1. Introduction
Breast cancer (BC) is one of the most commonly diagnosed cancers, which is a very significant clinical problem. It is estimated that there were nearly 2,300,000 new cases of this cancer in 2020, which is around 6300 new diagnoses per day. Unfortunately, these numbers are intimidating and treatment is becoming more and more challenging for clinicians due to increasing chemoresistance and the high mortality rate associated with it [
1]. For this reason, there is a constant need for improving methods involving early cancer diagnostics or creating new drugs with precise molecular action and high anticancer activity, including overcoming multidrug resistance (MDR).
Oncologic surgery is the main therapeutic procedure for the treatment of BC, and systemic administration of cytostatic drugs is based on a strategy of neoadjuvant therapy (before surgery, to support surgical treatment), adjuvant therapy (after surgery, as an adjunct to operation), or induction therapy (to reduce tumor mass so that surgery can be performed) [
2]. In 1978, the U.S. Food and Drug Administration (FDA) approved cisplatin for oncology treatment and today it is one of the most commonly used drugs during chemotherapy, especially in triple-negative BC (TNBC). Therapy with this agent is burdened with many harsh side effects that limit its effective dose and clinical use, but this is not the most serious medical problem [
3]. The major difficulty is the increasing MDR of cancer cells to this substance [
4]. From the available evidence, long-term treatment of BC with cisplatin leads to the development of resistance and a decrease in its therapeutic efficacy, despite the initial inhibition of tumor growth [
5]. Therefore, based on these two premises, the search for anticancer agents with a high therapeutic index, exhibiting the potential to overcome MDR, is ongoing.
Selenium (Se) belongs to the group of trace elements. Despite its relatively small quantities in the human body, it exerts many important functions in it. It manifests its activity mainly through selenoproteins, into which it is incorporated in the form of selenomethionine (SeMet) or selenocysteine (Sec). These selenoamino acids are found in two important antioxidant enzymes—glutathione peroxidase (GPx) and thioredoxin reductase (TrxR)—which protect cells from oxidative stress. Moreover, Se is involved in the metabolism of thyroid hormones (it is incorporated into the enzyme—iodothyronine deiodinase (DIO)) and the recovery of vitamin C and E from their metabolites. Its indispensable role is also seen in the aging process, fertility, and the immune system, and recent reports indicate its preventive effect against cancer [
6,
7,
8]. Meta-analyses and systematic reviews of clinical trials with Se confirm its chemopreventive activity in lung [
9], thyroid [
10], and breast cancer [
11], among others. This effect is probably due to not only the neutralization of free radicals but also the inhibition of neoangiogenesis and induction of apoptosis in cancer cells [
6], which would indicate that compounds containing Se in their structure could be potential chemopreventive or anticancer agents.
There are many Se compounds among which there are two main chemical groups: inorganic and organic derivatives. Inorganic Se compounds—selenites, selenates—exhibit genotoxic effects; hence, organic Se-containing compounds devoid of this toxicity to DNA have gained more attention. The group of organoselenium compounds consists of more than 10 chemical classes—including selenoesters [
12]. These compounds are characterized by rapid action as a result of their molecule hydrolysis in the intracellular environment and release of reactive Se-containing forms with anticancer activity [
13]. In the course of research performed by many teams all over the world, it was shown that selenoesters possess high cytotoxic potential, even in nanomolar concentrations, against many types of cancers (lung, colorectal, hepatic, pancreatic, ovarian, cervical, skin) [
14,
15,
16,
17]. Moreover, they exhibit the ability to overcome MDR [
18,
19,
20], which provides a promising premise for further studies on this class of compounds and to explore their molecular mechanism of anticancer effects. Therefore, the aim of our study was an in-depth evaluation of the anticancer activity of novel selenoesters (EDA-71, E-NS-4) in MCF-7 and MDA-MB-231 human breast cancer cells.
2. Materials and Methods
2.1. Materials
Cisplatin, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), dimethyl sulfoxide (DMSO), formaldehyde, glycine, methanol, sodium hydroxide, sodium dodecyl sulfate (SDS), and Tris were purchased from Sigma-Aldrich (St. Louis, MO, USA). Ethanol and sodium chloride were obtained from Avantor Performance Materials (Poland), while hydrochloric acid and trichloroacetic acid (TCA) were from Chempur (Poland). Stock cultures of human breast cancer cells (MCF-7 and MDA-MB-231) and normal human breast epithelial cells (MCF-10A) were provided by the American Type Culture Collection (ATCC, Manassas, VA, USA). Dulbecco’s Minimal Eagle Medium (DMEM), fetal bovine serum (FBS), phosphate-buffered saline (PBS) used in a cell culture, trypsin, glutamine, penicillin, and streptomycin were from Gibco (San Diego, CA, USA). An MEGM Mammary Epithelial Cell Growth Medium BulletKit was purchased from Lonza Bioscience (Basel, Switzerland). [3H]-thymidine (7 Ci/mmol) was received from Moravek Biochemicals (Brea, CA, USA), and Scintillation Coctail Ultima Gold XR from PerkinElmer (Waltham, MA, USA). DNase-free RNase A Solution was a product of Promega (Madison, WI, USA). FITC Annexin V Apoptosis Detection Kit II, JC-1 MitoScreen Kit and Stain Buffer were from BD Pharmigen (San Diego, CA, USA). An Autophagy Assay kit, FAM-FLICA® Caspase-3/7 Assay kit, FAM-FLICA® Caspase-8 Assay kit, FAM-FLICA® Caspase-9 Assay kit, FAM-FLICA® Caspase-10 Assay kit, and propidium iodide were purchased from ImmunoChemistry Technologies (Bloomington, MN, USA). Cyclin E1 Mouse mAb, Cyclin A2 Rabbit mAb, mTOR Rabbit mAb (Alexa Fluor® 647 Conjugate), NF-κB p65 XP® Rabbit mAb (Alexa Fluor® 647 Conjugate), Alexa Fluor® 647-labeled secondary anti-mouse antibody, Alexa Fluor® 488-labeled secondary anti-rabbit antibody were products of Cell Signaling Technology (Beverly, MA, USA).
