |
| Stage II | 57 (10.75) | 10 (9.90) | 4 (19.05%) | |
| Stage III | 123 (23.21) | 13 (12.87) | 2 (9.52%) | |
| Stage IV | 82 (15.47) | 12 (11.88) | 2 (9.52%) | |
| Not Available | 3 (0.57) | 0 (0.00) | 0 (0.00) |
| TCGA Training Cohort | ||||||
|---|---|---|---|---|---|---|
| Univariate | Multivariate | |||||
| Factors | HR (95% CI) | p Value | HR (95% CI) | p Value | ||
| FAMGS Risk Score | 3.729 (2.752–5.053) | <0.001 | 2.647 (1.911–3.673) | <0.001 | ||
| Age | 1.825 (1.333–2.5) | <0.001 | 1.624 (1.18–2.234) | 0.003 | ||
| Gender | 0.941 (0.691–1.283) | 0.7 | ||||
| Grade | G1 + G2 | 1 | G1 + G2 | 1 | ||
| G3 | 1.947 (1.339–2.832) | <0.001 | G3 | 1.222 (0.823–1.813) | 0.321 | |
| G4 | 5.235 (3.521–7.787) | <0.001 | G4 | 1.616 (1.01–2.587) | 0.045 | |
| AJCC Stage | Stage I + II | 1 | Stage I + II | 1 | ||
| Stage III | 2.51 (1.713–3.678) | <0.001 | Stage III | 1.75 (1.172–2.611) | 0.006 | |
| Stage IV | 6.192 (4.341–8.833) | <0.001 | Stage IV | 3.618 (2.403–5.448) | <0.001 | |
| EMTAB Validation Cohort | ||||||
| Univariate | Multivariate | |||||
| Factors | HR (95% CI) | p value | HR (95% CI) | p value | ||
| FAMGS Risk Score | 4.419 (1.872–10.431) | <0.001 | 2.964 (1.073–8.184) | 0.036 | ||
| Age | 2.262 (0.891–5.747) | 0.086 | 1.717 (0.604–4.823) | 0.313 | ||
| Gender | 0.441 (0.131–1.486) | 0.187 | ||||
| Grade | G1 + G2 | 1 | G1 + G2 | 1 | ||
| G3 | 3.015 (1.247–7.288) | 0.014 | G3 | 1.193 (0.425–3.347) | 0.738 | |
| G4 | 12.378 (3.222–47.557) | <0.001 | G4 | 3.277 (0.699–15.351) | 0.132 | |
| AJCC Stage | Stage I + II | 1 | Stage I + II | 1 | ||
| Stage III | 5.651 (1.985–16.081) | 0.001 | Stage III | 3.284 (1.084–9.948) | 0.036 | |
| Stage IV | 9.298 (3.551–24.341) | <0.001 | Stage IV | 6.246 (2.116–18.438) | <0.001 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Zhang, D.; Hu, X. A Potential Fatty Acid Metabolism-Related Gene Signature for Prognosis in Clear Cell Renal Cell Carcinoma. Cancers 2022, 14, 4943. https://doi.org/10.3390/cancers14194943
Zhang H, Zhang D, Hu X. A Potential Fatty Acid Metabolism-Related Gene Signature for Prognosis in Clear Cell Renal Cell Carcinoma. Cancers. 2022; 14(19):4943. https://doi.org/10.3390/cancers14194943
Chicago/Turabian StyleZhang, He, Di Zhang, and Xiaopeng Hu. 2022. "A Potential Fatty Acid Metabolism-Related Gene Signature for Prognosis in Clear Cell Renal Cell Carcinoma" Cancers 14, no. 19: 4943. https://doi.org/10.3390/cancers14194943
APA StyleZhang, H., Zhang, D., & Hu, X. (2022). A Potential Fatty Acid Metabolism-Related Gene Signature for Prognosis in Clear Cell Renal Cell Carcinoma. Cancers, 14(19), 4943. https://doi.org/10.3390/cancers14194943