
The Role of RKIP in the Regulation of EMT in the Tumor Microenvironment



Abstract
Simple Summary
Abstract
1. Introduction
2. Epithelial vs. Mesenchymal Cells
2.1. Epithelial Cells
2.2. Mesenchymal Cells
3. Overview on Oncogenic EMT: EMT-Associated Biomarkers
3.1. Clinically Relevant EMT-Inducing Biomarkers and Their Regulation
3.1.1. Vimentin
- Transcriptional Regulation
- Epigenetic Regulation
- Post-Transcriptional Regulation
- Post Translational Regulation
3.1.2. N-Cadherin
- Transcriptional Regulation
- Epigenetic Regulation
- Post-Transcriptional Regulation
- Post-Translational Regulation
3.1.3. EMT-Inducing Transcription Factor (EMT-TF) Snail
- Transcriptional Regulation
- Epigenetic Regulation
- Post-Transcriptional Regulation
- Post-Translational Regulation
3.1.4. Epithelial Cell Adhesion Molecule (EpCAM)
- Transcriptional Regulation
- Epigenetic Regulation
- Post-Transcriptional
- Post-Translational Regulation
4. Clinically Relevant EMT-Suppressing Biomarkers and Their Regulation
4.1. E-Cadherin
- Transcriptional
- Epigenetics
- Post-Transcriptional
- Post-Translational
4.2. Laminin
5. RKIP: A Novel Oncogenic EMT Suppressor
5.1. RKIP-Mediated Signaling in Cancer
5.2. RKIP Expression in Human Cancers
5.3. Regulation of RKIP Expression
- Epigenetic Regulation
- Transcriptional Regulation
- Post-Transcriptional Regulation
- Post-Translational Regulation
6. Crosstalk between RKIP and Known EMT Regulators
6.1. RKIP/Snail
6.2. RKIP/Vimentin
6.3. RKIP/N-Cadherin
6.4. RKIP/E-Cadherin
6.5. RKIP/EPCAM
6.6. RKIP/Laminin
7. Association Patterns between RKIP Expression and Known EMT Regulators Assessed by Bioinformatic Analyses
- (1)
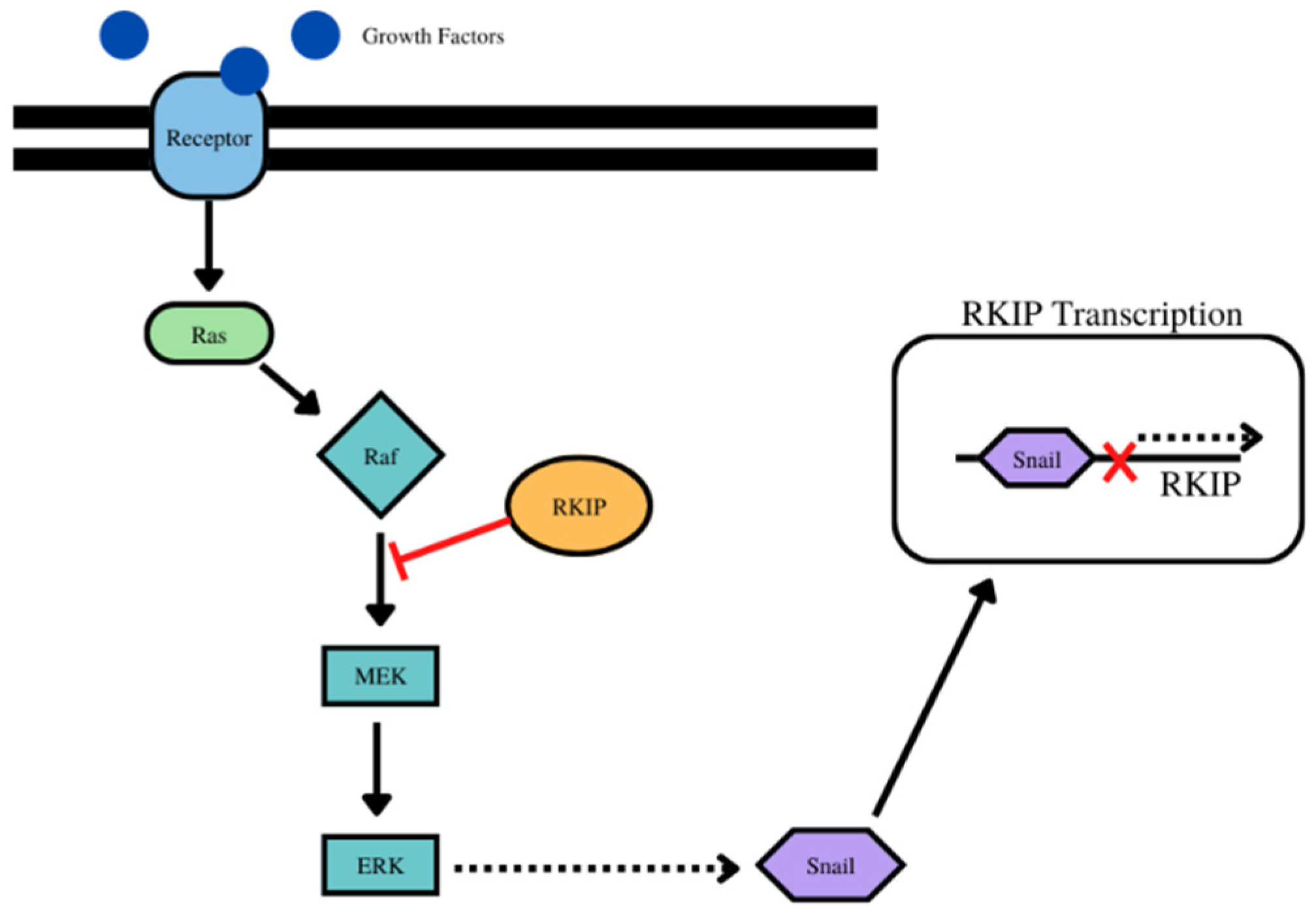
- For RKIP and Snail, for example, while Snail is a transcriptional repressor of RKIP, in turn, RKIP inhibits NF-kB and downstream its target Snail. IN addition, the inhibition of the Raf/Mek/Erk pathway by RKIP results in the inhibition of downstream effector Snail. (Figure 1) Bioinformatic analyses using the pair-wise Pearsons correlations across 23 different types of human cancers and normal tissues revealed that RKIP was negatively correlated with Snail in 9 cancer types and positively correlated in 2 cancer types (Table 1, Supplementary Figure S1).
- (2)
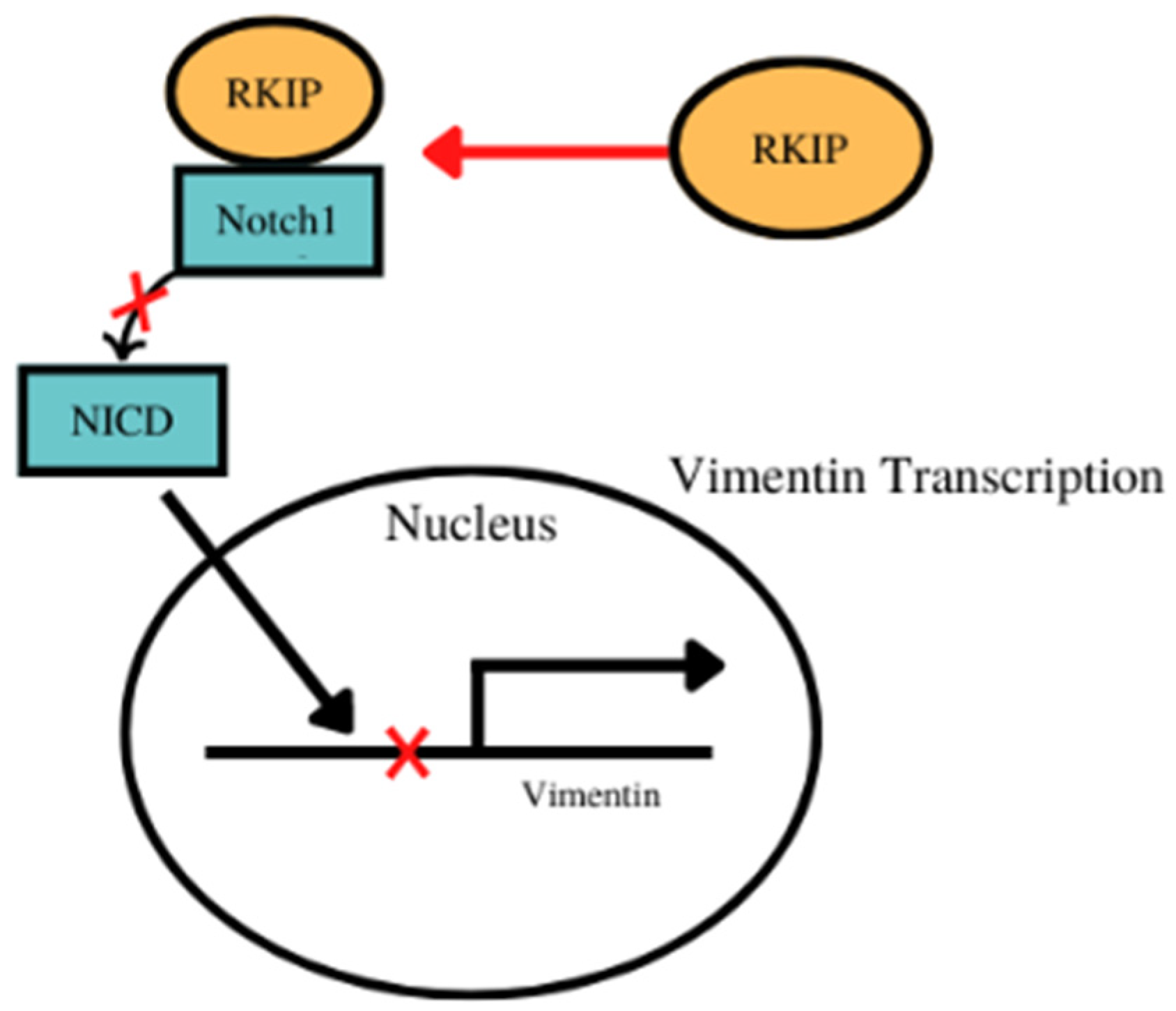
- For vimentin, there was an inverse relationship between the expressions of RKIP and vimentin experimentally in various cancers. For example, the activation of Notch 1 which promotes the expression of vimentin and RKIP’s interaction with Notch 1 prevents its nuclear localization and the expression of vimentin amongst other EMT proteins (Figure 2). Bioinformatic analyses showed that RKIP was negatively correlated with vimentin in 10 cancer types (as expected) and positively correlated with 4 cancer types (Table 2, Supplementary Figure S2).