2.2. Tested Compounds
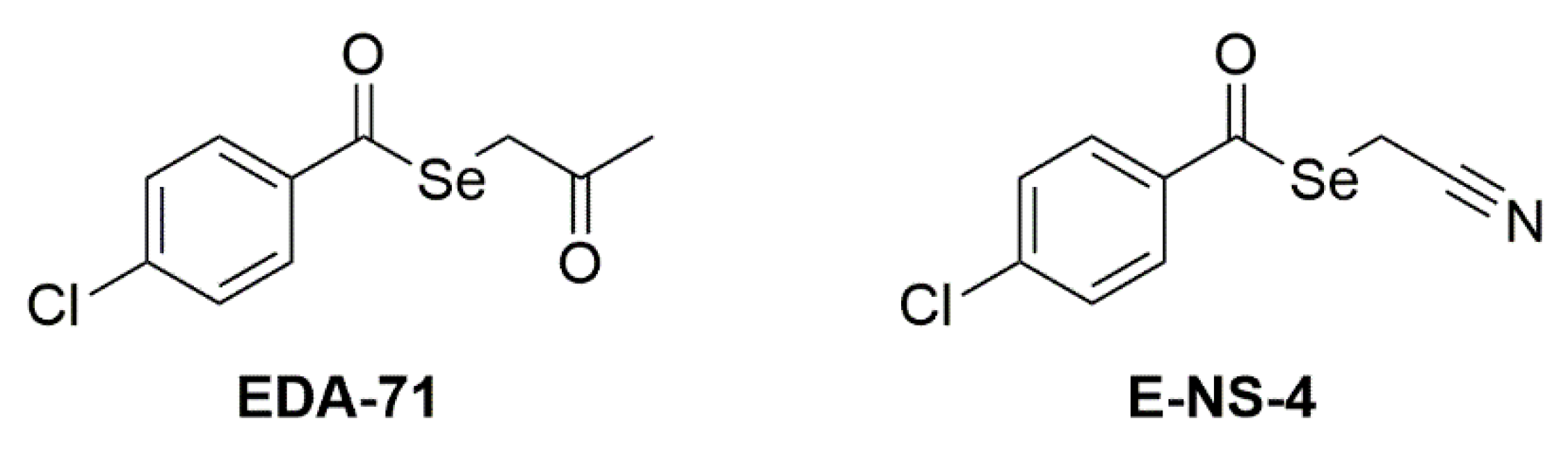
The two tested compounds (
Figure 1) are EDA-71 (Se-(2-oxopropyl) 4-chlorobenzoselenoate) and E-NS-4 (Se-cyanomethyl 4-chlorobenzoselenoate). They share the selenoester functional group (-(C=O)-Se-) and the aryl moiety (4-chlorophenyl) bound to the carbonyl of the selenoester. The difference lies in the alkyl moiety bound to the selenium atom, which contains a ketone group in EDA-71 (2-oxopropyl) and a nitrile group in E-NS-4 (2-cyanomethyl). The synthesis and characterization of the two selenocompounds have been described in previous works: EDA-71 in [
14] and E-NS-4 in [
21]. Briefly, 4-chlorobenzoyl chloride is added over a solution of sodium hydrogen selenide generated
in situ by the reduction of elemental grey selenium with sodium borohydride. After completion of the reaction, the crude of reaction is filtered to remove the salts generated in the process, and over the filtrate, chloroacetone or chloroacetonitrile is added, to form EDA-71 and E-NS-4, respectively. The desired compound precipitates from the reaction media and it is filtered, dried, and purified as previously described [
14,
21].
2.3. Cell Culture of MCF-7, MDA-MB-231, and MCF-10A Cells
Human breast cancer cell lines (MCF-7 and MDA-MB-231) and normal human breast epithelial cells (MCF-10A) were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). MCF-7 and MDA-MB-231 cells were cultured in Dulbecco’s Modified Eagle Medium (Gibco, San Diego, CA, USA), MCF-10A cells were cultured in Mammary Epithelial Cell Growth Medium with supplements: BPE, hEGF, insulin, hydrocortisone, GA-1000 (Lonza, Basel, Switzerland). All media were complemented by 10% of fetal bovine serum (FBS) and 1% of antibiotics: penicillin and streptomycin (both Gibco, San Diego, CA, USA). The cells were maintained in an incubator that provides the optimal growth conditions for the cell culture: 5% CO2, 37 °C, and humidity in a range of 90–95%. The cells were cultured in 100 mm plates (Sarstedt, Newton, NC, USA). Subsequently after obtaining a subconfluent cell culture, the cells were detached with 0.05% trypsin with 0.02% EDTA (Gibco, San Diego, CA, USA). Then, utilizing a Scepter 3.0 handheld automated cell counter (Milipore, Burlington, MA, USA), the number of cells was quantified and seeded at a density of 5 × 105 cells per well in six-well plates (“Nunc”) in 2 mL of the growth medium (Dulbecco’s Modified Eagle Medium or Mammary Epithelial Cell Growth Medium, respectively). In the present study, cells that obtained 80% of confluence were used.
