
Oxidative Stress and Autophagy Mediate Anti-Cancer Properties of Cannabis Derivatives in Human Oral Cancer Cells

 , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Methods and Materials
2.1. Cell Culture
2.2. Drugs
2.3. Cell Viability Assay
2.4. LDH Assay
2.5. Determination of ROS Levels by Flow Cytometry
2.6. Measurement of Mitochondrial Superoxide
2.7. Mitochondrial Membrane Potential (ΔΨm) Assay
2.8. Intracellular Reduced Glutathione (GSH) Assay
2.9. Quantification of Cellular Autophagy
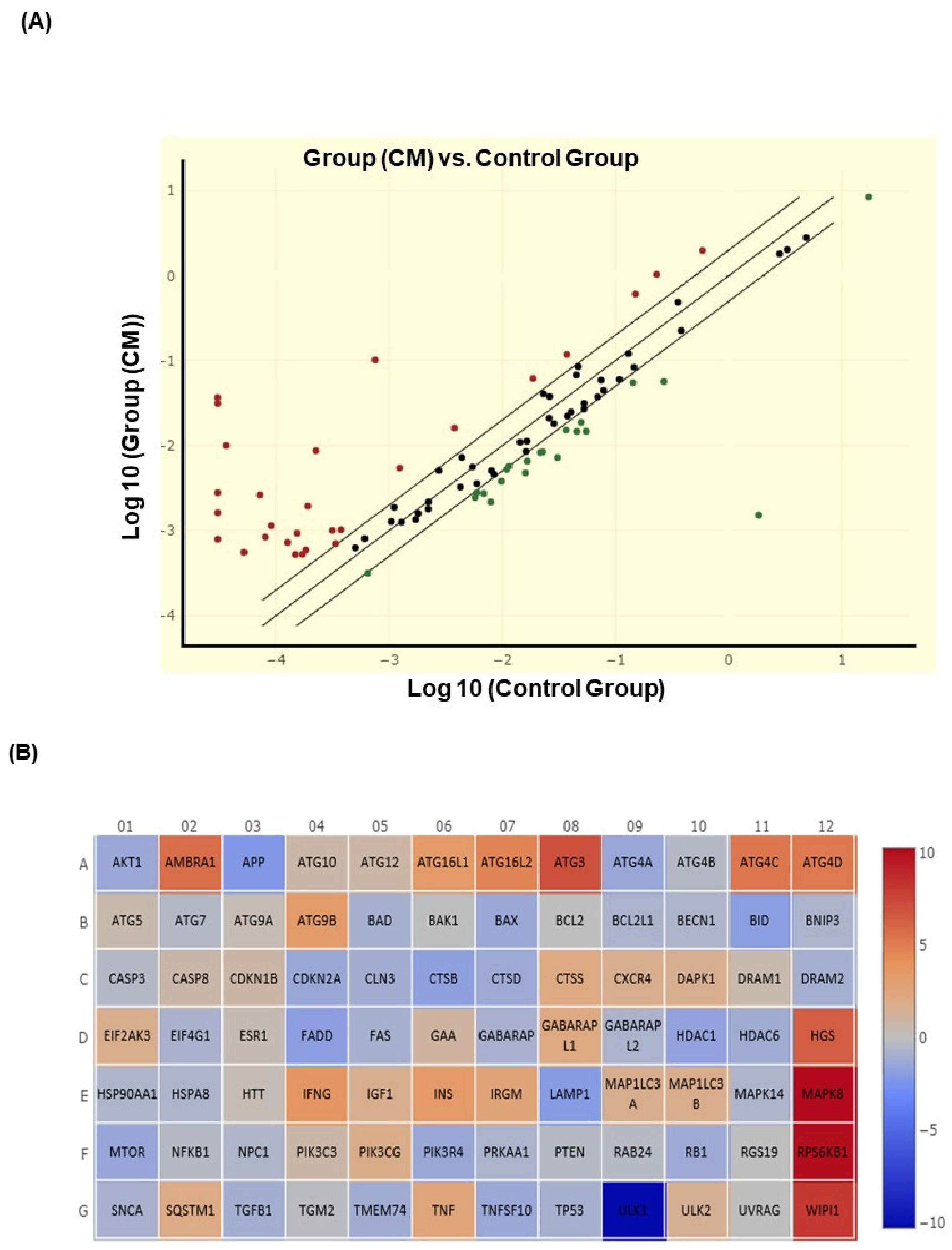
2.10. Autophagy Gene Expression by Using RT2 Profiler PCR Array
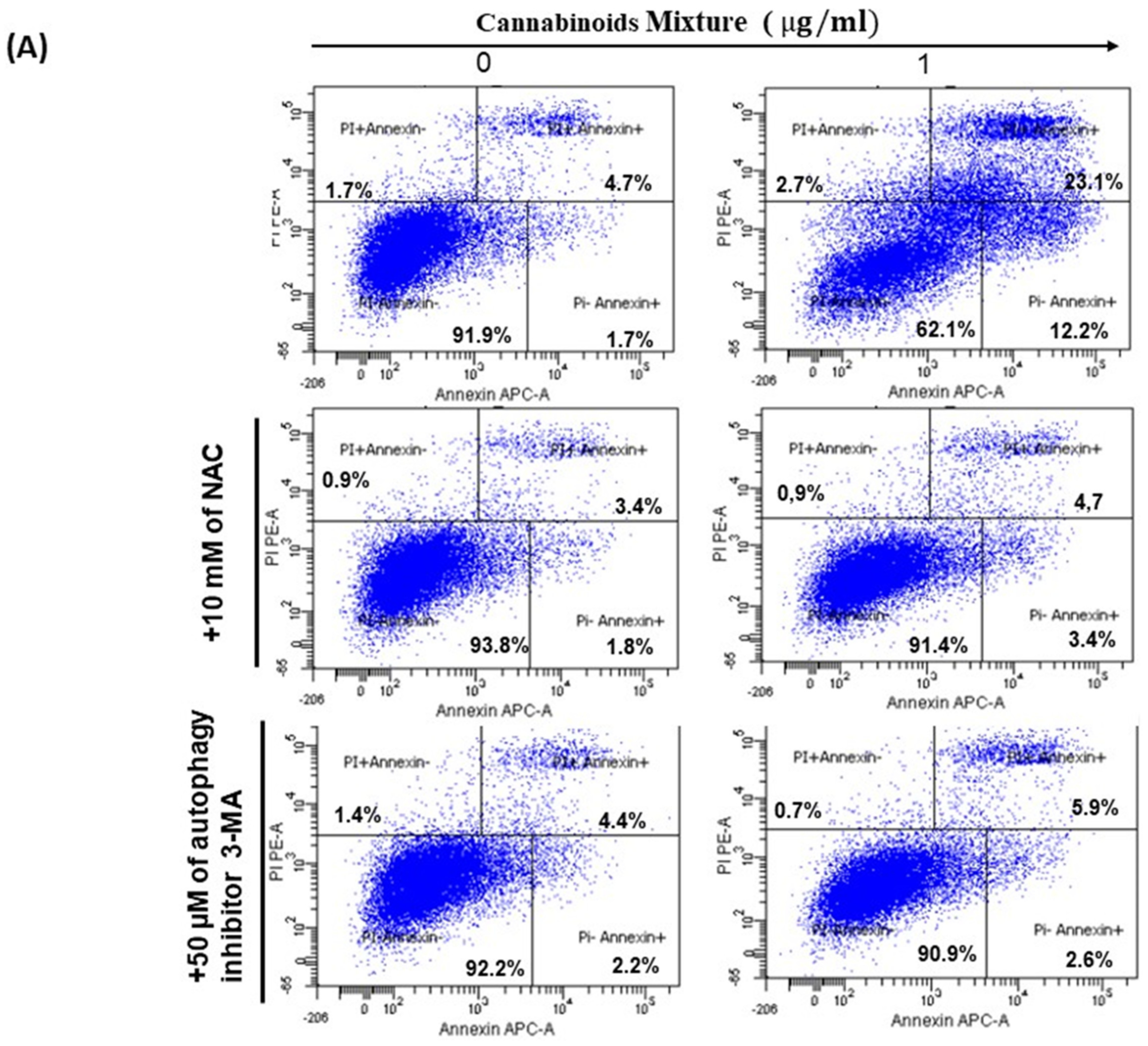
2.11. Cell Apoptosis and Programmed Cell Death by Flow Cytometry with Annexin V Assay
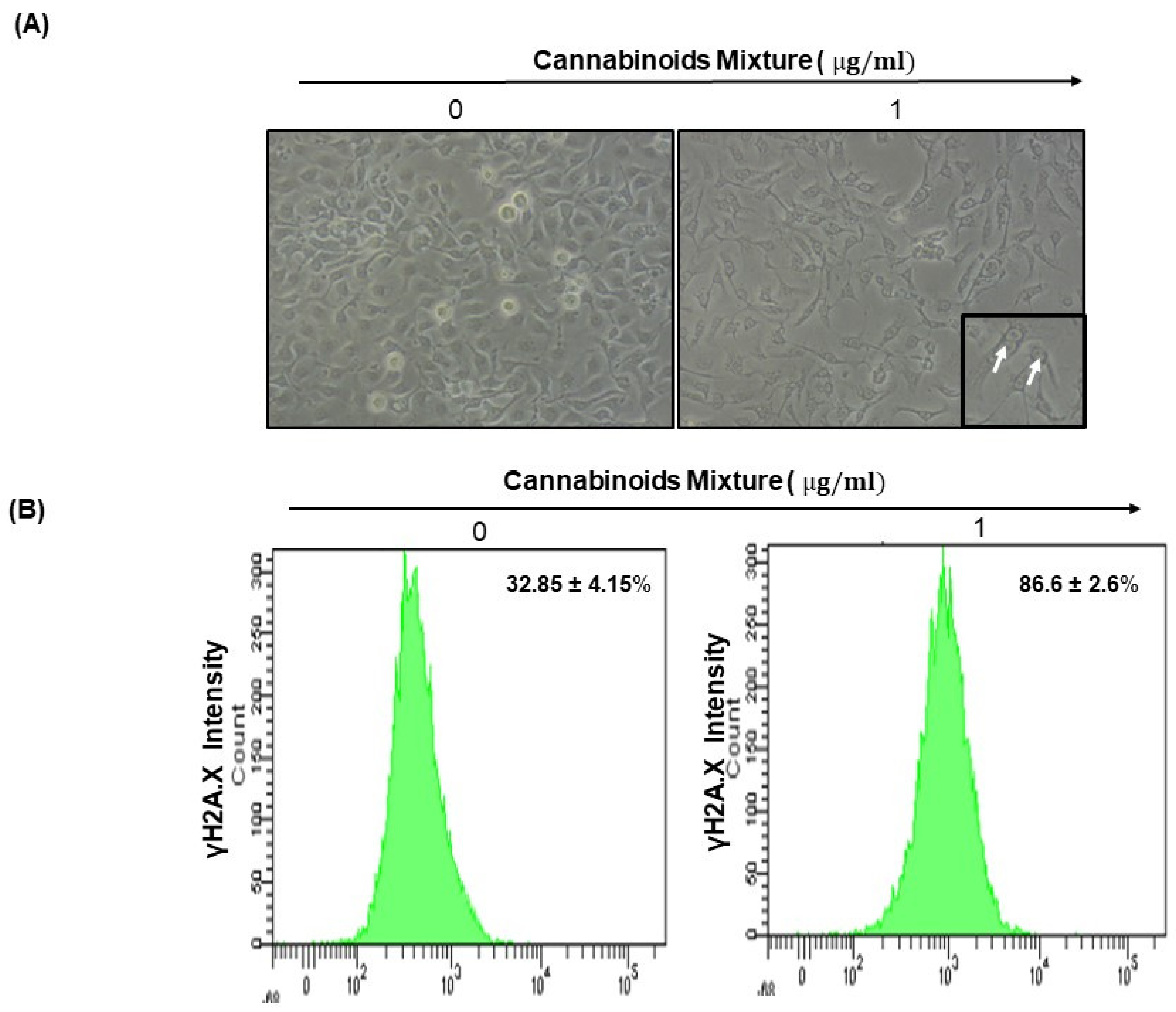
