

Preclinical Evaluation of Novel Tyrosine-Kinase Inhibitors in Medullary Thyroid Cancer
 , , , , and
, , , , and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Line Culture
2.2. RNA Isolation
2.3. Polymerase Chain Reaction (PCR)
2.4. Drug Preparation
2.5. Cell Viability Assay
2.6. Cell Cycle Analysis
2.7. Flow Cytometric Analysis of Apoptosis
2.8. Wound-Healing Assay
2.9. In Vivo Assay for Tumor-Induced Angiogenesis
2.10. Statistical Analysis
3. Results
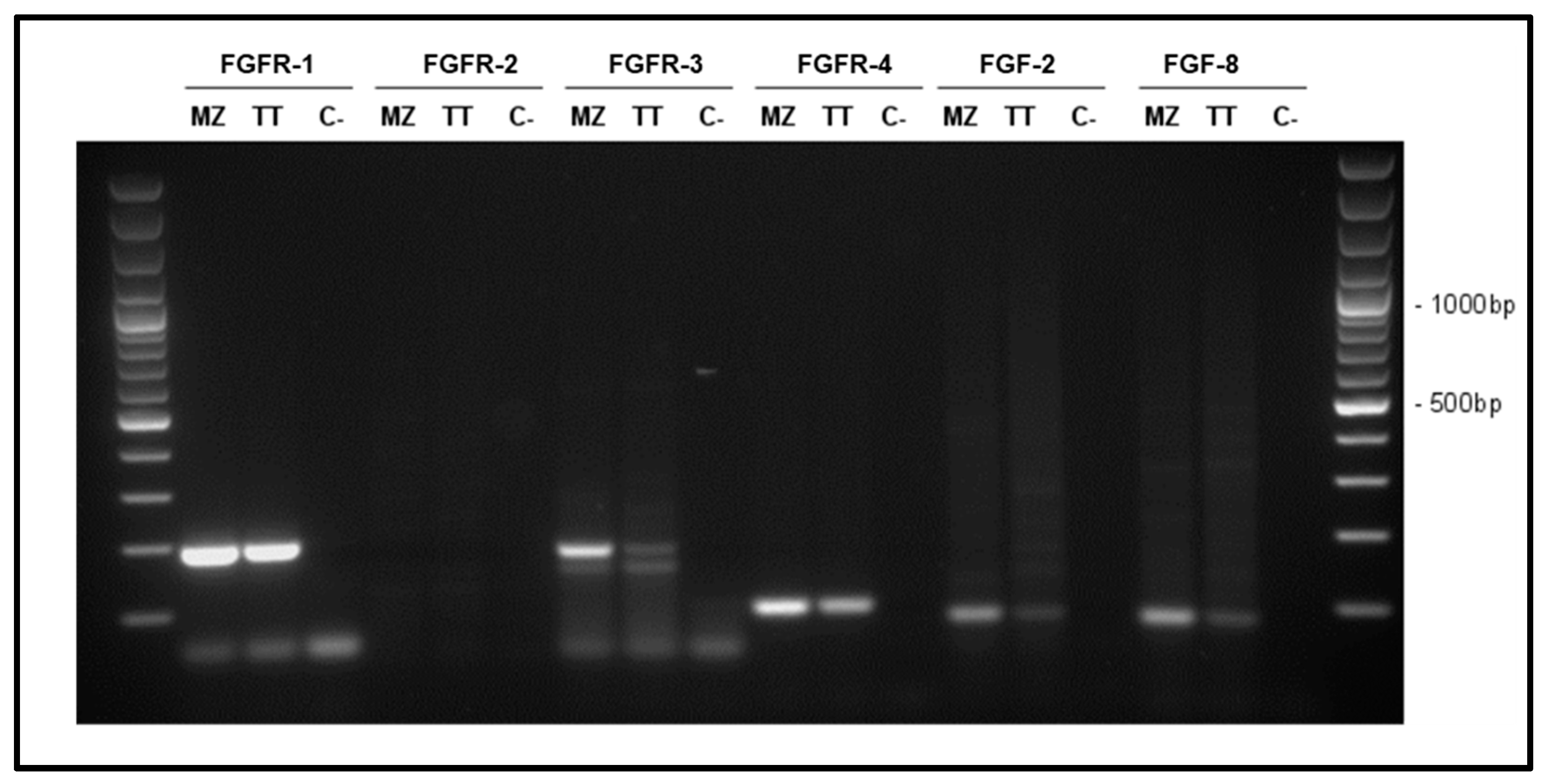
3.1. Characterization of FGF System in MTC Cells
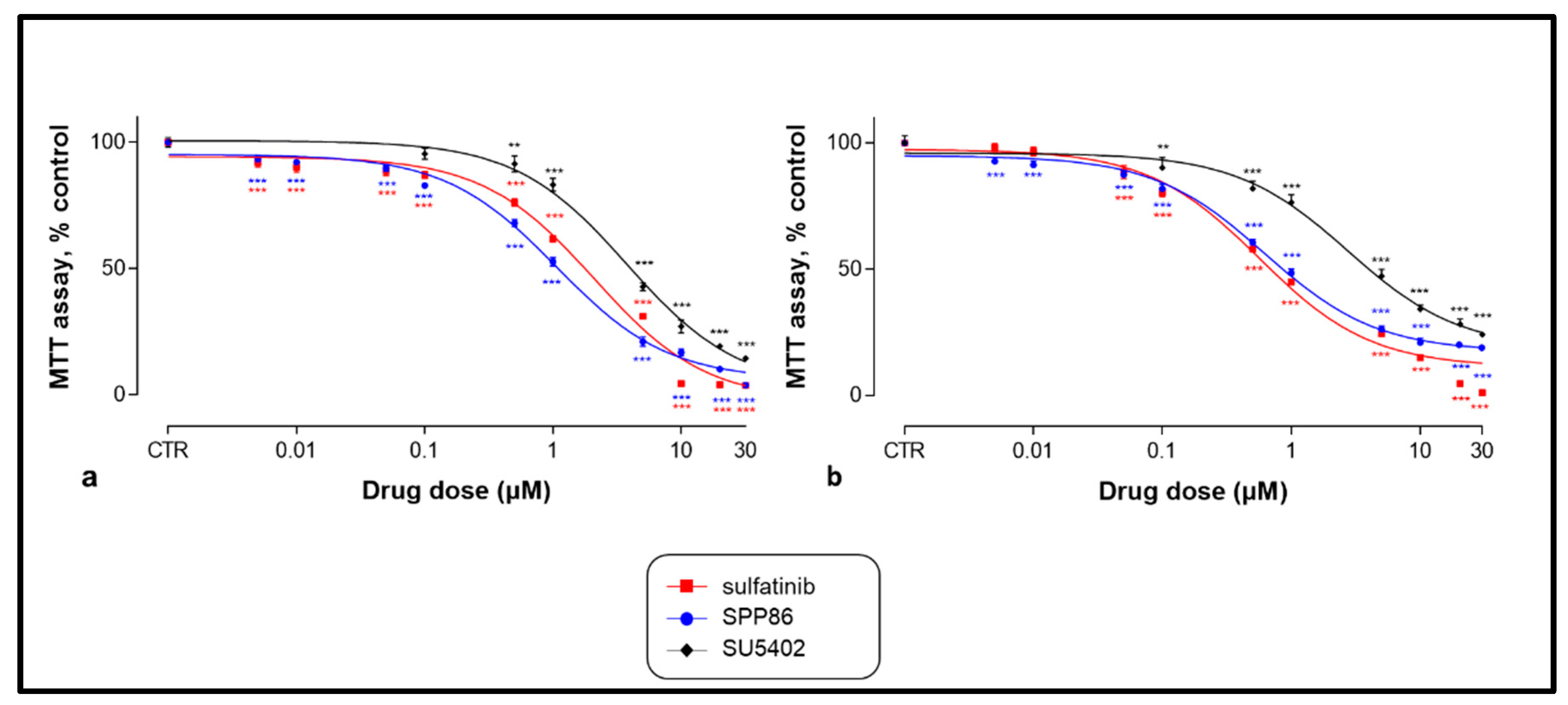
3.2. Effects of TKIs on Cell Viability
3.3. Effects of TKIs on Cell Cycle
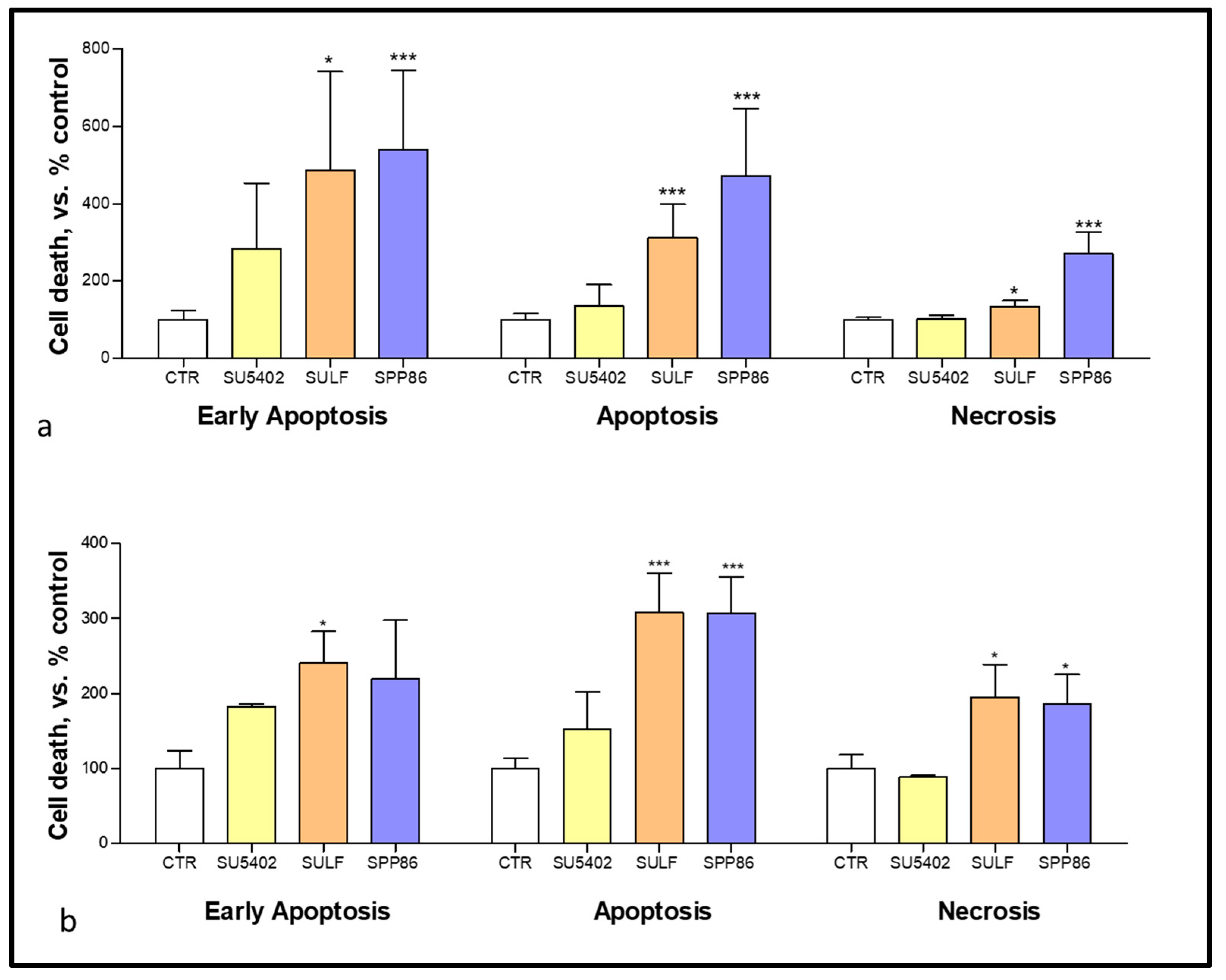
3.4. Effects of TKIs on Apoptosis and Necrosis
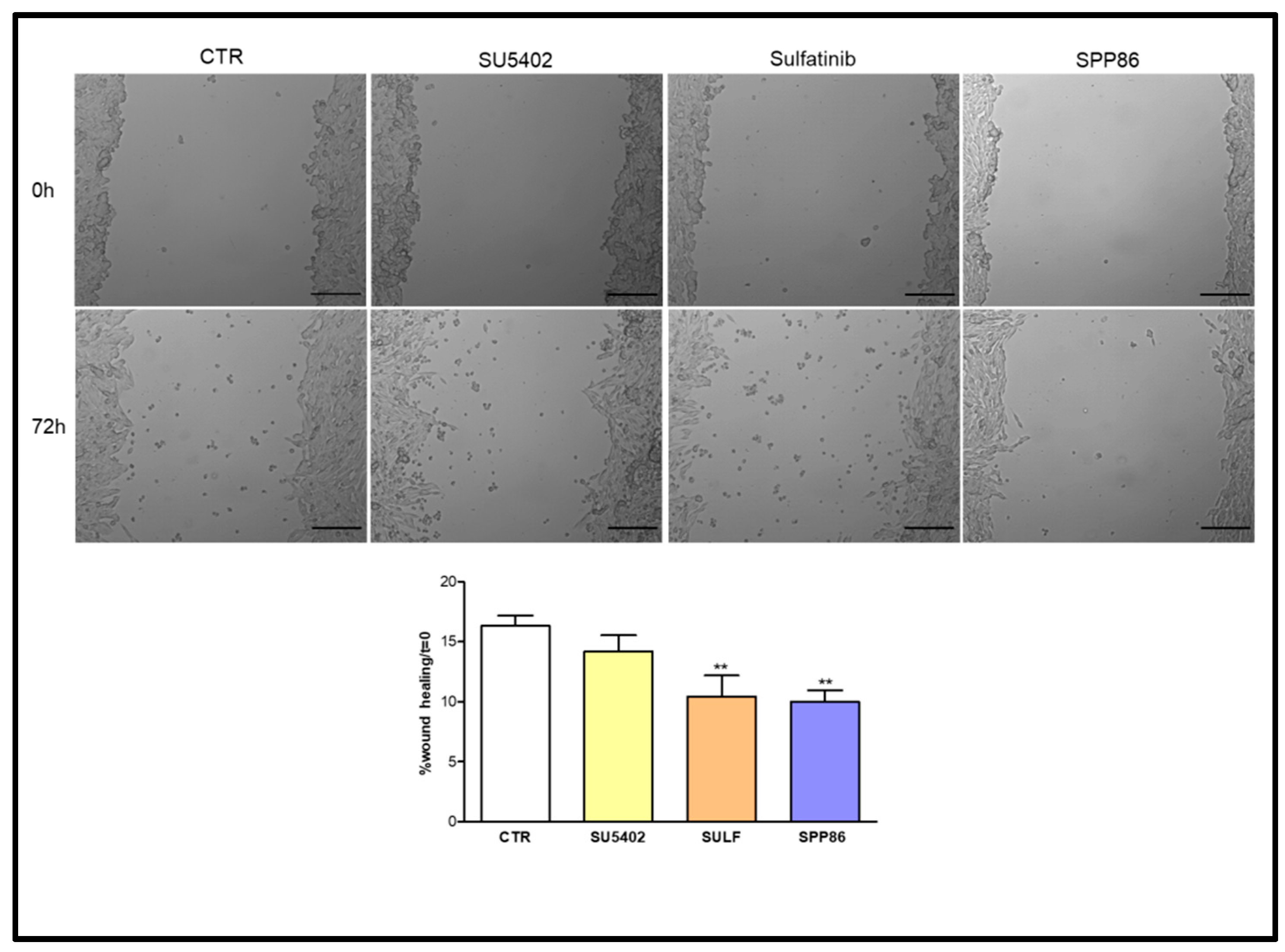
3.5. Effects of TKIs on Cell Migration
3.6. Effects of TKIs on TT Cell Line-Induced Angiogenesis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cancela-Nieto, M.G.; Sánchez-Sobrino, P.; Velo-García, A. Procalcitonin as a Marker of Medullary Thyroid Carcinoma. Minerva Endocrinol. 2021, 46, 481–482. [Google Scholar] [CrossRef]
- Thomas, C.M.; Asa, S.L.; Ezzat, S.; Sawka, A.M.; Goldstein, D. Diagnosis and Pathologic Characteristics of Medullary Thyroid Carcinoma-Review of Current Guidelines. Curr. Oncol. 2019, 26, 338–344. [Google Scholar] [CrossRef]
- Araque, K.A.; Gubbi, S.; Klubo-Gwiezdzinska, J. Updates on the Management of Thyroid Cancer. Horm. Metab. Res. 2020, 52, 562–577. [Google Scholar] [CrossRef] [PubMed]
- Larouche, V.; Akirov, A.; Thomas, C.M.; Krzyzanowska, M.K.; Ezzat, S. A Primer on the Genetics of Medullary Thyroid Cancer. Curr. Oncol. 2019, 26, 389–394. [Google Scholar] [CrossRef]
- Murakumo, Y.; Jijiwa, M.; Asai, N.; Ichihara, M.; Takahashi, M. RET and Neuroendocrine Tumors. Pituitary 2006, 9, 179–192. [Google Scholar] [CrossRef]
- Cabanillas, M.E.; Habra, M.A. Lenvatinib: Role in Thyroid Cancer and Other Solid Tumors. Cancer Treat. Rev. 2016, 42, 47–55. [Google Scholar] [CrossRef]
- Vitale, G.; Fonderico, F.; Martignetti, A.; Caraglia, M.; Ciccarelli, A.; Nuzzo, V.; Abbruzzese, A.; Lupoli, G. Pamidronate Improves the Quality of Life and Induces Clinical Remission of Bone Metastases in Patients with Thyroid Cancer. Br. J. Cancer 2001, 84, 1586–1590. [Google Scholar] [CrossRef]
- Lupoli, G.; Cascone, E.; Arlotta, F.; Vitale, G.; Celentano, L.; Salvatore, M.; Lombardi, G. Treatment of Advanced Medullary Thyroid Carcinoma with a Combination of Recombinant Interferon Alpha-2b and Octreotide. Cancer 1996, 78, 1114–1118. [Google Scholar] [CrossRef]