2.4. Cell Viability Assay
The cytotoxic activity of the tested compounds against breast cancer (MCF-7 and MDA-MB-231) and normal human breast epithelial (MCF-10A) cells was determined by MTT assay according to Carmichael’s method. The evaluation of cell viability was based on measuring their ability to enzymatically reduce yellow tetrazolium salt (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, MTT) to purple formazan under the influence of the enzyme—mitochondrial dehydrogenase, which is only found in living cells [
22]. All cultured cell lines were treated with different concentrations of the tested compounds in medium (0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, and 5 µM) and incubated for 24 h in an incubator providing optimal conditions for cell culture growth (37 °C temperature, 5% CO
2, and 90–95% humidity) in six-well plates (baseline seeding density: 5 × 10
5 cells/well). After this time, the medium was removed and the cell monolayer was washed with warm phosphate-buffered saline (PBS) without calcium and magnesium. In the next step, PBS and MTT solution in PBS (5 mg/mL) were added to each well to obtain a final MTT concentration of 0.5 mg/mL, and then the cells were incubated for 4 h (37 °C temperature, 5% CO
2, 90–95% humidity). After the incubation period, the content of each well was removed, 1 mL of DMSO was added to dissolve the formazan, and then the plate with the added DMSO was mixed for 5–10 min on a microplate shaker (Boeco, Hamburg, Germany). Then, 10 µL of Sorensen’s glycine buffer (a solution of 0.1 M glycine and 0.1 M sodium chloride adjusted to pH 10.5 with 0.1 M sodium hydroxide) was added and the absorbance of the obtained solution was immediately measured at a wavelength of λ = 570 nm using a Thermo Scientific Evolution 201 UV-VIS spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). The absorbance result obtained in the control cells (without the addition of the tested compounds) was taken as 100%, while the viability of cells incubated with the tested compounds was presented as a percentage of the control value.
2.5. [3H]-Thymidine Incorporation Assay
The antiproliferative activity of the tested compounds was evaluated by measuring the incorporation of [3H]-thymidine into the cancer cell DNA after 24 h of incubation. MCF-7 and MDA-MB-231 breast cancer cells were seeded into six-well plates (density 5 ×105 cells/well) and cultured for 24 h in an incubator under optimal conditions for cell growth (37 °C, 5% CO2, 90–95% humidity). Then, they were incubated under the same conditions for 24 h with a growth medium containing the tested compounds at different concentrations (0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 4.5, and 5 µM). After 24 h, the medium was removed, the cells were washed with PBS (Corning, Kennebunk, ME, USA), and a fresh medium was added. After that, the cells were treated with 0.5 µCi of tritium-labeled thymidine (specific activity 7 Ci/mmol) by incubating them with this compound for 4 h under the same conditions as the cell culture. In the first step, the medium was removed and the cell monolayer was washed twice with 1 mL of 0.05 M Tris-HCl buffer pH 7.4 containing 0.11 M NaCl. To denature proteins, the cells were washed twice with 1 mL of 5% trichloroacetic acid (TCA) solution. Finally, cell lysis was performed by adding 1 mL of 0.1 M sodium hydroxide (NaOH) solution containing 1% sodium dodecyl sulfate (SDS) to each well. After five minutes, the obtained cell lysates were transferred to scintillation vials with 2 mL of scintillation fluid added to them beforehand. Radioactivity was measured using the Scintillation Counter 1900 TR, TRI-CARB (Packard, Perkin Elmer, Inc., San Jose, CA, USA). The intensity of DNA biosynthesis in the analyzed cells was expressed as dpm/well. The result of radioactivity measurement in the control cells (without the addition of tested compounds) was taken as 100%, while the values of cells incubated with the tested compounds were presented as a percentage of the control value.