- (3)
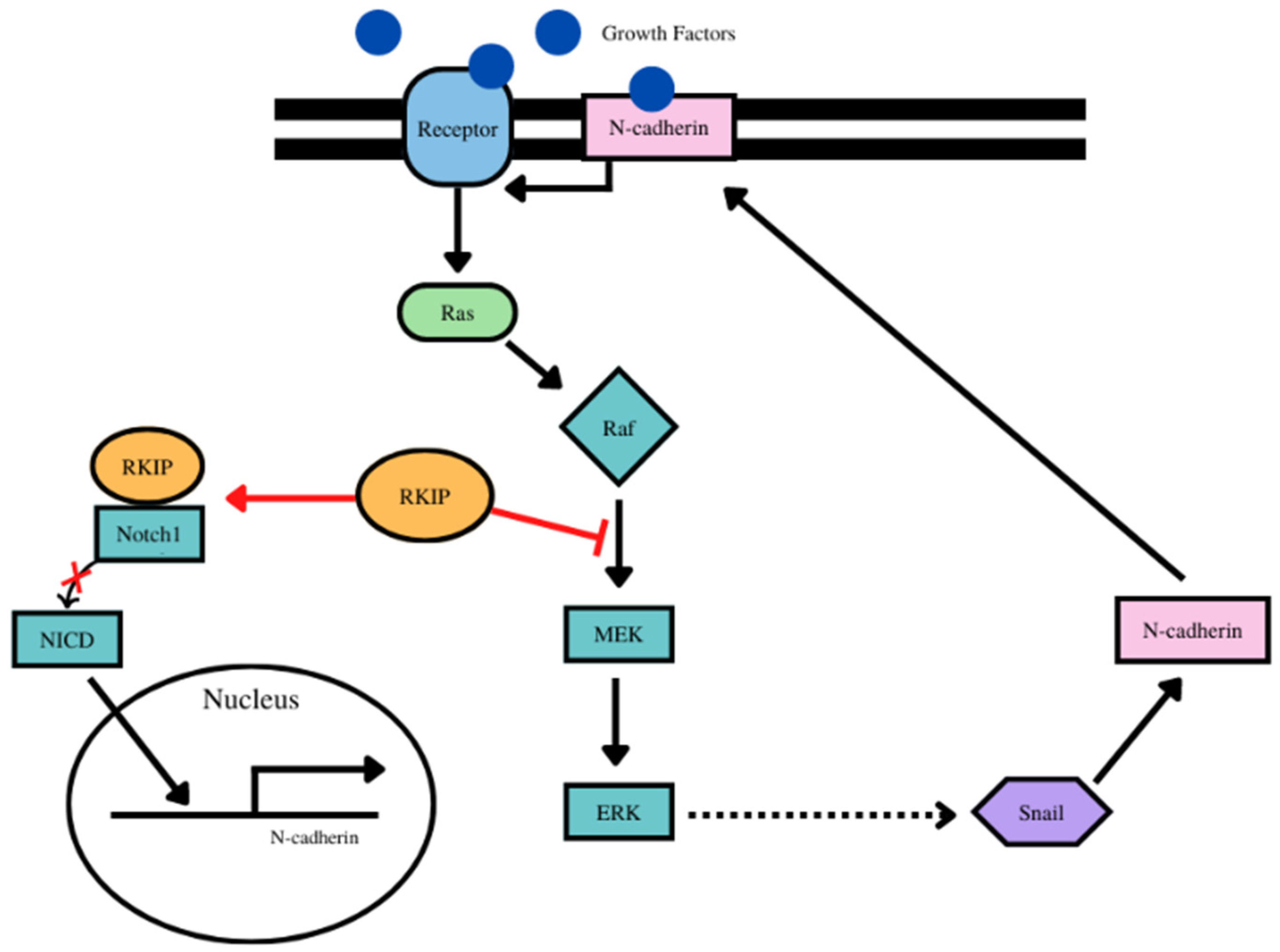
- For N-cadherin, there was an inverse relationship between RKIP and N-cadherin expressions. N-cadherin is a target of upstream Snail and Snail is inhibited by RKIP, hence inhibition f N-cadherin by RKIP. Additionally, vimentin activates the Erk pathway which regulates Snail and thus the inhibition by RKIP of the ERK/Snail/N-cadherin axis. (Figure 3). Bioinformatic analyses demonstrated that RKIP was negatively correlated with only one cancer type and positively correlated with 7 cancer types (Table 3, Supplementary Figure S3). Interestingly, these data are not predicted nor expected and reveal that each cancer type signaling network is different and complex and the various cross-talks are being modulated by different factors inherent with the cancer type.
- (4)
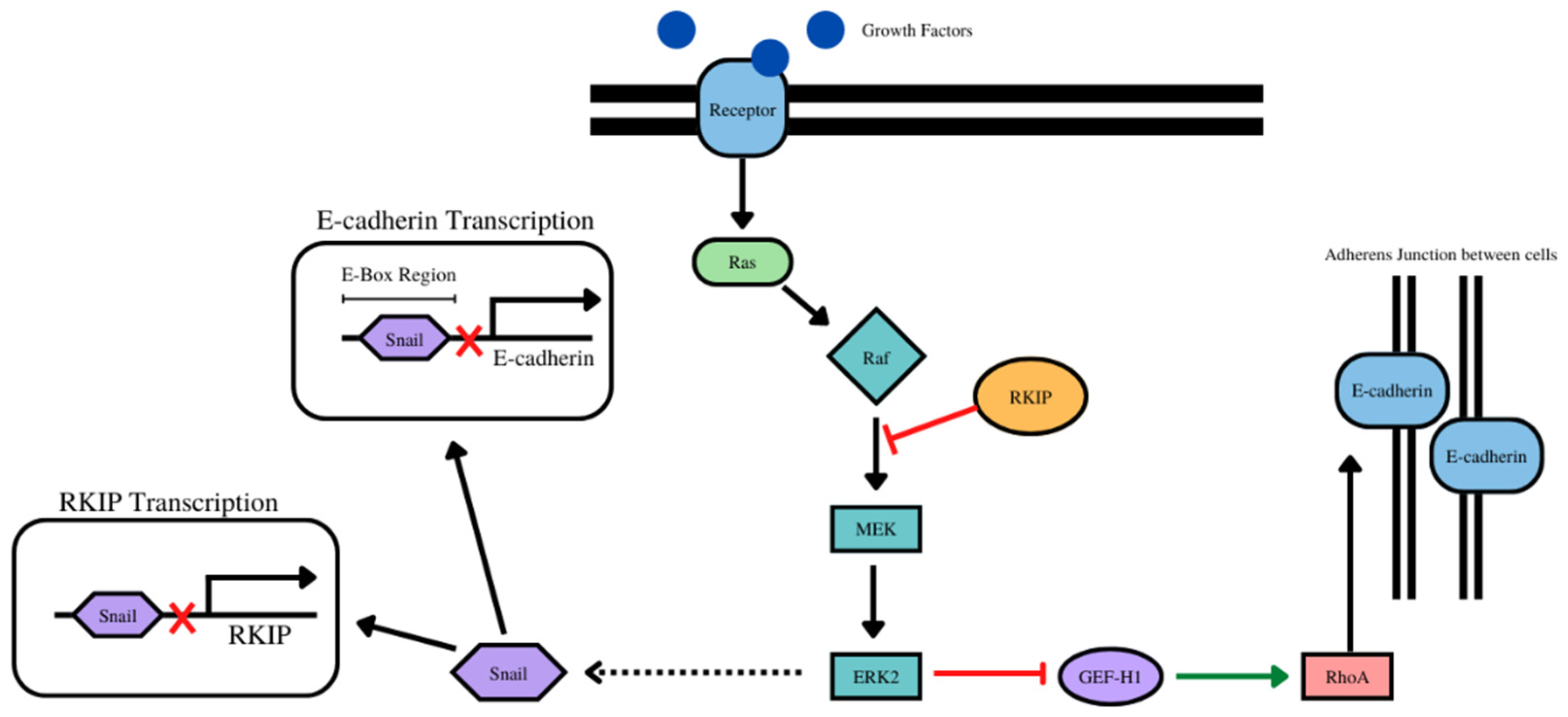
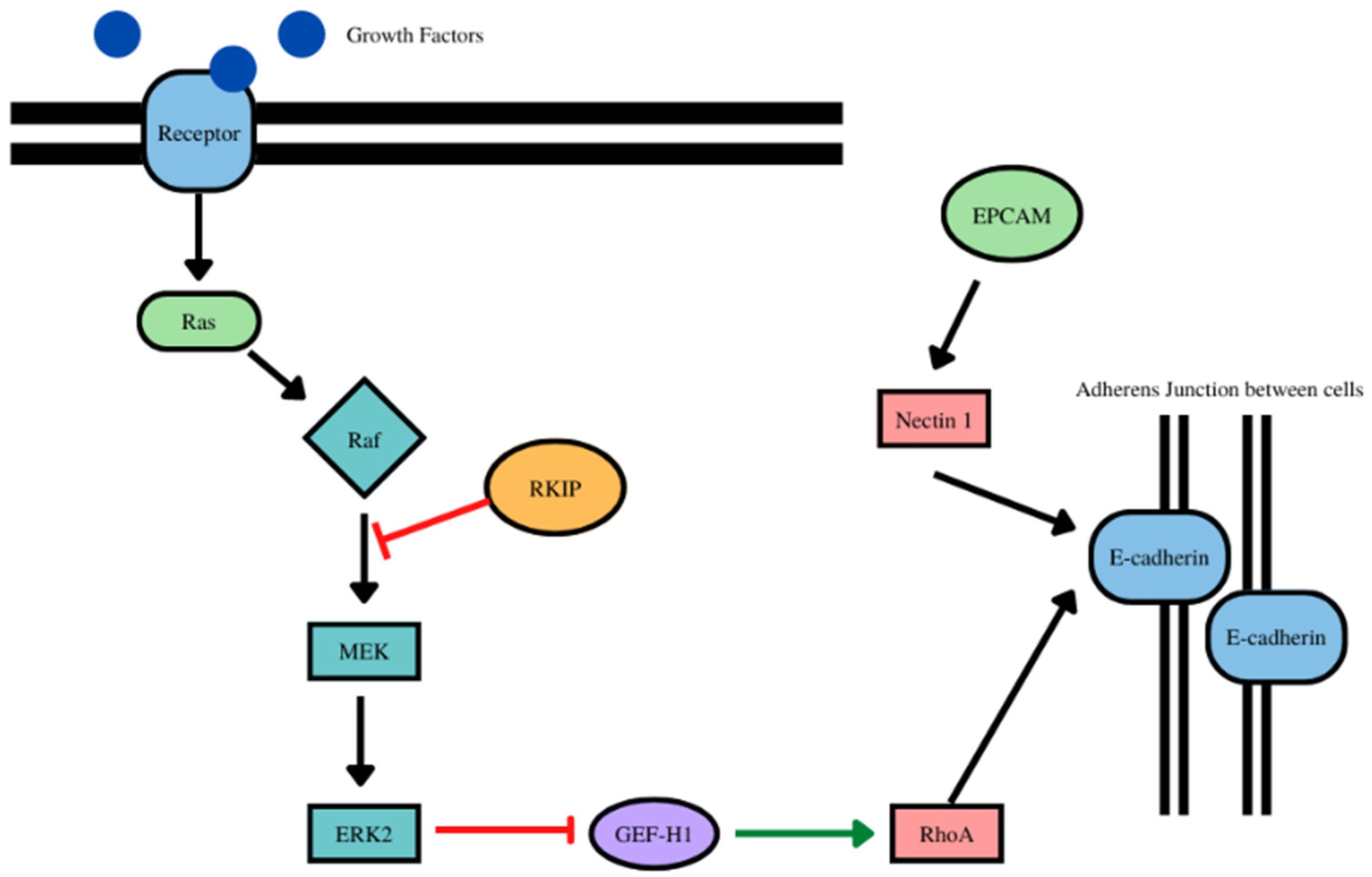
- The overexpression of RKIP results in the inhibition of NF-kB and downstream the RKIP repressor Snail Figure 4). With E-cadherin, there was a positive correlation with 14 cancer types (as expected) and negative correlation with one cancer type. (Table 4, Supplementary Figure S4).