- Vitale, G.; Tagliaferri, P.; Caraglia, M.; Rampone, E.; Ciccarelli, A.; Raffaele Bianco, A.; Abbruzzese, A.; Lupoli, G. Slow Release Lanreotide in Combination with Interferon-2b in the Treatment of Symptomatic Advanced Medullary Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2000, 85, 983–988. [Google Scholar] [CrossRef]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a Novel MET and VEGFR2 Inhibitor, Simultaneously Suppresses Metastasis, Angiogenesis, and Tumor Growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef] [Green Version]
- Bentzien, F.; Zuzow, M.; Heald, N.; Gibson, A.; Shi, Y.; Goon, L.; Yu, P.; Engst, S.; Zhang, W.; Huang, D.; et al. In Vitro and In Vivo Activity of Cabozantinib (XL184), an Inhibitor of RET, MET, and VEGFR2, in a Model of Medullary Thyroid Cancer. Thyroid 2013, 23, 1569–1577. [Google Scholar] [CrossRef]
- Capp, C.; Wajner, S.M.; Siqueira, D.R.; Brasil, B.A.; Meurer, L.; Maia, A.L. Increased Expression of Vascular Endothelial Growth Factor and Its Receptors, VEGFR-1 and VEGFR-2, in Medullary Thyroid Carcinoma. Thyroid 2010, 20, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Ton, G.T.N.; Banaszynski, M.E.; Kolesar, J.M. Vandetanib: A Novel Targeted Therapy for the Treatment of Metastatic or Locally Advanced Medullary Thyroid Cancer. Am. J. Health Syst. Pharm. 2013, 70, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Wells, S.A.; Robinson, B.G.; Gagel, R.F.; Dralle, H.; Fagin, J.A.; Santoro, M.; Baudin, E.; Elisei, R.; Jarzab, B.; Vasselli, J.R.; et al. Vandetanib in Patients with Locally Advanced or Metastatic Medullary Thyroid Cancer: A Randomized, Double-Blind Phase III Trial. J. Clin. Oncol. 2012, 30, 134–141. [Google Scholar] [CrossRef]
- Viola, D.; Valerio, L.; Molinaro, E.; Agate, L.; Bottici, V.; Biagini, A.; Lorusso, L.; Cappagli, V.; Pieruzzi, L.; Giani, C.; et al. Treatment of Advanced Thyroid Cancer with Targeted Therapies: Ten Years of Experience. Endocr.-Relat. Cancer 2016, 23, R185–R205. [Google Scholar] [CrossRef]
- Holden, S.N.; Eckhardt, S.G.; Basser, R.; de Boer, R.; Rischin, D.; Green, M.; Rosenthal, M.A.; Wheeler, C.; Barge, A.; Hurwitz, H.I. Clinical Evaluation of ZD6474, an Orally Active Inhibitor of VEGF and EGF Receptor Signaling, in Patients with Solid, Malignant Tumors. Ann. Oncol. 2005, 16, 1391–1397. [Google Scholar] [CrossRef]
- Elisei, R.; Schlumberger, M.J.; Müller, S.P.; Schöffski, P.; Brose, M.S.; Shah, M.H.; Licitra, L.; Jarzab, B.; Medvedev, V.; Kreissl, M.C.; et al. Cabozantinib in Progressive Medullary Thyroid Cancer. J. Clin. Oncol. 2013, 31, 3639–3646. [Google Scholar] [CrossRef] [PubMed]
- Efstathiadou, Z.A.; Tsentidis, C.; Bargiota, A.; Daraki, V.; Kotsa, K.; Ntali, G.; Papanastasiou, L.; Tigas, S.; Toulis, K.; Pazaitou-Panayiotou, K.; et al. Benefits and Limitations of TKIs in Patients with Medullary Thyroid Cancer: A Systematic Review and Meta-Analysis. Eur. Thyroid. J. 2021, 10, 125–139. [Google Scholar] [CrossRef]