2.6. Flow Cytometry Assessment of Annexin V and Propidium Iodide Binding
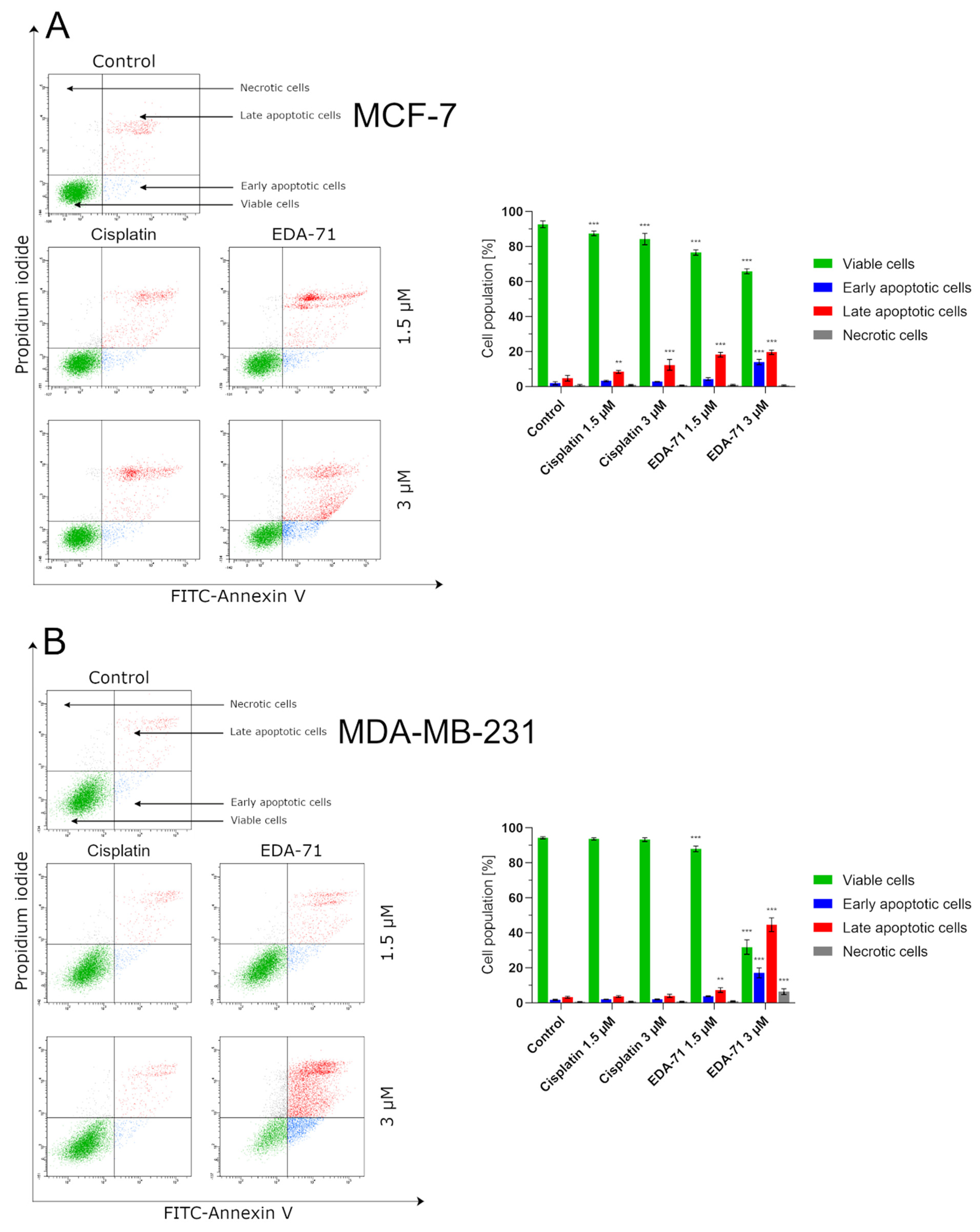
The apoptosis induction by the tested compounds was assessed by the exposure of phosphatidylserine on the cell membrane, to which fluorescein isothiocyanate (FITC)-labeled annexin V binds with high affinity in the presence of Ca2+ calcium ions. The FITC Annexin V Apoptosis Detection Kit II (BD Pharmingen, San Diego, CA, USA) and a flow cytometer (BD FACSCanto II, BD Biosciences Systems, San Jose, CA, USA) were used for this detection. The assay was performed according to the manufacturer’s instructions. Breast cancer cells (MCF-7 and MDA-MB-231) were incubated for 24 h (37 °C, 5% CO2, 90–95% humidity) with the most active compound (EDA-71) and the reference drug (cisplatin) at concentrations of 1.5 and 3 µM. Flow cytometer calibration was performed by preparing two controls—a positive and a negative control. The positive control was cells in which apoptosis was induced using 2 µL of 3% formaldehyde in buffer and placing them on ice for 30 min. The negative control was cells that were not treated with any of the proapoptotic agents. First, in cells treated with the tested compounds as well as the controls, the medium was removed and the cells were washed twice with cold PBS. Subsequently, the cells were resuspended in Binding Buffer included in the kit at a concentration of 1 × 106 cells/mL. From each sample, 100 µL of cell suspension was taken and transferred to test tubes, to which 5 µL each of FITC Annexin V and propidium iodide (PI) was then added. The contents of the test tubes were gently vortexed and incubated for 15 min at room temperature, protected from light. After the required time, the contents of the test tubes were made up to 500 µL with Binding Buffer and immediately analyzed in a flow cytometer (10,000 events measured). After the flow cytometer readout, the results were analyzed using FACSDiva software (BD Biosciences Systems, San Jose, CA, USA). The equipment was calibrated with BD Cytometer Setup and Tracking Beads (BD Biosciences, San Diego, CA, USA).
2.7. Assessment of Changes in Mitochondrial Membrane Potential
In the early phase of apoptotic death occurring via the intrinsic pathway, there is an increase in mitochondrial membrane permeability, resulting in a decrease in mitochondrial membrane potential (MMP, ΔΨm). To detect these changes, the carbocyanine lipophilic cationic fluorochrome JC-1 included in the JC-1 MitoScreen kit (BD Pharmigen, San Diego, CA, USA) and a flow cytometer (BD FACSCanto II) with the appropriate software to analyze the obtained results (FACSDiva; both from BD Biosciences Systems) were used. The entire cell staining and cytometric analysis procedure was performed according to the instructions provided with the kit. MCF-7 and MDA-MB-231 cells were incubated for 24 h (37 °C, 5% CO2, 90–95% humidity) with cisplatin and compound EDA-71 (concentrations of 1.5 and 3 µM). After the incubation time with the tested compounds, the cells (1 × 106 cells/sample) were washed and resuspended in 0.5 mL of buffer containing 10 µg/mL JC-1 dye. Incubation was carried out for 15 min at room temperature, protected from light. Afterwards, the cells were washed twice with buffer, resuspended in 0.5 mL PBS and immediately analyzed using a flow cytometer (BD FACSCanto II; 10,000 events measured) and FACSDiva software to count the percentage of cells with reduced ΔΨm. The equipment was calibrated with BD Cytometer Setup and Tracking Beads (BD Biosciences, San Diego, CA, USA).
2.8. Caspases 3/7, 8, 9, and 10 Enzymatic Activity Assay
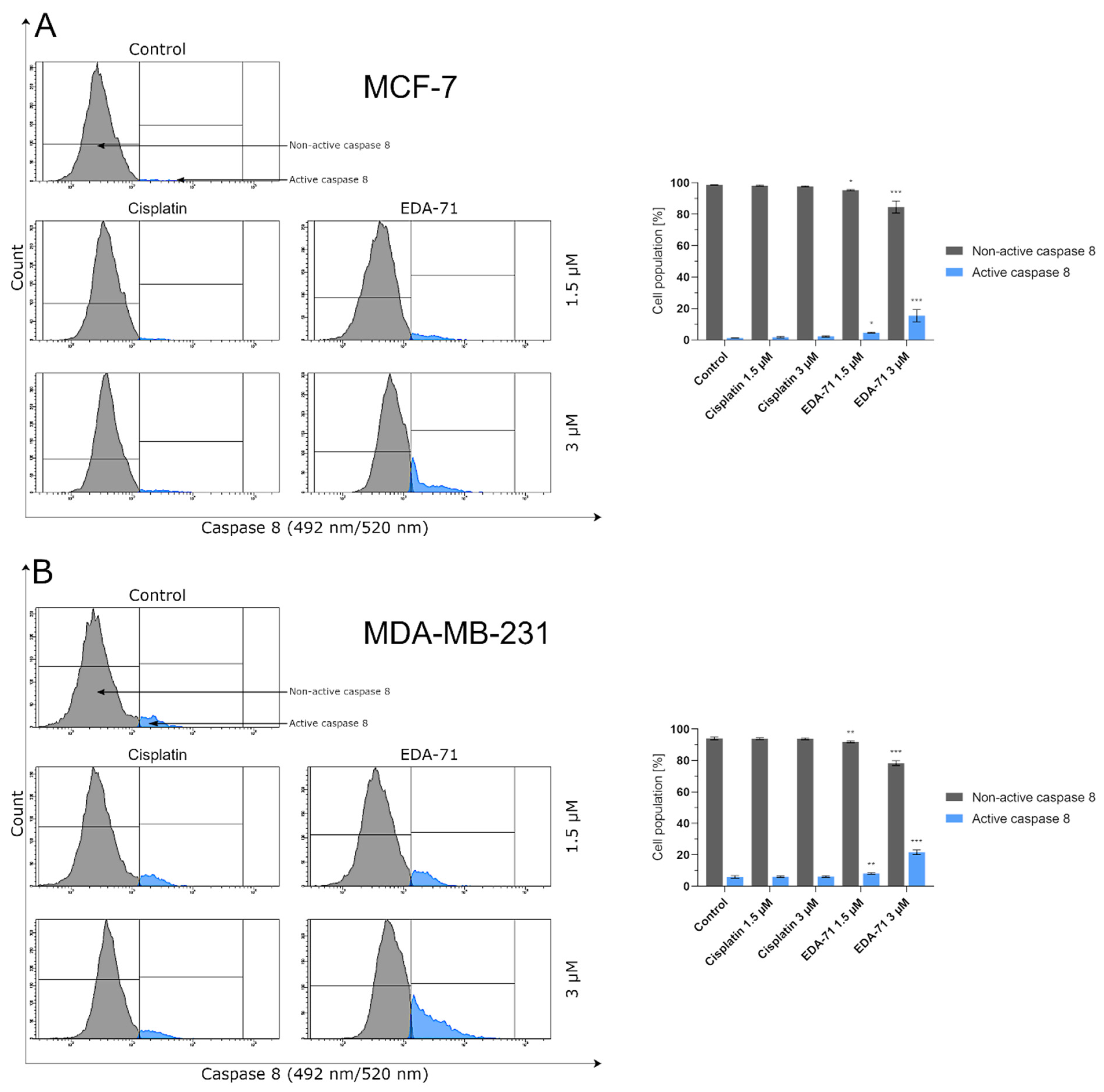