- (5)
- RKIP and EPCAM play a role in the stabilization of E-cadherin at the adherens junctions (Figure 5). With EPCAM, there were positive correlations with 14 cancer types (as expected) and negatively correlated with one cancer type (Table 5, Supplementary Figure S5).
- (6)
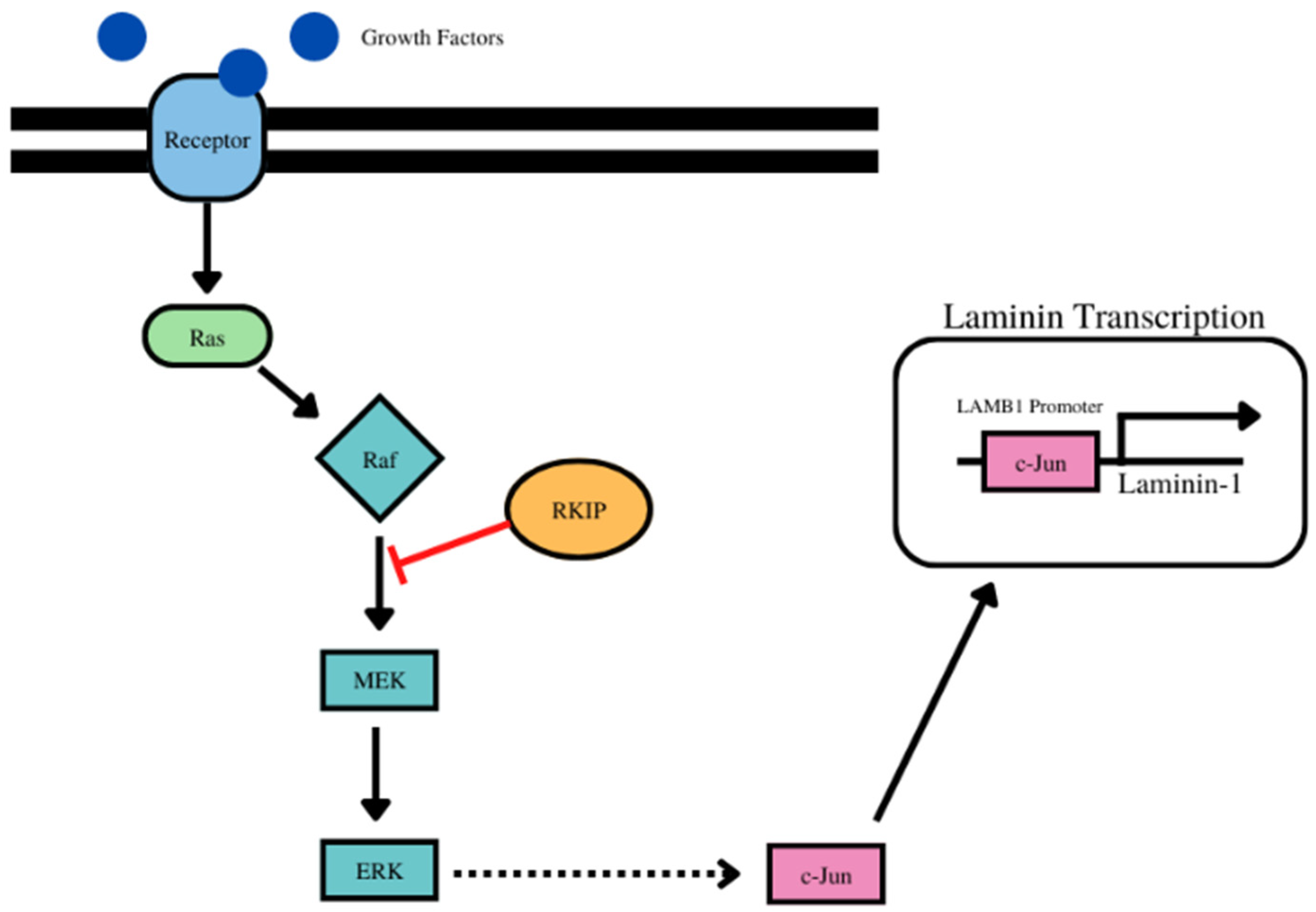
- RKIP regulates laminin alpha 1 via the activation of c-jun (Figure 6). For laminin subunit alpha 1, there were 6 positive correlations with 6 cancer types and inverse correlations with 5 cancer types (Table 6, Supplementary Figure S6).
- (7)
- With laminin subunit beta 1, there was negative correlations with 7 cancer types and positive correlations with 5 cancer types (Table 7, Supplementary Figure S7).
8. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| RKIP | Raf Kinase Inhibitor Protein |
| EMT | Epithelial Mesenchymal Transition |
| MET | Mesenchymal Epithelial Transition |
| EPCAM | Epithelial Cellular Adhesion Molecule |
| PCP | Planar Cell Polarity |
| NF-κB | Nuclear Factor kappa B |
| CTC | Circulating Tumor Cell |
| OS | Overall Survival |
| TGFβ | Transforming Growth Factor Beta |
| SFK | Src family of kinases |
| GPCR | G protein coupled receptor |
| PKC | Protein Kinase C |
| NICD | Notch Intracellular domain |
References
- Thiery, J.P. Epithelial–mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Yilmaz, M.; Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef]
- Greenburg, G.; Hay, E.D. Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J. Cell Biol. 1982, 95, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Shook, D.; Keller, R. Mechanisms, mechanics and function of epithelial–mesenchymal transitions in early development. Mech. Dev. 2003, 120, 1351–1383. [Google Scholar] [CrossRef]
- Radisky, D.C. Epithelial-mesenchymal transition. J. Cell Sci. 2005, 118, 4325–4326. [Google Scholar] [CrossRef]
- Gooding, A.J.; Schiemann, W.P. Epithelial–Mesenchymal Transition Programs and Cancer Stem Cell Phenotypes: Mediators of Breast Cancer Therapy Resistance. Mol. Cancer Res. 2020, 18, 1257–1270. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. Epithelial Plasticity: A Common Theme in Embryonic and Cancer Cells. Science 2013, 342, 1234850. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Bakir, B.; Chiarella, A.M.; Pitarresi, J.R.; Rustgi, A.K. EMT, MET, plasticity and tumor metastasis. Trends Cell Biol. 2020, 30, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Dedhar, S.; Kalluri, R.; Thompson, E.W. The epithelial–mesenchymal transition: New insights in signaling, development, and disease. J. Cell Biol. 2006, 172, 973–981. [Google Scholar] [CrossRef]
- Gillard, G.; Röper, K. Control of cell shape during epithelial morphogenesis: Recent advances. Curr. Opin. Genet. Dev. 2020, 63, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Weinberg, R.A. Epithelial-to-mesenchymal transition in cancer: Complexity and opportunities. Front. Med. 2018, 12, 361–373. [Google Scholar] [CrossRef]
- Janda, E.; Lehmann, K.; Killisch, I.; Jechlinger, M.; Herzig, M.; Downward, J.; Beug, H.; Grünert, S. Ras and TGFβ cooperatively regulate epithelial cell plasticity and metastasis. J. Cell Biol. 2002, 156, 299–314. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B.P. The Role of Snail in EMT and Tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 963–972. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Díaz-López, A.; Moreno-Bueno, G.; Cano, A. Role of microRNA in epithelial to mesenchymal transition and metastasis and clinical perspectives. Cancer Manag. Res. 2014, 6, 205–216. [Google Scholar] [CrossRef]
- Batlle, E.; Sancho, E.; Francí, C.; Domínguez, D.; Monfar, M.; Baulida, J.; García de Herreros, A. The transcription factor Snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial–mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef]
- Klymkowsky, M.W.; Savagner, P. Epithelial-Mesenchymal Transition: A Cancer Researcher’s Conceptual Friend and Foe. Am. J. Pathol. 2009, 174, 1588–1593. [Google Scholar] [CrossRef]
- Simons, M.; Mlodzik, M. Planar Cell Polarity Signaling: From Fly Development to Human Disease. Annu. Rev. Genet. 2008, 42, 517–540. [Google Scholar] [CrossRef] [PubMed]
- Martin-Belmonte, F.; Perez-Moreno, M. Epithelial cell polarity, stem cells and cancer. Nat. Rev. Cancer 2012, 12, 23–38. [Google Scholar] [CrossRef]
- Rodriguez-Boulan, E.; Macara, I.G. Organization and execution of the epithelial polarity programme. Nat. Rev. Mol. Cell Biol. 2014, 15, 225–242. [Google Scholar] [CrossRef]
- Butler, M.T.; Wallingford, J.B. Planar cell polarity in development and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 375–388. [Google Scholar] [CrossRef]
- Vladar, E.K.; Bayly, R.D.; Sangoram, A.; Scott, M.P.; Axelrod, J.D. Microtubules Enable the Planar Cell Polarity of Airway Cilia. Curr. Biol. 2012, 22, 2203–2212. [Google Scholar] [CrossRef]
- Chausovsky, A.; Bershadsky, A.D.; Borisy, G.G. Cadherin-mediated regulation of microtubule dynamics. Nat. Cell Biol. 2000, 2, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Marshall, W.F.; Kintner, C. Cilia Orientation and the Fluid Mechanics of Development. Curr. Opin. Cell Biol. 2008, 20, 48–52. [Google Scholar] [CrossRef]
- VanderVorst, K.; Hatakeyama, J.; Berg, A.; Lee, H.; Carraway, K.L. Cellular and molecular mechanisms underlying planar cell polarity pathway contributions to cancer malignancy. Semin. Cell Dev. Biol. 2018, 81, 78–87. [Google Scholar] [CrossRef]
- Kim, S.H.; Li, Z.; Sacks, D.B. E-cadherin-mediated Cell-Cell Attachment Activates Cdc42. J. Biol. Chem. 2000, 275, 36999–37005. [Google Scholar] [CrossRef]
- Shin, K.; Fogg, V.C.; Margolis, B. Tight Junctions and Cell Polarity. Annu. Rev. Cell Dev. Biol. 2006, 22, 207–235. [Google Scholar] [CrossRef]
- Weber, G.F.; Bjerke, M.A.; DeSimone, D.W. Integrins and cadherins join forces to form adhesive networks. J. Cell Sci. 2011, 124, 1183–1193. [Google Scholar] [CrossRef]
- Hutterer, A.; Betschinger, J.; Petronczki, M.; Knoblich, J.A. Sequential Roles of Cdc42, Par-6, aPKC, and Lgl in the Establishment of Epithelial Polarity during Drosophila Embryogenesis. Dev. Cell 2004, 6, 845–854. [Google Scholar] [CrossRef]
- Warner, S.J.; Longmore, G.D. Cdc42 antagonizes Rho1 activity at adherens junctions to limit epithelial cell apical tension. J. Cell Biol. 2009, 187, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Nejsum, L.N.; Nelson, W.J. Epithelial cell surface polarity: The early steps. Front. Biosci. 2009, 14, 1088–1098. [Google Scholar] [CrossRef] [PubMed][Green Version]
- St Johnston, D.; Ahringer, J. Cell Polarity in Eggs and Epithelia: Parallels and Diversity. Cell 2010, 141, 757–774. [Google Scholar] [CrossRef]
- Wood, W.; Martin, P. Structures in focus—filopodia. Int. J. Biochem. Cell Biol. 2002, 34, 726–730. [Google Scholar] [CrossRef]
- Khurana, S.; George, S.P. The role of actin bundling proteins in the assembly of filopodia in epithelial cells. Cell Adhes. Migr. 2011, 5, 409–420. [Google Scholar] [CrossRef]
- Frisch, S.; Francis, H. Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 1994, 124, 619–626. [Google Scholar] [CrossRef]
- Nguyen, H.A.; Vu, S.H.; Jung, S.; Lee, B.S.; Nguyen, T.N.Q.; Lee, H.; Lee, H.; Myagmarjav, D.; Jo, T.; Choi, Y.; et al. SERTAD1 Sensitizes Breast Cancer Cells to Doxorubicin and Promotes Lysosomal Protein Biosynthesis. Biomedicines 2022, 10, 1148. [Google Scholar] [CrossRef]
- Shapiro, L.; Fannon, A.M.; Kwong, P.D.; Thompson, A.; Lehmann, M.S.; Grübel, G.; Legrand, J.-F.; Als-Nielsen, J.; Colman, D.R.; Hendrickson, W.A. Structural basis of cell-cell adhesion by cadherins. Nature 1995, 374, 327–337. [Google Scholar] [CrossRef]
- Vasioukhin, V.; Bauer, C.; Yin, M.; Fuchs, E. Directed Actin Polymerization Is the Driving Force for Epithelial Cell–Cell Adhesion. Cell 2000, 100, 209–219. [Google Scholar] [CrossRef]
- Gumbiner, B.M. Cell Adhesion: The Molecular Basis of Tissue Architecture and Morphogenesis. Cell 1996, 84, 345–357. [Google Scholar] [CrossRef]
