

Targeting Glucose Metabolism Enzymes in Cancer Treatment: Current and Emerging Strategies

 , ,
, ,  , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
2. Drugs That Target Glucose Metabolism Enzymes
2.1. Drugs Targeting Glucose Transferase (GLUT)
2.2. Drugs Targeting Hexokinase (HK)
2.3. Drugs Targeting Phosphofructokinase (PFK)
2.4. Drugs Targeting Pyruvate Kinase (PK)
2.5. Drugs Targeting Lactate Dehydrogenase (LDH)
2.6. Drugs Targeting Aldolase (ALDO)
2.7. Drugs Targeting Phosphoglycerate Kinase 1 (PGK1)
2.8. Drugs Targeting Phosphoglycerate Mutase 1 (PGAM1)
2.9. Drugs Targeting Enolase (ENO)
2.10. Drugs Targeting Monocarboxylate Transporters (MCTs)
2.11. Drugs Targeting Isocitrate Dehydrogenase (IDH)
3. Combinational Strategies Using Glucose Metabolism Enzyme Inhibitors
4. Limitations of Drugs Targeting Glucose Metabolism Enzymes
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Ben-Haim, S.; Ell, P. 18F-FDG PET and PET/CT in the evaluation of cancer treatment response. J. Nucl. Med. 2009, 50, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Bravatà, V.; Stefano, A.; Cammarata, F.P.; Minafra, L.; Russo, G.; Nicolosi, S.; Pulizzi, S.; Gelfi, C.; Gilardi, M.C.; Messa, C. Genotyping analysis and 18FDG uptake in breast cancer patients: A preliminary research. J. Exp. Clin. Cancer Res. 2013, 32, 23. [Google Scholar] [CrossRef]
- Bomanji, J.B.; Costa, D.C.; Ell, P.J. Clinical role of positron emission tomography in oncology. Lancet Oncol. 2001, 2, 157–164. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Wu, Q.; Hitomi, M.; Rahim, N.; Kim, Y.; Sloan, A.E.; Weil, R.J.; Nakano, I.; Sarkaria, J.N.; Stringer, B.W.; et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nat. Neurosci. 2013, 16, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Isaac, R.; Yan, W.; Ruan, X.; Jiang, L.; Wan, Y.; Wang, J.; Wang, E.; Caron, C.; Neben, S.; et al. Cancer-cell-secreted extracellular vesicles suppress insulin secretion through miR-122 to impair systemic glucose homeostasis and contribute to tumour growth. Nat. Cell Biol. 2022, 24, 954–967. [Google Scholar] [CrossRef] [PubMed]
- Szablewski, L. Expression of glucose transporters in cancers. Biochim. Biophys. Acta 2013, 1835, 164–169. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Marsh, J.; Shelton, L.M.; Huysentruyt, L.C.; Mukherjee, P. Is the restricted ketogenic diet a viable alternative to the standard of care for managing malignant brain cancer? Epilepsy Res. 2012, 100, 310–326. [Google Scholar] [CrossRef]
- Luo, Y.; Li, Y.; Huang, Z.; Li, X.; Wang, Y.; Hou, J.; Zhou, S. A Nanounit Strategy Disrupts Energy Metabolism and Alleviates Immunosuppression for Cancer Therapy. Nano Lett. 2022, 22, 6418–6427. [Google Scholar] [CrossRef]
- Yu, J.; Wei, Z.; Li, Q.; Wan, F.; Chao, Z.; Zhang, X.; Lin, L.; Meng, H.; Tian, L. Advanced Cancer Starvation Therapy by Simultaneous Deprivation of Lactate and Glucose Using a MOF Nanoplatform. Adv. Sci. 2021, 8, e2101467. [Google Scholar] [CrossRef]
- Zhang, J.; Liang, C.; Wei, Z.; Yang, W.; Ge, W.; Qu, X.; Si, W.; Wang, W.; Mou, X.; Dong, X. TME-triggered MnSiO3@Met@GOx nanosystem for ATP dual-inhibited starvation/chemodynamic synergistic therapy. Biomaterials 2022, 287, 121682. [Google Scholar] [CrossRef]
- Champ, C.E.; Palmer, J.D.; Volek, J.S.; Werner-Wasik, M.; Andrews, D.W.; Evans, J.J.; Glass, J.; Kim, L.; Shi, W. Targeting metabolism with a ketogenic diet during the treatment of glioblastoma multiforme. J. Neurooncol. 2014, 117, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Sainero-Alcolado, L.; Liano-Pons, J.; Ruiz-Perez, M.V.; Arsenian-Henriksson, M. Targeting mitochondrial metabolism for precision medicine in cancer. Cell Death Differ. 2022, 29, 1304–1317. [Google Scholar] [CrossRef] [PubMed]
- Volland, J.M.; Kaupp, J.; Schmitz, W.; Wunsch, A.C.; Balint, J.; Mollmann, M.; El-Mesery, M.; Frackmann, K.; Peter, L.; Hartmann, S.; et al. Mass Spectrometric Metabolic Fingerprinting of 2-Deoxy-D-Glucose (2-DG)-Induced Inhibition of Glycolysis and Comparative Analysis of Methionine Restriction versus Glucose Restriction under Perfusion Culture in the Murine L929 Model System. Int. J. Mol. Sci. 2022, 23, 9220. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Ducker, G.S.; Rabinowitz, J.D. One-carbon metabolism in health and disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef]
- Angelin, A.; Gil-de-Gómez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J., III; Kopinski, P.K.; Wang, L. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab. 2017, 25, 1282–1293. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S. Inhibitory effect of tumor cell–derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Ivashkiv, L.B. The hypoxia–lactate axis tempers inflammation. Nat. Rev. Immunol. 2020, 20, 85–86. [Google Scholar] [CrossRef]
- Ippolito, L.; Morandi, A.; Giannoni, E.; Chiarugi, P. Lactate: A metabolic driver in the tumour landscape. Trends Biochem. Sci. 2019, 44, 153–166. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Ohshima, K.; Morii, E. Metabolic reprogramming of cancer cells during tumor progression and metastasis. Metabolites 2021, 11, 28. [Google Scholar] [CrossRef]
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 1–10. [Google Scholar] [CrossRef]
- Stein, E.M.; DiNardo, C.D.; Fathi, A.T.; Mims, A.S.; Pratz, K.W.; Savona, M.R.; Stein, A.S.; Stone, R.M.; Winer, E.S.; Seet, C.S.; et al. Ivosidenib or enasidenib combined with intensive chemotherapy in patients with newly diagnosed AML: A phase 1 study. Blood 2021, 137, 1792–1803. [Google Scholar] [CrossRef]
- Chang, S.M.; Vander Heiden, M.G. Inhibiting GLUTtony in cancer. Cell Chem. Biol. 2022, 29, 353–355. [Google Scholar] [CrossRef]
- Thorens, B.; Mueckler, M. Glucose transporters in the 21st Century. Am. J. Physiol.-Endocrinol. Metab. 2010, 298, E141–E145. [Google Scholar] [CrossRef]
- Mueckler, M.; Thorens, B. The SLC2 (GLUT) family of membrane transporters. Mol. Asp. Med. 2013, 34, 121–138. [Google Scholar] [CrossRef]
- Bao, Y.Y.; Zhong, J.T.; Shen, L.F.; Dai, L.B.; Zhou, S.H.; Fan, J.; Yao, H.T.; Lu, Z.J. Effect of Glut-1 and HIF-1alpha double knockout by CRISPR/CAS9 on radiosensitivity in laryngeal carcinoma via the PI3K/Akt/mTOR pathway. J. Cell Mol. Med. 2022, 26, 2881–2894. [Google Scholar] [CrossRef]
- Sharma, V.; Singh, T.G.; Mannan, A. Therapeutic implications of glucose transporters (GLUT) in cerebral ischemia. Neurochem Res. 2022, 47, 2173–2186. [Google Scholar] [CrossRef] [PubMed]
- Barron, C.C.; Bilan, P.J.; Tsakiridis, T.; Tsiani, E. Facilitative glucose transporters: Implications for cancer detection, prognosis and treatment. Metabolism 2016, 65, 124–139. [Google Scholar] [CrossRef] [PubMed]
- Bukkuri, A.; Gatenby, R.A.; Brown, J.S. GLUT1 production in cancer cells: A tragedy of the commons. NPJ Syst. Biol. Appl. 2022, 8, 22. [Google Scholar] [CrossRef]
- Guo, Z.; Cheng, Z.; Wang, J.; Liu, W.; Peng, H.; Wang, Y.; Rao, A.S.; Li, R.j.; Ying, X.; Korangath, P. Discovery of a potent GLUT inhibitor from a library of rapafucins by using 3D microarrays. Angew. Chem. 2019, 131, 17318–17322. [Google Scholar] [CrossRef]
- Granchi, C.; Tuccinardi, T.; Minutolo, F. Design, synthesis, and evaluation of GLUT inhibitors. Glucose Transp. 2018, 1713, 93–108. [Google Scholar]
- Siebeneicher, H.; Bauser, M.; Buchmann, B.; Heisler, I.; Mueller, T.; Neuhaus, R.; Rehwinkel, H.; Telser, J.; Zorn, L. Identification of novel GLUT inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 1732–1737. [Google Scholar] [CrossRef]
- Wei, C.; Bajpai, R.; Sharma, H.; Heitmeier, M.; Jain, A.D.; Matulis, S.M.; Nooka, A.K.; Mishra, R.K.; Hruz, P.W.; Schiltz, G.E. Development of GLUT4-selective antagonists for multiple myeloma therapy. Eur. J. Med. Chem. 2017, 139, 573–586. [Google Scholar] [CrossRef]
- Chen, W.-L.; Wang, Y.-Y.; Zhao, A.; Xia, L.; Xie, G.; Su, M.; Zhao, L.; Liu, J.; Qu, C.; Wei, R. Enhanced fructose utilization mediated by SLC2A5 is a unique metabolic feature of acute myeloid leukemia with therapeutic potential. Cancer Cell 2016, 30, 779–791. [Google Scholar] [CrossRef]
- Gonzalez, P.S.; O’Prey, J.; Cardaci, S.; Barthet, V.J.; Sakamaki, J.-i.; Beaumatin, F.; Roseweir, A.; Gay, D.M.; Mackay, G.; Malviya, G. Mannose impairs tumour growth and enhances chemotherapy. Nature 2018, 563, 719–723. [Google Scholar] [CrossRef]
- Robichaud, T.; Appleyard, A.N.; Herbert, R.B.; Henderson, P.J.; Carruthers, A. Determinants of ligand binding affinity and cooperativity at the GLUT1 endofacial site. Biochemistry 2011, 50, 3137–3148. [Google Scholar] [CrossRef]
- Chan, D.A.; Sutphin, P.D.; Nguyen, P.; Turcotte, S.; Lai, E.W.; Banh, A.; Reynolds, G.E.; Chi, J.-T.; Wu, J.; Solow-Cordero, D.E. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci. Transl. Med. 2011, 3, 94ra70. [Google Scholar] [CrossRef]
- Kraus, D.; Reckenbeil, J.; Veit, N.; Kuerpig, S.; Meisenheimer, M.; Beier, I.; Stark, H.; Winter, J.; Probstmeier, R. Targeting glucose transport and the NAD pathway in tumor cells with STF-31: A re-evaluation. Cell Oncol. 2018, 41, 485–494. [Google Scholar] [CrossRef]
- Pliszka, M.; Szablewski, L. Glucose Transporters as a Target for Anticancer Therapy. Cancers 2021, 13, 4184. [Google Scholar] [CrossRef]
- Zhao, Y.; Butler, E.B.; Tan, M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013, 4, e532. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, Y.; Zhang, W.; Bergmeier, S.; Qian, Y.; Akbar, H.; Colvin, R.; Ding, J.; Tong, L.; Wu, S. A Small-Molecule Inhibitor of Glucose Transporter 1 Downregulates Glycolysis, Induces Cell-Cycle Arrest, and Inhibits Cancer Cell Growth In Vitro and In VivoA Glut1 Inhibitor Reduces Cancer Growth In Vitro and In Vivo. Mol. Cancer Ther. 2012, 11, 1672–1682. [Google Scholar] [CrossRef]
- Ojelabi, O.A.; Lloyd, K.P.; Simon, A.H.; De Zutter, J.K.; Carruthers, A. WZB117 (2-Fluoro-6-(m-hydroxybenzoyloxy) Phenyl m-Hydroxybenzoate) inhibits GLUT1-mediated sugar transport by binding reversibly at the exofacial sugar binding site. J. Biol. Chem. 2016, 291, 26762–26772. [Google Scholar] [CrossRef]