Activation of the caspase cascade occurs as a result of the initiation of the apoptotic process in the cell and is induced by the cytotoxic activity of the compound. In this regard, assessment of initiator (caspases 8, 9, and 10) and executioner (caspases 3 and 7) caspase activity was performed using FAM-FLICA® Caspase Assay kits (all from ImmunoChemistry Technologies, Bloomington, MN, USA) according to the manufacturer’s instructions. After 24 h incubation of MCF-7 and MDA-MB-231, breast cancer cells with the tested compounds (concentrations of 1.5 and 3 µM), the cells were collected, washed twice with cold PBS, and resuspended in Apoptosis Wash Buffer to a final concentration of 5 × 105 cells/mL. In the next step, 290 µL each of cell suspension was taken and transferred into tubes. Then, 10 µL of FLICA solution diluted immediately before use (1:5 v/v, using PBS) was added to the cells, mixed by pipetting, and incubated in the dark for 1 h at 37 °C. After this time, the cells were washed twice with 2 mL Apoptosis Wash Buffer, centrifuged, and resuspended in 300 µL of the buffer. Thus, prepared samples were immediately analyzed using a BD FACSCanto II flow cytometer (10,000 events) with FACSDiva software (both from BD Biosciences Systems, San Jose, CA, USA). The equipment calibration was performed using BD Cytometer Setup and Tracking Beads (BD Biosciences, San Diego, CA, USA).
2.9. Measuring the Number of Autophagosomes and Autolysosomes by Autophagy Assay
An autophagy assay was performed to evaluate whether the tested compounds induce the autophagy process in MCF-7 and MDA-MB-231 breast cancer cells. The stain included in the Autophagy Assay, Red kit (ImmunoChemistry Technologies, Bloomington, MN, USA) was an aliphatic molecule with the ability to enter the cell and fluoresce brightly upon binding to the lipid bilayer membranes of autophagosomes and autolysosomes. The entire assay was performed according to the manufacturer’s instructions. Cells were exposed to 1.5 and 3 µM of the tested compound (EDA-71) and cisplatin for 24 h. After drug treatment, unfixed cells were washed and resuspended in PBS at a concentration of 5 × 105 cells/mL. Then, 490 µL each of cell suspension was taken, transferred to test tubes, and 10 µL of Autophagy Probe, Red solution (previously diluted 1:5 in PBS) was added and incubated (30 min, 37 °C, in the dark). After incubation, the cells were washed and resuspended in Cellular Assay Buffer, finally adding Fixative at a ratio of 1:5 (v/v). After this step, the prepared samples were immediately measured using a flow cytometer (BD FACSCanto II; 10,000 events measured), and the percentage of cells with an occurring autophagy process was calculated using FACSDiva software (both from BD Biosciences Systems, San Jose, CA, USA). The equipment calibration was performed using BD Cytometer Setup and Tracking Beads (BD Biosciences, San Diego, CA, USA).
2.10. Cell Cycle Analysis Using Flow Cytometry
Seeded at a density of 5 × 105 cells/well (six-well plate) and then cultured for 24 h under optimal conditions for cell culture growth (37 °C, 5% CO2, 90–95% humidity), MCF-7 and MDA-MB-231 breast cancer cells were treated for 24 h with 1.5 and 3 µM EDA-71 and cisplatin. After 24 h, cells were collected and fixed in 1 mL of 70% ethanol. The thus prepared samples were placed in a freezer at -20 °C overnight. On the next day, the fixed cells were washed with cold PBS, centrifuged for 10 min at 2000 rpm, and the supernatant was removed. The cell pellet was then resuspended in PBS containing 50 μg/mL DNase-free RNase A Solution (Promega, Madison, WI, USA) and stained with 100 μg/mL PI (ImmunoChemistry Technologies, Bloomington, MN, USA). Incubation was performed for 30 min at 37 °C in the dark. In the next step, the cells were washed and resuspended in PBS. Cell cycle phase distribution was analyzed using the FACSCanto II flow cytometer (10,000 events measured) with FACSDiva software (both from BD Biosciences Systems, San Jose, CA, USA) and then FCS Express software (De Novo Software, Pasadena, CA, USA). The equipment was calibrated with BD Cytometer Setup and Tracking Beads (BD Biosciences, San Diego, CA, USA).