- Klezovitch, O.; Vasioukhin, V. Cadherin signaling: Keeping cells in touch. F1000Research 2015, 4, 550. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-S.; Yi, B.-R.; Kim, N.-H.; Choi, K.-C. Role of the epithelial–mesenchymal transition and its effects on embryonic stem cells. Exp. Mol. Med. 2014, 46, e108. [Google Scholar] [CrossRef] [PubMed]
- Cunha, G.R.; Baskin, L. Mesenchymal-epithelial interaction techniques. Differentiation 2016, 91, 20–27. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef]
- Lauffenburger, D.A.; Horwitz, A.F. Cell Migration: A Physically Integrated Molecular Process. Cell 1996, 84, 359–369. [Google Scholar] [CrossRef]
- Vallenius, T. Actin stress fibre subtypes in mesenchymal-migrating cells. Open Biol. 2013, 3, 130001. [Google Scholar] [CrossRef]
- Vicente-Manzanares, M.; Koach, M.A.; Whitmore, L.; Lamers, M.L.; Horwitz, A.F. Segregation and activation of myosin IIB creates a rear in migrating cells. J. Cell Biol. 2008, 183, 543–554. [Google Scholar] [CrossRef]
- Dobra, K.; Andäng, M.; Syrokou, A.; Karamanos, N.K.; Hjerpe, A. Differentiation of Mesothelioma Cells Is Influenced by the Expression of Proteoglycans. Exp. Cell Res. 2000, 258, 12–22. [Google Scholar] [CrossRef]
- Scheau, C.; Badarau, I.A.; Costache, R.; Caruntu, C.; Mihai, G.L.; Didilescu, A.C.; Constantin, C.; Neagu, M. The Role of Matrix Metalloproteinases in the Epithelial-Mesenchymal Transition of Hepatocellular Carcinoma. Anal. Cell. Pathol. 2019, 2019, e9423907. [Google Scholar] [CrossRef]
- Szatmári, T.; Dobra, K. The Role of Syndecan-1 in Cellular Signaling and its Effects on Heparan Sulfate Biosynthesis in Mesenchymal Tumors. Front. Oncol. 2013, 3, 310. [Google Scholar] [CrossRef]
- Choi, S.S.; Diehl, A.M. Epithelial-to-Mesenchymal Transitions in the Liver. Hepatology 2009, 50, 2007–2013. [Google Scholar] [CrossRef]
- Bischoff, J. Endothelial-to-Mesenchymal Transition. Circ. Res. 2019, 124, 1163–1165. [Google Scholar] [CrossRef]
- Lachat, C.; Peixoto, P.; Hervouet, E. Epithelial to Mesenchymal Transition History: From Embryonic Development to Cancers. Biomolecules 2021, 11, 782. [Google Scholar] [CrossRef]
- Lei, Y.; Chen, L.; Zhang, G.; Shan, A.; Ye, C.; Liang, B.; Sun, J.; Liao, X.; Zhu, C.; Chen, Y.; et al. MicroRNAs target the Wnt/β-catenin signaling pathway to regulate epithelial-mesenchymal transition in cancer. Oncol. Rep. 2020, 44, 1299–1313. [Google Scholar] [CrossRef]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef]
- Song, L.-B.; Li, J.; Liao, W.-T.; Feng, Y.; Yu, C.-P.; Hu, L.-J.; Kong, Q.-L.; Xu, L.-H.; Zhang, X.; Liu, W.-L.; et al. The polycomb group protein Bmi-1 represses the tumor suppressor PTEN and induces epithelial-mesenchymal transition in human nasopharyngeal epithelial cells. J. Clin. Investig. 2009, 119, 3626–3636. [Google Scholar] [CrossRef]
- Lau, M.-T.; So, W.-K.; Leung, P.C.K. Fibroblast Growth Factor 2 Induces E-Cadherin Down-Regulation via PI3K/Akt/mTOR and MAPK/ERK Signaling in Ovarian Cancer Cells. PLoS ONE 2013, 8, e59083. [Google Scholar] [CrossRef]
- Qi, Y.; Liu, J.; Chao, J.; Scheuerman, M.P.; Rahimi, S.A.; Lee, L.Y.; Li, S. PTEN suppresses epithelial–mesenchymal transition and cancer stem cell activity by downregulating Abi1. Sci. Rep. 2020, 10, 12685. [Google Scholar] [CrossRef]
- Santamaria, P.G.; Moreno-Bueno, G.; Portillo, F.; Cano, A. EMT: Present and future in clinical oncology. Mol. Oncol. 2017, 11, 718–738. [Google Scholar] [CrossRef]
- Zhang, Y.; Wen, Z.; Shi, X.; Liu, Y.-J.; Eriksson, J.E.; Jiu, Y. The diverse roles and dynamic rearrangement of vimentin during viral infection. J. Cell Sci. 2020, 134, jcs250597. [Google Scholar] [CrossRef]
- Eckes, B.; Dogic, D.; Colucci-Guyon, E.; Wang, N.; Maniotis, A.; Ingber, D.; Merckling, A.; Langa, F.; Aumailley, M.; Delouvée, A.; et al. Impaired mechanical stability, migration and contractile capacity in vimentin-deficient fibroblasts. J. Cell Sci. 1998, 11, 1897–1907. [Google Scholar] [CrossRef]
- Satelli, A.; Li, S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell. Mol. Life Sci. 2011, 68, 3033–3046. [Google Scholar] [CrossRef]
- Battaglia, R.A.; Delic, S.; Herrmann, H.; Snider, N.T. Vimentin on the move: New developments in cell migration. F1000Research 2018, 7, 1796. [Google Scholar] [CrossRef]
- Wu, S.; Du, Y.; Beckford, J.; Alachkar, H. Upregulation of the EMT marker vimentin is associated with poor clinical outcome in acute myeloid leukemia. J. Transl. Med. 2018, 16, 170. [Google Scholar] [CrossRef]
- Liu, C.-Y.; Lin, H.-H.; Tang, M.-J.; Wang, Y.-K. Vimentin contributes to epithelial-mesenchymal transition cancer cell mechanics by mediating cytoskeletal organization and focal adhesion maturation. Oncotarget 2015, 6, 15966–15983. [Google Scholar] [CrossRef]
- Bindels, S.; Mestdagt, M.; Vandewalle, C.; Jacobs, N.; Volders, L.; Noël, A.; van Roy, F.; Berx, G.; Foidart, J.-M.; Gilles, C. Regulation of vimentin by SIP1 in human epithelial breast tumor cells. Oncogene 2006, 25, 4975–4985. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, X.; Salmon, M.; Lin, X.; Zehner, Z.E. TGFβ1 regulation of vimentin gene expression during differentiation of the C2C12 skeletal myogenic cell line requires Smads, AP-1 and Sp1 family members. Biochim. Biophys. Acta 2007, 1773, 427–439. [Google Scholar] [CrossRef]
- Sommerova, L.; Ondrouskova, E.; Vojtesek, B.; Hrstka, R. Suppression of AGR2 in a TGF-β-induced Smad regulatory pathway mediates epithelial-mesenchymal transition. BMC Cancer 2017, 17, 546. [Google Scholar] [CrossRef] [PubMed]
- Shirahata, A.; Sakata, M.; Sakuraba, K.; Goto, T.; Mizukami, H.; Saito, M.; Ishibashi, K.; Kigawa, G.; Nemoto, H.; Sanada, Y.; et al. Vimentin Methylation as a Marker for Advanced Colorectal Carcinoma. Anticancer. Res. 2009, 29, 279–281. [Google Scholar] [PubMed]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol. Cancer 2016, 15, 18. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-Y.; Lee, C.-H.; Li, Y.-S.; Huang, J.-T.; Lan, S.-H.; Wang, Y.-F.; Lai, W.-W.; Wang, Y.-C.; Lin, Y.-J.; Liu, H.-S.; et al. MicroRNA-146a suppresses tumor malignancy via targeting vimentin in esophageal squamous cell carcinoma cells with lower fibronectin membrane assembly. J. Biomed. Sci. 2020, 27, 102. [Google Scholar] [CrossRef]
- Kim, T.W.; Lee, Y.S.; Yun, N.H.; Shin, C.H.; Hong, H.K.; Kim, H.H.; Cho, Y.B. MicroRNA-17-5p regulates EMT by targeting vimentin in colorectal cancer. Br. J. Cancer 2020, 123, 1123–1130. [Google Scholar] [CrossRef]
- Lin, S.-L.; Lin, Y.-H.; Chi, H.-C.; Lin, T.-K.; Chen, W.-J.; Yeh, C.-T.; Lin, K.-H. A Novel Long Non-Coding RNA-01488 Suppressed Metastasis and Tumorigenesis by Inducing miRNAs That Reduce Vimentin Expression and Ubiquitination of Cyclin E. Cells 2020, 9, 1504. [Google Scholar] [CrossRef]
- Satelli, A.; Batth, I.S.; Brownlee, Z.; Rojas, C.; Meng, Q.H.; Kopetz, S.; Li, S. Potential role of nuclear PD-L1 expression in cell-surface vimentin positive circulating tumor cells as a prognostic marker in cancer patients. Sci. Rep. 2016, 6, 28910. [Google Scholar] [CrossRef]
- Paulin, D.; Lilienbaum, A.; Kardjian, S.; Agbulut, O.; Li, Z. Vimentin: Regulation and pathogenesis. Biochimie 2022, 197, 96–112. [Google Scholar] [CrossRef]
- Loh, C.-Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef]
- Rosso, M.; Majem, B.; Devis, L.; Lapyckyj, L.; Besso, M.J.; Llauradó, M.; Abascal, M.F.; Matos, M.L.; Lanau, L.; Castellví, J.; et al. E-cadherin: A determinant molecule associated with ovarian cancer progression, dissemination and aggressiveness. PLoS ONE 2017, 12, e0184439. [Google Scholar] [CrossRef]
- Wang, M.; Ren, D.; Guo, W.; Huang, S.; Wang, Z.; Li, Q.; Du, H.; Song, L.; Peng, X. N-cadherin promotes epithelial-mesenchymal transition and cancer stem cell-like traits via ErbB signaling in prostate cancer cells. Int. J. Oncol. 2016, 48, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Parker Kerrigan, B.C.; Yang, D.; Hu, L.; Shmulevich, I.; Sood, A.K.; Xue, F.; Zhang, W. Post-transcriptional regulatory network of epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions. J. Hematol. Oncol. 2014, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Kamikihara, T.; Ishigami, S.; Arigami, T.; Matsumoto, M.; Okumura, H.; Uchikado, Y.; Kita, Y.; Kurahara, H.; Kijima, Y.; Ueno, S.; et al. Clinical implications of N-cadherin expression in gastric cancer. Pathol. Int. 2012, 62, 161–166. [Google Scholar] [CrossRef]