- Yakisich, J.S.; Azad, N.; Kaushik, V.; Iyer, A.K.V. The Biguanides Metformin and Buformin in Combination with 2-Deoxy-glucose or WZB-117 Inhibit the Viability of Highly Resistant Human Lung Cancer Cells. Stem Cells Int. 2019, 2019, 6254269. [Google Scholar] [CrossRef]
- Siebeneicher, H.; Cleve, A.; Rehwinkel, H.; Neuhaus, R.; Heisler, I.; Müller, T.; Bauser, M.; Buchmann, B. Identification and optimization of the first highly selective GLUT1 inhibitor BAY-876. ChemMedChem 2016, 11, 2261–2271. [Google Scholar] [CrossRef]
- Wu, Q.; Deblois, G.; Cruickshank, J.; Duan, S.; Lima-Fernandes, E.; Haight, J.; Tonekaboni, S.A.M.; Fortier, A.-M.; Kuasne, H.; McKee, T.D. GLUT1 inhibition blocks growth of RB1-positive triple negative breast cancer. Nat. Commun. 2020, 11, 4205. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, W.; Idowu, M.O.; Oh, U.; Wang, X.-Y.; Temkin, S.M.; Fang, X. Ovarian cancer relies on glucose transporter 1 to fuel glycolysis and growth: Anti-tumor activity of BAY-876. Cancers 2018, 11, 33. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Qu, G.; Choi, S.-C.; Cornaby, C.; Titov, A.; Kanda, N.; Teng, X.; Wang, H.; Morel, L. Targeting T cell activation and lupus autoimmune phenotypes by inhibiting glucose transporters. Front. Immunol. 2019, 10, 833. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.-H.; Ho, C.-T.; Chen, Z.-F.; Chen, L.-C.; Whang-Peng, J.; Lin, T.-N.; Ho, Y.-S. The apple polyphenol phloretin inhibits breast cancer cell migration and proliferation via inhibition of signals by type 2 glucose transporter. J. Food Drug Anal. 2018, 26, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Ung, P.M.-U.; Song, W.; Cheng, L.; Zhao, X.; Hu, H.; Chen, L.; Schlessinger, A. Inhibitor discovery for the human GLUT1 from homology modeling and virtual screening. ACS Chem. Biol. 2016, 11, 1908–1916. [Google Scholar] [CrossRef] [PubMed]
- Karageorgis, G.; Reckzeh, E.S.; Ceballos, J.; Schwalfenberg, M.; Sievers, S.; Ostermann, C.; Pahl, A.; Ziegler, S.; Waldmann, H. Chromopynones are pseudo natural product glucose uptake inhibitors targeting glucose transporters GLUT-1 and -3. Nat. Chem. 2018, 10, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, K.; Mandal, S.; Roy, A. Recent advancements in therapeutic targeting of the Warburg effect in refractory ovarian cancer: A promise towards disease remission. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188563. [Google Scholar] [CrossRef] [PubMed]
- Karageorgis, G.; Foley, D.J.; Laraia, L.; Waldmann, H. Principle and design of pseudo-natural products. Nat. Chem. 2020, 12, 227–235. [Google Scholar] [CrossRef]
- Casiraghi, A.; Bensimon, A.; Superti-Furga, G. Recent developments in ligands and chemical probes targeting solute carrier transporters. Curr. Opin. Chem. Biol. 2021, 62, 53–63. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, Y.; Dong, L.; Huang, Y.; Yuan, J.; Ben, W.; Yang, Y.; Ning, N.; Lu, M.; Guan, Y. Therapeutic role of EF 24 targeting glucose transporter 1-mediated metabolism and metastasis in ovarian cancer cells. Cancer Sci. 2013, 104, 1690–1696. [Google Scholar] [CrossRef]
- George Thompson, A.M.; Iancu, C.V.; Nguyen, T.T.H.; Kim, D.; Choe, J.-y. Inhibition of human GLUT1 and GLUT5 by plant carbohydrate products; insights into transport specificity. Sci. Rep. 2015, 5, 12804. [Google Scholar] [CrossRef]
- Tilekar, K.; Upadhyay, N.; Iancu, C.V.; Pokrovsky, V.; Choe, J.-y.; Ramaa, C. Power of two: Combination of therapeutic approaches involving glucose transporter (GLUT) inhibitors to combat cancer. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2020, 1874, 188457. [Google Scholar] [CrossRef]
- Kapoor, K.; Finer-Moore, J.S.; Pedersen, B.P.; Caboni, L.; Waight, A.; Hillig, R.C.; Bringmann, P.; Heisler, I.; Müller, T.; Siebeneicher, H. Mechanism of inhibition of human glucose transporter GLUT1 is conserved between cytochalasin B and phenylalanine amides. Proc. Natl. Acad. Sci. USA 2016, 113, 4711–4716. [Google Scholar] [CrossRef]
- Almahmoud, S.; Jin, W.; Geng, L.; Wang, J.; Wang, X.; Vennerstrom, J.L.; Zhong, H.A. Ligand-based design of GLUT inhibitors as potential antitumor agents. Bioorg. Med. Chem. 2020, 28, 115395. [Google Scholar] [CrossRef]
- Tilekar, K.; Upadhyay, N.; Schweipert, M.; Hess, J.D.; Macias, L.H.; Mrowka, P.; Meyer-Almes, F.-J.; Aguilera, R.J.; Iancu, C.V.; Choe, J.-y. Permuted 2, 4-thiazolidinedione (TZD) analogs as GLUT inhibitors and their in-vitro evaluation in leukemic cells. Eur. J. Pharm. Sci. 2020, 154, 105512. [Google Scholar] [CrossRef]
- Meng, Y.; Xu, X.; Luan, H.; Li, L.; Dai, W.; Li, Z.; Bian, J. The progress and development of GLUT1 inhibitors targeting cancer energy metabolism. Future Med. Chem. 2019, 11, 2333–2352. [Google Scholar] [CrossRef]
- Tilekar, K.; Upadhyay, N.; Hess, J.D.; Macias, L.H.; Mrowka, P.; Aguilera, R.J.; Meyer-Almes, F.-J.; Iancu, C.V.; Choe, J.-y.; Ramaa, C. Structure guided design and synthesis of furyl thiazolidinedione derivatives as inhibitors of GLUT 1 and GLUT 4, and evaluation of their anti-leukemic potential. Eur. J. Med. Chem. 2020, 202, 112603. [Google Scholar] [CrossRef]
- Ceballos, J.; Schwalfenberg, M.; Karageorgis, G.; Reckzeh, E.S.; Sievers, S.; Ostermann, C.; Pahl, A.; Sellstedt, M.; Nowacki, J.; Carnero Corrales, M.A. Synthesis of Indomorphan Pseudo-Natural Product Inhibitors of Glucose Transporters GLUT-1 and -3. Angew. Chem. 2019, 131, 17172–17181. [Google Scholar] [CrossRef]
- Lin, S.-T.; Tu, S.-H.; Yang, P.-S.; Hsu, S.-P.; Lee, W.-H.; Ho, C.-T.; Wu, C.-H.; Lai, Y.-H.; Chen, M.-Y.; Chen, L.-C. Apple polyphenol phloretin inhibits colorectal cancer cell growth via inhibition of the type 2 glucose transporter and activation of p53-mediated signaling. J. Agric. Food Chem. 2016, 64, 6826–6837. [Google Scholar] [CrossRef]
- Ji, Z.; Huo, C.; Yang, P. Genistein inhibited the proliferation of kidney cancer cells via CDKN2a hypomethylation: Role of abnormal apoptosis. Int. Urol. Nephrol. 2020, 52, 1049–1055. [Google Scholar] [CrossRef]
- Hirata, H.; Ueno, K.; Nakajima, K.; Tabatabai, Z.; Hinoda, Y.; Ishii, N.; Dahiya, R. Genistein downregulates onco-miR-1260b and inhibits Wnt-signalling in renal cancer cells. Br. J. Cancer 2013, 108, 2070–2078. [Google Scholar] [CrossRef]
- Wood, T.E.; Dalili, S.; Simpson, C.D.; Hurren, R.; Mao, X.; Saiz, F.S.; Gronda, M.; Eberhard, Y.; Minden, M.D.; Bilan, P.J. A novel inhibitor of glucose uptake sensitizes cells to FAS-induced cell death. Mol. Cancer Ther. 2008, 7, 3546–3555. [Google Scholar] [CrossRef]
- Ocana, M.C.; Martinez-Poveda, B.; Mari-Beffa, M.; Quesada, A.R.; Medina, M.A. Fasentin diminishes endothelial cell proliferation, differentiation and invasion in a glucose metabolism-independent manner. Sci. Rep. 2020, 10, 6132. [Google Scholar] [CrossRef]
- Li, B.; Jiang, J.; Assaraf, Y.G.; Xiao, H.; Chen, Z.S.; Huang, C. Surmounting cancer drug resistance: New insights from the perspective of N(6)-methyladenosine RNA modification. Drug. Resist. Updates 2020, 53, 100720. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Melstrom, L.G.; Salabat, M.R.; Ding, X.-Z.; Strouch, M.J.; Grippo, P.J.; Mirzoeva, S.; Pelling, J.C.; Bentrem, D.J. Apigenin down-regulates the hypoxia response genes: HIF-1α, GLUT-1, and VEGF in human pancreatic cancer cells. J. Surg. Res. 2011, 167, 173–181. [Google Scholar] [CrossRef]
- Melstrom, L.G.; Salabat, M.R.; Ding, X.-Z.; Milam, B.M.; Strouch, M.; Pelling, J.C.; Bentrem, D.J. Apigenin inhibits the GLUT-1 glucose transporter and the phosphoinositide 3-kinase/Akt pathway in human pancreatic cancer cells. Pancreas 2008, 37, 426–431. [Google Scholar] [CrossRef]
- Fang, J.; Bao, Y.Y.; Zhou, S.H.; Fan, J. Apigenin inhibits the proliferation of adenoid cystic carcinoma via suppression of glucose transporter-1. Mol. Med. Rep. 2015, 12, 6461–6466. [Google Scholar] [CrossRef] [PubMed]
- Allavena, G.; Del Bello, B.; Tini, P.; Volpi, N.; Valacchi, G.; Miracco, C.; Pirtoli, L.; Maellaro, E. Trehalose inhibits cell proliferation and amplifies long-term temozolomide- and radiation-induced cytotoxicity in melanoma cells: A role for autophagy and premature senescence. J. Cell. Physiol. 2019, 234, 11708–11721. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Digel, M.; Küch, E.M.; Stremmel, W.; Füllekrug, J. Silybin and dehydrosilybin decrease glucose uptake by inhibiting GLUT proteins. J. Cell. Biochem. 2011, 112, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Gunnink, L.K.; Alabi, O.D.; Kuiper, B.D.; Gunnink, S.M.; Schuiteman, S.J.; Strohbehn, L.E.; Hamilton, K.E.; Wrobel, K.E.; Louters, L.L. Curcumin directly inhibits the transport activity of GLUT1. Biochimie 2016, 125, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Zambrano, A.; Molt, M.; Uribe, E.; Salas, M. Glut 1 in cancer cells and the inhibitory action of resveratrol as a potential therapeutic strategy. Int. J. Mol. Sci. 2019, 20, 3374. [Google Scholar] [CrossRef]
- Park, J.B. Flavonoids are potential inhibitors of glucose uptake in U937 cells. Biochem. Biophys. Res. Commun. 1999, 260, 568–574. [Google Scholar] [CrossRef]
- Hamilton, K.E.; Rekman, J.F.; Gunnink, L.K.; Busscher, B.M.; Scott, J.L.; Tidball, A.M.; Stehouwer, N.R.; Johnecheck, G.N.; Looyenga, B.D.; Louters, L.L. Quercetin inhibits glucose transport by binding to an exofacial site on GLUT1. Biochimie 2018, 151, 107–114. [Google Scholar] [CrossRef]
- Kwon, O.; Eck, P.; Chen, S.; Corpe, C.P.; Lee, J.H.; Kruhlak, M.; Levine, M. Inhibition of the intestinal glucose transporter GLUT2 by flavonoids. FASEB J. 2007, 21, 366–377. [Google Scholar] [CrossRef]
- Azevedo, C.; Correia-Branco, A.; Araújo, J.R.; Guimaraes, J.T.; Keating, E.; Martel, F. The chemopreventive effect of the dietary compound kaempferol on the MCF-7 human breast cancer cell line is dependent on inhibition of glucose cellular uptake. Nutr. Cancer 2015, 67, 504–513. [Google Scholar] [CrossRef]