2.11. Determination of NF-κB and mTOR Using Flow Cytometry
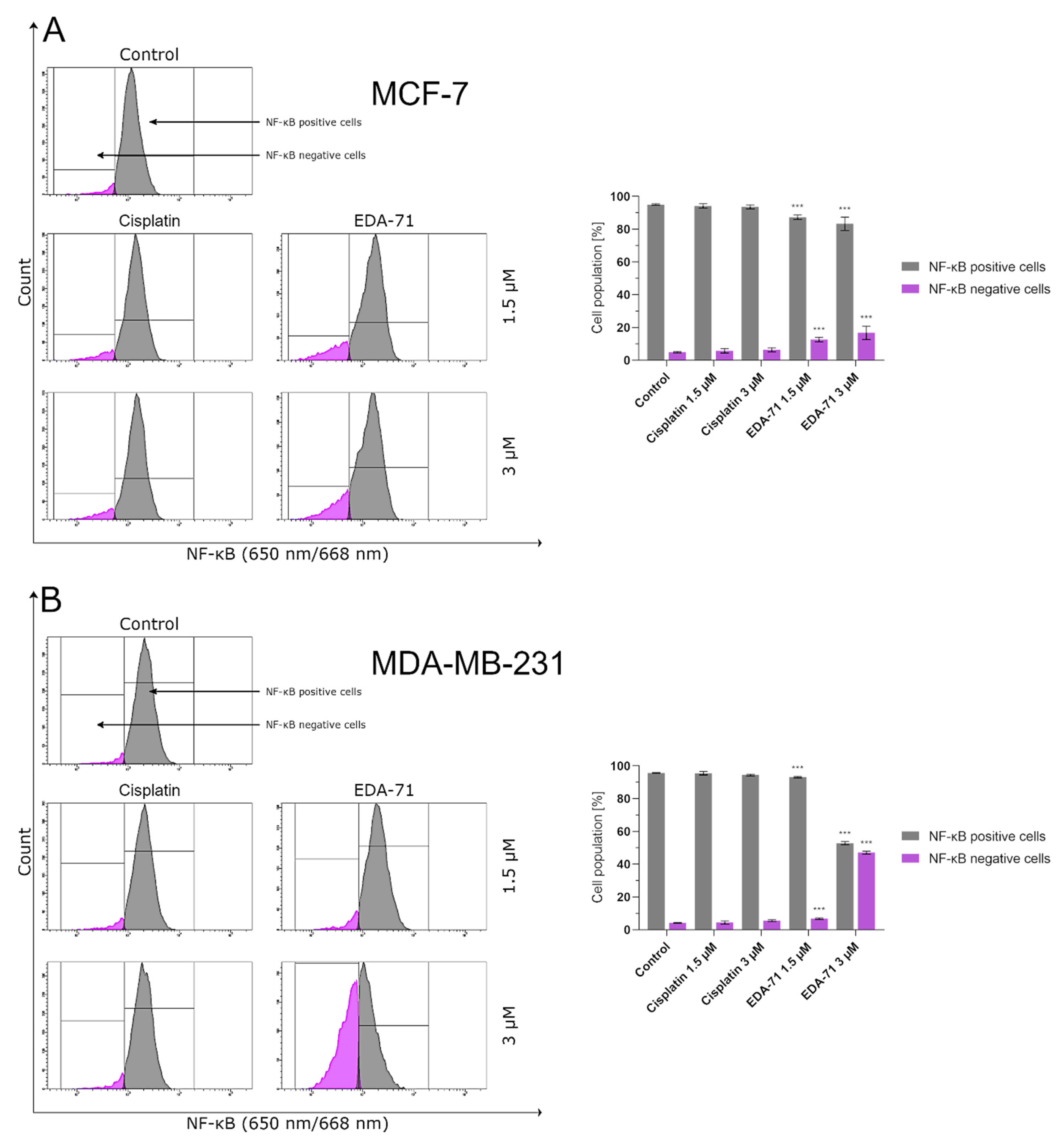
The protein levels of NF-κB and mTOR in MCF-7 and MDA-MB-231 breast cancer cells after 24 h exposure to the tested compounds (EDA-71 and cisplatin; concentrations of 1.5 and 3 µM) were evaluated by flow cytometry. For this purpose, an mTOR antibody and an NF-κB antibody conjugated to Alexa Fluor® 647 (all from Cell Signaling Technology, Beverly, MA, USA) were used. Each assay was performed according to the manufacturer’s protocol available on their website. After incubation with the tested compounds, the cells were centrifuged, resuspended in 4% formaldehyde (100 µL/1 × 106 cells), and fixed for 15 min at room temperature. Afterwards, to remove the formaldehyde, the cells were washed by centrifugation with excess PBS and began the permeabilization step. For this purpose, ice-cold 90% methanol was added to the pre-chilled cell pellet, gently vortexed, and incubated for 1 h on ice. Later, the cells were washed by centrifugation with excess PBS again, resuspended in 100 µL of 1:100 diluted primary antibody (Stain Buffer was used for dilution), and incubated for 1 h at room temperature in the dark. After incubation, the cells were washed, resuspended in 300 µL PBS and immediately tested with a flow cytometer (BD FACSCanto II, BD Biosciences Systems, San Jose, CA, USA; 10,000 events measured). The obtained results were analyzed using FACSDiva software (BD Biosciences Systems, San Jose, CA, USA). The equipment calibration was performed using BD Cytometer Setup and Tracking Beads (BD Biosciences, San Diego, CA, USA).