- Journal, E.; Saradamba, A.; Buch, P.R.; Murawala, H.A.; Balakrishnan, S. Scholars Research Library SU5402, a Pharmacological Inhibitor of Fibroblast Growth Factor Receptor (FGFR), Effectively Hampers the Initiation and Progression of Fin Regeneration in Teleost Fish. Eur. J. Zool. Res. 2013, 4, 1–9. [Google Scholar]
- Tang, D.; He, Y.; Li, W.; Li, H. Wnt/β-Catenin Interacts with the FGF Pathway to Promote Proliferation and Regenerative Cell Proliferation in the Zebrafish Lateral Line Neuromast. Exp. Mol. Med. 2019, 51, 1–16. [Google Scholar] [CrossRef]
- Oshima, N.; Onimaru, H.; Yamagata, A.; Ito, S.; Imakiire, T.; Kumagai, H. Rostral Ventrolateral Medulla Neuron Activity Is Suppressed by Klotho and Stimulated by FGF23 in Newborn Wistar Rats. Auton. Neurosci. Basic Clin. 2020, 224, 102640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudenzi, G.; Albertelli, M.; Dicitore, A.; Würth, R.; Gatto, F.; Barbieri, F.; Cotelli, F.; Florio, T.; Ferone, D.; Persani, L.; et al. Patient-Derived Xenograft in Zebrafish Embryos: A New Platform for Translational Research in Neuroendocrine Tumors. Endocrine 2017, 57, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Cirello, V.; Gaudenzi, G.; Grassi, E.S.; Colombo, C.; Vicentini, L.; Ferrero, S.; Persani, L.; Vitale, G.; Fugazzola, L. Tumor and Normal Thyroid Spheroids: From Tissues to Zebrafish. Minerva Endocrinol. 2018, 43, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zabel, M.; Grzeszkowiak, J. Characterisation of Thyroid Medullary Carcinoma TT Cell Line. Histol. Histopathol. 1997, 12, 283–289. [Google Scholar]
- Cooley, L.D.; Elder, F.F.B.; Knuth, A.; Gagel, R.F. Cytogenetic Characterization of Three Human and Three Rat Medullary Thyroid Carcinoma Cell Lines. Cancer Genet. Cytogenet. 1995, 80, 138–149. [Google Scholar] [CrossRef]
- Cuccuru, G.; Lanzi, C.; Cassinelli, G.; Pratesi, G.; Tortoreto, M.; Petrangolini, G.; Seregni, E.; Martinetti, A.; Laccabue, D.; Zanchi, C.; et al. Cellular Effects and Antitumor Activity of RET Inhibitor RPI-1 on MEN2A-Associated Medullary Thyroid Carcinoma. JNCI J. Natl. Cancer Inst. 2004, 96, 1006–1014. [Google Scholar] [CrossRef]
- Vitale, G.; Gaudenzi, G.; Dicitore, A.; Cotelli, F.; Ferone, D.; Persani, L. Zebrafish as an Innovative Model for Neuroendocrine Tumors. Endocr. Relat. Cancer 2014, 21, R67–R83. [Google Scholar] [CrossRef]
- Carra, S.; Gaudenzi, G. New Perspectives in Neuroendocrine Neoplasms Research from Tumor Xenografts in Zebrafish Embryos. Minerva Endocrinol. 2020, 45, 393–394. [Google Scholar] [CrossRef]