- Correia-Branco, A.; Azevedo, C.F.; Araujo, J.R.; Guimaraes, J.T.; Faria, A.; Keating, E.; Martel, F. Xanthohumol impairs glucose uptake by a human first-trimester extravillous trophoblast cell line (HTR-8/SVneo cells) and impacts the process of placentation. Mhr Basic Sci. Reprod. Med. 2015, 21, 803–815. [Google Scholar] [CrossRef]
- Sage, J.M.; Cura, A.J.; Lloyd, K.P.; Carruthers, A. Caffeine inhibits glucose transport by binding at the GLUT1 nucleotide-binding site. Am. J. Physiol.-Cell Physiol. 2015, 308, C827–C834. [Google Scholar] [CrossRef]
- Hyun, D.H. Insights into the New Cancer Therapy through Redox Homeostasis and Metabolic Shifts. Cancers 2020, 12, 1822. [Google Scholar] [CrossRef]
- Ojeda, P.; Pérez, A.; Ojeda, L.; Vargas-Uribe, M.; Rivas, C.I.; Salas, M.; Vera, J.C.; Reyes, A.M. Noncompetitive blocking of human GLUT1 hexose transporter by methylxanthines reveals an exofacial regulatory binding site. Am. J. Physiol.-Cell Physiol. 2012, 303, C530–C539. [Google Scholar] [CrossRef]
- Ren, Y.; Yuan, C.; Qian, Y.; Chai, H.-B.; Chen, X.; Goetz, M.; Kinghorn, A.D. Constituents of an extract of Cryptocarya rubra housed in a repository with cytotoxic and glucose transport inhibitory effects. J. Nat. Prod. 2014, 77, 550–556. [Google Scholar] [CrossRef]
- Hevia, D.; González-Menéndez, P.; Quiros-González, I.; Miar, A.; Rodríguez-García, A.; Tan, D.X.; Reiter, R.J.; Mayo, J.C.; Sainz, R.M. Melatonin uptake through glucose transporters: A new target for melatonin inhibition of cancer. J. Pineal Res. 2015, 58, 234–250. [Google Scholar] [CrossRef]
- Shahruzaman, S.H.; Mustafa, M.F.; Ramli, S.; Maniam, S.; Fakurazi, S.; Maniam, S. The Cytotoxic Properties of Baeckea frutescens Branches Extracts in Eliminating Breast Cancer Cells. Evid. Based Complement Altern. Med. 2019, 2019, 9607590. [Google Scholar] [CrossRef]
- Cheng, X.; Yan, H.; Pang, S.; Ya, M.; Qiu, F.; Qin, P.; Zeng, C.; Lu, Y. Liposomes as Multifunctional Nano-Carriers for Medicinal Natural Products. Front. Chem. 2022, 10, 963004. [Google Scholar] [CrossRef] [PubMed]
- Feitosa, R.C.; Ishikawa, E.S.A.; Silva, M.; Silva-Junior, A.A.D.; Oliveira-Nascimento, L. Five decades of doxycycline: Does nanotechnology improve its properties? Int. J. Pharm. 2022, 618, 121655. [Google Scholar] [CrossRef]
- Seker Karatoprak, G.; Kupeli Akkol, E.; Yucel, C.; Bahadir Acikara, O.; Sobarzo-Sanchez, E. Advances in Understanding the Role of Aloe Emodin and Targeted Drug Delivery Systems in Cancer. Oxid Med. Cell Longev. 2022, 2022, 7928200. [Google Scholar] [CrossRef]
- Choi, J.; Mathew, S.; Oerter, S.; Appelt-Menzel, A.; Hansmann, J.; Schmitz, T. Online Measurement System for Dynamic Flow Bioreactors to Study Barrier Integrity of hiPSC-Based Blood-Brain Barrier In Vitro Models. Bioengineering 2022, 9, 39. [Google Scholar] [CrossRef]
- Figueiras, A.; Domingues, C.; Jarak, I.; Santos, A.I.; Parra, A.; Pais, A.; Alvarez-Lorenzo, C.; Concheiro, A.; Kabanov, A.; Cabral, H.; et al. New Advances in Biomedical Application of Polymeric Micelles. Pharmaceutics 2022, 14, 1700. [Google Scholar] [CrossRef]
- Jannat, T.; Hossain, M.J.; El-Shehawi, A.M.; Kuddus, M.R.; Rashid, M.A.; Albogami, S.; Jafri, I.; El-Shazly, M.; Haque, M.R. Chemical and Pharmacological Profiling of Wrightia coccinea (Roxb. Ex Hornem.) Sims Focusing Antioxidant, Cytotoxic, Antidiarrheal, Hypoglycemic, and Analgesic Properties. Molecules 2022, 27, 4024. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.E. Isozymes of mammalian hexokinase: Structure, subcellular localization and metabolic function. J. Exp. Biol. 2003, 206, 2049–2057. [Google Scholar] [CrossRef]
- Wolf, A.; Agnihotri, S.; Micallef, J.; Mukherjee, J.; Sabha, N.; Cairns, R.; Hawkins, C.; Guha, A. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J. Exp. Med. 2011, 208, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Mathupala, S.P.; Rempel, A.; Pedersen, P.L. Glucose catabolism in cancer cells: Identification and characterization of a marked activation response of the type II hexokinase gene to hypoxic conditions. J. Biol. Chem. 2001, 276, 43407–43412. [Google Scholar] [CrossRef]
- Mathupala, S.P.; Heese, C.; Pedersen, P.L. Glucose catabolism in cancer cells. The type II hexokinase promoter contains functionally active response elements for the tumor suppressor p53. J. Biol. Chem. 1997, 272, 22776–22780. [Google Scholar] [CrossRef]
- Hu, J.; Cao, J.; Topatana, W.; Juengpanich, S.; Li, S.; Zhang, B.; Shen, J.; Cai, L.; Cai, X.; Chen, M. Targeting mutant p53 for cancer therapy: Direct and indirect strategies. J. Hematol. Oncol. 2021, 14, 157. [Google Scholar] [CrossRef]
- Liu, S.; Yan, B.; Lai, W.; Chen, L.; Xiao, D.; Xi, S.; Jiang, Y.; Dong, X.; An, J.; Chen, X.; et al. As a novel p53 direct target, bidirectional gene HspB2/alphaB-crystallin regulates the ROS level and Warburg effect. Biochim. Biophys. Acta 2014, 1839, 592–603. [Google Scholar] [CrossRef]
- Kaghad, M.; Bonnet, H.; Yang, A.; Creancier, L.; Biscan, J.C.; Valent, A.; Minty, A.; Chalon, P.; Lelias, J.M.; Dumont, X.; et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 1997, 90, 809–819. [Google Scholar] [CrossRef]
- Yang, A.; Kaghad, M.; Wang, Y.; Gillett, E.; Fleming, M.D.; Dotsch, V.; Andrews, N.C.; Caput, D.; McKeon, F. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol. Cell 1998, 2, 305–316. [Google Scholar] [CrossRef]
- Venkatanarayan, A.; Raulji, P.; Norton, W.; Chakravarti, D.; Coarfa, C.; Su, X.; Sandur, S.K.; Ramirez, M.S.; Lee, J.; Kingsley, C.V.; et al. IAPP-driven metabolic reprogramming induces regression of p53-deficient tumours in vivo. Nature 2015, 517, 626–630. [Google Scholar] [CrossRef]
- Viticchie, G.; Agostini, M.; Lena, A.M.; Mancini, M.; Zhou, H.; Zolla, L.; Dinsdale, D.; Saintigny, G.; Melino, G.; Candi, E. p63 supports aerobic respiration through hexokinase II. Proc. Natl. Acad. Sci. USA 2015, 112, 11577–11582. [Google Scholar] [CrossRef]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 2013, 24, 213–228. [Google Scholar] [CrossRef]
- DeWaal, D.; Nogueira, V.; Terry, A.R.; Patra, K.C.; Jeon, S.-M.; Guzman, G.; Au, J.; Long, C.P.; Antoniewicz, M.R.; Hay, N. Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat. Commun. 2018, 9, 446. [Google Scholar] [CrossRef]
- Neary, C.L.; Pastorino, J.G. Akt inhibition promotes hexokinase 2 redistribution and glucose uptake in cancer cells. J. Cell. Physiol. 2013, 228, 1943–1948. [Google Scholar] [CrossRef]
- Garcia, S.N.; Guedes, R.C.; Marques, M.M. Unlocking the potential of HK2 in cancer metabolism and therapeutics. Curr. Med. Chem. 2019, 26, 7285–7322. [Google Scholar] [CrossRef]
- Nawaz, M.H.; Ferreira, J.C.; Nedyalkova, L.; Zhu, H.; Carrasco-Lopez, C.; Kirmizialtin, S.; Rabeh, W.M. The catalytic inactivation of the N-half of human hexokinase 2 and structural and biochemical characterization of its mitochondrial conformation. Biosci. Rep. 2018, 38, BSR20171666. [Google Scholar] [CrossRef]
- Shan, W.; Zhou, Y.; Tam, K.Y. The development of small-molecule inhibitors targeting hexokinase 2. Drug. Discov. Today 2022, 27, 2574–2585. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, Q.; Peng, S.; Liu, X. The combination of the glycolysis inhibitor 2-DG and sorafenib can be effective against sorafenib-tolerant persister cancer cells. OncoTargets Ther. 2019, 12, 5359. [Google Scholar] [CrossRef] [PubMed]
- Muley, P.; Olinger, A.; Tummala, H. 2-Deoxyglucose induces cell cycle arrest and apoptosisin colorectal cancer cells independent of its glycolysis inhibition. Nutr. Cancer 2015, 67, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Galluzzi, L.; Kroemer, G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M.C.; Asbury, C.R.; Daniels, D.; Du, J.; Aykin-Burns, N.; Smith, B.J.; Li, L.; Spitz, D.R.; Cullen, J.J. 2-deoxy-D-glucose causes cytotoxicity, oxidative stress, and radiosensitization in pancreatic cancer. Free. Radic. Biol. Med. 2008, 44, 322–331. [Google Scholar] [CrossRef]
- Simons, A.L.; Ahmad, I.M.; Mattson, D.M.; Dornfeld, K.J.; Spitz, D.R. 2-Deoxy-D-glucose combined with cisplatin enhances cytotoxicity via metabolic oxidative stress in human head and neck cancer cells. Cancer Res. 2007, 67, 3364–3370. [Google Scholar] [CrossRef]
- Maschek, G.; Savaraj, N.; Priebe, W.; Braunschweiger, P.; Hamilton, K.; Tidmarsh, G.F.; De Young, L.R.; Lampidis, T.J. 2-deoxy-D-glucose increases the efficacy of adriamycin and paclitaxel in human osteosarcoma and non-small cell lung cancers in vivo. Cancer Res. 2004, 64, 31–34. [Google Scholar] [CrossRef]
- Pajak, B.; Siwiak, E.; Sołtyka, M.; Priebe, A.; Zieliński, R.; Fokt, I.; Ziemniak, M.; Jaśkiewicz, A.; Borowski, R.; Domoradzki, T. 2-Deoxy-d-glucose and its analogs: From diagnostic to therapeutic agents. Int. J. Mol. Sci. 2019, 21, 234. [Google Scholar] [CrossRef]
- Li, L.; Fath, M.A.; Scarbrough, P.M.; Watson, W.H.; Spitz, D.R. Combined inhibition of glycolysis, the pentose cycle, and thioredoxin metabolism selectively increases cytotoxicity and oxidative stress in human breast and prostate cancer. Redox Biol. 2015, 4, 127–135. [Google Scholar] [CrossRef]
- Raez, L.E.; Papadopoulos, K.; Ricart, A.D.; Chiorean, E.G.; DiPaola, R.S.; Stein, M.N.; Rocha Lima, C.M.; Schlesselman, J.J.; Tolba, K.; Langmuir, V.K. A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2013, 71, 523–530. [Google Scholar] [CrossRef]
- Belzacq, A.-S.; El Hamel, C.; Vieira, H.L.; Cohen, I.; Haouzi, D.; Métivier, D.; Marchetti, P.; Brenner, C.; Kroemer, G. Adenine nucleotide translocator mediates the mitochondrial membrane permeabilization induced by lonidamine, arsenite and CD437. Oncogene 2001, 20, 7579–7587. [Google Scholar] [CrossRef]
- Nath, K.; Guo, L.; Nancolas, B.; Nelson, D.S.; Shestov, A.A.; Lee, S.-C.; Roman, J.; Zhou, R.; Leeper, D.B.; Halestrap, A.P. Mechanism of antineoplastic activity of lonidamine. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2016, 1866, 151–162. [Google Scholar] [CrossRef]
- Di Cosimo, S.; Ferretti, G.; Papaldo, P.; Carlini, P.; Fabi, A.; Cognetti, F. Lonidamine: Efficacy and safety in clinical trials for the treatment of solid tumors. Drugs Today 2003, 39, 157–174. [Google Scholar] [CrossRef]