2.12. Determination of Cyclin E1 and Cyclin A2 Using Flow Cytometry
Apart from analyzing the distribution of cell cycle phases, in this study, cyclins responsible for regulating the cell division cycle (cyclin E1 and cyclin A2) were also assayed. Up-regulation of cyclin E1 and its binding to a complex with cyclin-dependent kinase 2 (Cdk2) enables cell cycle transition from the G1 to the S phase, while cyclin A2/Cdk1 complex stimulates cell transition from the G2 to the M phase (mitosis). Identification of these proteins was performed after 24 h treatment of MCF-7 and MDA-MB-231 cells with the tested compounds (EDA-71 and cisplatin; concentrations of 1.5 and 3 µM). Cyclin E1 and cyclin A2 antibodies (both from Cell Signaling Technology, Beverly, MA, USA) were used in this study, and each assay was performed according to the manufacturer’s protocol available on their website. After incubation with the tested compounds, the cells were centrifuged, resuspended in 4% formaldehyde (100 µL/1 × 106 cells), and fixed for 15 min at room temperature. Afterwards, to remove the formaldehyde, the cells were washed by centrifugation with excess PBS and began the permeabilization step. For this purpose, ice-cold 90% methanol was added to the pre-chilled cell pellet, gently vortexed, and incubated for 1 h on ice. Later, the cells were washed by centrifugation with excess PBS again, resuspended in 100 µL of 1:100 diluted primary antibody (Stain Buffer was used for dilution), and incubated for 1 h at room temperature in the dark. After the required incubation period, the cells were washed, resuspended in 100 µL of secondary antibody diluted in Stain Buffer (1:100 ratio; anti-mouse IgG Alexa Fluor® 647 Conjugate in the case of cyclin E1 and anti-rabbit IgG Alexa Fluor® 488 Conjugate in the case of cyclin A2; both from Cell Signaling Technology, Beverly, MA, USA) and incubated for 30 min at room temperature, protected from light. Subsequently, the cells were washed, resuspended in 300 µL PBS and immediately tested with a flow cytometer (BD FACSCanto II, BD Biosciences Systems, San Jose, CA, USA; 10,000 events measured). The obtained results were analyzed using FACSDiva software (BD Biosciences Systems, San Jose, CA, USA). The equipment calibration was performed using BD Cytometer Setup and Tracking Beads (BD Biosciences, San Diego, CA, USA).
2.13. Statistical Analysis
All results are presented as mean ± standard deviation (SD) from at least three independent experiments in triplicate. Statistical analysis was performed using GraphPad Prism 8 software (GraphPad Software, San Diego, CA, USA). Statistical differences between the experimental (treated cells) and control (untreated cells) groups were determined using one-way ANOVA followed by Dunnett’s test. Differences were considered statistically significant when p < 0.05.
4. Discussion
The steadily increasing incidence of cancer appears to be a worrying and global health problem. This is why numerous research teams are still taking steps to reduce this disease entity. One of the widely used therapeutic approaches in the treatment of cancer is chemotherapy, and the search for an effective anticancer drug is setting new directions of research in the field of oncology. An important position in the biological and biomedical processes of cancer is now occupied by Se compounds. Recent reports suggest an important role for Se in the chemoprevention of many cancers, including breast cancer. The mechanism would be based mainly on the maintenance of the oxidative–antioxidant balance in the body, the inhibition of angiogenesis, and the induction of the apoptotic process in cancer cells. Therefore, it is believed that compounds containing this element could be potential chemopreventive or anticancer agents [
6].
Previously, it was shown that selenium compounds have been known to be highly cytotoxic toward a variety of cancer cell lines [
12,
19,
40,
41,
42]. In this study, we demonstrated that both EDA-71 and E-NS-4 selenium compounds exhibited cytotoxic activity on breast cancer cell lines (MCF-7 and MDA-MB-231), with IC
50 values in terms of low micromolar concentrations (1.4 to 4.52 µM). We utilized a cell line that was more sensitive to the compound MCF-7 and one that was most resistant to MDA-MB-231 in order to determine if the compounds elicited different responses that may display their resistance or sensitivity to the compounds. Unfortunately, the study showed that the tested compounds did not show selectivity to the analyzed breast cancer cells. Nevertheless, it is significant that they show high cytotoxic activity at lower concentrations than cisplatin. Moreover, tested compounds EDA-71 and E-NS-4 exhibited comparable cytotoxicity in both normal breast epithelial and breast cancer cells. Similar observations were reported by the team of Manda et al. [
43] who studied the effects of another selenocompound, sodium selenite, in A549 lung cancer cell line and BEAS-2B normal human bronchial epithelial cells. However, the broad toxicity of these compounds may be overcome if the novel selenoesters were to be used in future clinical trials. Shortcomings frequently encountered with anticancer compounds, such as normal tissue toxicity, as well as the high incidence rate of drug resistance, may be overcome through the use of lipid-based nanocarriers or other nanomaterials. Importantly, these systems are biocompatible and based on physiological and morphological differences in cancer cells, and the additional functionalization of their surface enables targeting against these cells, sparing normal cells [
44,
45]. In a study conducted by Tang et al. in 2022 [
46], it was observed that the use of estrone-conjugated PEGylated liposome containing carboplatin increased the efficacy of this anticancer drug against ovarian cancer xenografts and significantly reduced acute toxicity in mice.
High cytotoxic activity can lead to cell death by several significantly different death pathways—apoptosis, entosis, necrosis, necroptosis (regulated necrosis), and autophagy—and the type of cell death depends on the properties of the chemical compound used or the dose applied [
12,
16,
40,
42]. The study by Shigemi et al. [
47] showed that Se compounds direct cancer cells mainly to the apoptosis pathway. The studies of our team seem to confirm this thesis, as they demonstrated a high capacity of EDA-71 to induce apoptosis in the concentration range oscillating around 1.5–3.0 µM, with a simultaneous negligible ability to activate necrosis. Due to the fact that EDA-71 showed much higher cytotoxic activity in comparison with E-NS-4, we focused our further attention only on this compound. The results obtained by flow cytometry after 24 h of incubation with the tested compound showed a correlation with the results obtained by MTT. The results obtained with both methods indicate that the type of cell death shifts towards apoptosis as the concentration of the tested compound increases. This is probably related to the fact that EDA-71 leads to very severe cell dysfunction and the activation of proteins involved in the process of apoptosis or autophagy. Additionally, the high proapoptotic activity of EDA-71 was also present in normal human breast epithelial cells (MCF-10A). Despite this unfavorable fact, it should be noted that the selenium structure investigated in this study may provide an excellent molecular framework for a novel group of anticancer drugs in the future. Compounds with high proapoptotic activity are still considered an excellent basis for future anticancer drugs [
48] and the high toxicity of compound EDA-71 can be reduced by using, for example, nanocarriers [
45] as we discussed above.