2.12. Cell Morphology
2.13. DNA Damage Assessment by Flow Cytometry
2.14. Cell Migration Determined by Wound-Healing Assay
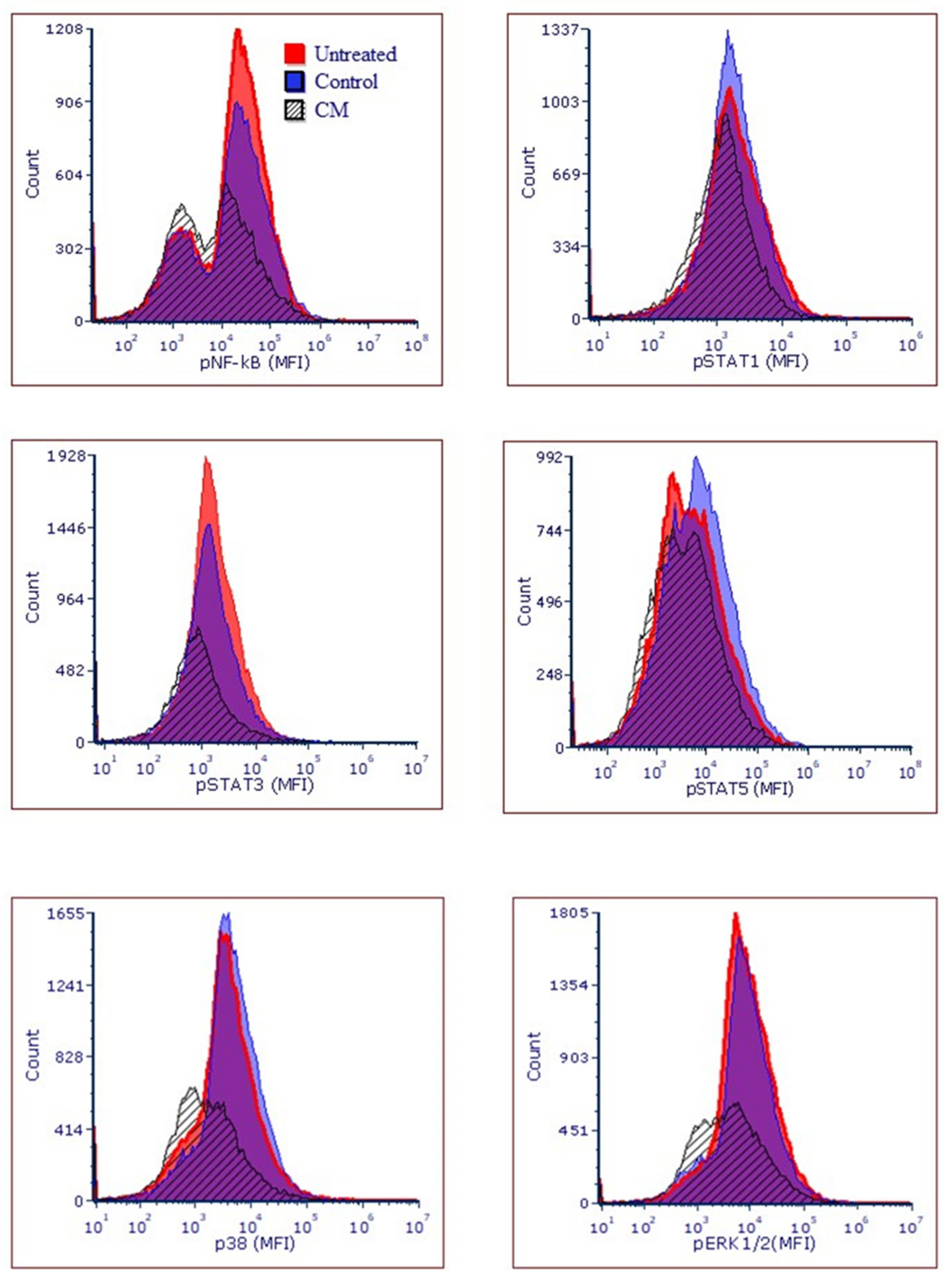
2.15. Flow Cytometric Analysis for Studies on Cancer Pathways
2.16. Statistical Analysis
3. Results
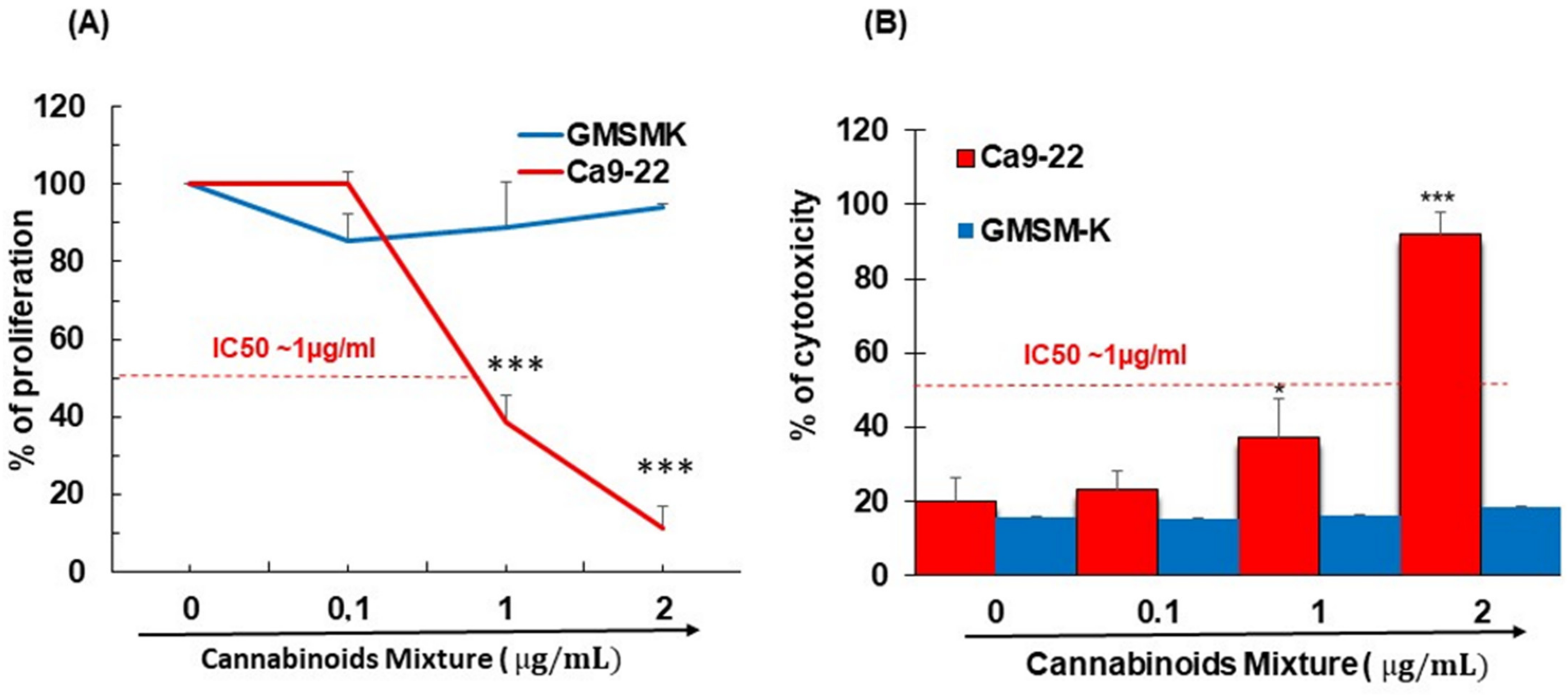
3.1. CM Selectively Inhibits Oral Cancer Cell Proliferation and Promote Their Cytotoxicity