- Gadducci, A.; Brunetti, I.; Muttini, M.; Fanucchi, A.; Dargenio, F.; Giannessi, P.; Conte, P. Epidoxorubicin and lonidamine in refractory or recurrent epithelial ovarian cancer. Eur. J. Cancer 1994, 30, 1432–1435. [Google Scholar] [CrossRef]
- De Lena, M.; Lorusso, V.; Bottalico, C.; Brandi, M.; De Mitrio, A.; Catino, A.; Guida, M.; Latorre, A.; Leone, B.; Vallejo, C. Revertant and potentiating activity of lonidamine in patients with ovarian cancer previously treated with platinum. J. Clin. Oncol. 1997, 15, 3208–3213. [Google Scholar] [CrossRef]
- De Marinis, F.; Rinaldi, M.; Ardizzoni, A.; Bruzzi, P.; Pennucci, M.C.; Portalone, L.; D’Aprile, M.; Ripanti, P.; Romano, F.; Belli, M. The role of vindesine and lonidamine in the treatment of elderly patients with advanced non-small cell lung cancer: A phase III randomized fonicap trial. Tumori J. 1999, 85, 177–182. [Google Scholar] [CrossRef]
- Berruti, A.; Bitossi, R.; Gorzegno, G.; Bottini, A.; Alquati, P.; De Matteis, A.; Nuzzo, F.; Giardina, G.; Danese, S.; De Lena, M. Time to progression in metastatic breast cancer patients treated with epirubicin is not improved by the addition of either cisplatin or lonidamine: Final results of a phase III study with a factorial design. J. Clin. Oncol. 2002, 20, 4150–4159. [Google Scholar] [CrossRef]
- Price, G.S.; Page, R.L.; Riviere, J.E.; Cline, J.M.; Thrall, D. Pharmacokinetics and toxicity of oral and intravenous lonidamine in dogs. Cancer Chemother. Pharmacol. 1996, 38, 129–135. [Google Scholar] [CrossRef]
- Nath, K.; Nelson, D.S.; Heitjan, D.F.; Leeper, D.B.; Zhou, R.; Glickson, J.D. Lonidamine induces intracellular tumor acidification and ATP depletion in breast, prostate and ovarian cancer xenografts and potentiates response to doxorubicin. NMR Biomed. 2015, 28, 281–290. [Google Scholar] [CrossRef]
- Amadori, D.; Frassineti, G.L.; De Matteis, A.; Mustacchi, G.; Santoro, A.; Cariello, S.; Ferrari, M.; Nascimben, O.; Nanni, O.; Lombardi, A. Modulating effect of lonidamine on response to doxorubicin in metastatic breast cancer patients: Results from a multicenter prospective randomized trial. Breast Cancer Res. Treat. 1998, 49, 209–217. [Google Scholar] [CrossRef]
- Liu, X.; Li, Y.; Wang, K.; Chen, Y.; Shi, M.; Zhang, X.; Pan, W.; Li, N.; Tang, B. GSH-responsive nanoprodrug to inhibit glycolysis and alleviate immunosuppression for cancer therapy. Nano Lett. 2021, 21, 7862–7869. [Google Scholar] [CrossRef]
- Geschwind, J.-F.; Georgiades, C.S.; Ko, Y.H.; Pedersen, P.L. Recently elucidated energy catabolism pathways provide opportunities for novel treatments in hepatocellular carcinoma. Expert Rev. Anticancer Ther. 2004, 4, 449–457. [Google Scholar] [CrossRef]
- Ihrlund, L.S.; Hernlund, E.; Khan, O.; Shoshan, M.C. 3-Bromopyruvate as inhibitor of tumour cell energy metabolism and chemopotentiator of platinum drugs. Mol. Oncol. 2008, 2, 94–101. [Google Scholar] [CrossRef]
- Cao, X.; Jia, G.; Zhang, T.; Yang, M.; Wang, B.; Wassenaar, P.A.; Cheng, H.; Knopp, M.V.; Sun, D. Non-invasive MRI tumor imaging and synergistic anticancer effect of HSP90 inhibitor and glycolysis inhibitor in RIP1-Tag2 transgenic pancreatic tumor model. Cancer Chemother. Pharmacol. 2008, 62, 985–994. [Google Scholar] [CrossRef]
- Guo, Y.; Wei, L.; Zhou, Y.; Lu, N.; Tang, X.; Li, Z.; Wang, X. Flavonoid GL-V9 induces apoptosis and inhibits glycolysis of breast cancer via disrupting GSK-3β-modulated mitochondrial binding of HKII. Free Radic. Biol. Med. 2020, 146, 119–129. [Google Scholar] [CrossRef]
- Li, S.; Li, J.; Dai, W.; Zhang, Q.; Feng, J.; Wu, L.; Liu, T.; Yu, Q.; Xu, S.; Wang, W. Genistein suppresses aerobic glycolysis and induces hepatocellular carcinoma cell death. Br. J. Cancer 2017, 117, 1518–1528. [Google Scholar] [CrossRef]
- Tao, L.; Wei, L.; Liu, Y.; Ding, Y.; Liu, X.; Zhang, X.; Wang, X.; Yao, Y.; Lu, J.; Wang, Q. Gen-27, a newly synthesized flavonoid, inhibits glycolysis and induces cell apoptosis via suppression of hexokinase II in human breast cancer cells. Biochem. Pharmacol. 2017, 125, 12–25. [Google Scholar] [CrossRef]
- Wei, L.; Zhou, Y.; Dai, Q.; Qiao, C.; Zhao, L.; Hui, H.; Lu, N.; Guo, Q. Oroxylin A induces dissociation of hexokinase II from the mitochondria and inhibits glycolysis by SIRT3-mediated deacetylation of cyclophilin D in breast carcinoma. Cell Death Dis. 2013, 4, e601. [Google Scholar] [CrossRef] [PubMed]
- Goldin, N.; Arzoine, L.; Heyfets, A.; Israelson, A.; Zaslavsky, Z.; Bravman, T.; Bronner, V.; Notcovich, A.; Shoshan-Barmatz, V.; Flescher, E. Methyl jasmonate binds to and detaches mitochondria-bound hexokinase. Oncogene 2008, 27, 4636–4643. [Google Scholar] [CrossRef]
- Klippel, S.; Jakubikova, J.; Delmore, J.; Ooi, M.; McMillin, D.; Kastritis, E.; Laubach, J.; Richardson, P.G.; Anderson, K.C.; Mitsiades, C.S. Methyljasmonate displays in vitro and in vivo activity against multiple myeloma cells. Br. J. Haematol. 2012, 159, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Arbini, A.A.; Moro, L. Mitochondria and cancer chemoresistance. Biochim. Biophys. Acta (BBA)-Bioenerg. 2017, 1858, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zheng, M.; Wu, S.; Gao, S.; Yang, M.; Li, Z.; Min, Q.; Sun, W.; Chen, L.; Xiang, G. Benserazide, a dopadecarboxylase inhibitor, suppresses tumor growth by targeting hexokinase 2. J. Exp. Clin. Cancer Res. 2017, 36, 58. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Wu, C.; Yang, K.; Yang, Y.; Liu, Y.; Gao, S.; Wang, Q.; Li, C.; Chen, L.; Li, H. Novel selective hexokinase 2 inhibitor Benitrobenrazide blocks cancer cells growth by targeting glycolysis. Pharmacol. Res. 2021, 164, 105367. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-N.; Yang, L.; Ling, J.-Y.; Czajkowsky, D.M.; Wang, J.-F.; Zhang, X.-W.; Zhou, Y.-M.; Ge, F.; Yang, M.-K.; Xiong, Q. Systematic identification of arsenic-binding proteins reveals that hexokinase-2 is inhibited by arsenic. Proc. Natl. Acad. Sci. USA 2015, 112, 15084–15089. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, R.A.; Garcia-Smith, R.; Dorsey, J.; Griffith, J.K.; Bisoffi, M.; Trujillo, K.A. Tumor necrosis factor alpha induces Warburg-like metabolism and is reversed by anti-inflammatory curcumin in breast epithelial cells. Int. J. Cancer 2013, 133, 2504–2510. [Google Scholar] [CrossRef]
- Gao, F.; Li, M.; Liu, W.-B.; Zhou, Z.-S.; Zhang, R.; Li, J.-L.; Zhou, K.-C. Epigallocatechin gallate inhibits human tongue carcinoma cells via HK2-mediated glycolysis. Oncol. Rep. 2015, 33, 1533–1539. [Google Scholar] [CrossRef]
- Dai, W.; Wang, F.; Lu, J.; Xia, Y.; He, L.; Chen, K.; Li, J.; Li, S.; Liu, T.; Zheng, Y. By reducing hexokinase 2, resveratrol induces apoptosis in HCC cells addicted to aerobic glycolysis and inhibits tumor growth in mice. Oncotarget 2015, 6, 13703. [Google Scholar] [CrossRef]
- Li, H.; Hu, S.; Pang, Y.; Li, M.; Chen, L.; Liu, F.; Liu, M.; Wang, Z.; Cheng, X. Bufalin inhibits glycolysis-induced cell growth and proliferation through the suppression of Integrin β2/FAK signaling pathway in ovarian cancer. Am. J. Cancer Res. 2018, 8, 1288. [Google Scholar]
- Yang, Y.; Cao, Y.; Chen, L.; Liu, F.; Qi, Z.; Cheng, X.; Wang, Z. Cryptotanshinone suppresses cell proliferation and glucose metabolism via STAT3/SIRT3 signaling pathway in ovarian cancer cells. Cancer Med. 2018, 7, 4610–4618. [Google Scholar] [CrossRef]
- Li, W.; Ma, X.; Li, N.; Liu, H.; Dong, Q.; Zhang, J.; Yang, C.; Liu, Y.; Liang, Q.; Zhang, S. Resveratrol inhibits Hexokinases II mediated glycolysis in non-small cell lung cancer via targeting Akt signaling pathway. Exp. Cell Res. 2016, 349, 320–327. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, Q.; Zheng, S.; Liu, T.; Yang, L.; Han, X.; Lu, X. Shikonin inhibits tumor growth of ESCC by suppressing PKM2 mediated aerobic glycolysis and STAT3 phosphorylation. J. Cancer 2021, 12, 4830. [Google Scholar] [CrossRef]
- Huang, Y.P.; Chang, N.W. Proteomic analysis of oral cancer reveals new potential therapeutic targets involved in the Warburg effect. Clin. Exp. Pharmacol. Physiol. 2017, 44, 880–887. [Google Scholar] [CrossRef]
- Chen, G.-Q.; Tang, C.-F.; Shi, X.-K.; Lin, C.-Y.; Fatima, S.; Pan, X.-H.; Yang, D.-J.; Zhang, G.; Lu, A.-P.; Lin, S.-H. Halofuginone inhibits colorectal cancer growth through suppression of Akt/mTORC1 signaling and glucose metabolism. Oncotarget 2015, 6, 24148. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, X.; Wang, Y.; Sun, Q.; Chen, M.; Liu, S.; Zou, X. Licochalcone A suppresses hexokinase 2-mediated tumor glycolysis in gastric cancer via downregulation of the Akt signaling pathway. Oncol. Rep. 2018, 39, 1181–1190. [Google Scholar] [CrossRef]
- Gao, X.; Han, H. Jolkinolide B inhibits glycolysis by downregulating hexokinase 2 expression through inactivating the Akt/mTOR pathway in non-small cell lung cancer cells. J. Cell. Biochem. 2018, 119, 4967–4974. [Google Scholar] [CrossRef]
- Zhou, Y.; Zheng, X.; Lu, J.; Chen, W.; Li, X.; Zhao, L. Ginsenoside 20 (S)-Rg3 inhibits the warburg effect via modulating DNMT3A/MiR-532-3p/HK2 pathway in ovarian cancer cells. Cell. Physiol. Biochem. 2018, 45, 2548–2559. [Google Scholar] [CrossRef]
- Agnihotri, S.; Mansouri, S.; Burrell, K.; Li, M.; Mamatjan, Y.; Liu, J.; Nejad, R.; Kumar, S.; Jalali, S.; Singh, S.K. Ketoconazole and Posaconazole Selectively Target HK2-expressing Glioblastoma CellsAzoles Inhibit HK2 and GBM Growth. Clin. Cancer Res. 2019, 25, 844–855. [Google Scholar] [CrossRef]
- Li, W.; Hao, J.; Zhang, L.; Cheng, Z.; Deng, X.; Shu, G. Astragalin reduces hexokinase 2 through increasing miR-125b to inhibit the proliferation of hepatocellular carcinoma cells in vitro and in vivo. J. Agric. Food Chem. 2017, 65, 5961–5972. [Google Scholar] [CrossRef] [PubMed]
- Chesney, J. 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase and tumor cell glycolysis. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 535–539. [Google Scholar] [CrossRef]