- Verrienti, A.; Tallini, G.; Colato, C.; Boichard, A.; Checquolo, S.; Pecce, V.; Sponziello, M.; Rosignolo, F.; de Biase, D.; Rhoden, K.; et al. RET Mutation and Increased Angiogenesis in Medullary Thyroid Carcinomas. Endocr.-Relat. Cancer 2016, 23, 665–676. [Google Scholar] [CrossRef]
- Matrone, A.; Gambale, C.; Prete, A.; Elisei, R. Sporadic Medullary Thyroid Carcinoma: Towards a Precision Medicine. Front. Endocrinol. 2022, 13, 864253. [Google Scholar] [CrossRef]
- Alao, J.P.; Michlikova, S.; Dinér, P.; Grøtli, M.; Sunnerhagen, P. Selective Inhibition of RET Mediated Cell Proliferation in Vitro by the Kinase Inhibitor SPP86. BMC Cancer 2014, 14, 853. [Google Scholar] [CrossRef] [PubMed]
- Pacini, F.; Elisei, R.; Romei, C.; Pinchera, A. RET Proto-Oncogene Mutations in Thyroid Carcinomas: Clinical Relevance. J. Endocrinol. Investig. 2000, 23, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Wohllkt, N.; Cote, G.J.; Bugalhot, M.M.J.; Ordonez, N.; Evans, D.B.; Goepfert, H.; Khorana, S.; Schultz, P.; Richards, C.S.; Gagel, R.F. Relevance of RET Proto-Oncogene Mutations in Sporadic Medullary Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 1996, 81, 3740–3745. [Google Scholar]
- Thein, K.Z.; Velcheti, V.; Mooers, B.H.M.; Wu, J.; Subbiah, V. Precision Therapy for RET-Altered Cancers with RET Inhibitors. Trends Cancer 2021, 7, 1074–1088. [Google Scholar] [CrossRef]
- Kim, M.; Kim, B.H. Current Guidelines for Management of Medullary Thyroid Carcinoma. Endocrinol. Metab. 2021, 36, 514–524. [Google Scholar] [CrossRef]
- Thornton, K.; Kim, G.; Maher, V.E.; Chattopadhyay, S.; Tang, S.; Moon, Y.J.; Song, P.; Marathe, A.; Balakrishnan, S.; Zhu, H.; et al. Vandetanib for the Treatment of Symptomatic or Progressive Medullary Thyroid Cancer in Patients with Unresectable Locally Advanced or Metastatic Disease: U.S. Food and Drug Administration Drug Approval Summary. Clin. Cancer Res. 2012, 18, 3722–3730. [Google Scholar] [CrossRef]
- Milling, R.V.; Grimm, D.; Krüger, M.; Grosse, J.; Kopp, S.; Bauer, J.; Infanger, M.; Wehland, M. Pazopanib, Cabozantinib, and Vandetanib in the Treatment of Progressive Medullary Thyroid Cancer with a Special Focus on the Adverse Effects on Hypertension. Int. J. Mol. Sci. 2018, 19, 3258. [Google Scholar] [CrossRef]
- Cappagli, V.; Moriconi, D.; Bonadio, A.G.; Giannese, D.; la Manna, G.; Egidi, M.F.; Comai, G.; Vischini, G.; Bottici, V.; Elisei, R.; et al. Proteinuria Is a Late-Onset Adverse Event in Patients Treated with Cabozantinib. J. Endocrinol. Investig. 2021, 44, 95–103. [Google Scholar] [CrossRef]