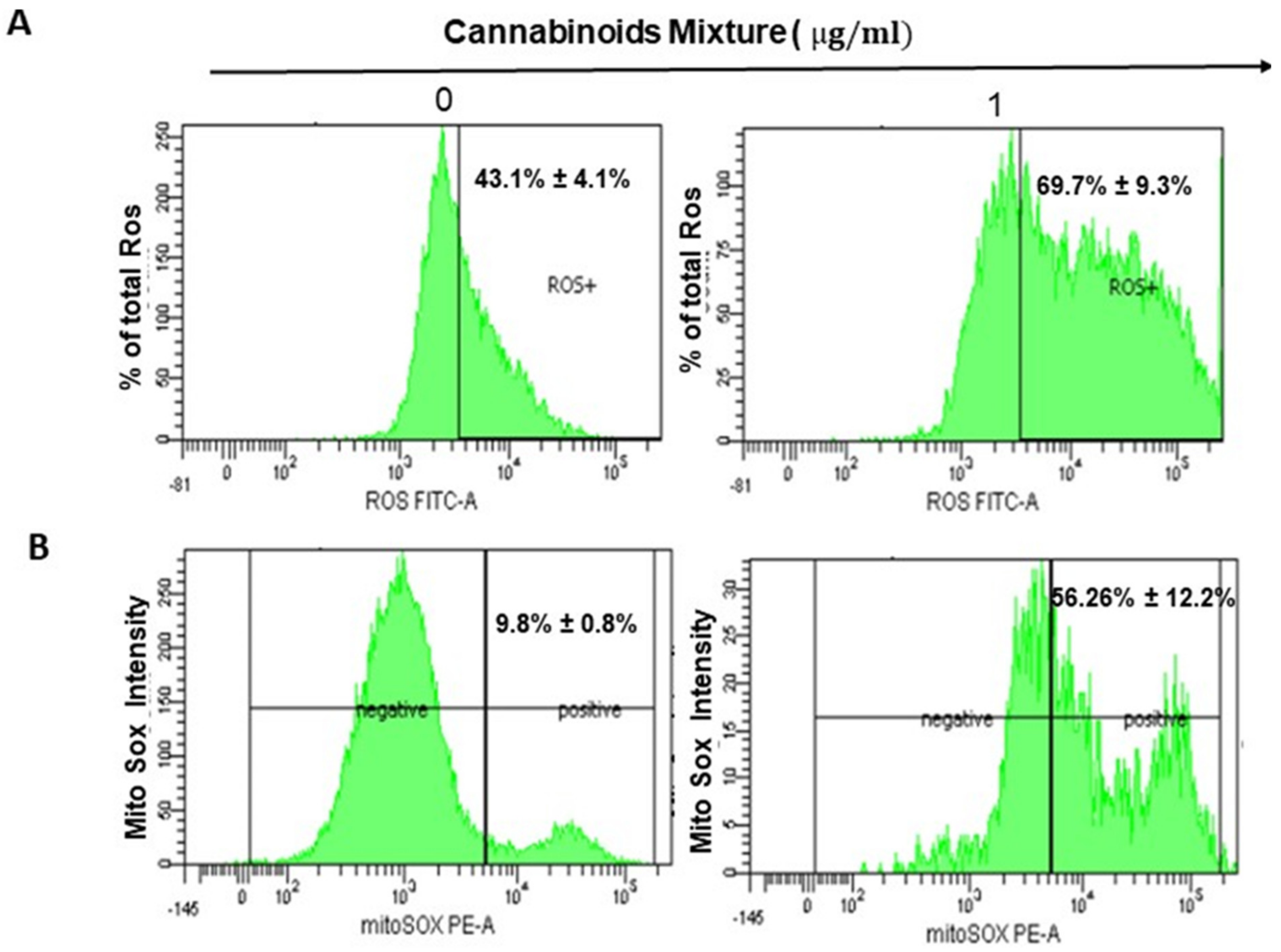
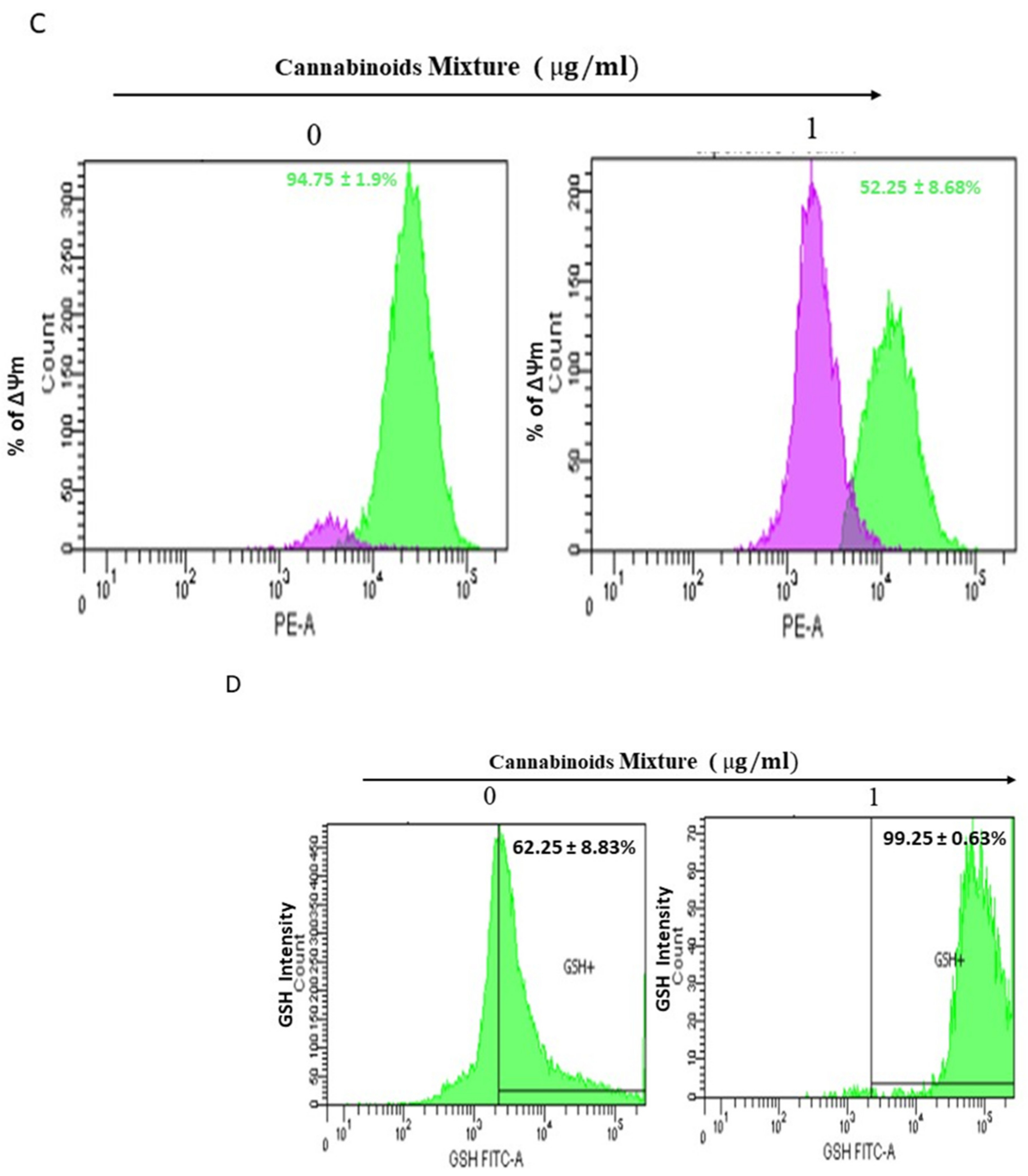
3.2. CM Exposure Results in Both Total Intracellular ROS and Mitochondrial ROS Induction and Provokes Mitochondrial Membrane Potential Inhibition
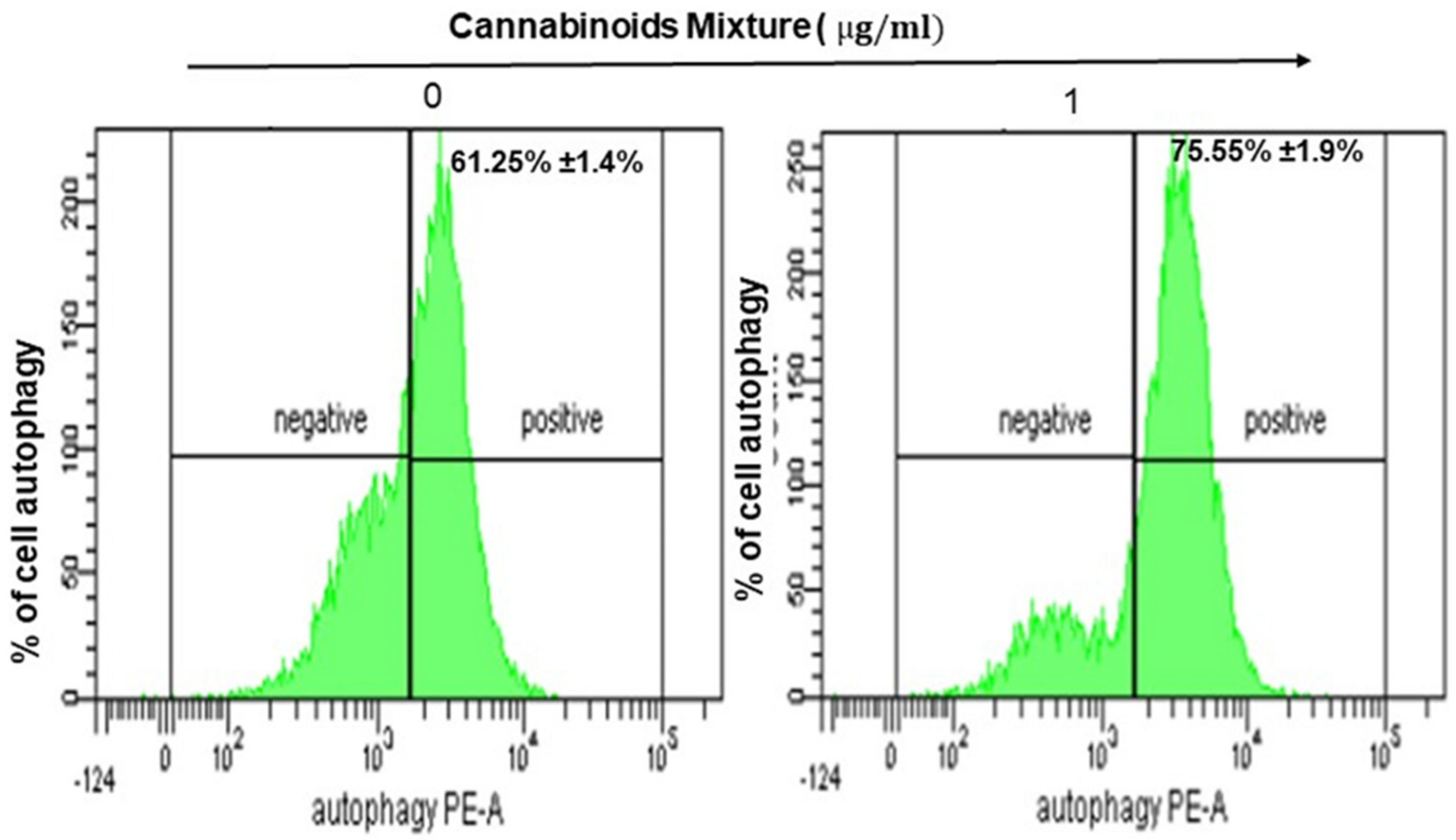
3.3. CM Stimulates Autophagy in Oral Cancer Cells
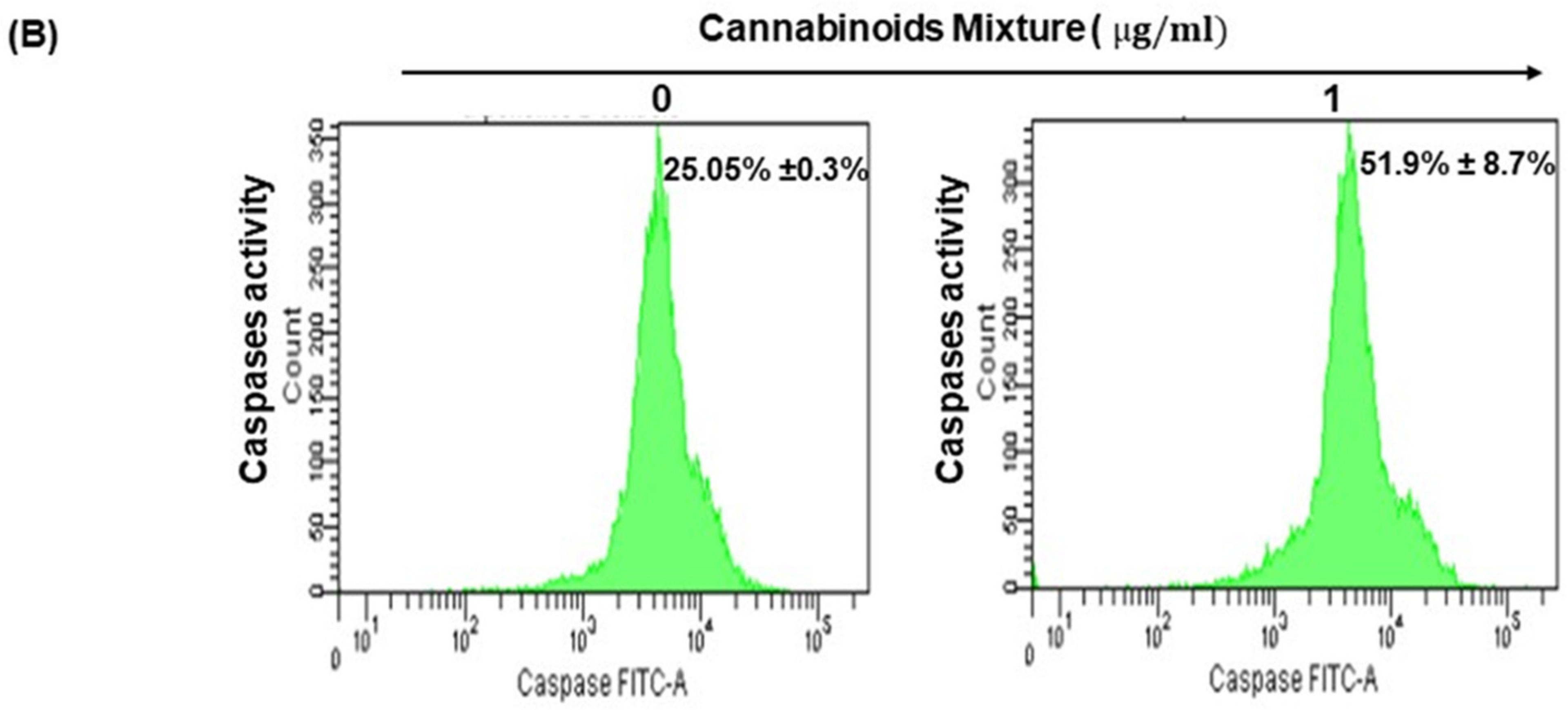