- Cao, Z.-Q.; Wang, Z.; Leng, P. Aberrant N-cadherin expression in cancer. Biomed. Pharmacother. 2019, 118, 109320. [Google Scholar] [CrossRef]
- Mrozik, K.M.; Blaschuk, O.W.; Cheong, C.M.; Zannettino, A.C.W.; Vandyke, K. N-cadherin in cancer metastasis, its emerging role in haematological malignancies and potential as a therapeutic target in cancer. BMC Cancer 2018, 18, 939. [Google Scholar] [CrossRef]
- Araki, K.; Shimura, T.; Suzuki, H.; Tsutsumi, S.; Wada, W.; Yajima, T.; Kobayahi, T.; Kubo, N.; Kuwano, H. E/N-cadherin switch mediates cancer progression via TGF-β-induced epithelial-to-mesenchymal transition in extrahepatic cholangiocarcinoma. Br. J. Cancer 2011, 105, 1885–1893. [Google Scholar] [CrossRef]
- Kim, K.; Lu, Z.; Hay, E.D. Direct Evidence for a Role of Β-Catenin/Lef-1 Signaling Pathway in Induction of Emt. Cell Biol. Int. 2002, 26, 463–476. [Google Scholar] [CrossRef]
- Aban, C.E.; Lombardi, A.; Neiman, G.; Biani, M.C.; La Greca, A.; Waisman, A.; Moro, L.N.; Sevlever, G.; Miriuka, S.; Luzzani, C. Downregulation of E-cadherin in pluripotent stem cells triggers partial EMT. Sci. Rep. 2021, 11, 2048. [Google Scholar] [CrossRef]
- Hao, L.; Ha, J.R.; Kuzel, P.; Garcia, E.; Persad, S. Cadherin switch from E- to N-cadherin in melanoma progression is regulated by the PI3K/PTEN pathway through Twist and Snail. Br. J. Dermatol. 2012, 166, 1184–1197. [Google Scholar] [CrossRef]
- Nowak, E.; Bednarek, I. Aspects of the Epigenetic Regulation of EMT Related to Cancer Metastasis. Cells 2021, 10, 3435. [Google Scholar] [CrossRef]
- Cheng, B.; Zhu, Q.; Lin, W.; Wang, L. MicroRNA-122 inhibits epithelial-mesenchymal transition of hepatic stellate cells induced by the TGF-β1/Smad signaling pathway. Exp. Ther. Med. 2019, 17, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.-B.; He, X.-J.; Xia, Y.-J.; Hu, W.-J.; Luo, J.-G.; Zhang, J.; Tao, H.-Q. MicroRNA-145-5p inhibits gastric cancer invasiveness through targeting N-cadherin and ZEB2 to suppress epithelial–mesenchymal transition. OncoTargets Ther. 2016, 9, 2305–2315. [Google Scholar] [CrossRef]
- Zhang, X.-F.; Zhang, X.-Q.; Chang, Z.-X.; Wu, C.-C.; Guo, H. microRNA-145 modulates migration and invasion of bladder cancer cells by targeting N-cadherin. Mol. Med. Rep. 2018, 17, 8450–8456. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Liang, C. MicroRNA-148b inhibits proliferation and the epithelial-mesenchymal transition and increases radiosensitivity in non-small cell lung carcinomas by regulating ROCK1. Exp. Ther. Med. 2018, 15, 3609–3616. [Google Scholar] [CrossRef]
- Zhou, S.; Liu, F.; Zhang, A.; Liang, H.; Wang, Y.; Ma, R.; Jiang, Y.; Sun, N. MicroRNA-199b-5p attenuates TGF-β1-induced epithelial–mesenchymal transition in hepatocellular carcinoma. Br. J. Cancer 2017, 117, 233–244. [Google Scholar] [CrossRef]
- Wan, Y.; Liu, H.; Zhang, M.; Huang, Z.; Zhou, H.; Zhu, Y.; Tao, Y.; Xie, N.; Liu, X.; Hou, J.; et al. Prognostic value of epithelial-mesenchymal transition-inducing transcription factors in head and neck squamous cell carcinoma: A meta-analysis. Head Neck 2020, 42, 1067–1076. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, B.P. Snail: More than EMT. Cell Adhes. Migr. 2010, 4, 199. [Google Scholar] [CrossRef]
- Martin, T.A.; Goyal, A.; Watkins, G.; Jiang, W.G. Expression of the Transcription Factors Snail, Slug, and Twist and Their Clinical Significance in Human Breast Cancer. Ann. Surg. Oncol. 2005, 12, 488–496. [Google Scholar] [CrossRef]
- Villarejo, A.; Cortés-Cabrera, Á.; Molina-Ortíz, P.; Portillo, F.; Cano, A. Differential Role of Snail1 and Snail2 Zinc Fingers in E-cadherin Repression and Epithelial to Mesenchymal Transition. J. Biol. Chem. 2014, 289, 930–941. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Fitzgerald, T.L.; Yang, L.V.; Lertpiriyapong, K.; Steelman, L.S.; Abrams, S.L.; Montalto, G.; Cervello, M.; Neri, L.M.; Cocco, L.; et al. Roles of GSK-3 and microRNAs on epithelial mesenchymal transition and cancer stem cells. Oncotarget 2016, 8, 14221–14250. [Google Scholar] [CrossRef]
- Wu, L.; Feng, H.; Hu, J.; Tian, X.; Zhang, C. Valproic acid (VPA) promotes the epithelial mesenchymal transition of hepatocarcinoma cells via transcriptional and post-transcriptional up regulation of Snail. Biomed. Pharmacother. 2016, 84, 1029–1035. [Google Scholar] [CrossRef]
- Lin, Y.; Dong, C.; Zhou, B.P. Epigenetic Regulation of EMT: The Snail Story. Curr. Pharm. Des. 2014, 20, 1698–1705. [Google Scholar] [CrossRef]
- Lin, X.; Chai, G.; Wu, Y.; Li, J.; Chen, F.; Liu, J.; Luo, G.; Tauler, J.; Du, J.; Lin, S.; et al. RNA m6A methylation regulates the epithelial mesenchymal transition of cancer cells and translation of Snail. Nat. Commun. 2019, 10, 2065. [Google Scholar] [CrossRef] [PubMed]
- Solé, C.; Lawrie, C.H. MicroRNAs and Metastasis. Cancers 2020, 12, 96. [Google Scholar] [CrossRef]
- Xiong, Y.; Wang, Y.; Wang, L.; Huang, Y.; Xu, Y.; Xu, L.; Guo, Y.; Lu, J.; Li, X.; Zhu, M.; et al. MicroRNA-30b targets Snail to impede epithelial-mesenchymal transition in pancreatic cancer stem cells. J. Cancer 2018, 9, 2147–2159. [Google Scholar] [CrossRef]
- Yu, B.; Jiang, K.; Zhang, J. MicroRNA-124 suppresses growth and aggressiveness of osteosarcoma and inhibits TGF-β-mediated AKT/GSK-3β/SNAIL-1 signaling. Mol. Med. Rep. 2018, 17, 6736–6744. [Google Scholar] [CrossRef]
- Zuo, Q.-F.; Cao, L.-Y.; Yu, T.; Gong, L.; Wang, L.-N.; Zhao, Y.-L.; Xiao, B.; Zou, Q.-M. MicroRNA-22 inhibits tumor growth and metastasis in gastric cancer by directly targeting MMP14 and Snail. Cell Death Dis. 2015, 6, e2000. [Google Scholar] [CrossRef]
- Qin, Z.; Wang, T.; Su, S.; Shen, L.; Zhu, G.; Liu, Q.; Zhang, L.; Liu, K.; Zhang, Y.; Zhou, Z.; et al. BRD4 Promotes Gastric Cancer Progression and Metastasis through Acetylation-Dependent Stabilization of Snail. Cancer Res. 2019, 79, 4869–4881. [Google Scholar] [CrossRef]
- Dong, L.; Zhang, X.; Xiang, W.; Ni, J.; Zhou, W.; Li, H. Post-transcription mediated Snail stabilization is involved in radiation exposure induced invasion and migration of hepatocarcinoma cells. Biomed. Pharmacother. 2018, 103, 767–772. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, X.; Zhao, Y.; Jin, Y.; Zheng, J. G-protein-coupled estrogen receptor suppresses the migration of osteosarcoma cells via post-translational regulation of Snail. J. Cancer Res. Clin. Oncol. 2019, 145, 87–96. [Google Scholar] [CrossRef]
- Thorpe, H.; Asiri, A.; Akhlaq, M.; Ilyas, M. Cten promotes epithelial-mesenchymal transition through the post-transcriptional stabilization of Snail. Mol. Carcinog. 2017, 56, 2601–2609. [Google Scholar] [CrossRef]
- Gires, O.; Pan, M.; Schinke, H.; Canis, M.; Baeuerle, P.A. Expression and function of epithelial cell adhesion molecule EpCAM: Where are we after 40 years? Cancer Metastasis Rev. 2020, 39, 969–987. [Google Scholar] [CrossRef]
- Schnell, U.; Cirulli, V.; Giepmans, B.N.G. EpCAM: Structure and function in health and disease. Biochim. Biophys. Acta (BBA)—Biomembr. 2013, 1828, 1989–2001. [Google Scholar] [CrossRef]
- Slanchev, K.; Carney, T.J.; Stemmler, M.P.; Koschorz, B.; Amsterdam, A.; Schwarz, H.; Hammerschmidt, M. The Epithelial Cell Adhesion Molecule EpCAM Is Required for Epithelial Morphogenesis and Integrity during Zebrafish Epiboly and Skin Development. PLoS Genet. 2009, 5, e1000563. [Google Scholar] [CrossRef]
- Fagotto, F. EpCAM as Modulator of Tissue Plasticity. Cells 2020, 9, 2128. [Google Scholar] [CrossRef]