- Carra, S.; Gaudenzi, G.; Dicitore, A.; Saronni, D.; Cantone, M.C.; Plebani, A.; Ghilardi, A.; Borghi, M.O.; Hofland, L.J.; Persani, L.; et al. Vandetanib versus Cabozantinib in Medullary Thyroid Carcinoma: A Focus on Anti-angiogenic Effects in Zebrafish Model. Int. J. Mol. Sci. 2021, 22, 3031. [Google Scholar] [CrossRef]
- Wirth, L.J.; Robinson, B.; Boni, V.; Tan, D.S.W.; McCoach, C.; Massarelli, E.; Hess, L.M.; Jen, M.H.; Kherani, J.; Olek, E.; et al. Patient-Reported Outcomes with Selpercatinib Treatment Among Patients with RET-Mutant Medullary Thyroid Cancer in the Phase I/II LIBRETTO-001. Trial. Oncol. 2021, 27, 13–21. [Google Scholar] [CrossRef]
- Jozaghi, Y.; Zafereo, M.; Williams, M.D.; Gule-Monroe, M.K.; Wang, J.; Grubbs, E.G.; Vaporciyan, A.; Hu, M.I.; Busaidy, N.; Dadu, R.; et al. Neoadjuvant Selpercatinib for Advanced Medullary Thyroid Cancer. Head Neck 2021, 43, E7–E12. [Google Scholar] [CrossRef] [PubMed]
- Locantore, P.; Novizio, R.; Corsello, A.; Paragliola, R.M.; Pontecorvi, A.; Corsello, S.M. Discovery, Preclinical Development, and Clinical Application of Pralsetinib in the Treatment of Thyroid Cancer. Expert. Opin. Drug Discov. 2022, 17, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Bradford, D.; Larkins, E.; Pai-Scherf, L.H.; Chatterjee, S.; Mishra-Kalyani, P.S.; Wearne, E.; Helms, W.S.; Ayyoub, A.; Bi, Y.; et al. FDA Approval Summary: Pralsetinib for the Treatment of Lung and Thyroid Cancers with RET Gene Mutations or Fusions. Clin. Cancer Res. 2021, 27, 5452–5456. [Google Scholar] [CrossRef] [PubMed]
- Wirth, L.J.; Sherman, E.; Robinson, B.; Solomon, B.; Kang, H.; Lorch, J.; Worden, F.; Brose, M.; Patel, J.; Leboulleux, S.; et al. Efficacy of Selpercatinib in RET -Altered Thyroid Cancers. N. Engl. J. Med. 2020, 383, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Shankar, A.; Kurzawinski, T.; Ross, E.; Stoneham, S.; Beale, T.; Proctor, I.; Hulse, T.; Simpson, K.; Gaze, M.N.; Cattaneo, E.; et al. Treatment Outcome with a Selective RET Tyrosine Kinase Inhibitor Selpercatinib in Children with Multiple Endocrine Neoplasia Type 2 and Advanced Medullary Thyroid Carcinoma. Eur. J. Cancer 2021, 158, 38–46. [Google Scholar] [CrossRef]
- Subbiah, V.; Hu, M.I.; Wirth, L.J.; Schuler, M.; Mansfield, A.S.; Curigliano, G.; Brose, M.S.; Zhu, V.W.; Leboulleux, S.; Bowles, D.W.; et al. Pralsetinib for Patients with Advanced or Metastatic RET-Altered Thyroid Cancer (ARROW): A Multi-Cohort, Open-Label, Registrational, Phase 1/2 Study. Lancet Diabetes Endocrinol. 2021, 9, 491–501. [Google Scholar] [CrossRef]
- Solomon, B.J.; Tan, L.; Lin, J.J.; Wong, S.Q.; Hollizeck, S.; Ebata, K.; Tuch, B.B.; Yoda, S.; Gainor, J.F.; Sequist, L.V.; et al. RET Solvent Front Mutations Mediate Acquired Resistance to Selective RET Inhibition in RET-Driven Malignancies. J. Thorac. Oncol. 2020, 15, 541–549. [Google Scholar] [CrossRef]
