

CREPT Disarms the Inhibitory Activity of HDAC1 on Oncogene Expression to Promote Tumorigenesis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfections
2.2. Plasmids and Reagents
2.3. Human Tumor Specimens and Staining
2.4. Co-Immunoprecipitation and Western Blotting
2.5. Immunofluorescence Analysis
2.6. Colony Formation Assay and Cell Viability Assay
2.7. Luciferase Assays
2.8. Real-Time Quantitative PCR (qRT-PCR) Analysis
2.9. Chromatin Immunoprecipitation (ChIP) Assay
2.10. Experimental Animals
2.11. Tumor Formation
2.12. Statistical Analysis
3. Results
3.1. CREPT Is Positively Correlated with HDAC1 in Human Tumors
3.2. HDAC1 Represses the Activity of CREPT in Promoting Tumorigenesis
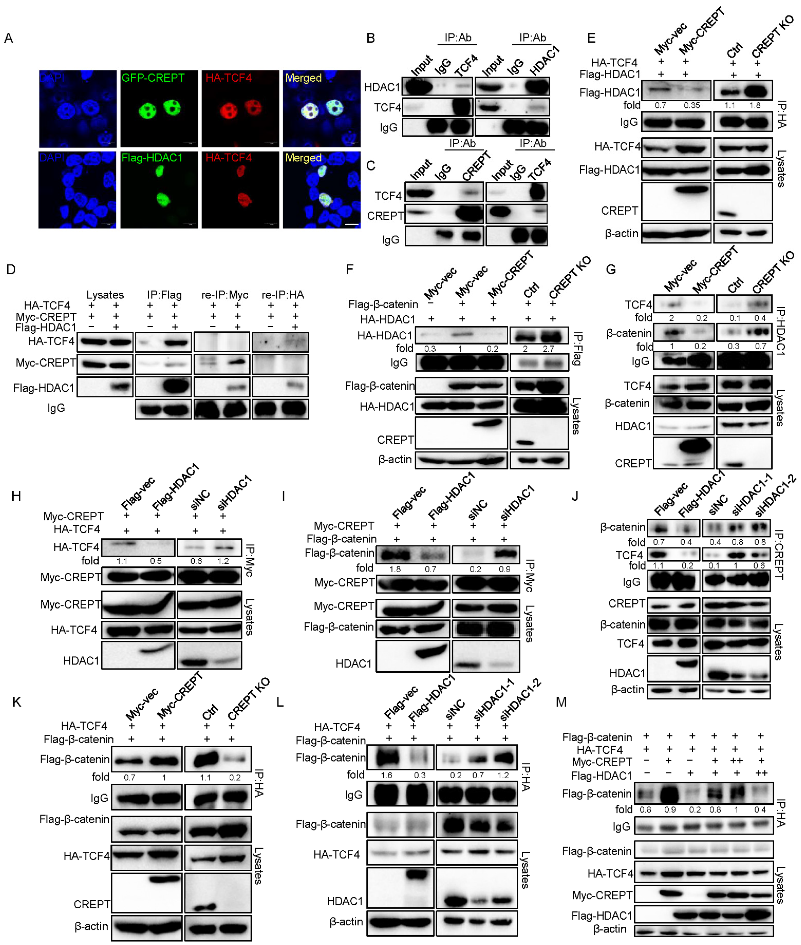
3.3. HDAC1 Specifically Interacts with CREPT
3.4. HDAC1 and CREPT Exclusively Interact with TCF4 and β-Catenin
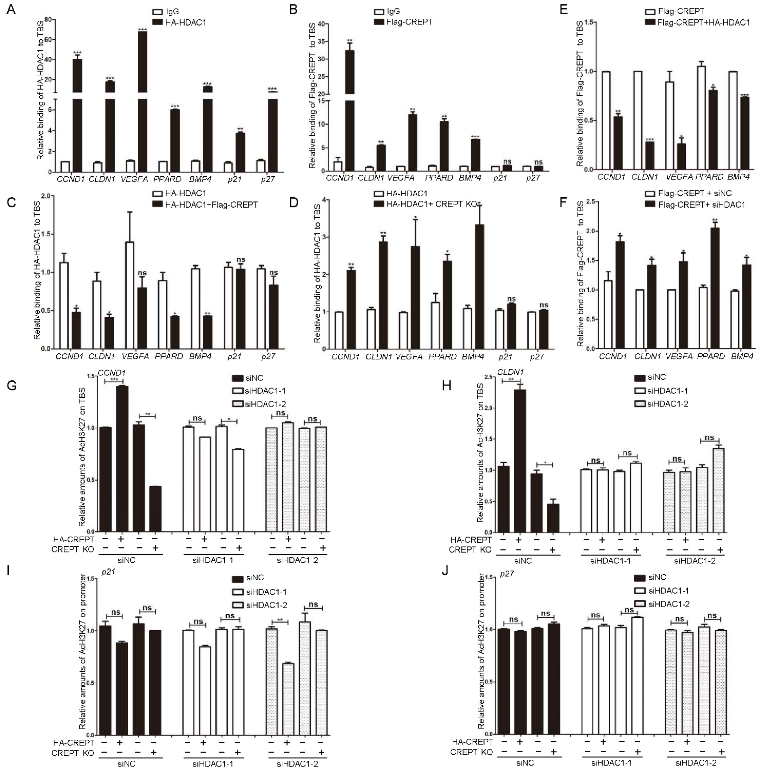
3.5. CREPT Blocks the Occupancy of HDAC1 at the TBS Region of Target Genes
3.6. Wnt3a Influences the Binding of HDAC1/CREPT to the TBS Region
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Nebbioso, A.; Tambaro, F.P.; Dell’Aversana, C.; Altucci, L. Cancer epigenetics: Moving forward. PLoS Genet. 2018, 14, e1007362. [Google Scholar] [CrossRef] [PubMed]
- Suva, M.L.; Riggi, N.; Bernstein, B.E. Epigenetic reprogramming in cancer. Science 2013, 339, 1567–1570. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Allis, C.D.; Wang, G.G. The language of chromatin modification in human cancers. Nat. Rev. Cancer 2021, 21, 413–430. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Shen, Y.; Wei, W.; Zhou, D.X. Histone Acetylation Enzymes Coordinate Metabolism and Gene Expression. Trends Plant Sci. 2015, 20, 614–621. [Google Scholar] [CrossRef]
- Wang, L.; Tang, Y.; Cole, P.A.; Marmorstein, R. Structure and chemistry of the p300/CBP and Rtt109 histone acetyltransferases: Implications for histone acetyltransferase evolution and function. Curr. Opin. Struct. Biol. 2008, 18, 741–747. [Google Scholar] [CrossRef][Green Version]
- Hildmann, C.; Riester, D.; Schwienhorst, A. Histone deacetylases—An important class of cellular regulators with a variety of functions. Appl. Microbiol. Biotechnol. 2007, 75, 487–497. [Google Scholar] [CrossRef]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef]
- Weichert, W.; Roske, A.; Gekeler, V.; Beckers, T.; Ebert, M.P.; Pross, M.; Dietel, M.; Denkert, C.; Rocken, C. Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: A retrospective analysis. Lancet Oncol. 2008, 9, 139–148. [Google Scholar] [CrossRef]