3.4. CM Induces Apoptosis in Oral Cancer Cells Via Caspase Activation
3.5. CM Affects Ca9-22 Cell Morphology and Induces DNA Damage
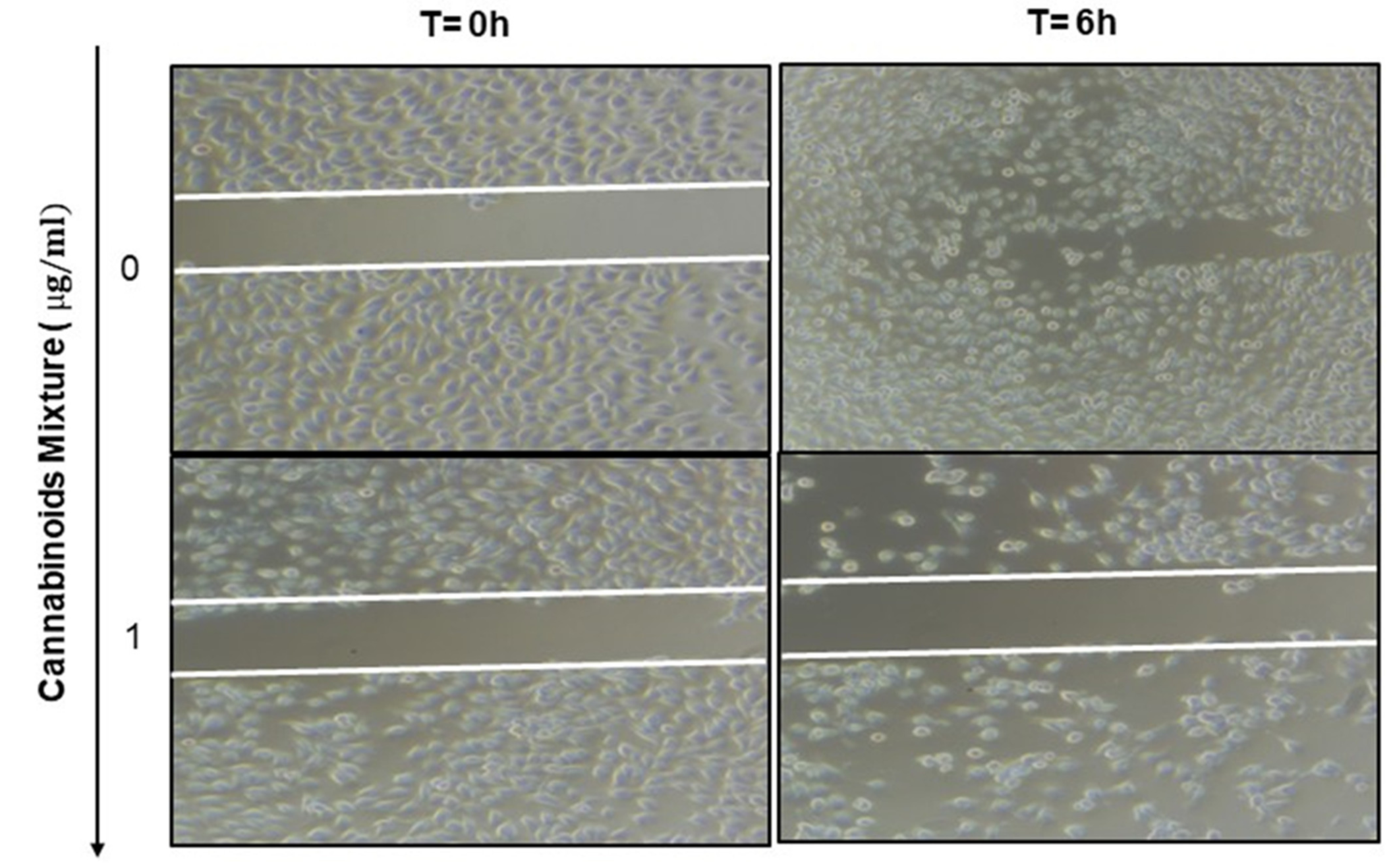
3.6. Anti-Migratory Effect of Cannabinoid Mixture in Ca9-22 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA-Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Zhang, S.Z.; Xie, L.; Shang, Z.J. Burden of Oral Cancer on the 10 Most Populous Countries from 1990 to 2019: Estimates from the Global Burden of Disease Study 2019. Int. J. Environ. Res. Public Health 2022, 19, 875. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Key Statistics for Oral Cavity and Oropharyngeal Cancers. 2022, pp. 1–14. Available online: https://www.cancer.org/ (accessed on 1 September 2022).