- Houddane, A.; Bultot, L.; Novellasdemunt, L.; Johanns, M.; Gueuning, M.-A.; Vertommen, D.; Coulie, P.G.; Bartrons, R.; Hue, L.; Rider, M.H. Role of Akt/PKB and PFKFB isoenzymes in the control of glycolysis, cell proliferation and protein synthesis in mitogen-stimulated thymocytes. Cell. Signal. 2017, 34, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Chen, Y.; Zhu, Y. The molecular basis of targeting PFKFB3 as a therapeutic strategy against cancer. Oncotarget 2017, 8, 62793. [Google Scholar] [CrossRef] [PubMed]
- Brooke, D.G.; van Dam, E.M.; Watts, C.K.; Khoury, A.; Dziadek, M.A.; Brooks, H.; Graham, L.-J.K.; Flanagan, J.U.; Denny, W.A. Targeting the Warburg Effect in cancer; relationships for 2-arylpyridazinones as inhibitors of the key glycolytic enzyme 6-phosphofructo-2-kinase/2, 6-bisphosphatase 3 (PFKFB3). Bioorg. Med. Chem. 2014, 22, 1029–1039. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, A.; Telang, S.; Clem, B.; Chesney, J. Regulation of glucose metabolism by 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatases in cancer. Exp. Mol. Pathol. 2009, 86, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Bartrons, R.; Rodríguez-García, A.; Simon-Molas, H.; Castaño, E.; Manzano, A.; Navarro-Sabaté, À. The potential utility of PFKFB3 as a therapeutic target. Expert Opin. Ther. Targets 2018, 22, 659–674. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Meng, Q.; Xi, Q.; Wang, H.; Wu, G. PFKFB3 was overexpressed in gastric cancer patients and promoted the proliferation and migration of gastric cancer cells. Cancer Biomark. 2017, 18, 249–256. [Google Scholar] [CrossRef]
- Li, J.; Zhang, S.; Liao, D.; Zhang, Q.; Chen, C.; Yang, X.; Jiang, D.; Pang, J. Overexpression of PFKFB3 promotes cell glycolysis and proliferation in renal cell carcinoma. BMC Cancer 2022, 22, 83. [Google Scholar] [CrossRef]
- Clem, B.; Telang, S.; Clem, A.; Yalcin, A.; Meier, J.; Simmons, A.; Rasku, M.A.; Arumugam, S.; Dean, W.L.; Eaton, J. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol. Cancer Ther. 2008, 7, 110–120. [Google Scholar] [CrossRef]
- Clem, B.F.; O’Neal, J.; Tapolsky, G.; Clem, A.L.; Imbert-Fernandez, Y.; Kerr, D.A.; Klarer, A.C.; Redman, R.; Miller, D.M.; Trent, J.O. Targeting 6-Phosphofructo-2-Kinase (PFKFB3) as a Therapeutic Strategy against CancerTargeting PFKFB3 in Cancer. Mol. Cancer Ther. 2013, 12, 1461–1470. [Google Scholar] [CrossRef]
- Shi, L.; Pan, H.; Liu, Z.; Xie, J.; Han, W. Roles of PFKFB3 in cancer. Signal Transduct. Target. Ther. 2017, 2, 17044. [Google Scholar] [CrossRef]
- Mondal, S.; Roy, D.; Sarkar Bhattacharya, S.; Jin, L.; Jung, D.; Zhang, S.; Kalogera, E.; Staub, J.; Wang, Y.; Xuyang, W. Therapeutic targeting of PFKFB3 with a novel glycolytic inhibitor PFK158 promotes lipophagy and chemosensitivity in gynecologic cancers. Int. J. Cancer 2019, 144, 178–189. [Google Scholar] [CrossRef]
- Seo, M.; Kim, J.-D.; Neau, D.; Sehgal, I.; Lee, Y.-H. Structure-based development of small molecule PFKFB3 inhibitors: A framework for potential cancer therapeutic agents targeting the Warburg effect. PLoS ONE 2011, 6, e24179. [Google Scholar] [CrossRef]
- Li, F.-L.; Liu, J.-P.; Bao, R.-X.; Yan, G.; Feng, X.; Xu, Y.-P.; Sun, Y.-P.; Yan, W.; Ling, Z.-Q.; Xiong, Y. Acetylation accumulates PFKFB3 in cytoplasm to promote glycolysis and protects cells from cisplatin-induced apoptosis. Nat. Commun. 2018, 9, 508. [Google Scholar] [CrossRef]
- Feng, Y.; Wu, L. mTOR up-regulation of PFKFB3 is essential for acute myeloid leukemia cell survival. Biochem. Biophys. Res. Commun. 2017, 483, 897–903. [Google Scholar] [CrossRef]
- Yan, S.; Zhou, N.; Zhang, D.; Zhang, K.; Zheng, W.; Bao, Y.; Yang, W. PFKFB3 inhibition attenuates oxaliplatin-induced autophagy and enhances its cytotoxicity in colon cancer cells. Int. J. Mol. Sci. 2019, 20, 5415. [Google Scholar] [CrossRef]
- Zhu, Y.; Lu, L.; Qiao, C.; Shan, Y.; Li, H.; Qian, S.; Hong, M.; Zhao, H.; Li, J.; Yang, Z. Targeting PFKFB3 sensitizes chronic myelogenous leukemia cells to tyrosine kinase inhibitor. Oncogene 2018, 37, 2837–2849. [Google Scholar] [CrossRef]
- Telang, S.; Yaddanadupi, K.; Tapolsky, G.; Redman, R.; Chesney, J. Taking the sweet out of Th17 cells to potentiate immuno-oncology drugs. Cancer Res. 2016, 76, 557. [Google Scholar] [CrossRef]
- Gustafsson, N.; Färnegårdh, K.; Bonagas, N.; Ninou, A.H.; Groth, P.; Wiita, E.; Jönsson, M.; Hallberg, K.; Lehto, J.; Pennisi, R. Targeting PFKFB3 radiosensitizes cancer cells and suppresses homologous recombination. Nat. Commun. 2018, 9, 3873. [Google Scholar] [CrossRef]
- Lea, M.A.; Guzman, Y.; Desbordes, C. Inhibition of growth by combined treatment with inhibitors of lactate dehydrogenase and either phenformin or inhibitors of 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase 3. Anticancer Res. 2016, 36, 1479–1488. [Google Scholar] [PubMed]
- Wang, Y.; Qu, C.; Liu, T.; Wang, C. PFKFB3 inhibitors as potential anticancer agents: Mechanisms of action, current developments, and structure-activity relationships. Eur. J. Med. Chem. 2020, 203, 112612. [Google Scholar] [CrossRef]
- Kotowski, K.; Rosik, J.; Machaj, F.; Supplitt, S.; Wiczew, D.; Jabłońska, K.; Wiechec, E.; Ghavami, S.; Dzięgiel, P. Role of PFKFB3 and PFKFB4 in cancer: Genetic basis, impact on disease development/progression, and potential as therapeutic targets. Cancers 2021, 13, 909. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-H.; Li, X.-F.; Liu, J.-T.; Wang, H.; Fan, L.-L.; Li, J.; Sun, G.-P. PKM2, a potential target for regulating cancer. Gene 2018, 668, 48–53. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, S.R.; Hwang, S.R.; Rolland, D.; Murga-Zamalloa, C.; Basrur, V.; Conlon, K.P.; Fermin, D.; Wolfe, T.; Raskind, A.; Ruan, C. Integrated phosphoproteomic and metabolomic profiling reveals NPM-ALK–mediated phosphorylation of PKM2 and metabolic reprogramming in anaplastic large cell lymphoma. Blood J. Am. Soc. Hematol. 2013, 122, 958–968. [Google Scholar] [CrossRef]
- Yang, X.; Qian, K. Protein O-GlcNAcylation: Emerging mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2017, 18, 452–465. [Google Scholar] [CrossRef]
- Li, J.; Li, S.; Guo, J.; Li, Q.; Long, J.; Ma, C.; Ding, Y.; Yan, C.; Li, L.; Wu, Z. Natural product micheliolide (MCL) irreversibly activates pyruvate kinase M2 and suppresses leukemia. J. Med. Chem. 2018, 61, 4155–4164. [Google Scholar] [CrossRef]
- Qi, H.; Ning, X.; Yu, C.; Ji, X.; Jin, Y.; McNutt, M.A.; Yin, Y. Succinylation-dependent mitochondrial translocation of PKM2 promotes cell survival in response to nutritional stress. Cell Death Dis. 2019, 10, 170. [Google Scholar] [CrossRef]
- Liu, F.; Ma, F.; Wang, Y.; Hao, L.; Zeng, H.; Jia, C.; Wang, Y.; Liu, P.; Ong, I.M.; Li, B. PKM2 methylation by CARM1 activates aerobic glycolysis to promote tumorigenesis. Nat. Cell Biol. 2017, 19, 1358–1370. [Google Scholar] [CrossRef]
- Martin, S.P.; Fako, V.; Dang, H.; Dominguez, D.A.; Khatib, S.; Ma, L.; Wang, H.; Zheng, W.; Wang, X.W. PKM2 inhibition may reverse therapeutic resistance to transarterial chemoembolization in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2020, 39, 99. [Google Scholar] [CrossRef]
- Wang, Y.; Hao, F.; Nan, Y.; Qu, L.; Na, W.; Jia, C.; Chen, X. PKM2 inhibitor shikonin overcomes the cisplatin resistance in bladder cancer by inducing necroptosis. Int. J. Biol. Sci. 2018, 14, 1883. [Google Scholar] [CrossRef]
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.-k.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, D.S. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 2011, 334, 1278–1283. [Google Scholar] [CrossRef]
- Goldberg, M.S.; Sharp, P.A. Pyruvate kinase M2-specific siRNA induces apoptosis and tumor regression. J. Exp. Med. 2012, 209, 217–224. [Google Scholar] [CrossRef]
- Chen, J.; Xie, J.; Jiang, Z.; Wang, B.; Wang, Y.; Hu, X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene 2011, 30, 4297–4306. [Google Scholar] [CrossRef]
- Tao, T.; Su, Q.; Xu, S.; Deng, J.; Zhou, S.; Zhuang, Y.; Huang, Y.; He, C.; He, S.; Peng, M. Down-regulation of PKM2 decreases FASN expression in bladder cancer cells through AKT/mTOR/SREBP-1c axis. J. Cell. Physiol. 2019, 234, 3088–3104. [Google Scholar] [CrossRef]
- Tang, J.-C.; Zhao, J.; Long, F.; Chen, J.-y.; Mu, B.; Jiang, Z.; Ren, Y.; Yang, J. Efficacy of Shikonin against Esophageal Cancer Cells and its possible mechanisms in vitro and in vivo. J. Cancer 2018, 9, 32. [Google Scholar] [CrossRef]
- Boulos, J.C.; Rahama, M.; Hegazy, M.-E.F.; Efferth, T. Shikonin derivatives for cancer prevention and therapy. Cancer Lett. 2019, 459, 248–267. [Google Scholar] [CrossRef]
- Li, H.; Tong, Y.; Bai, L.; Ye, L.; Zhong, L.; Duan, X.; Zhu, Y. Lactoferrin functionalized PEG-PLGA nanoparticles of shikonin for brain targeting therapy of glioma. Int. J. Biol. Macromol. 2018, 107, 204–211. [Google Scholar] [CrossRef]
- Tian, H.; Zhang, T.; Qin, S.; Huang, Z.; Zhou, L.; Shi, J.; Nice, E.C.; Xie, N.; Huang, C.; Shen, Z. Enhancing the therapeutic efficacy of nanoparticles for cancer treatme nt using versatile targeted strategies. J. Hematol. Oncol. 2022, 15, 132. [Google Scholar] [CrossRef]
- Kontogiannopoulos, K.N.; Assimopoulou, A.N.; Hatziantoniou, S.; Karatasos, K.; Demetzos, C.; Papageorgiou, V.P. Chimeric advanced drug delivery nano systems (chi-aDDnSs) for shikonin combining dendritic and liposomal technology. Int. J. Pharm. 2012, 422, 381–389. [Google Scholar] [CrossRef]
- Kontogiannopoulos, K.N.; Tsermentseli, S.K.; Assimopoulou, A.N.; Papageorgiou, V.P. Sterically stabilized liposomes as a potent carrier for shikonin. J. Liposome Res. 2014, 24, 230–240. [Google Scholar] [CrossRef]
- Li, J.; Zhou, S.; Yu, J.; Cai, W.; Yang, Y.; Kuang, X.; Liu, H.; He, Z.; Wang, Y. Low dose shikonin and anthracyclines coloaded liposomes induce robust immunogenetic cell death for synergistic chemo-immunotherapy. J. Control Release 2021, 335, 306–319. [Google Scholar] [CrossRef]
- Assimopoulou, A.N.; Papageorgiou, V.P. Preparation and release studies of alkannin-containing microcapsules. J. Microencapsul. 2004, 21, 161–173. [Google Scholar] [CrossRef]