- Minamiya, Y.; Ono, T.; Saito, H.; Takahashi, N.; Ito, M.; Mitsui, M.; Motoyama, S.; Ogawa, J. Expression of histone deacetylase 1 correlates with a poor prognosis in patients with adenocarcinoma of the lung. Lung Cancer 2011, 74, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.S.; Wang, L.; Abrams, J.; Wang, G. Histone deacetylases (HDACs) in XPC gene silencing and bladder cancer. J. Hematol. Oncol. 2011, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Jin, L.W.; Wang, I.F.; Wei, W.Y.; Ho, P.C.; Liu, Y.C.; Tsai, K.J. HDAC1 dysregulation induces aberrant cell cycle and DNA damage in progress of TDP-43 proteinopathies. EMBO Mol. Med. 2020, 12, e10622. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Zeng, J.; Liu, H.; Wang, T.; Yu, Z.; Chen, J. Role of HDAC1 in the progression of gastric cancer and the correlation with lncRNAs. Oncol. Lett. 2019, 17, 3296–3304. [Google Scholar] [CrossRef]
- Yang, W.B.; Hsu, C.C.; Hsu, T.I.; Liou, J.P.; Chang, K.Y.; Chen, P.Y.; Liu, J.J.; Yang, S.T.; Wang, J.Y.; Yeh, S.H.; et al. Increased activation of HDAC1/2/6 and Sp1 underlies therapeutic resistance and tumor growth in glioblastoma. Neuro. Oncol. 2020, 22, 1439–1451. [Google Scholar] [CrossRef]
- Zhang, L.; Bu, L.; Hu, J.; Xu, Z.; Ruan, L.; Fang, Y.; Wang, P. HDAC1 knockdown inhibits invasion and induces apoptosis in non-small cell lung cancer cells. Biol. Chem. 2018, 399, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Wilting, R.H.; Yanover, E.; Heideman, M.R.; Jacobs, H.; Horner, J.; van der Torre, J.; DePinho, R.A.; Dannenberg, J.H. Overlapping functions of Hdac1 and Hdac2 in cell cycle regulation and haematopoiesis. EMBO J. 2010, 29, 2586–2597. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Cubizolles, F.; Zhang, Y.; Reichert, N.; Kohler, H.; Seiser, C.; Matthias, P. Histone deacetylases 1 and 2 act in concert to promote the G1-to-S progression. Genes Dev. 2010, 24, 455–469. [Google Scholar] [CrossRef]
- Jiang, N.; Niu, G.; Pan, Y.H.; Pan, W.; Zhang, M.F.; Zhang, C.Z.; Shen, H. CBX4 transcriptionally suppresses KLF6 via interaction with HDAC1 to exert oncogenic activities in clear cell renal cell carcinoma. EBioMedicine 2020, 53, 102692. [Google Scholar] [CrossRef]
- Zeng, Y.; Kotake, Y.; Pei, X.H.; Smith, M.D.; Xiong, Y. p53 binds to and is required for the repression of Arf tumor suppressor by HDAC and polycomb. Cancer Res. 2011, 71, 2781–2792. [Google Scholar] [CrossRef]
- Zhang, Q.; Song, Y.; Chen, W.; Wang, X.; Miao, Z.; Cao, L.; Li, F.; Wang, G. By recruiting HDAC1, MORC2 suppresses p21 Waf1/Cip1 in gastric cancer. Oncotarget 2015, 6, 16461–16470. [Google Scholar] [CrossRef] [PubMed]
- Tong, Z.T.; Cai, M.Y.; Wang, X.G.; Kong, L.L.; Mai, S.J.; Liu, Y.H.; Zhang, H.B.; Liao, Y.J.; Zheng, F.; Zhu, W.; et al. EZH2 supports nasopharyngeal carcinoma cell aggressiveness by forming a co-repressor complex with HDAC1/HDAC2 and Snail to inhibit E-cadherin. Oncogene 2012, 31, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Alalem, M.; Ray, B.K. Loss of epigenetic Kruppel-like factor 4 histone deacetylase (KLF-4-HDAC)-mediated transcriptional suppression is crucial in increasing vascular endothelial growth factor (VEGF) expression in breast cancer. J. Biol. Chem. 2013, 288, 27232–27242. [Google Scholar] [CrossRef] [PubMed]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef]