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef]
- Pietrobon, G.; Tagliabue, M.; Stringa, L.M.; De Berardinis, R.; Chu, F.; Zocchi, J.; Carlotto, E.; Chiocca, S.; Ansarin, M. Leukoplakia in the Oral Cavity and Oral Microbiota: A Comprehensive Review. Cancers 2021, 13, 4439. [Google Scholar] [CrossRef]
- Manoharan, S.; Kolanjiappan, K.; Suresh, K.; Panjamurthy, K. Lipid peroxidation & antioxidants status in patients with oral squamous cell carcinoma. Indian J. Med. Res. 2005, 122, 529–534. [Google Scholar]
- Alexandra, T.; Marina, I.M.; Daniela, M.; Ioana, S.I.; Maria, B.; Radu, R.; Maria, T.A.; Tudor, S.; Maria, G. Autophagy-A Hidden but Important Actor on Oral Cancer Scene. Int. J. Mol. Sci. 2020, 21, 9325. [Google Scholar] [CrossRef]
- Patil, S.; Rao, R.S.; Raj, A.T. Dual Role of Autophagy in Oral Cancer. J. Int. Oral Health 2015, 7, i–ii. [Google Scholar] [PubMed]
- Halliwell, B. Biochemistry of oxidative stress. Biochem. Soc. Trans. 2007, 35 Pt 5, 1147–1150. [Google Scholar] [CrossRef]
- Halliwell, B. Oxidative stress and cancer: Have we moved forward? Biochem. J. 2007, 401, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Lu, P.; Beeraka, N.M.; Sukocheva, O.A.; Madhunapantula, S.V.; Liu, J.; Sinelnikov, M.Y.; Nikolenko, V.N.; Bulygin, K.V.; Mikhaleva, L.M.; et al. Mitochondrial mutations and mitoepigenetics: Focus on regulation of oxidative stress-induced responses in breast cancers. Semin. Cancer Biol. 2022, 83, 556–569. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, J.; Beeraka, N.M.; Tang, C.; Babayeva, Y.V.; Sinelnikov, M.Y.; Zhang, X.; Zhang, J.; Liu, J.; Reshetov, I.V.; et al. Advances in the Prevention and Treatment of Obesity-Driven Effects in Breast Cancers. Front. Oncol. 2022, 12, 820968. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, C.; Wang, X.; Sun, Y.; Zhang, J.; Chen, J.; Shi, Y. An Epigenetic Role of Mitochondria in Cancer. Cells 2022, 11, 2518. [Google Scholar] [CrossRef]
- Sauer, H.; Wartenberg, M.; Hescheler, J. Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cell. Physiol. Biochem. 2001, 11, 173–186. [Google Scholar] [CrossRef]
- Rivera, C. Essentials of oral cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 11884–11894. [Google Scholar]
- Li, C.; Li, T.; Huang, L.; Yang, M.; Zhu, G. Self-assembled Lipid Nanoparticles for Ratiometric Codelivery of Cisplatin and siRNA Targeting XPF to Combat Drug Resistance in Lung Cancer. Chem-Asian J. 2019, 14, 570–576. [Google Scholar] [CrossRef]
- Cancer SCd. Cannabis et Cannabinoïdes à des fins Médicales. 2018. Available online: https://cancer.ca/fr/treatments/complementary-therapies/medical-cannabis-and-cannabinoids (accessed on 1 September 2022).
- Andre, C.M.; Hausman, J.-F.; Guerriero, G. Cannabis sativa: The Plant of the Thousand and One Molecules. Front. Plant Sci. 2016, 7, 19. [Google Scholar] [CrossRef]
- Valjent, E.; Pagès, C.; Rogard, M.; Besson, M.-J.; Maldonado, R.; Caboche, J. Delta 9-tetrahydrocannabinol-induced MAPK/ERK and Elk-1 activation in vivo depends on dopaminergic transmission. Eur. J. Neurosci. 2001, 14, 342–352. [Google Scholar] [CrossRef]
- Herrera, B.; Carracedo, A.; Diez-Zaera, M.; Guzmán, M.; Velasco, G. P38 MAPK is involved in CB2 receptor-induced apoptosis of human leukaemia cells. FEBS Lett. 2005, 579, 5084–5088. [Google Scholar] [CrossRef]
- Kozela, E.; Pietr, M.; Juknat, A.; Rimmerman, N.; Levy, R.; Vogel, Z. Cannabinoids Delta(9)-tetrahydrocannabinol and cannabidiol differentially inhibit the lipopolysaccharide-activated NF-kappaB and interferon-beta/STAT proinflammatory pathways in BV-2 microglial cells. J. Biol. Chem. 2010, 285, 1616–1626. [Google Scholar] [CrossRef]
- Echigo, R.; Sugimoto, N.; Yachie, A.; Ohno-Shosaku, T. Cannabinoids inhibit peptidoglycan-induced phosphorylation of NF-kappaB and cell growth in U87MG human malignant glioma cells. Oncol. Rep. 2012, 28, 1176–1180. [Google Scholar] [CrossRef]
- Nalli, Y.; Dar, M.S.; Bano, N.; Rasool, J.U.; Sarkar, A.R.; Banday, J.; Bhat, A.Q.; Rafia, B.; Vishwakarma, R.A.; Ali, A. Analyzing the role of cannabinoids as modulators of Wnt/beta-catenin signaling pathway for their use in the management of neuropathic pain. Bioorg. Med. Chem. Lett. 2019, 29, 1043–1046. [Google Scholar] [CrossRef]
- Puighermanal, E.; Busquets-Garcia, A.; Gomis-González, M.; Marsicano, G.; Maldonado, R.; Ozaita, A. Dissociation of the Pharmacological Effects of THC by mTOR Blockade. Neuropsychopharmacology 2013, 38, 1334–1343. [Google Scholar] [CrossRef] [PubMed]
- Puighermanal, E.; Marsicano, G.; Busquets-Garcia, A.; Lutz, B.; Maldonado, R.; Ozaita, A. Cannabinoid modulation of hippocampal long-term memory is mediated by mTOR signaling. Nat. Neurosci. 2009, 12, 152-U18. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Bennett, A. Cannabinoids: A New Group of Agonists of PPARs. PPAR Res. 2007, 2007, 23513. [Google Scholar] [CrossRef]
- Vara, D.; Morell, C.M.; Rodríguez-Henche, N.; Díaz-Laviada, I. Involvement of PPARgamma in the antitumoral action of cannabinoids on hepatocellular carcinoma. Cell Death Dis. 2013, 4, e618. [Google Scholar] [CrossRef]
- Semlali, A.; Beji, S.; Ajala, I.; Rouabhia, M. Effects of tetrahydrocannabinols on human oral cancer cell proliferation, apoptosis, autophagy, oxidative stress, and DNA damage. Arch. Oral. Biol. 2021, 129, 105200. [Google Scholar] [CrossRef]
- Semlali, A.; Contant, C.; Al-Otaibi, B.; Al-Jammaz, I.; Chandad, F. The curcumin analog (PAC) suppressed cell survival and induced apoptosis and autophagy in oral cancer cells. Sci. Rep. 2021, 11, 11701. [Google Scholar] [CrossRef]
- Contant, C.; Rouabhia, M.; Loubaki, L.; Chandad, F.; Semlali, A. Anethole induces anti-oral cancer activity by triggering apoptosis, autophagy and oxidative stress and by modulation of multiple signaling pathways. Sci. Rep. 2021, 11, 13087. [Google Scholar] [CrossRef]