- Dinér, P.; Alao, J.P.; Söderlund, J.; Sunnerhagen, P.; Grøtli, M. Preparation of 3-Substituted-1-Isopropyl-1H-pyrazolo [3,4-d]pyrimidin-4-amines as RET Kinase Inhibitors. J. Med. Chem. 2012, 55, 4872–4876. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Giacomini, A.; Chiodelli, P.; Matarazzo, S.; Rusnati, M.; Presta, M.; Ronca, R. Blocking the FGF/FGFR System as a “Two-Compartment” Antiangiogenic/Antitumor Approach in Cancer Therapy. Pharmacol. Res. 2016, 107, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Ceolin, L.; da Silveira Duval, M.A.; Benini, A.F.; Ferreira, C.V.; Maia, A.L. Medullary Thyroid Carcinoma beyond Surgery: Advances, Challenges, and Perspectives. Endocr.-Relat. Cancer 2019, 26, R499–R518. [Google Scholar] [CrossRef] [PubMed]
- Atzori, M.G.; Ceci, C.; Ruffini, F.; Trapani, M.; Barbaccia, M.L.; Tentori, L.; D’Atri, S.; Lacal, P.M.; Graziani, G. Role of VEGFR-1 in Melanoma Acquired Resistance to the BRAF Inhibitor Vemurafenib. J. Cell. Mol. Med. 2020, 24, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Turczyk, L.; Kitowska, K.; Mieszkowska, M.; Mieczkowski, K.; Czaplinska, D.; Piasecka, D.; Kordek, R.; Skladanowski, A.C.; Potemski, P.; Romanska, H.M.; et al. FGFR2-Driven Signaling Counteracts Tamoxifen Effect on ERα-Positive Breast Cancer Cells. Neoplasia 2017, 19, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, N.; Fassl, A.; Mondani, G.; Generali, D.; Otto, T. Targeting Aberrant FGFR Signaling to Overcome CDK4/6 Inhibitor Resistance in Breast Cancer. Cells 2021, 10, 293. [Google Scholar] [CrossRef]
- Sun, Y.; Fan, X.; Zhang, Q.; Shi, X.; Xu, G.; Zou, C. Cancer-Associated Fibroblasts Secrete FGF-1 to Promote Ovarian Proliferation, Migration, and Invasion through the Activation of FGF-1/FGFR4 Signaling. Tumor Biol. 2017, 39, 1010428317712592. [Google Scholar] [CrossRef]
- Huang, S.; Liang, S.; Chen, G.; Chen, J.; You, K.; Ye, H.; Li, Z.; He, S. Overexpression of Glycosyltransferase 8 Domain Containing 2 Confers Ovarian Cancer to CDDP Resistance by Activating FGFR/PI3K Signalling Axis. Oncogenesis 2021, 10, 55. [Google Scholar] [CrossRef]
- Giacomini, A.; Grillo, E.; Rezzola, S.; Ribatti, D.; Rusnati, M.; Ronca, R.; Presta, M. The Fgf/Fgfr System in the Physiopathology of the Prostate Gland. Physiol. Rev. 2021, 101, 569–610. [Google Scholar] [CrossRef]
- Zhu, G.-J.; Liu, F.; Xu, Y.-G.; Zhao, C.-X.; Zhao, J.-G.; Sun, C. HMGN5 Promotes Invasion and Migration of Colorectal Cancer through Activating FGF/FGFR Pathway. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 1330–1338. [Google Scholar] [CrossRef]