- Chen, G.; Yang, Y.; Liu, W.; Huang, L.; Yang, L.; Lei, Y.; Wu, H.; Lei, Z.; Guo, J. EpCAM is essential for maintenance of the small intestinal epithelium architecture via regulation of the expression and localization of proteins that compose adherens junctions. Int. J. Mol. Med. 2021, 47, 621–632. [Google Scholar] [CrossRef]
- Yahyazadeh Mashhadi, S.M.; Kazemimanesh, M.; Arashkia, A.; Azadmanesh, K.; Meshkat, Z.; Golichenari, B.; Sahebkar, A. Shedding light on the EpCAM: An overview. J. Cell. Physiol. 2019, 234, 12569–12580. [Google Scholar] [CrossRef]
- Keller, L.; Werner, S.; Pantel, K. Biology and clinical relevance of EpCAM. Cell Stress 2019, 3, 165–180. [Google Scholar] [CrossRef]
- van der Gun, B.T.F.; Melchers, L.J.; Ruiters, M.H.J.; de Leij, L.F.M.H.; McLaughlin, P.M.J.; Rots, M.G. EpCAM in carcinogenesis: The good, the bad or the ugly. Carcinogenesis 2010, 31, 1913–1921. [Google Scholar] [CrossRef]
- van der Gun, B.T.F.; de Groote, M.L.; Kazemier, H.G.; Arendzen, A.J.; Terpstra, P.; Ruiters, M.H.J.; McLaughlin, P.M.J.; Rots, M.G. Transcription factors and molecular epigenetic marks underlying EpCAM overexpression in ovarian cancer. Br. J. Cancer 2011, 105, 312–319. [Google Scholar] [CrossRef]
- Mohtar, M.A.; Syafruddin, S.E.; Nasir, S.N.; Low, T.Y. Revisiting the Roles of Pro-Metastatic EpCAM in Cancer. Biomolecules 2020, 10, 255. [Google Scholar] [CrossRef]
- Gumbiner, B.M. Regulation of cadherin-mediated adhesion in morphogenesis. Nat. Rev. Mol. Cell Biol. 2005, 6, 622–634. [Google Scholar] [CrossRef]
- Wong, S.H.M.; Fang, C.M.; Chuah, L.-H.; Leong, C.O.; Ngai, S.C. E-cadherin: Its dysregulation in carcinogenesis and clinical implications. Crit. Rev. Oncol. Hematol. 2018, 121, 11–22. [Google Scholar] [CrossRef]
- Coopman, P.; Djiane, A. Adherens Junction and E-Cadherin complex regulation by epithelial polarity. Cell. Mol. Life Sci. 2016, 73, 3535–3553. [Google Scholar] [CrossRef]
- Beavon, I.R.G. The E-cadherin–catenin complex in tumour metastasis: Structure, function and regulation. Eur. J. Cancer 2000, 36, 1607–1620. [Google Scholar] [CrossRef]
- Yonemura, S.; Wada, Y.; Watanabe, T.; Nagafuchi, A.; Shibata, M. α-Catenin as a tension transducer that induces adherens junction development. Nat. Cell Biol. 2010, 12, 533–542. [Google Scholar] [CrossRef]
- Petrova, Y.I.; Schecterson, L.; Gumbiner, B.M. Roles for E-cadherin cell surface regulation in cancer. Mol. Biol. Cell 2016, 27, 3233–3244. [Google Scholar] [CrossRef]
- Mendonsa, A.M.; Na, T.-Y.; Gumbiner, B.M. E-cadherin in contact inhibition and cancer. Oncogene 2018, 37, 4769–4780. [Google Scholar] [CrossRef]
- Chua, H.L.; Bhat-Nakshatri, P.; Clare, S.E.; Morimiya, A.; Badve, S.; Nakshatri, H. NF-κB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: Potential involvement of ZEB-1 and ZEB-2. Oncogene 2007, 26, 711–724. [Google Scholar] [CrossRef]
- Odero-Marah, V.; Hawsawi, O.; Henderson, V.; Sweeney, J. Epithelial-Mesenchymal Transition (EMT) and Prostate Cancer. In Cell & Molecular Biology of Prostate Cancer: Updates, Insights and New Frontiers; Advances in Experimental Medicine and Biology; Schatten, H., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 101–110. ISBN 978-3-319-95693-0. [Google Scholar]
- Choi, J.; Park, S.Y.; Joo, C.-K. Transforming Growth Factor-β1 Represses E-Cadherin Production via Slug Expression in Lens Epithelial Cells. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2708–2718. [Google Scholar] [CrossRef]
- van Roy, F.; Berx, G. The cell-cell adhesion molecule E-cadherin. Cell. Mol. Life Sci. 2008, 65, 3756–3788. [Google Scholar] [CrossRef]
- Yu, T.-X.; Gu, B.-L.; Yan, J.-K.; Zhu, J.; Yan, W.-H.; Chen, J.; Qian, L.-X.; Cai, W. CUGBP1 and HuR regulate E-cadherin translation by altering recruitment of E-cadherin mRNA to processing bodies and modulate epithelial barrier function. Am. J. Physiol.-Cell Physiol. 2016, 310, C54–C65. [Google Scholar] [CrossRef]
- Fujita, Y.; Krause, G.; Scheffner, M.; Zechner, D.; Leddy, H.E.M.; Behrens, J.; Sommer, T.; Birchmeier, W. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat. Cell Biol. 2002, 4, 222–231. [Google Scholar] [CrossRef]
- Shrestha, H.; Ryu, T.; Seo, Y.-W.; Park, S.-Y.; He, Y.; Dai, W.; Park, E.; Simkhada, S.; Kim, H.; Lee, K.; et al. Hakai, an E3-ligase for E-cadherin, stabilizes δ-catenin through Src kinase. Cell. Signal. 2017, 31, 135–145. [Google Scholar] [CrossRef]
- Hu, P.; O’Keefe, E.J.; Rubenstein, D.S. Tyrosine Phosphorylation of Human Keratinocyte β-Catenin and Plakoglobin Reversibly Regulates their Binding to E-Cadherin and α-Catenin. J. Investig. Dermatol. 2001, 117, 1059–1067. [Google Scholar] [CrossRef]
- Advedissian, T.; Proux-Gillardeaux, V.; Nkosi, R.; Peyret, G.; Nguyen, T.; Poirier, F.; Viguier, M.; Deshayes, F. E-cadherin dynamics is regulated by galectin-7 at epithelial cell surface. Sci. Rep. 2017, 7, 17086. [Google Scholar] [CrossRef]
- Dollé, L.; Theise, N.D.; Schmelzer, E.; Boulter, L.; Gires, O.; van Grunsven, L.A. EpCAM and the biology of hepatic stem/progenitor cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G233–G250. [Google Scholar] [CrossRef]
- Ekblom, P.; Lonai, P.; Talts, J.F. Expression and biological role of laminin-1. Matrix Biol. 2003, 22, 35–47. [Google Scholar] [CrossRef]
- Ekblom, M.; Falk, M.; Salmivirta, K.; Durbeej, M.; Ekblom, P. Laminin Isoforms and Epithelial Development. Ann. N. Y. Acad. Sci. 1998, 857, 194–211. [Google Scholar] [CrossRef]
- Reuters, I.; Weber, M.; Schulze-Lohoff, E. Rho/Rho Kinase Pathway Regulates Maintenance of the Differentiated Tubular Epithelial Cell Phenotype on Laminin-1. Nephron Physiol. 2006, 104, p95–p106. [Google Scholar] [CrossRef]
- Lee, H.; Kim, W.-J.; Kang, H.-G.; Jang, J.-H.; Choi, I.J.; Chun, K.-H.; Kim, S.-J. Upregulation of LAMB1 via ERK/c-Jun Axis Promotes Gastric Cancer Growth and Motility. Int. J. Mol. Sci. 2021, 22, 626. [Google Scholar] [CrossRef]
- Gordon-Weeks, A.; Lim, S.Y.; Yuzhalin, A.; Lucotti, S.; Vermeer, J.A.F.; Jones, K.; Chen, J.; Muschel, R.J. Tumour-Derived Laminin α5 (LAMA5) Promotes Colorectal Liver Metastasis Growth, Branching Angiogenesis and Notch Pathway Inhibition. Cancers 2019, 11, 630. [Google Scholar] [CrossRef]
- Giannini, S.; Lee-Sundlov, M.M.; Rivadeneyra, L.; Di Buduo, C.A.; Burns, R.; Lau, J.T.; Falet, H.; Balduini, A.; Hoffmeister, K.M. β4GALT1 controls β1 integrin function to govern thrombopoiesis and hematopoietic stem cell homeostasis. Nat. Commun. 2020, 11, 356. [Google Scholar] [CrossRef]
- Lepucki, A.; Orlińska, K.; Mielczarek-Palacz, A.; Kabut, J.; Olczyk, P.; Komosińska-Vassev, K. The Role of Extracellular Matrix Proteins in Breast Cancer. J. Clin. Med. 2022, 11, 1250. [Google Scholar] [CrossRef]
- Karamitopoulou, E.; Zlobec, I.; Gloor, B.; Kondi-Pafiti, A.; Lugli, A.; Perren, A. Loss of Raf-1 kinase inhibitor protein (RKIP) is strongly associated with high-grade tumor budding and correlates with an aggressive phenotype in pancreatic ductal adenocarcinoma (PDAC). J. Transl. Med. 2013, 11, 311. [Google Scholar] [CrossRef]
- Zaravinos, A.; Bonavida, B.; Chatzaki, E.; Baritaki, S. RKIP: A Key Regulator in Tumor Metastasis Initiation and Resistance to Apoptosis: Therapeutic Targeting and Impact. Cancers 2018, 10, 287. [Google Scholar] [CrossRef]
- Bernier, I.; Jollés, P. Purification and characterization of a basic 23 kDa cytosolic protein from bovine brain. Biochim. Biophys. Acta (BBA)—Protein Struct. Mol. Enzymol. 1984, 790, 174–181. [Google Scholar] [CrossRef]
- Escara-Wilke, J.; Yeung, K.; Keller, E.T. Raf kinase inhibitor protein (RKIP) in cancer. Cancer Metastasis Rev. 2012, 31, 615–620. [Google Scholar] [CrossRef]
- Kousaku, O.; Kenichi, M. A human cDNA sequence homologue of bovine phosphatidylethanolamine-binding protein. Gene 1994, 140, 293–294. [Google Scholar] [CrossRef]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef]
- Guo, Y.-J.; Pan, W.-W.; Liu, S.-B.; Shen, Z.-F.; Xu, Y.; Hu, L.-L. ERK/MAPK signalling pathway and tumorigenesis (Review). Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef]