- Kontogiannopoulos, K.N.; Assimopoulou, A.N.; Tsivintzelis, I.; Panayiotou, C.; Papageorgiou, V.P. Electrospun fiber mats containing shikonin and derivatives with potential biomedical applications. Int. J. Pharm. 2011, 409, 216–228. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhu, Z.; Zhang, G.; Lin, F.; Liu, Y.; Zhang, Y.; Feng, J.; Chen, W.; Meng, Q.; Chen, L. AS1411 Aptamer/Hyaluronic Acid-Bifunctionalized Microemulsion Co-Loading Shikonin and Docetaxel for Enhanced Antiglioma Therapy. J. Pharm. Sci. 2019, 108, 3684–3694. [Google Scholar] [CrossRef] [PubMed]
- Jing, Q.; Ruan, H.; Li, J.; Wang, Z.; Pei, L.; Hu, H.; He, Z.; Wu, T.; Ruan, S.; Guo, T.; et al. Keratinocyte membrane-mediated nanodelivery system with dissolving microneedles for targeted therapy of skin diseases. Biomaterials 2021, 278, 121142. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhao, M.; Xu, Y.; Hu, X.; Dai, Y.; Wang, D. Hybrid micelles codelivering shikonin and IDO-1 siRNA enhance immunotherapy by remodeling immunosuppressive tumor microenvironment. Int. J. Pharm. 2021, 597, 120310. [Google Scholar] [CrossRef]
- Nindawat, S.; Agrawal, V. Fabrication of silver nanoparticles using Arnebia hispidissima (Lehm.) A. DC. root extract and unravelling their potential biomedical applications. Artif. Cells Nanomed. Biotechnol. 2019, 47, 166–180. [Google Scholar] [CrossRef]
- Tian, H.; Zhang, M.; Jin, G.; Jiang, Y.; Luan, Y. Cu-MOF chemodynamic nanoplatform via modulating glutathione and H2O2 in tumor microenvironment for amplified cancer therapy. J. Colloid Interface Sci. 2021, 587, 358–366. [Google Scholar] [CrossRef]
- Tian, H.; Zhou, L.; Wang, Y.; Nice, E.C.; Huang, C.; Zhang, H. A targeted nanomodulator capable of manipulating tumor microenvironment against metastasis. J. Control Release 2022, 348, 590–600. [Google Scholar] [CrossRef]
- Su, Q.; Luo, S.; Tan, Q.; Deng, J.; Zhou, S.; Peng, M.; Tao, T.; Yang, X. The role of pyruvate kinase M2 in anticancer therapeutic treatments. Oncol. Lett. 2019, 18, 5663–5672. [Google Scholar] [CrossRef]
- Shang, D.; Wu, J.; Guo, L.; Xu, Y.; Liu, L.; Lu, J. Metformin increases sensitivity of osteosarcoma stem cells to cisplatin by inhibiting expression of PKM2. Int. J. Oncol. 2017, 50, 1848–1856. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, Z.; Wang, H.; Chen, X.; Jin, C. Activation of AMPK by metformin promotes renal cancer cell proliferation under glucose deprivation through its interaction with PKM2. Int. J. Biol. Sci. 2019, 15, 617. [Google Scholar] [CrossRef]
- Ivanova, D.; Zhelev, Z.; Getsov, P.; Nikolova, B.; Aoki, I.; Higashi, T.; Bakalova, R. Vitamin K: Redox-modulation, prevention of mitochondrial dysfunction and anticancer effect. Redox Biol. 2018, 16, 352–358. [Google Scholar] [CrossRef]
- Wellington, K.W.; Hlatshwayo, V.; Kolesnikova, N.I.; Saha, S.T.; Kaur, M.; Motadi, L.R. Anticancer activities of vitamin K3 analogues. Investig. New Drugs 2020, 38, 378–391. [Google Scholar] [CrossRef]
- Chen, J.; Jiang, Z.; Wang, B.; Wang, Y.; Hu, X. Vitamin K3 and K5 are inhibitors of tumor pyruvate kinase M2. Cancer Lett. 2012, 316, 204–210. [Google Scholar] [CrossRef]
- Zhou, Y.; Huang, Z.; Su, J.; Li, J.; Zhao, S.; Wu, L.; Zhang, J.; He, Y.; Zhang, G.; Tao, J. Benserazide is a novel inhibitor targeting PKM2 for melanoma treatment. Int. J. Cancer 2020, 147, 139–151. [Google Scholar] [CrossRef]
- Shankar Babu, M.; Mahanta, S.; Lakhter, A.J.; Hato, T.; Paul, S.; Naidu, S.R. Lapachol inhibits glycolysis in cancer cells by targeting pyruvate kinase M2. PLoS ONE 2018, 13, e0191419. [Google Scholar] [CrossRef]
- Ning, X.; Qi, H.; Li, R.; Li, Y.; Jin, Y.; McNutt, M.A.; Liu, J.; Yin, Y. Discovery of novel naphthoquinone derivatives as inhibitors of the tumor cell specific M2 isoform of pyruvate kinase. Eur. J. Med. Chem. 2017, 138, 343–352. [Google Scholar] [CrossRef]
- Gao, C.L.; Hou, G.G.; Liu, J.; Ru, T.; Xu, Y.Z.; Zhao, S.Y.; Ye, H.; Zhang, L.Y.; Chen, K.X.; Guo, Y.W. Synthesis and Target Identification of Benzoxepane Derivatives as Potential Anti-Neuroinflammatory Agents for Ischemic Stroke. Angew. Chem. 2020, 132, 2450–2460. [Google Scholar] [CrossRef]
- Liu, J.; Wu, N.; Ma, L.; Liu, M.; Liu, G.; Zhang, Y.; Lin, X. Oleanolic acid suppresses aerobic glycolysis in cancer cells by switching pyruvate kinase type M isoforms. PLoS ONE 2014, 9, e91606. [Google Scholar] [CrossRef]
- Son, J.Y.; Yoon, S.; Tae, I.H.; Park, Y.J.; De, U.; Jeon, Y.; Park, Y.J.; Rhyu, I.J.; Lee, B.M.; Chung, K.H. Novel therapeutic roles of MC-4 in combination with everolimus against advanced renal cell carcinoma by dual targeting of Akt/pyruvate kinase muscle isozyme M2 and mechanistic target of rapamycin complex 1 pathways. Cancer Med. 2018, 7, 5083–5095. [Google Scholar] [CrossRef]
- Siddiqui, F.A.; Prakasam, G.; Chattopadhyay, S.; Rehman, A.U.; Padder, R.A.; Ansari, M.A.; Irshad, R.; Mangalhara, K.; Bamezai, R.N.; Husain, M. Curcumin decreases Warburg effect in cancer cells by down-regulating pyruvate kinase M2 via mTOR-HIF1α inhibition. Sci. Rep. 2018, 8, 8323. [Google Scholar] [CrossRef]
- Yang, R.; Fang, X.-L.; Zhen, Q.; Chen, Q.-Y.; Feng, C. Mitochondrial targeting nano-curcumin for attenuation on PKM2 and FASN. Colloids Surf. B Biointerfaces 2019, 182, 110405. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Han, L.; Jian, Y.; Ma, Y.; Yan, W.; Chen, X.; Xu, H.; Li, L. Resveratrol induces apoptosis in human melanoma cell through negatively regulating Erk/PKM2/Bcl-2 axis. OncoTargets Ther. 2018, 11, 8995. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wang, Y.; Wu, C.; Yang, P.; Li, H.; Li, Z. Resveratrol induces cancer cell apoptosis through MiR-326/PKM2-mediated ER stress and mitochondrial fission. J. Agric. Food Chem. 2016, 64, 9356–9367. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Wu, L.; Ji, J.; Chen, K.; Yu, Q.; Zhang, J.; Chen, J.; Mao, Y.; Wang, F.; Dai, W. PKM2 is the target of proanthocyanidin B2 during the inhibition of hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 204. [Google Scholar] [CrossRef]
- Zhu, S.; Guo, Y.; Zhang, X.; Liu, H.; Yin, M.; Chen, X.; Peng, C. Pyruvate kinase M2 (PKM2) in cancer and cancer therapeutics. Cancer Lett. 2021, 503, 240–248. [Google Scholar] [CrossRef]
- Zahra, K.; Dey, T.; Mishra, S.P.; Pandey, U. Pyruvate kinase M2 and cancer: The role of PKM2 in promoting tumorigenesis. Front. Oncol. 2020, 10, 159. [Google Scholar] [CrossRef]
- Israelsen, W.J.; Dayton, T.L.; Davidson, S.M.; Fiske, B.P.; Hosios, A.M.; Bellinger, G.; Li, J.; Yu, Y.; Sasaki, M.; Horner, J.W. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell 2013, 155, 397–409. [Google Scholar] [CrossRef]
- Anastasiou, D.; Yu, Y.; Israelsen, W.J.; Jiang, J.-K.; Boxer, M.B.; Hong, B.S.; Tempel, W.; Dimov, S.; Shen, M.; Jha, A. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat. Chem. Biol. 2012, 8, 839–847. [Google Scholar] [CrossRef]
- Mohammad, G.H.; Vassileva, V.; Acedo, P.; Olde Damink, S.W.; Malago, M.; Dhar, D.K.; Pereira, S.P. Targeting pyruvate kinase M2 and lactate dehydrogenase a is an effective combination strategy for the treatment of pancreatic cancer. Cancers 2019, 11, 1372. [Google Scholar] [CrossRef]
- Ding, Y.; Xue, Q.; Liu, S.; Hu, K.; Wang, D.; Wang, T.; Li, Y.; Guo, H.; Hao, X.; Ge, W.; et al. Identification of Parthenolide Dimers as Activators of Pyruvate Kinase M2 in Xenografts of Glioblastoma Multiforme in Vivo. J. Med. Chem. 2020, 63, 1597–1611. [Google Scholar] [CrossRef]
- Wubben, T.J.; Pawar, M.; Weh, E.; Smith, A.; Sajjakulnukit, P.; Zhang, L.; Dai, L.; Hager, H.; Pai, M.P.; Lyssiotis, C.A. Small molecule activation of metabolic enzyme pyruvate kinase muscle isozyme 2, PKM2, circumvents photoreceptor apoptosis. Sci. Rep. 2020, 10, 2990. [Google Scholar] [CrossRef]
- Kim, D.J.; Park, Y.S.; Kim, N.D.; Min, S.H.; You, Y.M.; Jung, Y.; Koo, H.; Noh, H.; Kim, J.A.; Park, K.C.; et al. A novel pyruvate kinase M2 activator compound that suppresses lung cancer cell viability under hypoxia. Mol. Cells 2015, 38, 373–379. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, B.; Wu, X.; Li, R.; Ning, X.; Liu, Y.; Liu, Z.; Ge, Z.; Li, R.; Yin, Y. New pyridin-3-ylmethyl carbamodithioic esters activate pyruvate kinase M2 and potential anticancer lead compounds. Bioorg. Med. Chem. 2015, 23, 4815–4823. [Google Scholar] [CrossRef]
- Li, Y.; Bao, M.; Yang, C.; Chen, J.; Zhou, S.; Sun, R.; Wu, C.; Li, X.; Bao, J. Computer-aided identification of a novel pyruvate kinase M2 activator compound. Cell Prolif. 2018, 51, e12509. [Google Scholar] [CrossRef]
- Li, R.Z.; Fan, X.X.; Shi, D.F.; Zhu, G.Y.; Wang, Y.W.; Luo, L.X.; Pan, H.D.; Yao, X.J.; Leung, E.L.; Liu, L. Identification of a new pyruvate kinase M2 isoform (PKM2) activator for the treatment of non-small-cell lung cancer (NSCLC). Chem. Biol. Drug Des. 2018, 92, 1851–1858. [Google Scholar] [CrossRef]
- Aslan, E.; Guler, C.; Adem, S. In vitro effects of some flavonoids and phenolic acids on human pyruvate kinase isoenzyme M2. J. Enzym. Inhib. Med. Chem. 2016, 31, 314–317. [Google Scholar] [CrossRef]
- Goetze, K.; Walenta, S.; Ksiazkiewicz, M.; Kunz-Schughart, L.A.; Mueller-Klieser, W. Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int. J. Oncol. 2011, 39, 453–463. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.R.; Cleveland, J.L. Targeting lactate metabolism for cancer therapeutics. J. Clin. Investig. 2013, 123, 3685–3692. [Google Scholar] [CrossRef] [PubMed]
- Gallo, M.; Sapio, L.; Spina, A.; Naviglio, D.; Calogero, A.; Naviglio, S. Lactic dehydrogenase and cancer: An overview. Front. Biosci.-Landmark 2015, 20, 1234–1249. [Google Scholar]
- San-Millán, I.; Brooks, G.A. Reexamining cancer metabolism: Lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis 2017, 38, 119–133. [Google Scholar] [CrossRef]
- Pathria, G.; Scott, D.A.; Feng, Y.; Sang Lee, J.; Fujita, Y.; Zhang, G.; Sahu, A.D.; Ruppin, E.; Herlyn, M.; Osterman, A.L. Targeting the Warburg effect via LDHA inhibition engages ATF 4 signaling for cancer cell survival. EMBO J. 2018, 37, e99735. [Google Scholar] [CrossRef]