- Ferguson, H.R.; Smith, M.P.; Francavilla, C. Fibroblast Growth Factor Receptors (FGFRs) and Noncanonical Partners in Cancer Signaling. Cells 2021, 10, 1201. [Google Scholar] [CrossRef]
- Kong, D.; Zhou, H.; Neelakantan, D.; Hughes, C.J.; Hsu, J.Y.; Srinivasan, R.R.; Lewis, M.T.; Ford, H.L. VEGF-C Mediates Tumor Growth and Metastasis through Promoting EMT-Epithelial Breast Cancer Cell Crosstalk. Oncogene 2021, 40, 964–979. [Google Scholar] [CrossRef]
- Prasad, C.B.; Singh, D.; Pandey, L.K.; Pradhan, S.; Singh, S.; Narayan, G. VEGFa/VEGFR2 Autocrine and Paracrine Signaling Promotes Cervical Carcinogenesis via β-Catenin and Snail. Int. J. Biochem. Cell Biol. 2022, 142, 106122. [Google Scholar] [CrossRef] [PubMed]
- Schoeffner, D.J.; Matheny, S.L.; Akahane, T.; Factor, V.; Berry, A.; Merlino, G.; Thorgeirsson, U.P. VEGF Contributes to Mammary Tumor Growth in Transgenic Mice through Paracrine and Autocrine Mechanisms. Lab. Investig. 2005, 85, 608–623. [Google Scholar] [CrossRef] [PubMed]
- Su, J.-L.; Yen, C.-J.; Chen, P.-S.; Chuang, S.-E.; Hong, C.-C.; Kuo, I.-H.; Chen, H.-Y.; Hung, M.-C.; Kuo, M.-L. The Role of the VEGF-C/VEGFR-3 Axis in Cancer Progression. Br. J. Cancer 2007, 96, 541–545. [Google Scholar] [CrossRef]
- Komorowski, J.; Pasieka, Z.; Jankiewicz-Wika, J.; Stepień, H. Matrix Metalloproteinases, Tissue Inhibitors of Matrix Metalloproteinases and Angiogenic Cytokines in Peripheral Blood of Patients with Thyroid Cancer. Thyroid 2002, 12, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, S.; Huang, P.; Dackiw, A.; Asa, S.L. Dual Inhibition of RET and FGFR4 Restrains Medullary Thyroid Cancer Cell Growth. Clin. Cancer Res. 2005, 11, 1336–1341. [Google Scholar] [CrossRef] [PubMed]
- Heilmann, A.M.; Subbiah, V.; Wang, K.; Sun, J.X.; Elvin, J.A.; Chmielecki, J.; Sherman, S.I.; Murthy, R.; Busaidy, N.L.; Subbiah, I.; et al. Comprehensive Genomic Profiling of Clinically Advanced Medullary Thyroid Carcinoma. Oncology 2016, 90, 339–346. [Google Scholar] [CrossRef]
- Rodríguez-Antona, C.; Pallares, J.; Montero-Conde, C.; Inglada-Pérez, L.; Castelblanco, E.; Landa, I.; Leskelä, S.; Leandro-García, L.J.; López-Jiménez, E.; Letón, R.; et al. Overexpression and Activation of EGFR and VEGFR2 in Medullary Thyroid Carcinomas Is Related to Metastasis. Endocr. Relat. Cancer 2010, 17, 7–16. [Google Scholar] [CrossRef]
- Schlumberger, M.; Jarzab, B.; Cabanillas, M.E.; Robinson, B.; Pacini, F.; Ball, D.W.; McCaffrey, J.; Newbold, K.; Allison, R.; Martins, R.G.; et al. A Phase II Trial of the Multitargeted Tyrosine Kinase Inhibitor Lenvatinib (E7080) in Advanced Medullary Thyroid Cancer. Clin. Cancer Res. 2016, 22, 44–53. [Google Scholar] [CrossRef]