- Yeung, K.; Seitz, T.; Li, S.; Janosch, P.; McFerran, B.; Kaiser, C.; Fee, F.; Katsanakis, K.D.; Rose, D.W.; Mischak, H.; et al. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature 1999, 401, 173–177. [Google Scholar] [CrossRef]
- Touboul, R.; Baritaki, S.; Zaravinos, A.; Bonavida, B. RKIP Pleiotropic Activities in Cancer and Inflammatory Diseases: Role in Immunity. Cancers 2021, 13, 6247. [Google Scholar] [CrossRef]
- Corbit, K.C.; Trakul, N.; Eves, E.M.; Diaz, B.; Marshall, M.; Rosner, M.R. Activation of Raf-1 Signaling by Protein Kinase C through a Mechanism Involving Raf Kinase Inhibitory Protein. J. Biol. Chem. 2003, 278, 13061–13068. [Google Scholar] [CrossRef]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-Activated Protein (MAP) Kinase Pathways: Regulation and Physiological Functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [CrossRef]
- Datar, I.; Feng, J.; Qiu, X.; Lewandowski, J.; Yeung, M.; Ren, G.; Aras, S.; Al-Mulla, F.; Cui, H.; Trumbly, R.; et al. RKIP Inhibits Local Breast Cancer Invasion by Antagonizing the Transcriptional Activation of MMP13. PLoS ONE 2015, 10, e0134494. [Google Scholar] [CrossRef]
- Lorenz, K.; Lohse, M.J.; Quitterer, U. Protein kinase C switches the Raf kinase inhibitor from Raf-1 to GRK-2. Nature 2003, 426, 574–579. [Google Scholar] [CrossRef]
- Evron, T.; Daigle, T.L.; Caron, M.G. GRK2: Multiple roles beyond G protein-coupled receptor desensitization. Trends Pharmacol. Sci. 2012, 33, 154–164. [Google Scholar] [CrossRef]
- Dong, Y.; Lin, X.; Kapoor, A.; Gu, Y.; Xu, H.; Major, P.; Tang, D. Insights of RKIP-Derived Suppression of Prostate Cancer. Cancers 2021, 13, 6388. [Google Scholar] [CrossRef]
- Skinner, J.J.; Rosner, M.R. RKIP Structure Drives Its Function: A Three-State Model for Regulation of RKIP. Crit. Rev. Oncog. 2014, 19, 483–488. [Google Scholar] [CrossRef]
- Yeung, K.C.; Rose, D.W.; Dhillon, A.S.; Yaros, D.; Gustafsson, M.; Chatterjee, D.; McFerran, B.; Wyche, J.; Kolch, W.; Sedivy, J.M. Raf Kinase Inhibitor Protein Interacts with NF-κB-Inducing Kinase and TAK1 and Inhibits NF-κB Activation. Mol. Cell. Biol. 2001, 21, 7207–7217. [Google Scholar] [CrossRef]
- Shvartsur, A.; Givechian, K.B.; Garban, H.; Bonavida, B. Overexpression of RKIP and its cross-talk with several regulatory gene products in multiple myeloma. J. Exp. Clin. Cancer Res. 2017, 36, 62. [Google Scholar] [CrossRef] [PubMed]
- Baritaki, S.; Huerta-Yepez, S.; da Lourdas Cabrava-Haimandez, M.; Sensi, M.; Canevari, S.; Libra, M.; Penichet, M.; Chen, H.; Berenson, J.R.; Bonavida, B. Unique Pattern of Overexpression of Raf-1 Kinase Inhibitory Protein in Its Inactivated Phosphorylated Form in Human Multiple Myeloma. For. Immunopathol. Dis. Therap. 2011, 2, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, K.; Rosner, M.R. Harnessing RKIP to Combat Heart Disease and Cancer. Cancers 2022, 14, 867. [Google Scholar] [CrossRef] [PubMed]
- Giovanini, G.; Barros, L.R.C.; Gama, L.R.; Tortelli, T.C.; Ramos, A.F. A Stochastic Binary Model for the Regulation of Gene Expression to Investigate Responses to Gene Therapy. Cancers 2022, 14, 633. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Dong, Z.; Lin, X.; Zhang, M.; Kuang, G.; Zhu, T. Decreased Expression and Aberrant Methylation of Raf Kinase Inhibitory Protein Gene in Esophageal Squamous Cell Carcinoma. Cancer Investig. 2012, 30, 703–711. [Google Scholar] [CrossRef]
- Yesilkanal, A.E.; Rosner, M.R. Targeting Raf Kinase Inhibitory Protein Regulation and Function. Cancers 2018, 10, 306. [Google Scholar] [CrossRef]
- Zaravinos, A.; Chatziioannou, M.; Lambrou, G.I.; Boulalas, I.; Delakas, D.; Spandidos, D.A. Implication of RAF and RKIP Genes in Urinary Bladder Cancer. Pathol. Oncol. Res. 2011, 17, 181–190. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, J.; Keller, J.M.; Yeung, K.; Keller, E.T.; Fu, Z. Transcriptional Regulation of RKIP Expression by Androgen in Prostate Cells. Cell Physiol. Biochem. 2012, 30, 1340–1350. [Google Scholar] [CrossRef]
- Beach, S.; Tang, H.; Park, S.; Dhillon, A.S.; Keller, E.T.; Kolch, W.; Yeung, K.C. Snail is a repressor of RKIP transcription in metastatic prostate cancer cells. Oncogene 2008, 27, 2243–2248. [Google Scholar] [CrossRef]
- Farooqi, A.A.; Li, Y.; Sarkar, F.H. The biological complexity of RKIP signaling in human cancers. Exp. Mol. Med. 2015, 47, e185. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Zhu, H.; Liu, X.; Wang, L.; Ning, J.; Xiao, C. MiR-543 Promotes Proliferation and Epithelial-Mesenchymal Transition in Prostate Cancer via Targeting RKIP. Cell. Physiol. Biochem. 2017, 41, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tian, Z.; Tan, Y.; Lian, G.; Chen, S.; Chen, S.; Li, J.; Li, X.; Huang, K.; Chen, Y. Bmi-1-induced miR-27a and miR-155 promote tumor metastasis and chemoresistance by targeting RKIP in gastric cancer. Mol. Cancer 2020, 19, 109. [Google Scholar] [CrossRef]
- Arthur, S.; Sundaram, U. Protein kinase C-mediated phosphorylation of RKIP regulates inhibition of Na-alanine cotransport by leukotriene D4 in intestinal epithelial cells. Am. J. Physiol.-Cell Physiol. 2014, 307, C1010–C1016. [Google Scholar] [CrossRef]
- Yang, X.; Shang, P.; Yu, B.; Jin, Q.; Liao, J.; Wang, L.; Ji, J.; Guo, X. Combination therapy with miR34a and doxorubicin synergistically inhibits Dox-resistant breast cancer progression via down-regulation of Snail through suppressing Notch/NF-κB and RAS/RAF/MEK/ERK signaling pathway. Acta Pharm. Sin. B 2021, 11, 2819–2834. [Google Scholar] [CrossRef]
- Bonavida, B. Linking Autophagy and the Dysregulated NFκB/SNAIL/YY1/RKIP/PTEN Loop in Cancer: Therapeutic Implications. Crit. Rev. Oncog. 2018, 23, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Bonavida, B. RKIP-mediated chemo-immunosensitization of resistant cancer cells via disruption of the NF-κB/Snail/YY1/RKIP resistance-driver loop. Crit. Rev. Oncog. 2014, 19, 431–445. [Google Scholar] [CrossRef] [PubMed]
- He, Q.-Y.; Yi, H.-M.; Yi, H.; Xiao, T.; Qu, J.-Q.; Yuan, L.; Zhu, J.-F.; Li, J.-Y.; Wang, Y.-Y.; Li, L.-N.; et al. Reduction of RKIP expression promotes nasopharyngeal carcinoma invasion and metastasis by activating Stat3 signaling. Oncotarget 2015, 6, 16422–16436. [Google Scholar]
- Noh, H.S.; Hah, Y.-S.; Ha, J.H.; Kang, M.Y.; Zada, S.; Rha, S.Y.; Kang, S.S.; Kim, H.J.; Park, J.-Y.; Byun, J.-H.; et al. Regulation of the epithelial to mesenchymal transition and metastasis by Raf kinase inhibitory protein-dependent Notch1 activity. Oncotarget 2016, 7, 4632–4646. [Google Scholar] [CrossRef]
- Raquel-Cunha, A.; Cardoso-Carneiro, D.; Reis, R.M.; Martinho, O. Current Status of Raf Kinase Inhibitor Protein (RKIP) in Lung Cancer: Behind RTK Signaling. Cells 2019, 8, 442. [Google Scholar] [CrossRef]
- Da, C.; Wu, K.; Yue, C.; Bai, P.; Wang, R.; Wang, G.; Zhao, M.; Lv, Y.; Hou, P. N-cadherin promotes thyroid tumorigenesis through modulating major signaling pathways. Oncotarget 2016, 8, 8131–8142. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Chen, X.; Zhang, Y.; Liu, C.; Wang, Z.; Xu, X.; Zhu, J.; Xue, T. LncRNA GATA6-AS1 Inhibits the Progression of Non-Small Cell Lung Cancer via Repressing microRNA-543 to Up-Regulating RKIP. Cancer Manag. Res. 2020, 12, 9327–9338. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Wang, Q.; Xie, J.; Shi, J.; Zhou, X.; Li, D.; Xiong, F.; Zhang, L. Expression and significance of RKIP and E-cadherin in lung squamous cell carcinoma. Pathol. Oncol. Res. 2013, 19, 19–26. [Google Scholar] [CrossRef]
- Kalpana, G.; Figy, C.; Feng, J.; Tipton, C.; De Castro, J.N.; Bach, V.N.; Borile, C.; LaSalla, A.; Odeh, H.N.; Yeung, M.; et al. The RhoA dependent anti-metastatic function of RKIP in breast cancer. Sci. Rep. 2021, 11, 17455. [Google Scholar] [CrossRef]
- Koelzer, V.H.; Karamitopoulou, E.; Dawson, H.; Kondi-Pafiti, A.; Zlobec, I.; Lugli, A. Geographic analysis of RKIP expression and its clinical relevance in colorectal cancer. Br. J. Cancer 2013, 108, 2088–2096. [Google Scholar] [CrossRef] [PubMed]