- Logan, C.Y.; Nusse, R. The Wnt signaling pathway in development and disease. Annu Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef]
- Lu, D.; Wu, Y.; Wang, Y.; Ren, F.; Wang, D.; Su, F.; Zhang, Y.; Yang, X.; Jin, G.; Hao, X.; et al. CREPT Accelerates Tumorigenesis by Regulating the Transcription of Cell-Cycle-Related Genes. Cancer Cell 2012, 21, 92–104. [Google Scholar] [CrossRef]
- Zhai, W.; Ye, X.; Wang, Y.; Feng, Y.; Wang, Y.; Lin, Y.; Ding, L.; Yang, L.; Wang, X.; Kuang, Y.; et al. CREPT/RPRD1B promotes tumorigenesis through STAT3-driven gene transcription in a p300-dependent manner. Br. J. Cancer 2021, 124, 1437–1448. [Google Scholar] [CrossRef]
- Ma, D.; Zou, Y.; Chu, Y.; Liu, Z.; Liu, G.; Chu, J.; Li, M.; Wang, J.; Sun, S.-y.; Chang, Z. A cell-permeable peptide-based PROTAC against the oncoprotein CREPT proficiently inhibits pancreatic cancer. Theranostics 2020, 10, 3708–3721. [Google Scholar] [CrossRef]
- Yang, L.; Yang, H.; Chu, Y.; Song, Y.; Ding, L.; Zhu, B.; Zhai, W.; Wang, X.; Kuang, Y.; Ren, F.; et al. CREPT is required for murine stem cell maintenance during intestinal regeneration. Nat. Commun. 2021, 12, 270. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, C.; Duan, X.; Ren, F.; Li, S.; Jin, Z.; Wang, Y.; Feng, Y.; Liu, Z.; Chang, Z. CREPT/RPRD1B, a Recently Identified Novel Protein Highly Expressed in Tumors, Enhances the β-Catenin·TCF4 Transcriptional Activity in Response to Wnt Signaling. J. Biol. Chem. 2014, 289, 22589–22599. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, S.; Kang, W.; Liu, C.; Dong, Y.; Ren, F.; Wang, Y.; Zhang, J.; Wang, G.; To, K.F.; et al. CREPT facilitates colorectal cancer growth through inducing Wnt/β-catenin pathway by enhancing p300-mediated β-catenin acetylation. Oncogene 2018, 37, 3485–3500. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, Y.; Li, J.; Wang, Y.; Ren, F.; Zhou, Y.; Wu, Y.; Feng, Y.; Zhou, Y.; Su, F.; et al. p15RS/RPRD1A (p15INK4b-related Sequence/Regulation of Nuclear Pre-mRNA Domain-containing Protein 1A) Interacts with HDAC2 in Inhibition of the Wnt/β-Catenin Signaling Pathway. J. Biol. Chem. 2015, 290, 9701–9713. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Wang, R.; Zhang, Y.; Liu, C.; Wang, Y.; Hu, J.; Zhang, L.; Chang, Z. Characterization of a Monoclonal Antibody Against CREPT, a Novel Protein Highly Expressed in Tumors. Monoclon. Antibodies Immunodiagn. Immunother. 2014, 33, 401–408. [Google Scholar] [CrossRef]
- Zheng, G.; Li, W.; Zuo, B.; Guo, Z.; Xi, W.; Wei, M.; Chen, P.; Wen, W.; Yang, A.G. High expression of CREPT promotes tumor growth and is correlated with poor prognosis in colorectal cancer. Biochem. Biophys. Res. Commun. 2016, 480, 436–442. [Google Scholar] [CrossRef]