- Wang, X.; Xu, L.; Wu, Q.; Liu, M.; Tang, F.; Cai, Y.; Fan, W.; Huang, H.; Gu, X. Inhibition of LDHA deliver potential anticancer performance in renal cell carcinoma. Urol. Int. 2017, 99, 237–244. [Google Scholar] [CrossRef]
- Feng, Y.; Xiong, Y.; Qiao, T.; Li, X.; Jia, L.; Han, Y. Lactate dehydrogenase A: A key player in carcinogenesis and potential target in cancer therapy. Cancer Med. 2018, 7, 6124–6136. [Google Scholar] [CrossRef]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef]
- Farabegoli, F.; Vettraino, M.; Manerba, M.; Fiume, L.; Roberti, M.; Di Stefano, G. Galloflavin, a new lactate dehydrogenase inhibitor, induces the death of human breast cancer cells with different glycolytic attitude by affecting distinct signaling pathways. Eur. J. Pharm. Sci. 2012, 47, 729–738. [Google Scholar] [CrossRef]
- Muramatsu, H.; Sumitomo, M.; Morinaga, S.; Kajikawa, K.; Kobayashi, I.; Nishikawa, G.; Kato, Y.; Watanabe, M.; Zennami, K.; Kanao, K. Targeting lactate dehydrogenase-A promotes docetaxel-induced cytotoxicity predominantly in castration-resistant prostate cancer cells. Oncol. Rep. 2019, 42, 224–230. [Google Scholar] [CrossRef]
- Zhou, M.; Zhao, Y.; Ding, Y.; Liu, H.; Liu, Z.; Fodstad, O.; Riker, A.I.; Kamarajugadda, S.; Lu, J.; Owen, L.B. Warburg effect in chemosensitivity: Targeting lactate dehydrogenase-A re-sensitizes taxol-resistant cancer cells to taxol. Mol. Cancer 2010, 9, 33. [Google Scholar] [CrossRef]
- Yu, H.; Yin, Y.; Yi, Y.; Cheng, Z.; Kuang, W.; Li, R.; Zhong, H.; Cui, Y.; Yuan, L.; Gong, F. Targeting lactate dehydrogenase A (LDHA) exerts antileukemic effects on T-cell acute lymphoblastic leukemia. Cancer Commun. 2020, 40, 501–517. [Google Scholar] [CrossRef]
- Koukourakis, M.; Tsolou, A.; Pouliliou, S.; Lamprou, I.; Papadopoulou, M.; Ilemosoglou, M.; Kostoglou, G.; Ananiadou, D.; Sivridis, E.; Giatromanolaki, A. Blocking LDHA glycolytic pathway sensitizes glioblastoma cells to radiation and temozolomide. Biochem. Biophys. Res. Commun. 2017, 491, 932–938. [Google Scholar] [CrossRef]
- Altinoz, M.A.; Ozpinar, A. Oxamate targeting aggressive cancers with special emphasis to brain tumors. Biomed. Pharmacother. 2022, 147, 112686. [Google Scholar] [CrossRef]
- Yang, X.; Lu, Y.; Hang, J.; Zhang, J.; Zhang, T.; Huo, Y.; Liu, J.; Lai, S.; Luo, D.; Wang, L. Lactate-Modulated Immunosuppression of Myeloid-Derived Suppressor Cells Contributes to the Radioresistance of Pancreatic CancerLactate-Activated MDSCs Promote Radioresistance in PDAC. Cancer Immunol. Res. 2020, 8, 1440–1451. [Google Scholar] [CrossRef]
- Maftouh, M.; Avan, A.; Sciarrillo, R.; Granchi, C.; Leon, L.; Rani, R.; Funel, N.; Smid, K.; Honeywell, R.; Boggi, U. Synergistic interaction of novel lactate dehydrogenase inhibitors with gemcitabine against pancreatic cancer cells in hypoxia. Br. J. Cancer 2014, 110, 172–182. [Google Scholar] [CrossRef]
- Kim, E.-Y.; Chung, T.-W.; Han, C.W.; Park, S.Y.; Park, K.H.; Jang, S.B.; Ha, K.-T. A novel lactate dehydrogenase inhibitor, 1-(phenylseleno)-4-(trifluoromethyl) benzene, suppresses tumor growth through apoptotic cell death. Sci. Rep. 2019, 9, 3969. [Google Scholar] [CrossRef]
- Schwab, M.; Thunborg, K.; Azimzadeh, O.; von Toerne, C.; Werner, C.; Shevtsov, M.; Di Genio, T.; Zdralevic, M.; Pouyssegur, J.; Renner, K. Targeting cancer metabolism breaks radioresistance by impairing the stress response. Cancers 2021, 13, 3762. [Google Scholar] [CrossRef]
- Fu, D.; Li, J.; Wei, J.; Zhang, Z.; Luo, Y.; Tan, H.; Ren, C. HMGB2 is associated with malignancy and regulates Warburg effect by targeting LDHB and FBP1 in breast cancer. Cell Commun. Signal. 2018, 16, 8. [Google Scholar] [CrossRef]
- Hu, H.; Juvekar, A.; Lyssiotis, C.A.; Lien, E.C.; Albeck, J.G.; Oh, D.; Varma, G.; Hung, Y.P.; Ullas, S.; Lauring, J. Phosphoinositide 3-kinase regulates glycolysis through mobilization of aldolase from the actin cytoskeleton. Cell 2016, 164, 433–446. [Google Scholar] [CrossRef]
- Li, X.; Jiang, F.; Ge, Z.; Chen, B.; Yu, J.; Xin, M.; Wang, J.; An, L.; Wei, J.; Wu, L. Fructose-bisphosphate aldolase a regulates hypoxic adaptation in hepatocellular carcinoma and involved with tumor malignancy. Dig. Dis. Sci. 2019, 64, 3215–3227. [Google Scholar] [CrossRef]
- Ma, D.; Chen, X.; Zhang, P.; Zhang, H.; Wei, L.; Hu, S.; Tang, J.; Zhou, M.; Xie, C.; Ou, R. Upregulation of the ALDOA/DNA-PK/p53 pathway by dietary restriction suppresses tumor growth. Oncogene 2018, 37, 1041–1048. [Google Scholar] [CrossRef]
- Grandjean, G.; de Jong, P.R.; James, B.P.; Koh, M.Y.; Lemos, R.; Kingston, J.; Aleshin, A.; Bankston, L.A.; Miller, C.P.; Cho, E.J. Definition of a Novel Feed-Forward Mechanism for Glycolysis-HIF1α Signaling in Hypoxic Tumors Highlights Aldolase A as a Therapeutic TargetTargeting a Glycolysis HIF-1 Feed-Forward Loop. Cancer Res. 2016, 76, 4259–4269. [Google Scholar] [CrossRef]
- Chang, Y.-C.; Chiou, J.; Yang, Y.-F.; Su, C.-Y.; Lin, Y.-F.; Yang, C.-N.; Lu, P.-J.; Huang, M.-S.; Yang, C.-J.; Hsiao, M. Therapeutic Targeting of Aldolase A Interactions Inhibits Lung Cancer Metastasis and Prolongs SurvivalTargeting of ALDOA Inhibits Lung Cancer Metastasis. Cancer Res. 2019, 79, 4754–4766. [Google Scholar] [CrossRef]
- Yu, T.; Zhao, Y.; Hu, Z.; Li, J.; Chu, D.; Zhang, J.; Li, Z.; Chen, B.; Zhang, X.; Pan, H. MetaLnc9 Facilitates Lung Cancer Metastasis via a PGK1-Activated AKT/mTOR PathwayMetaLnc9 Interacts with PGK1 and NONO in NSCLC. Cancer Res. 2017, 77, 5782–5794. [Google Scholar] [CrossRef]
- Huang, B.; Cai, W.; Wang, Q.; Liu, F.; Xu, M.; Zhang, Y. Gankyrin drives malignant transformation of gastric cancer and alleviates oxidative stress via mTORC1 activation. Oxid. Med. Cell. Longev. 2018, 2018, 9480316. [Google Scholar] [CrossRef]
- Hu, H.; Zhu, W.; Qin, J.; Chen, M.; Gong, L.; Li, L.; Liu, X.; Tao, Y.; Yin, H.; Zhou, H.; et al. Acetylation of PGK1 promotes liver cancer cell proliferation and tumorigenesis. Hepatology 2017, 65, 515–528. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Luo, Y.; Zhang, D.; Wang, X.; Zhang, P.; Li, H.; Ejaz, S.; Liang, S. PGK1-mediated cancer progression and drug resistance. Am. J. Cancer Res. 2019, 9, 2280–2302. [Google Scholar] [PubMed]
- Hitosugi, T.; Zhou, L.; Fan, J.; Elf, S.; Zhang, L.; Xie, J.; Wang, Y.; Gu, T.-L.; Alečković, M.; LeRoy, G. Tyr26 phosphorylation of PGAM1 provides a metabolic advantage to tumours by stabilizing the active conformation. Nat. Commun. 2013, 4, 1790. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Wu, H.; Lei, Y.; Zhang, H.; Liu, R.; Zhao, Y.; Chen, X.; Zeng, D.; Tong, A.; Chen, L. Quantitative proteomics identification of phosphoglycerate mutase 1 as a novel therapeutic target in hepatocellular carcinoma. Mol. Cancer 2010, 9, 81. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Gong, F.; Chen, Y.; Qiu, M.; Cheng, K.; Tang, J.; Ge, J.; Chen, N.; Zeng, H.; Liu, J. Proteomics identification of PGAM1 as a potential therapeutic target for urothelial bladder cancer. J. Proteom. 2016, 132, 85–92. [Google Scholar] [CrossRef]
- Chen, G.; Gharib, T.G.; Wang, H.; Huang, C.-C.; Kuick, R.; Thomas, D.G.; Shedden, K.A.; Misek, D.E.; Taylor, J.M.; Giordano, T.J. Protein profiles associated with survival in lung adenocarcinoma. Proc. Natl. Acad. Sci. USA 2003, 100, 13537–13542. [Google Scholar] [CrossRef]
- Evans, M.J.; Saghatelian, A.; Sorensen, E.J.; Cravatt, B.F. Target discovery in small-molecule cell-based screens by in situ proteome reactivity profiling. Nat. Biotechnol. 2005, 23, 1303–1307. [Google Scholar] [CrossRef]
- Hitosugi, T.; Zhou, L.; Elf, S.; Fan, J.; Kang, H.-B.; Seo, J.H.; Shan, C.; Dai, Q.; Zhang, L.; Xie, J. Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell 2012, 22, 585–600. [Google Scholar] [CrossRef]
- Fu, Q.-F.; Liu, Y.; Fan, Y.; Hua, S.-N.; Qu, H.-Y.; Dong, S.-W.; Li, R.-L.; Zhao, M.-Y.; Zhen, Y.; Yu, X.-L. Alpha-enolase promotes cell glycolysis, growth, migration, and invasion in non-small cell lung cancer through FAK-mediated PI3K/AKT pathway. J. Hematol. Oncol. 2015, 8, 22. [Google Scholar] [CrossRef]
- Yin, H.; Wang, L.; Liu, H.-L. ENO1 overexpression in pancreatic cancer patients and its clinical and diagnostic significance. Gastroenterol. Res. Pract. 2018, 2018, 3842198. [Google Scholar] [CrossRef]
- Qian, X.; Xu, W.; Xu, J.; Shi, Q.; Li, J.; Weng, Y.; Jiang, Z.; Feng, L.; Wang, X.; Zhou, J. Enolase 1 stimulates glycolysis to promote chemoresistance in gastric cancer. Oncotarget 2017, 8, 47691. [Google Scholar] [CrossRef]
- Scatena, R.; Bottoni, P.; Pontoglio, A.; Mastrototaro, L.; Giardina, B. Glycolytic enzyme inhibitors in cancer treatment. Expert Opin. Investig. Drugs 2008, 17, 1533–1545. [Google Scholar] [CrossRef]
- Chan, A.K.; Bruce, J.I.; Siriwardena, A.K. Glucose metabolic phenotype of pancreatic cancer. World J. Gastroenterol. 2016, 22, 3471. [Google Scholar] [CrossRef]
- Muller, F.L.; Colla, S.; Aquilanti, E.; Manzo, V.E.; Genovese, G.; Lee, J.; Eisenson, D.; Narurkar, R.; Deng, P.; Nezi, L. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature 2012, 488, 337–342. [Google Scholar] [CrossRef]
- Capello, M.; Ferri-Borgogno, S.; Riganti, C.; Chattaragada, M.S.; Principe, M.; Roux, C.; Zhou, W.; Petricoin, E.F.; Cappello, P.; Novelli, F. Targeting the Warburg effect in cancer cells through ENO1 knockdown rescues oxidative phosphorylation and induces growth arrest. Oncotarget 2016, 7, 5598. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.-W.; Kim, W.-H.; Park, S.-H.; Lee, J.; Kim, J.; Su, D.; Ha, H.-H.; Chang, Y.-T.; Williams, D.R. A unique small molecule inhibitor of enolase clarifies its role in fundamental biological processes. ACS Chem. Biol. 2013, 8, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, C.; Longatto-Filho, A.; Azevedo-Silva, J.; Casal, M.; Schmitt, F.C.; Baltazar, F. Role of monocarboxylate transporters in human cancers: State of the art. J. Bioenerg. Biomembr. 2012, 44, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Bola, B.M.; Chadwick, A.L.; Michopoulos, F.; Blount, K.G.; Telfer, B.A.; Williams, K.J.; Smith, P.D.; Critchlow, S.E.; Stratford, I.J. Inhibition of Monocarboxylate Transporter-1 (MCT1) by AZD3965 Enhances Radiosensitivity by Reducing Lactate TransportInhibition of MCT1 Potentiates Radiotherapy. Mol. Cancer Ther. 2014, 13, 2805–2816. [Google Scholar] [CrossRef]