- Roufas, C.; Chasiotis, D.; Makris, A.; Efstathiades, C.; Dimopoulos, C.; Zaravinos, A. The Expression and Prognostic Impact of Immune Cytolytic Activity-Related Markers in Human Malignancies: A Comprehensive Meta-analysis. Front. Oncol. 2018, 8, 27. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNAIL vs. RKIP | ||||
|---|---|---|---|---|
| Cancer | Gene vs. RKIP | R Values | Significance | Relationship |
| BLCA | SNAI1 | −0.14 | S | Inverse |
| BRCA | SNAI1 | −0.2 | S | Inverse |
| CESC | SNAI1 | −0.008 | NS | Inverse |
| CHOL | SNAI1 | −0.013 | NS | Inverse |
| COAD | SNAI1 | −0.017 | NS | Inverse |
| ESCA | SNAI1 | 0.0056 | NS | Direct |
| HNSC | SNAI1 | −0.013 | NS | Inverse |
| KICH | SNAI1 | −0.29 | S | Inverse |
| KIRC | SNAI1 | −0.18 | S | Inverse |
| KIRP | SNAI1 | −0.19 | S | Inverse |
| LICH | SNAI1 | −0.26 | S | Inverse |
| LUAD | SNAI1 | −0.18 | S | Inverse |
| LUSC | SNAI1 | −0.055 | NS | Inverse |
| PAAD | SNAI1 | 0.071 | NS | Direct |
| PCPG | SNAI1 | −0.11 | NS | Inverse |
| PRAD | SNAI1 | −0.15 | S | Inverse |
| READ | SNAI1 | 0.15 | NS | Direct |
| SARC | SNAI1 | −0.038 | NS | Inverse |
| SKCM | SNAI1 | −0.15 | S | Inverse |
| STAD | SNAI1 | 0.11 | S | Direct |
| THCA | SNAI1 | 0.44 | NS | Direct |
| THYM | SNAI1 | 0.21 | S | Direct |
| UCEC | SNAI1 | −0.085 | NS | Inverse |
| VIM vs. RKIP | ||||
|---|---|---|---|---|
| Cancer | Gene vs. RKIP | R Values | Significance | Relationship |
| BLCA | VIM | −0.15 | S | Inverse |
| BRCA | VIM | −0.25 | S | Inverse |
| CESC | VIM | 0.25 | S | Direct |
| CHOL | VIM | −0.068 | NS | Inverse |
| COAD | VIM | −0.15 | S | Inverse |
| ESCA | VIM | −0.2 | S | Inverse |
| HNSC | VIM | −0.011 | NS | Inverse |
| KICH | VIM | −0.16 | NS | Inverse |
| KIRC | VIM | −0.13 | S | Inverse |
| KIRP | VIM | 0.037 | NS | Direct |
| LICH | VIM | −0.29 | S | Inverse |
| LUAD | VIM | −0.1 | S | Inverse |
| LUSC | VIM | −0.025 | NS | Inverse |
| PAAD | VIM | 0.068 | NS | Direct |
| PCPG | VIM | −0.16 | S | Inverse |
| PRAD | VIM | −0.11 | S | Inverse |
| READ | VIM | −0.12 | NS | Inverse |
| SARC | VIM | −0.059 | NS | Inverse |
| SKCM | VIM | 0.2 | S | Direct |
| STAD | VIM | −0.11 | S | Inverse |
| THCA | VIM | 0.21 | S | Direct |
| THYM | VIM | 0.47 | S | Direct |
| UCEC | VIM | 0.13 | NS | Direct |
| CDH2 vs. RKIP | ||||
|---|---|---|---|---|
| Cancer | Gene vs. RKIP | R Values | Significance | Relationship |
| BLCA | CDH2 | −0.067 | NS | Inverse |
| BRCA | CDH2 | −0.044 | NS | Inverse |
| CESC | CDH2 | 0.11 | NS | Direct |
| CHOL | CDH2 | 0.24 | NS | Direct |
| COAD | CDH2 | −0.11 | NS | Inverse |
| ESCA | CDH2 | 0.064 | NS | Direct |
| HNSC | CDH2 | −0.015 | NS | Inverse |
| KICH | CDH2 | −0.17 | NS | Inverse |
| KIRC | CDH2 | 0.32 | S | Direct |
| KIRP | CDH2 | 0.26 | S | Direct |
| LICH | CDH2 | 0.039 | NS | Direct |
| LUAD | CDH2 | −0.017 | NS | Inverse |
| LUSC | CDH2 | 0.16 | S | Direct |
| PAAD | CDH2 | 0.33 | S | Direct |
| PCPG | CDH2 | 0.17 | S | Direct |
| PRAD | CDH2 | −0.0097 | NS | Inverse |
| READ | CDH2 | 0.3 | S | Direct |
| SARC | CDH2 | 0.12 | NS | Direct |
| SKCM | CDH2 | −0.13 | S | Inverse |
| STAD | CDH2 | 0.059 | NS | Direct |
| THCA | CDH2 | 0.34 | S | Direct |
| THYM | CDH2 | 0.058 | NS | Direct |
| UCEC | CDH2 | −0.01 | NS | Inverse |
| CDH1 vs. RKIP | ||||
|---|---|---|---|---|
| Cancer | Gene vs. RKIP | R Values | Significance | Relationship |
| BLCA | CDH1 | 0.28 | S | Direct |
| BRCA | CDH1 | 0.23 | S | Direct |
| CESC | CDH1 | 0.068 | NS | Direct |
| CHOL | CDH1 | 0.39 | S | Direct |
| COAD | CDH1 | 0.2 | S | Direct |
| ESCA | CDH1 | −0.036 | NS | Inverse |
| HNSC | CDH1 | 0.19 | S | Direct |
| KICH | CDH1 | 0.11 | NS | Direct |
| KIRC | CDH1 | 0.12 | S | Direct |
| KIRP | CDH1 | 0.018 | NS | Direct |
| LICH | CDH1 | −0.16 | S | Inverse |
| LUAD | CDH1 | 0.087 | NS | Direct |
| LUSC | CDH1 | 0.13 | S | Direct |
| PAAD | CDH1 | −0.053 | NS | Inverse |
| PCPG | CDH1 | −0.098 | NS | Inverse |
| PRAD | CDH1 | 0.3 | S | Direct |
| READ | CDH1 | 0.23 | S | Direct |
| SARC | CDH1 | 0.014 | NS | Direct |
| SKCM | CDH1 | 0.28 | S | Direct |
| STAD | CDH1 | 0.099 | S | Direct |
| THCA | CDH1 | 0.42 | S | Direct |
| THYM | CDH1 | 0.7 | S | Direct |
| UCEC | CDH1 | 0.33 | S | Direct |
| EPCAM vs. RKIP | ||||
|---|---|---|---|---|
| Cancer | Gene vs. RKIP | R Values | Significance | Relationship |
| BLCA | EPCAM | 0.3 | S | Direct |
| BRCA | EPCAM | 0.098 | S | Direct |
| CESC | EPCAM | 0.25 | S | Direct |
| CHOL | EPCAM | 0.18 | NS | Direct |
| COAD | EPCAM | 0.098 | NS | Direct |
| ESCA | EPCAM | 0.26 | S | Direct |
| HNSC | EPCAM | 0.45 | S | Direct |
| KICH | EPCAM | 0.28 | S | Direct |
| KIRC | EPCAM | 0.13 | S | Direct |
| KIRP | EPCAM | 0.028 | NS | Direct |
| LICH | EPCAM | −0.24 | S | Inverse |
| LUAD | EPCAM | 0.19 | S | Direct |
| LUSC | EPCAM | 0.23 | S | Direct |
| PAAD | EPCAM | −0.022 | NS | Inverse |
| PCPG | EPCAM | −0.0053 | NS | Inverse |
| PRAD | EPCAM | 0.18 | S | Direct |
| READ | EPCAM | 0.012 | NS | Direct |
| SARC | EPCAM | 0.043 | NS | Direct |
| SKCM | EPCAM | 0.0039 | NS | Direct |
| STAD | EPCAM | 0.018 | S | Direct |
| THCA | EPCAM | 0.26 | S | Direct |
| THYM | EPCAM | 0.57 | S | Direct |
| UCEC | EPCAM | 0.23 | S | Direct |
| LAMA1 vs. RKIP | ||||
|---|---|---|---|---|
| Cancer | Gene vs. RKIP | R Values | Significance | Relationship |
| BLCA | LAMA1 | −0.037 | NS | Inverse |
| BRCA | LAMA1 | −0.0905 | S | Inverse |
| CESC | LAMA1 | 0.15 | S | Direct |
| CHOL | LAMA1 | 0.0048 | NS | Direct |
| COAD | LAMA1 | −0.17 | S | Inverse |
| ESCA | LAMA1 | 0.15 | S | Direct |
| HNSC | LAMA1 | 0.11 | S | Direct |
| KICH | LAMA1 | 0.018 | NS | Direct |
| KIRC | LAMA1 | 0.27 | S | Direct |
| KIRP | LAMA1 | −0.064 | NS | Inverse |
| LICH | LAMA1 | −0.17 | S | Inverse |
| LUAD | LAMA1 | 0.032 | NS | Direct |
| LUSC | LAMA1 | 0.085 | NS | Direct |
| PAAD | LAMA1 | −0.15 | NS | Inverse |
| PCPG | LAMA1 | −0.16 | S | Inverse |
| PRAD | LAMA1 | 0.037 | NS | Direct |
| READ | LAMA1 | −0.17 | NS | Inverse |
| SARC | LAMA1 | −0.02 | NS | Inverse |
| SKCM | LAMA1 | 0.21 | S | Direct |
| STAD | LAMA1 | 0.12 | S | Direct |
| THCA | LAMA1 | −0.17 | S | Inverse |
| THYM | LAMA1 | 0.045 | NS | Direct |
| UCEC | LAMA1 | 0.056 | NS | Direct |
| LAMB1 vs. RKIP | ||||
|---|---|---|---|---|
| Cancer | Gene vs. RKIP | R Values | Significance | Relationship |
| BLCA | LAMB1 | −0.09 | NS | Inverse |
| BRCA | LAMB1 | −0.12 | S | Inverse |
| CESC | LAMB1 | 0.13 | S | Direct |
| CHOL | LAMB1 | −0.074 | NS | Inverse |
| COAD | LAMB1 | 0.0095 | NS | Direct |
| ESCA | LAMB1 | 0.088 | NS | Direct |
| HNSC | LAMB1 | 0.031 | NS | Direct |
| KICH | LAMB1 | 0.25 | S | Direct |
| KIRC | LAMB1 | −0.13 | S | Inverse |
| KIRP | LAMB1 | 0.03 | NS | Direct |
| LICH | LAMB1 | −0.038 | S | Inverse |
| LUAD | LAMB1 | −0.14 | S | Inverse |
| LUSC | LAMB1 | 0.027 | NS | Direct |
| PAAD | LAMB1 | 0.22 | S | Direct |
| PCPG | LAMB1 | −0.021 | S | Inverse |
| PRAD | LAMB1 | −0.094 | S | Inverse |
| READ | LAMB1 | −0.069 | NS | Inverse |
| SARC | LAMB1 | −0.037 | NS | Inverse |
| SKCM | LAMB1 | −0.15 | S | Inverse |
| STAD | LAMB1 | −0.034 | NS | Inverse |
| THCA | LAMB1 | 0.2 | S | Direct |
| THYM | LAMB1 | 0.33 | S | Direct |
| UCEC | LAMB1 | −0.027 | NS | Inverse |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cessna, H.; Baritaki, S.; Zaravinos, A.; Bonavida, B. The Role of RKIP in the Regulation of EMT in the Tumor Microenvironment. Cancers 2022, 14, 4596. https://doi.org/10.3390/cancers14194596
Cessna H, Baritaki S, Zaravinos A, Bonavida B. The Role of RKIP in the Regulation of EMT in the Tumor Microenvironment. Cancers. 2022; 14(19):4596. https://doi.org/10.3390/cancers14194596
Chicago/Turabian StyleCessna, Hannah, Stavroula Baritaki, Apostolos Zaravinos, and Benjamin Bonavida. 2022. "The Role of RKIP in the Regulation of EMT in the Tumor Microenvironment" Cancers 14, no. 19: 4596. https://doi.org/10.3390/cancers14194596
APA StyleCessna, H., Baritaki, S., Zaravinos, A., & Bonavida, B. (2022). The Role of RKIP in the Regulation of EMT in the Tumor Microenvironment. Cancers, 14(19), 4596. https://doi.org/10.3390/cancers14194596