- Yang, H.; Salz, T.; Zajac-Kaye, M.; Liao, D.; Huang, S.; Qiu, Y. Overexpression of histone deacetylases in cancer cells is controlled by interplay of transcription factors and epigenetic modulators. FASEB J. 2014, 28, 4265–4279. [Google Scholar] [CrossRef]
- Weichert, W.; Roske, A.; Niesporek, S.; Noske, A.; Buckendahl, A.C.; Dietel, M.; Gekeler, V.; Boehm, M.; Beckers, T.; Denkert, C. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: Specific role of class I histone deacetylases in vitro and in vivo. Clin. Cancer Res. 2008, 14, 1669–1677. [Google Scholar] [CrossRef]
- Santoro, F.; Botrugno, O.A.; Dal Zuffo, R.; Pallavicini, I.; Matthews, G.M.; Cluse, L.; Barozzi, I.; Senese, S.; Fornasari, L.; Moretti, S.; et al. A dual role for Hdac1: Oncosuppressor in tumorigenesis, oncogene in tumor maintenance. Blood 2013, 121, 3459–3468. [Google Scholar] [CrossRef]
- Qiao, W.; Liu, H.; Liu, R.; Liu, Q.; Zhang, T.; Guo, W.; Li, P.; Deng, M. Prognostic and clinical significance of histone deacetylase 1 expression in breast cancer: A meta-analysis. Clin. Chim. Acta 2018, 483, 209–215. [Google Scholar] [CrossRef]
- Seo, J.; Min, S.K.; Park, H.R.; Kim, D.H.; Kwon, M.J.; Kim, L.S.; Ju, Y.S. Expression of Histone Deacetylases HDAC1, HDAC2, HDAC3, and HDAC6 in Invasive Ductal Carcinomas of the Breast. J. Breast Cancer 2014, 17, 323–331. [Google Scholar] [CrossRef]
- Sudo, T.; Mimori, K.; Nishida, N.; Kogo, R.; Iwaya, T.; Tanaka, F.; Shibata, K.; Fujita, H.; Shirouzu, K.; Mori, M. Histone deacetylase 1 expression in gastric cancer. Oncol. Rep. 2011, 26, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Quint, K.; Agaimy, A.; Di Fazio, P.; Montalbano, R.; Steindorf, C.; Jung, R.; Hellerbrand, C.; Hartmann, A.; Sitter, H.; Neureiter, D.; et al. Clinical significance of histone deacetylases 1, 2, 3, and 7: HDAC2 is an independent predictor of survival in HCC. Virchows Arch. 2011, 459, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Chen, H.; Zuo, Q.; Huang, C.; Zhao, R.; Yu, X.; Wang, Y.; Zhang, Y.; Chang, Z.; Li, B. CREPT and p15RS regulate cell proliferation and cycling in chicken DF-1 cells through the Wnt/beta-catenin pathway. J. Cell Biochem. 2018, 119, 1083–1092. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Richardson, W.D. Genetics meets epigenetics: HDACs and Wnt signaling in myelin development and regeneration. Nat. Neurosci. 2009, 12, 815–817. [Google Scholar] [CrossRef] [PubMed]
- Mei, K.; Jin, Z.; Ren, F.; Wang, Y.; Chang, Z.; Wang, X. Structural basis for the recognition of RNA polymerase II C-terminal domain by CREPT and p15RS. Sci. China Life Sci. 2014, 57, 97–106. [Google Scholar] [CrossRef]
- Ye, F.; Chen, Y.; Hoang, T.; Montgomery, R.L.; Zhao, X.H.; Bu, H.; Hu, T.; Taketo, M.M.; van Es, J.H.; Clevers, H.; et al. HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta-catenin-TCF interaction. Nat. Neurosci. 2009, 12, 829–838. [Google Scholar] [CrossRef]