- Tazi, N.; Semlali, A.; Loubaki, L.; Alamri, A.; Rouabhia, M. Cannabis smoke condensate induces human gingival epithelial cell damage through apoptosis, autophagy, and oxidative stress. Arch. Oral Biol. 2022, 141, 105498. [Google Scholar] [CrossRef]
- Rip, J.; de Bruijn, M.J.W.; Kaptein, A.; Hendriks, R.W.; Corneth, O.B.J. Phosphoflow Protocol for Signaling Studies in Human and Murine B Cell Subpopulations. J. Immunol. 2020, 204, 2852–2863. [Google Scholar] [CrossRef]
- Alsamri, H.; Alneyadi, A.; Muhammad, K.; Ayoub, M.A.; Eid, A.; Iratni, R. Carnosol Induces p38-Mediated ER Stress Response and Autophagy in Human Breast Cancer Cells. Front. Oncol. 2022, 12, 911615. [Google Scholar] [CrossRef] [PubMed]
- Dewaele, M.; Maes, H.; Agostinis, P. ROS-mediated mechanisms of autophagy stimulation and their relevance in cancer therapy. Autophagy 2010, 6, 838–854. [Google Scholar] [CrossRef] [PubMed]
- Rathore, S.; Datta, G.; Kaur, I.; Malhotra, P.; Mohmmed, A. Disruption of cellular homeostasis induces organelle stress and triggers apoptosis like cell-death pathways in malaria parasite. Cell Death Dis. 2015, 6, e1803. [Google Scholar] [CrossRef]
- Park, S.-W.; Kim, J.-E.; Oh, S.-M.; Cha, W.-J.; Hah, J.-H.; Sung, M.-W. Anticancer effects of anandamide on head and neck squamous cell carcinoma cells via the production of receptor-independent reactive oxygen species. Head Neck 2015, 37, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Schieke, S.M.; Ma, M.; Cao, L.; McCoy, J.P., Jr.; Liu, C.; Hensel, N.F.; Barrett, A.J.; Boehm, M.; Finkel, T. Mitochondrial metabolism modulates differentiation and teratoma formation capacity in mouse embryonic stem cells. J. Biol. Chem. 2008, 283, 28506–28512. [Google Scholar] [CrossRef]
- Ye, T.; Zhu, S.; Zhu, Y.; Feng, Q.; He, B.; Xiong, Y.; Zhao, L.; Zhang, Y.; Yu, L.; Yang, L. Cryptotanshinone induces melanoma cancer cells apoptosis via ROS-mitochondrial apoptotic pathway and impairs cell migration and invasion. Biomed. Pharmacother. 2016, 82, 319–326. [Google Scholar] [CrossRef]
- Zhou, Y.J.; Zhang, S.P.; Liu, C.W.; Cai, Y.Q. The protection of selenium on ROS mediated-apoptosis by mitochondria dysfunction in cadmium-induced LLC-PK(1) cells. Toxicol In Vitro 2009, 23, 288–294. [Google Scholar] [CrossRef]
- Deponte, M. The Incomplete Glutathione Puzzle: Just Guessing at Numbers and Figures? Antioxid Redox Sign. 2017, 27, 1130–1161. [Google Scholar] [CrossRef]
- Kwon, D.H.; Cha, H.-J.; Lee, H.; Hong, S.-H.; Park, C.; Park, S.-H.; Kim, G.-Y.; Kim, S.; Kim, H.-S.; Hwang, H.-J.; et al. Protective Effect of Glutathione against Oxidative Stress-induced Cytotoxicity in RAW 264.7 Macrophages through Activating the Nuclear Factor Erythroid 2-Related Factor-2/Heme Oxygenase-1 Pathway. Antioxidants 2019, 8, 82. [Google Scholar] [CrossRef]
- Chaudhari, N.; Talwar, P.; Parimisetty, A.; Lefebvre d’Hellencourt, C.; Ravanan, P. A molecular web: Endoplasmic reticulum stress, inflammation, and oxidative stress. Front. Cell Neurosci. 2014, 8, 213. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Elazar, Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007, 17, 422–427. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, J.-H.; Jin, L.; Lin, S.-M.; Yang, Y.; Sui, Y.-X.; Shi, H. Over-expression of the Beclin1 gene upregulates chemosensitivity to anti-cancer drugs by enhancing therapy-induced apoptosis in cervix squamous carcinoma CaSki cells. Cancer Lett. 2010, 294, 204–210. [Google Scholar] [CrossRef]
- Li, Z.X.; Yang, J.; Huang, H.F. Oxidative stress induces H2AX phosphorylation in human spermatozoa. Febs. Lett. 2006, 580, 6161–6168. [Google Scholar] [CrossRef]
- Peng, S.-Y.; Tang, J.-Y.; Li, R.-N.; Huang, H.-W.; Wu, C.-Y.; Chiu, C.-C.; Chang, F.-R.; Zhang, H.-W.; Lee, Y.-J.; Sheu, J.-H.; et al. Oxidative Stress-Dependent Synergistic Antiproliferation, Apoptosis, and DNA Damage of Ultraviolet-C and Coral-Derived Sinularin Combined Treatment for Oral Cancer Cells. Cancers 2021, 13, 2450. [Google Scholar] [CrossRef]
- Junttila, M.R.; Ala-Aho, R.; Jokilehto, T.; Peltonen, J.; Kallajoki, M.; Grenman, R.; Jaakkola, P.; Westermarck, J.; Kähäri, V.-M. p38alpha and p38delta mitogen-activated protein kinase isoforms regulate invasion and growth of head and neck squamous carcinoma cells. Oncogene 2007, 26, 5267–5279. [Google Scholar] [CrossRef]
- Pua, L.J.W.; Mai, C.-W.; Chung, F.F.-L.; Khoo, A.S.-B.; Leong, C.-O.; Lim, W.-M.; Hii, L.-W. Functional Roles of JNK and p38 MAPK Signaling in Nasopharyngeal Carcinoma. Int. J. Mol. Sci. 2022, 23, 1108. [Google Scholar] [CrossRef]
- Verzella, D.; Pescatore, A.; Capece, D.; Vecchiotti, D.; Ursini, M.V.; Franzoso, G.; Alesse, E.; Zazzeroni, F. Life, death, and autophagy in cancer: NF-kappaB turns up everywhere. Cell Death Dis. 2020, 11, 210. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loubaki, L.; Rouabhia, M.; Zahrani, M.A.; Amri, A.A.; Semlali, A. Oxidative Stress and Autophagy Mediate Anti-Cancer Properties of Cannabis Derivatives in Human Oral Cancer Cells. Cancers 2022, 14, 4924. https://doi.org/10.3390/cancers14194924
Loubaki L, Rouabhia M, Zahrani MA, Amri AA, Semlali A. Oxidative Stress and Autophagy Mediate Anti-Cancer Properties of Cannabis Derivatives in Human Oral Cancer Cells. Cancers. 2022; 14(19):4924. https://doi.org/10.3390/cancers14194924
Chicago/Turabian StyleLoubaki, Lionel, Mahmoud Rouabhia, Mohamed Al Zahrani, Abdullah Al Amri, and Abdelhabib Semlali. 2022. "Oxidative Stress and Autophagy Mediate Anti-Cancer Properties of Cannabis Derivatives in Human Oral Cancer Cells" Cancers 14, no. 19: 4924. https://doi.org/10.3390/cancers14194924
APA StyleLoubaki, L., Rouabhia, M., Zahrani, M. A., Amri, A. A., & Semlali, A. (2022). Oxidative Stress and Autophagy Mediate Anti-Cancer Properties of Cannabis Derivatives in Human Oral Cancer Cells. Cancers, 14(19), 4924. https://doi.org/10.3390/cancers14194924