- Chen, J.; Ji, Q.; Bai, C.; Zheng, X.; Zhang, Y.; Shi, F.; Li, X.; Tang, P.; Xu, Z.; Huang, R.; et al. Surufatinib in Chinese Patients with Locally Advanced or Metastatic Differentiated Thyroid Cancer and Medullary Thyroid Cancer: A Multicenter, Open-Label, Phase II Trial. Thyroid 2020, 30, 1245–1253. [Google Scholar] [CrossRef]
- Li, D.; Chi, Y.; Chen, X.; Ge, M.; Zhang, Y.; Guo, Z.; Wang, J.; Chen, J.; Zhang, J.; Cheng, Y.; et al. Anlotinib in Locally Advanced or Metastatic Medullary Thyroid Carcinoma: A Randomized, Double-Blind Phase IIB Trial. Clin. Cancer Res. 2021, 27, 3567–3575. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Type | Primer Sequence | Ta | Length |
|---|---|---|---|---|
| FGFR-1 | Forward Reverse | GGGCTGGAATACTGCTACAA GCCAAAGTCTGCTATCTTCATC | 60 °C | 192 bp |
| FGFR-2 | Forward Reverse | GGATAACAACACGCCTCTCTT GCCCAAAGCAACCTTCTC | 56 °C | 144 bp |
| FGFR-3 | Forward Reverse | TGGTGTCCTGTGCCTACC CCGTTGGTCGTCTTCTTGT | 56 °C | 181 bp |
| FGFR-4 | Forward Reverse | AACCGCATTGGAGGCATT TCTACCAGGCAGGTGTATGT | 56 °C | 98 bp |
| FGF-2 | Forward Reverse | TGTGTCTATCAAAGGAGTGTG CCGTAACACATTTAGAAGCCA | 56 °C | 83 bp |
| FGF-8 | Forward Reverse | TCTCCCAACAGCATGTGAG CTGTAGAGTTGGTAGGTCCG | 56 °C | 82 bp |
| Cell Line | TKI | EC50 | Maximal Inhibition |
|---|---|---|---|
| SU5402 | 3.6 µM | −96.5% | |
| TT | Sulfatinib | 2 µM | −100% |
| SPP86 | 1.3 µM | −100% | |
| SU5402 | 2.6 µM | −80.6% | |
| MZ-CRC-1 | Sulfatinib | 0.6 µM | −89.5% |
| SPP86 | 0.6 µM | −82.5% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saronni, D.; Gaudenzi, G.; Dicitore, A.; Carra, S.; Cantone, M.C.; Borghi, M.O.; Barbieri, A.; Mignani, L.; Hofland, L.J.; Persani, L.; et al. Preclinical Evaluation of Novel Tyrosine-Kinase Inhibitors in Medullary Thyroid Cancer. Cancers 2022, 14, 4442. https://doi.org/10.3390/cancers14184442
Saronni D, Gaudenzi G, Dicitore A, Carra S, Cantone MC, Borghi MO, Barbieri A, Mignani L, Hofland LJ, Persani L, et al. Preclinical Evaluation of Novel Tyrosine-Kinase Inhibitors in Medullary Thyroid Cancer. Cancers. 2022; 14(18):4442. https://doi.org/10.3390/cancers14184442
Chicago/Turabian StyleSaronni, Davide, Germano Gaudenzi, Alessandra Dicitore, Silvia Carra, Maria Celeste Cantone, Maria Orietta Borghi, Andrea Barbieri, Luca Mignani, Leo J. Hofland, Luca Persani, and et al. 2022. "Preclinical Evaluation of Novel Tyrosine-Kinase Inhibitors in Medullary Thyroid Cancer" Cancers 14, no. 18: 4442. https://doi.org/10.3390/cancers14184442
APA StyleSaronni, D., Gaudenzi, G., Dicitore, A., Carra, S., Cantone, M. C., Borghi, M. O., Barbieri, A., Mignani, L., Hofland, L. J., Persani, L., & Vitale, G. (2022). Preclinical Evaluation of Novel Tyrosine-Kinase Inhibitors in Medullary Thyroid Cancer. Cancers, 14(18), 4442. https://doi.org/10.3390/cancers14184442