- Losson, H.; Schnekenburger, M.; Dicato, M.; Diederich, M. Natural Compound Histone Deacetylase Inhibitors (HDACi): Synergy with Inflammatory Signaling Pathway Modulators and Clinical Applications in Cancer. Molecules 2016, 21, 1608. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Zupkovitz, G.; Grausenburger, R.; Brunmeir, R.; Senese, S.; Tischler, J.; Jurkin, J.; Rembold, M.; Meunier, D.; Egger, G.; Lagger, S.; et al. The cyclin-dependent kinase inhibitor p21 is a crucial target for histone deacetylase 1 as a regulator of cellular proliferation. Mol. Cell Biol. 2010, 30, 1171–1181. [Google Scholar] [CrossRef]
- Staal, F.J.; Luis, T.C.; Tiemessen, M.M. WNT signalling in the immune system: WNT is spreading its wings. Nat. Rev. Immunol. 2008, 8, 581–593. [Google Scholar] [CrossRef]
- Dovey, O.M.; Foster, C.T.; Cowley, S.M. Histone deacetylase 1 (HDAC1), but not HDAC2, controls embryonic stem cell differentiation. Proc. Natl. Acad. Sci. USA 2010, 107, 8242–8247. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Schultz, R.M. HDAC1 and HDAC2 in mouse oocytes and preimplantation embryos: Specificity versus compensation. Cell Death Differ 2016, 23, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, S.A.; Singh, S.; Ugale, R.; Ranjan, V.; Kanojia, R.; Saha, S.; Tripathy, S.; Kumar, S.; Mehrotra, S.; Modi, D.R.; et al. Regulation of HDAC1 and HDAC2 during consolidation and extinction of fear memory. Brain Res. Bull. 2019, 150, 86–101. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, G.; Rakshit, S.; Sarkar, K. HDAC inhibitors: Targets for tumor therapy, immune modulation and lung diseases. Transl. Oncol. 2022, 16, 101312. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb Perspect Med. 2016, 6, a026831. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, Y.; Ning, B.; Tian, Y.; Lan, T.; Chu, Y.; Ren, F.; Wang, Y.; Meng, Q.; Li, J.; Jia, B.; et al. CREPT Disarms the Inhibitory Activity of HDAC1 on Oncogene Expression to Promote Tumorigenesis. Cancers 2022, 14, 4797. https://doi.org/10.3390/cancers14194797
Cao Y, Ning B, Tian Y, Lan T, Chu Y, Ren F, Wang Y, Meng Q, Li J, Jia B, et al. CREPT Disarms the Inhibitory Activity of HDAC1 on Oncogene Expression to Promote Tumorigenesis. Cancers. 2022; 14(19):4797. https://doi.org/10.3390/cancers14194797
Chicago/Turabian StyleCao, Yajun, Bobin Ning, Ye Tian, Tingwei Lan, Yunxiang Chu, Fangli Ren, Yinyin Wang, Qingyu Meng, Jun Li, Baoqing Jia, and et al. 2022. "CREPT Disarms the Inhibitory Activity of HDAC1 on Oncogene Expression to Promote Tumorigenesis" Cancers 14, no. 19: 4797. https://doi.org/10.3390/cancers14194797
APA StyleCao, Y., Ning, B., Tian, Y., Lan, T., Chu, Y., Ren, F., Wang, Y., Meng, Q., Li, J., Jia, B., & Chang, Z. (2022). CREPT Disarms the Inhibitory Activity of HDAC1 on Oncogene Expression to Promote Tumorigenesis. Cancers, 14(19), 4797. https://doi.org/10.3390/cancers14194797