- Beloueche-Babari, M.; Casals Galobart, T.; Delgado-Goni, T.; Wantuch, S.; Parkes, H.G.; Tandy, D.; Harker, J.A.; Leach, M.O. Monocarboxylate transporter 1 blockade with AZD3965 inhibits lipid biosynthesis and increases tumour immune cell infiltration. Br. J. Cancer 2020, 122, 895–903. [Google Scholar] [CrossRef]
- Huang, T.; Feng, Q.; Wang, Z.; Li, W.; Sun, Z.; Wilhelm, J.; Huang, G.; Vo, T.; Sumer, B.D.; Gao, J. Tumor-Targeted Inhibition of Monocarboxylate Transporter 1 Improves T-Cell Immunotherapy of Solid Tumors. Adv. Healthc. Mater. 2021, 10, 2000549. [Google Scholar] [CrossRef]
- Ždralević, M.; Vučetić, M.; Daher, B.; Marchiq, I.; Parks, S.K.; Pouysségur, J. Disrupting the ‘Warburg effect’re-routes cancer cells to OXPHOS offering a vulnerability point via ‘ferroptosis’-induced cell death. Adv. Biol. Regul. 2018, 68, 55–63. [Google Scholar] [CrossRef]
- Draoui, N.; Schicke, O.; Seront, E.; Bouzin, C.; Sonveaux, P.; Riant, O.; Feron, O. Antitumor activity of 7-aminocarboxycoumarin derivatives, a new class of potent inhibitors of lactate influx but not efflux. Mol. Cancer Ther. 2014, 13, 1410–1418. [Google Scholar] [CrossRef]
- Pertega-Gomes, N.; Felisbino, S.; Massie, C.E.; Vizcaino, J.R.; Coelho, R.; Sandi, C.; Simoes-Sousa, S.; Jurmeister, S.; Ramos-Montoya, A.; Asim, M. A glycolytic phenotype is associated with prostate cancer progression and aggressiveness: A role for monocarboxylate transporters as metabolic targets for therapy. J. Pathol. 2015, 236, 517–530. [Google Scholar] [CrossRef]
- Fang, Y.; Liu, W.; Tang, Z.; Ji, X.; Zhou, Y.; Song, S.; Tian, M.; Tao, C.; Huang, R.; Zhu, G. Monocarboxylate transporter 4 inhibition potentiates hepatocellular carcinoma immunotherapy through enhancing T cell infiltration and immune attack. Hepatology 2022. [Google Scholar] [CrossRef]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2012, 481, 380–384. [Google Scholar] [CrossRef]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Biological role and therapeutic potential of IDH mutations in cancer. Cancer Cell 2018, 34, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Romanidou, O.; Kotoula, V.; Fountzilas, G. Bridging cancer biology with the clinic: Comprehending and exploiting IDH gene mutations in gliomas. Cancer Genom. Proteom. 2018, 15, 421–436. [Google Scholar] [CrossRef] [PubMed]
- Rohle, D.; Popovici-Muller, J.; Palaskas, N.; Turcan, S.; Grommes, C.; Campos, C.; Tsoi, J.; Clark, O.; Oldrini, B.; Komisopoulou, E. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013, 340, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Levell, J.R.; Liu, G.; Caferro, T.; Sutton, J.; Shafer, C.M.; Costales, A.; Manning, J.R.; Zhao, Q.; Sendzik, M. Discovery and evaluation of clinical candidate IDH305, a brain penetrant mutant IDH1 inhibitor. ACS Med. Chem. Lett. 2017, 8, 1116–1121. [Google Scholar] [CrossRef] [PubMed]
- Lapa, B.; Goncalves, A.C.; Jorge, J.; Alves, R.; Pires, A.S.; Abrantes, A.M.; Coucelo, M.; Abrunhosa, A.; Botelho, M.F.; Nascimento-Costa, J.M.; et al. Acute myeloid leukemia sensitivity to metabolic inhibitors: Glycolysis showed to be a better therapeutic target. Med. Oncol. 2020, 37, 72. [Google Scholar] [CrossRef]
- Zdralevic, M.; Brand, A.; Di Ianni, L.; Dettmer, K.; Reinders, J.; Singer, K.; Peter, K.; Schnell, A.; Bruss, C.; Decking, S.M.; et al. Double genetic disruption of lactate dehydrogenases A and B is required to ablate the "Warburg effect" restricting tumor growth to oxidative metabolism. J. Biol. Chem. 2018, 293, 15947–15961. [Google Scholar] [CrossRef]
- Zhou, Y.; Guo, Y.; Tam, K.Y. Targeting glucose metabolism to develop anticancer treatments and therapeutic patents. Expert. Opin. Ther. Pat. 2022, 32, 441–453. [Google Scholar] [CrossRef]
- Korga, A.; Ostrowska, M.; Jozefczyk, A.; Iwan, M.; Wojcik, R.; Zgorka, G.; Herbet, M.; Vilarrubla, G.G.; Dudka, J. Apigenin and hesperidin augment the toxic effect of doxorubicin against HepG2 cells. BMC Pharmacol. Toxicol. 2019, 20, 22. [Google Scholar] [CrossRef]
- Zhang, D.; Li, J.; Wang, F.; Hu, J.; Wang, S.; Sun, Y. 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014, 355, 176–183. [Google Scholar] [CrossRef]
- Cheng, Y.; Diao, D.; Zhang, H.; Guo, Q.; Wu, X.; Song, Y.; Dang, C. High glucose-induced resistance to 5-fluorouracil in pancreatic cancer cells alleviated by 2-deoxy-D-glucose. Biomed. Rep. 2014, 2, 188–192. [Google Scholar] [CrossRef]
- Zhang, T.; Zhu, X.; Wu, H.; Jiang, K.; Zhao, G.; Shaukat, A.; Deng, G.; Qiu, C. Targeting the ROS/PI3K/AKT/HIF-1α/HK2 axis of breast cancer cells: Combined administration of Polydatin and 2-Deoxy-d-glucose. J. Cell. Mol. Med. 2019, 23, 3711–3723. [Google Scholar] [CrossRef]
- Takemura, A.; Che, X.-F.; Tabuchi, T.; Moriya, S.; Miyazawa, K.; Tomoda, A. Enhancement of cytotoxic and pro-apoptotic effects of 2-aminophenoxazine-3-one on the rat hepatocellular carcinoma cell line dRLh-84, the human hepatocellular carcinoma cell line HepG2, and the rat normal hepatocellular cell line RLN-10 in combination with 2-deoxy-D-glucose. Oncol. Rep. 2012, 27, 347–355. [Google Scholar]
- Sawayama, H.; Ogata, Y.; Ishimoto, T.; Mima, K.; Hiyoshi, Y.; Iwatsuki, M.; Baba, Y.; Miyamoto, Y.; Yoshida, N.; Baba, H. Glucose transporter 1 regulates the proliferation and cisplatin sensitivity of esophageal cancer. Cancer Sci. 2019, 110, 1705–1714. [Google Scholar] [CrossRef]
- Gong, Y.; Ji, P.; Yang, Y.-S.; Xie, S.; Yu, T.-J.; Xiao, Y.; Jin, M.-L.; Ma, D.; Guo, L.-W.; Pei, Y.-C. Metabolic-pathway-based subtyping of triple-negative breast cancer reveals potential therapeutic targets. Cell Metab. 2021, 33, 51–64.e9. [Google Scholar] [CrossRef]
- Zappasodi, R.; Serganova, I.; Cohen, I.J.; Maeda, M.; Shindo, M.; Senbabaoglu, Y.; Watson, M.J.; Leftin, A.; Maniyar, R.; Verma, S. CTLA-4 blockade drives loss of Treg stability in glycolysis-low tumours. Nature 2021, 591, 652–658. [Google Scholar] [CrossRef]
- Renner, K.; Bruss, C.; Schnell, A.; Koehl, G.; Becker, H.M.; Fante, M.; Menevse, A.N.; Kauer, N.; Blazquez, R.; Hacker, L.; et al. Restricting Glycolysis Preserves T Cell Effector Functions and Augments Checkpoint Therapy. Cell Rep. 2019, 29, 135–150.e9. [Google Scholar] [CrossRef]
- Ho, P.-C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.-C.; Cui, G.; Micevic, G.; Perales, J.C. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Zhan, L.; Yanxiang Guo, J. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef]
- Chaube, B.; Malvi, P.; Singh, S.V.; Mohammad, N.; Meena, A.S.; Bhat, M.K. Targeting metabolic flexibility by simultaneously inhibiting respiratory complex I and lactate generation retards melanoma progression. Oncotarget 2015, 6, 37281. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, D.; Sanin, D.E.; Pearce, E.J.; Pearce, E.L. Metabolic interventions in the immune response to cancer. Nat. Rev. Immunol. 2019, 19, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wenes, M.; Romero, P.; Huang, S.C.-C.; Fendt, S.-M.; Ho, P.-C. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 425–441. [Google Scholar] [CrossRef] [PubMed]








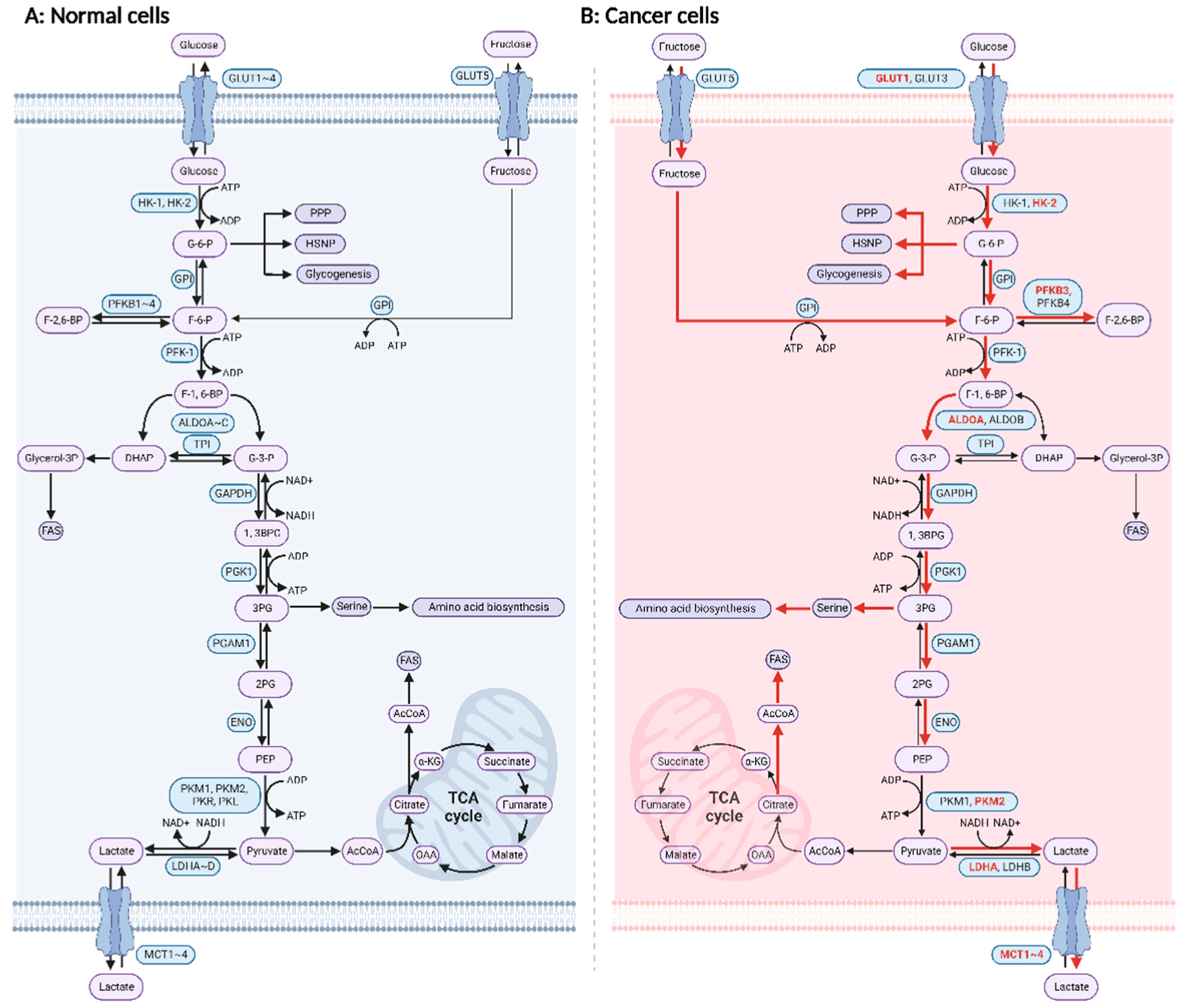{kind=link}
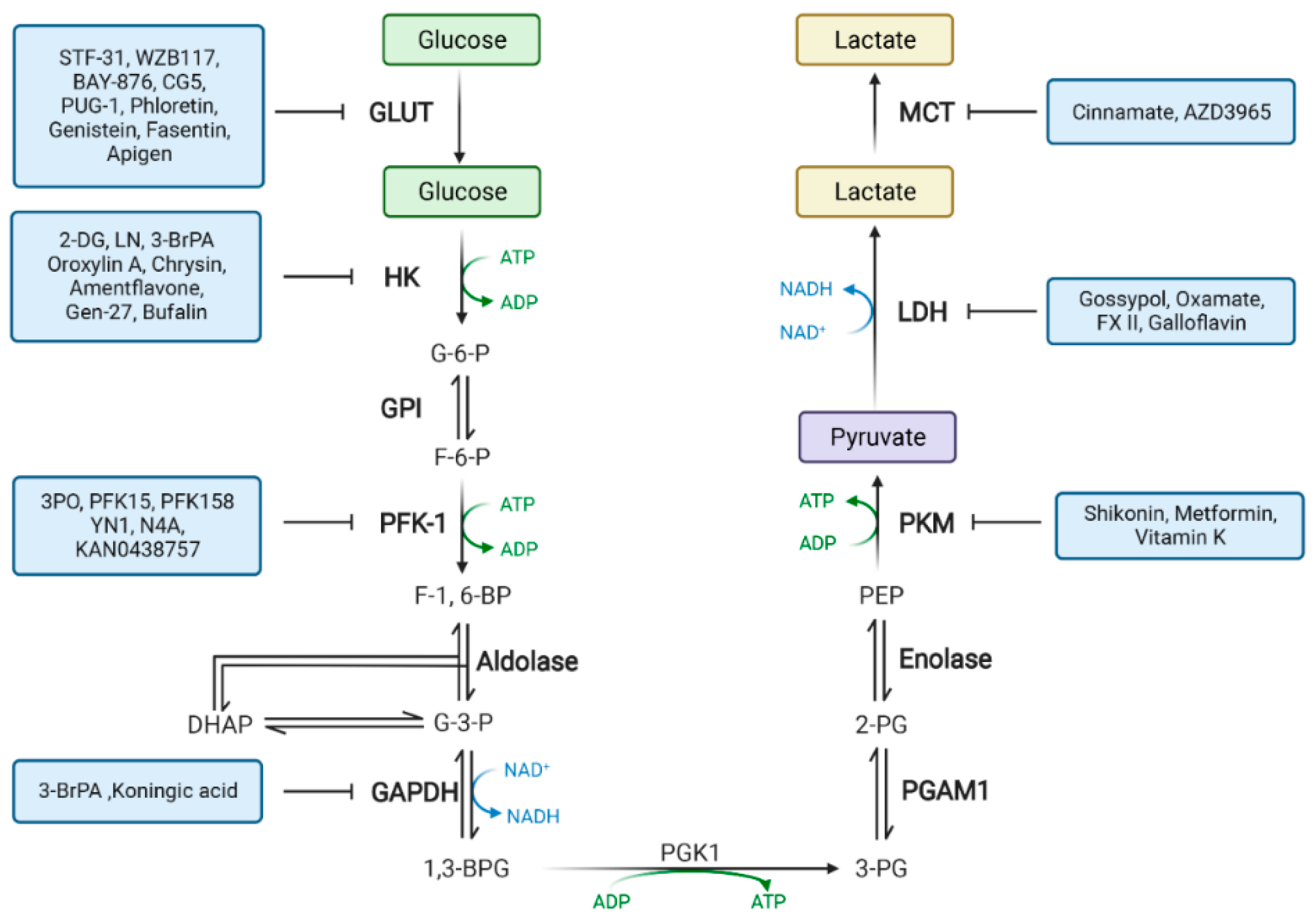{kind=link}
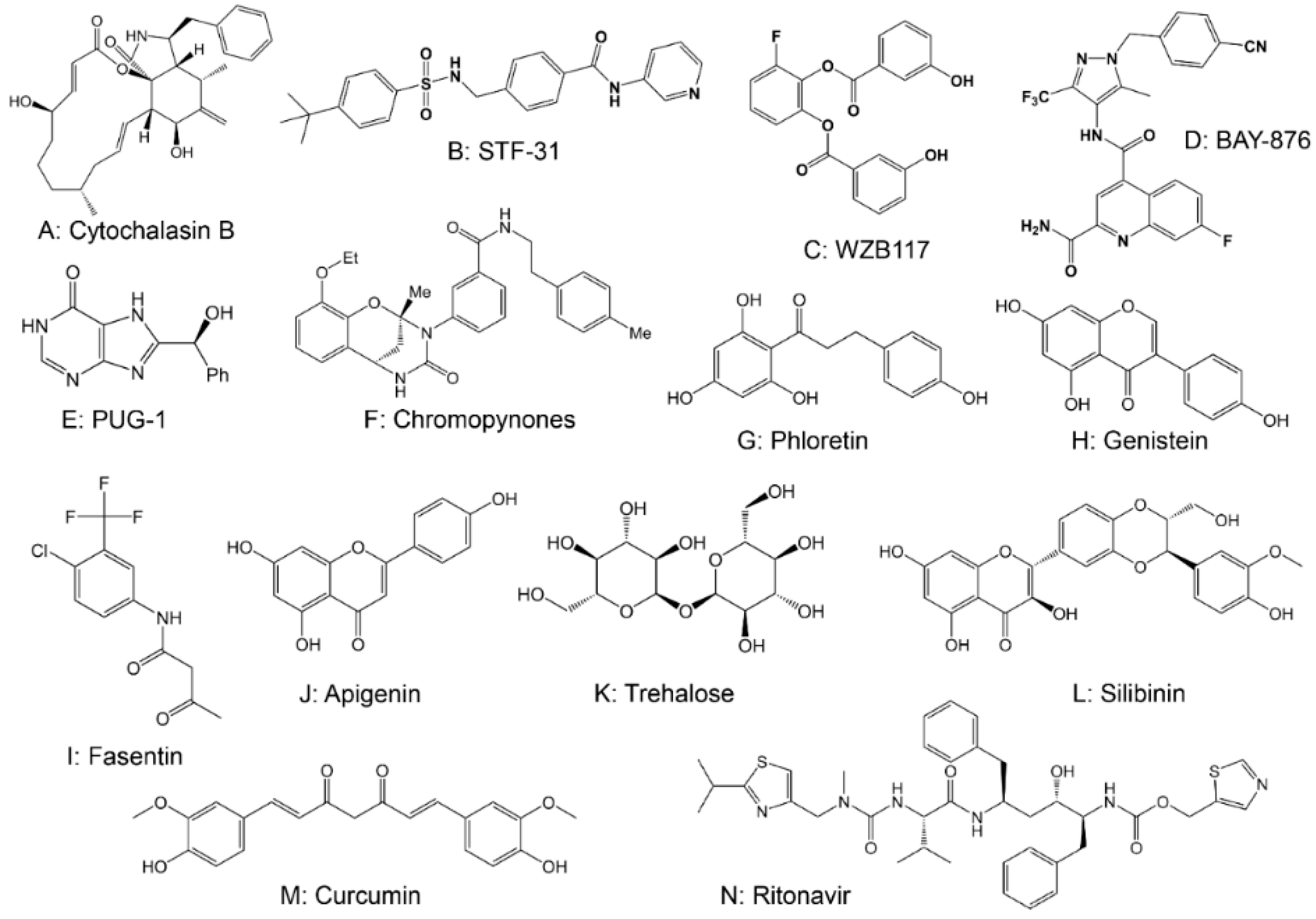{kind=link}
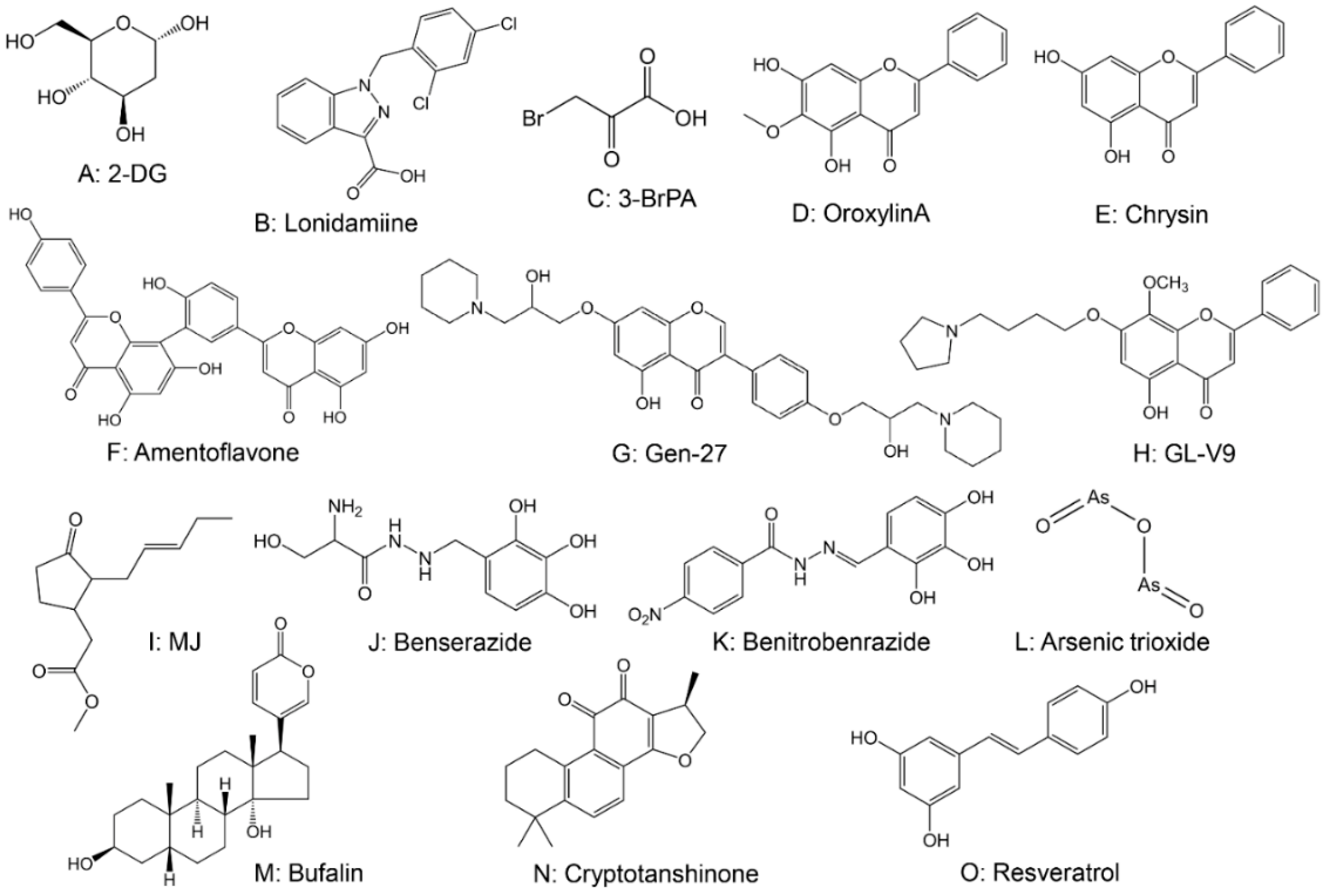{kind=link}
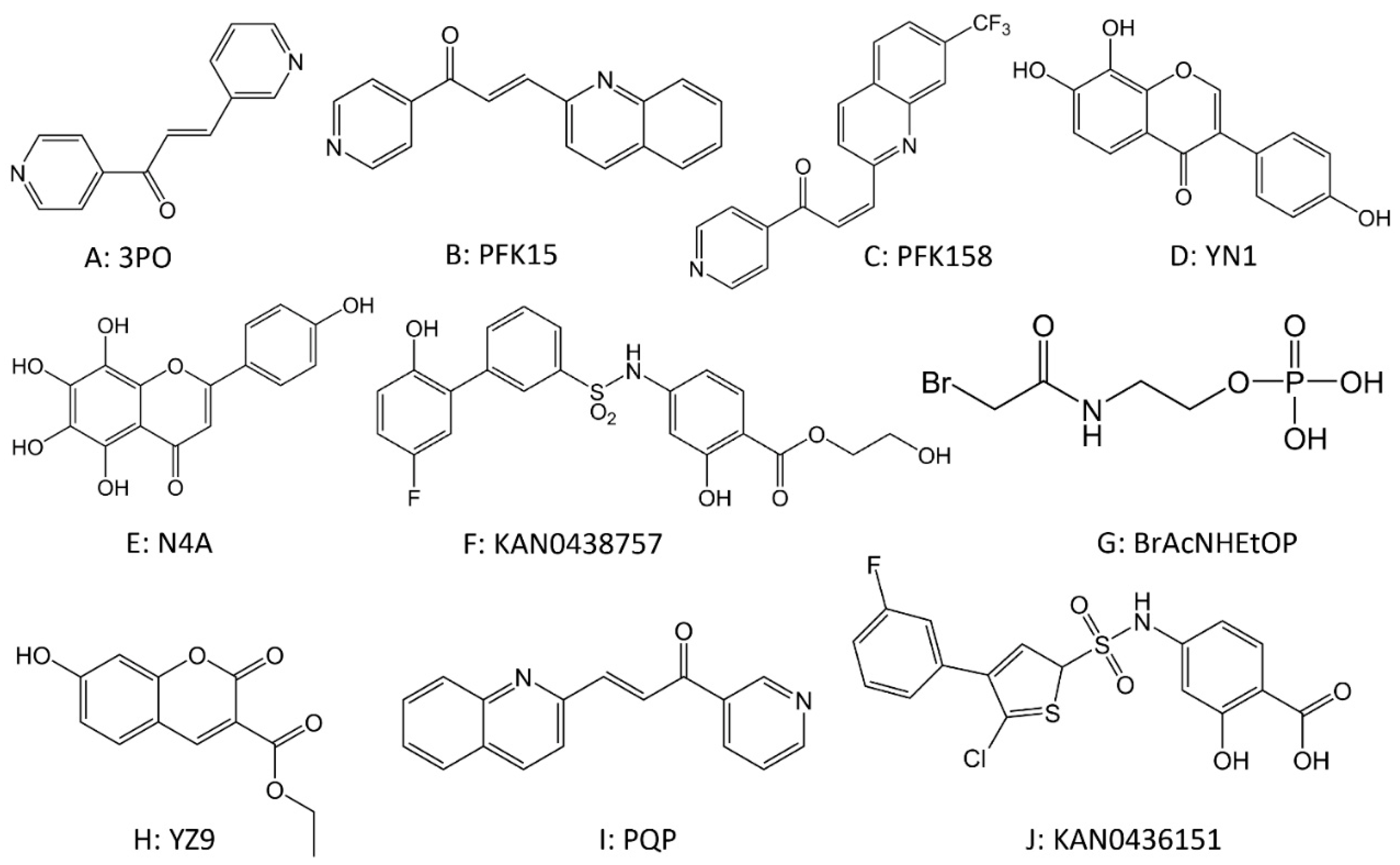{kind=link}
{kind=link}
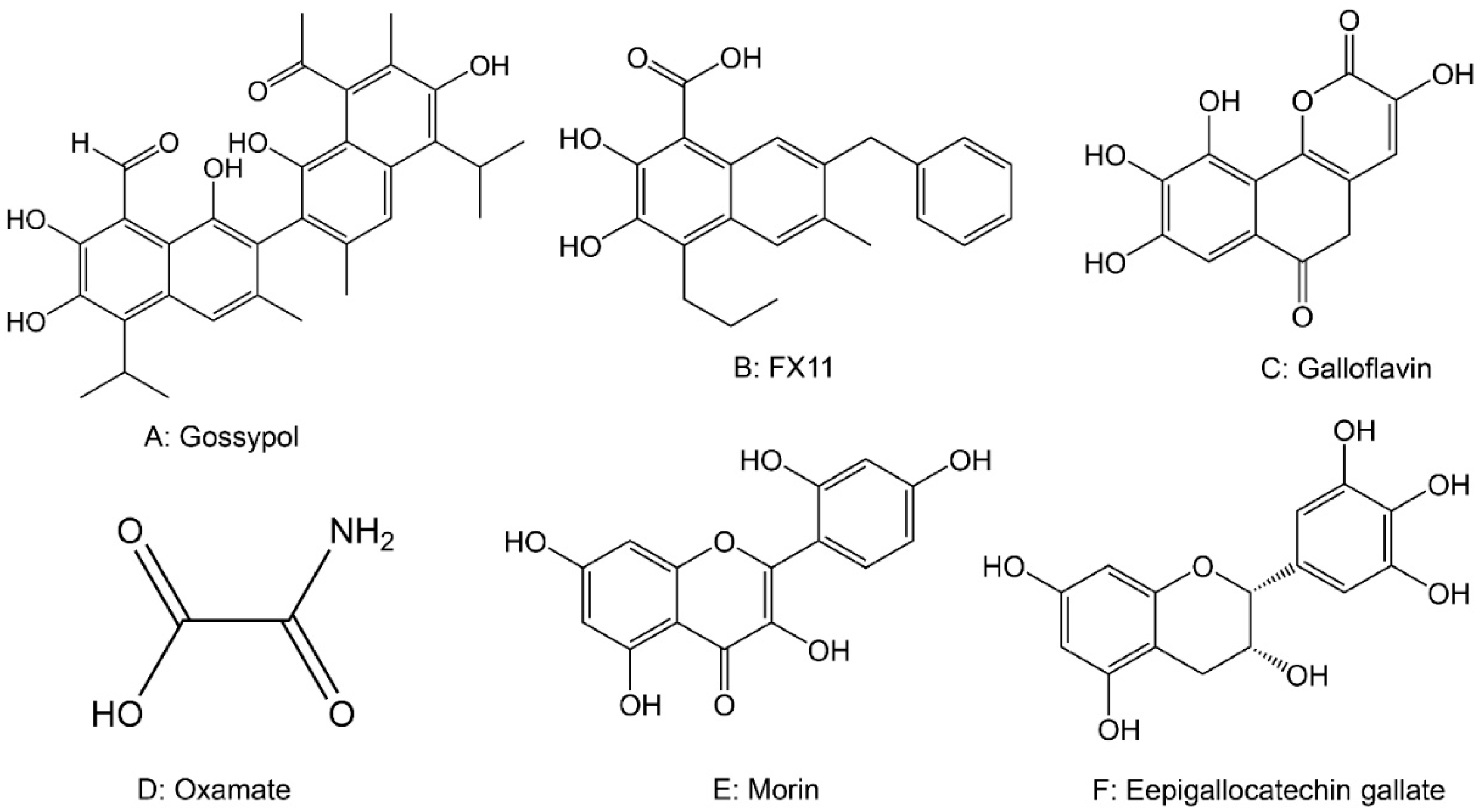{kind=link}
| Enzyme | Target | Agents | Tumor Type | Study Phase |
|---|---|---|---|---|
| GLUT | GLUT1 | STF-31 | RCC | Preclinical |
| WZB115, WZB117 | BC, LC | Preclinical | ||
| Fasentin | PC, Lymphoma | Preclinical | ||
| GLUT1/2 | Phloretin | HCC, BC, PC, LC, CC, etc. | Preclinical | |
| GLUT4 | Ritonavir | Multiple myeloma, BC, CLL, etc. | Phase I/II clinical trial | |
| GLUT5 | 2,5-AM | Acute myeloid leukemia | Preclinical | |
| HK | HK1/2 | Lonidamine | HCC, BC, LC, Melanoma, OC, etc. | Phase I/II clinical trial |
| HK2 | 2-DG | PC | Phase I/II clinical trial | |
| 3-BrPA | HCC, BC, Pancreatic cancer, etc. | Phase I/II clinical trial | ||
| PFK | PFKB3 | 3PO | LC, Pancreatic cancer, etc. | Phase I clinical trial |
| PFK15 | RCC, HCC, CC, Gastric Cancer, etc. | Phase I clinical trial | ||
| PFK158 | LC, OC, etc. | Phase I clinical trial | ||
| PK | PKM2 | Shikonin | BC, Skin cancer, Bladder cancer | Preclinical |
| Orlistat | OC | Preclinical | ||
| LDH | LDHA | AT-101 | Chronic lymphoblastic leukemia | Phase I/II clinical trial |
| Glloflavin | BC | Preclinical | ||
| Polyphenon E | BC, Colon cancer | Phase I/II clinical trial |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Li, Q.; Huang, Z.; Li, B.; Nice, E.C.; Huang, C.; Wei, L.; Zou, B. Targeting Glucose Metabolism Enzymes in Cancer Treatment: Current and Emerging Strategies. Cancers 2022, 14, 4568. https://doi.org/10.3390/cancers14194568
Zhang Y, Li Q, Huang Z, Li B, Nice EC, Huang C, Wei L, Zou B. Targeting Glucose Metabolism Enzymes in Cancer Treatment: Current and Emerging Strategies. Cancers. 2022; 14(19):4568. https://doi.org/10.3390/cancers14194568
Chicago/Turabian StyleZhang, Yi, Qiong Li, Zhao Huang, Bowen Li, Edouard C. Nice, Canhua Huang, Liuya Wei, and Bingwen Zou. 2022. "Targeting Glucose Metabolism Enzymes in Cancer Treatment: Current and Emerging Strategies" Cancers 14, no. 19: 4568. https://doi.org/10.3390/cancers14194568
APA StyleZhang, Y., Li, Q., Huang, Z., Li, B., Nice, E. C., Huang, C., Wei, L., & Zou, B. (2022). Targeting Glucose Metabolism Enzymes in Cancer Treatment: Current and Emerging Strategies. Cancers, 14(19), 4568. https://doi.org/10.3390/cancers14194568