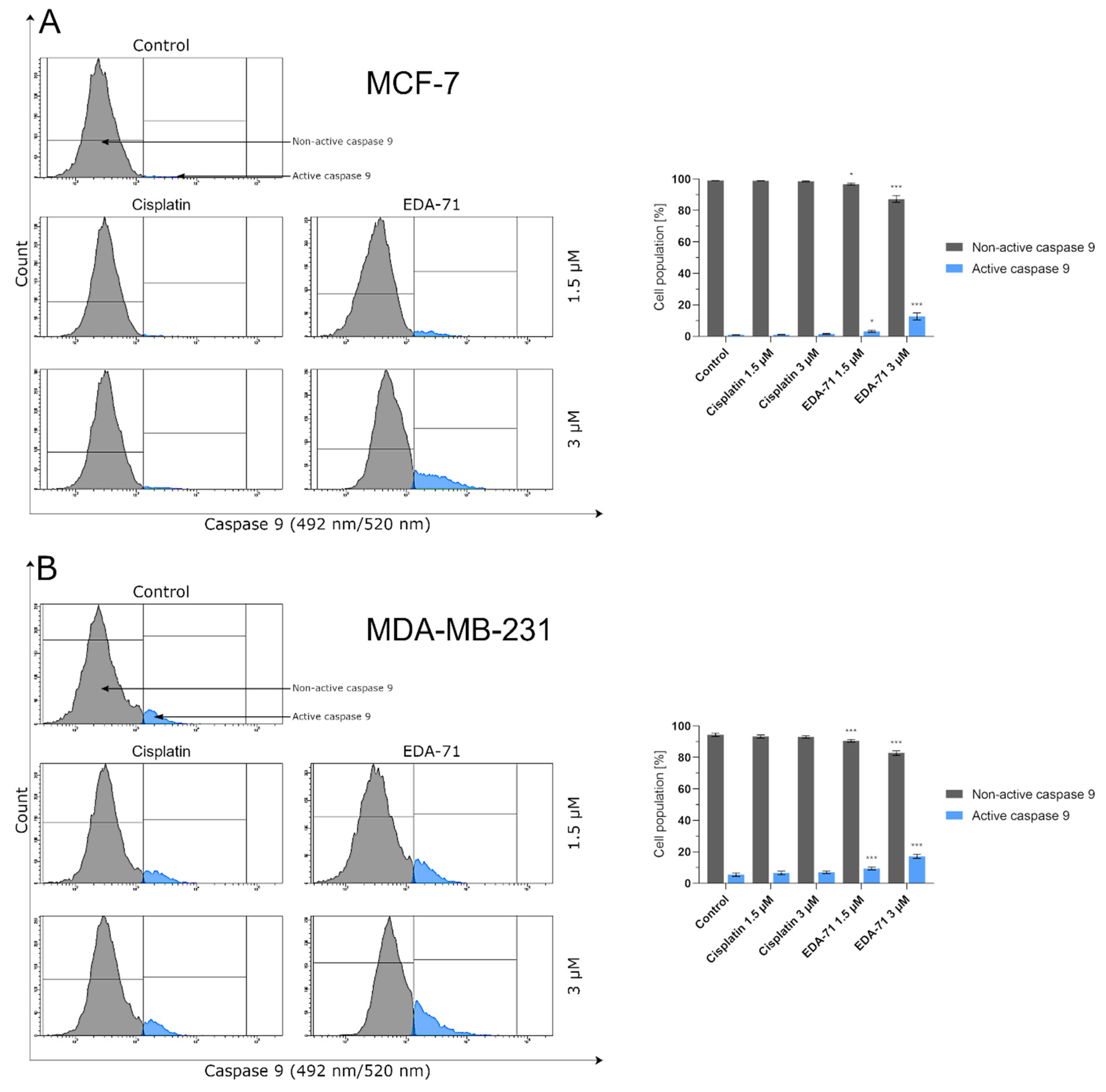
The process of Se-activated apoptosis can follow an intrinsic (mitochondrial) and extrinsic (receptor) pathway [
12,
40,
42,
47]. In the intrinsic pathway of apoptosis, a decrease in mitochondrial membrane potential, release of cytochrome c, and an increase in caspase 9 and caspase 3 activity can be observed [
23,
32,
49]. The mechanism of action of numerous selenoorganic compounds is related to the activation of the mitochondrial apoptotic pathway [
42,
47]. The study of Se derivative—ebselen—performed by Zhang et al. [
50] revealed a decrease in mitochondrial membrane potential, release of cytochrome c, and an increase in caspase 9 activity for human multiple myeloma U266 and RPMI8226 cells. To assess the ability of compound EDA-71 to direct cells to the apoptotic pathway associated with the activation of the intrinsic pathway, mitochondrial membrane potential change assay and a measurement of caspase 9 and caspase 3 activities were performed. Loss of mitochondrial transmembrane potential (ΔΨ
m) is considered an early marker of apoptosis [
51]. Sakallı Çetin et al. [
52] described a loss of ΔΨ
m that occurred in MCF-7 breast cancer cells treated with sodium selenite, while Zhang et al. [
53] demonstrated a decrease in ΔΨ
m in human epithelial cervical cancer HeLa cells treated with Se-containing phenylindolyl ketone derivative. The results we obtained for compound EDA-71 showed that the mitochondrial membrane potential decreased in a dose-dependent manner. The consequent loss of ΔΨ
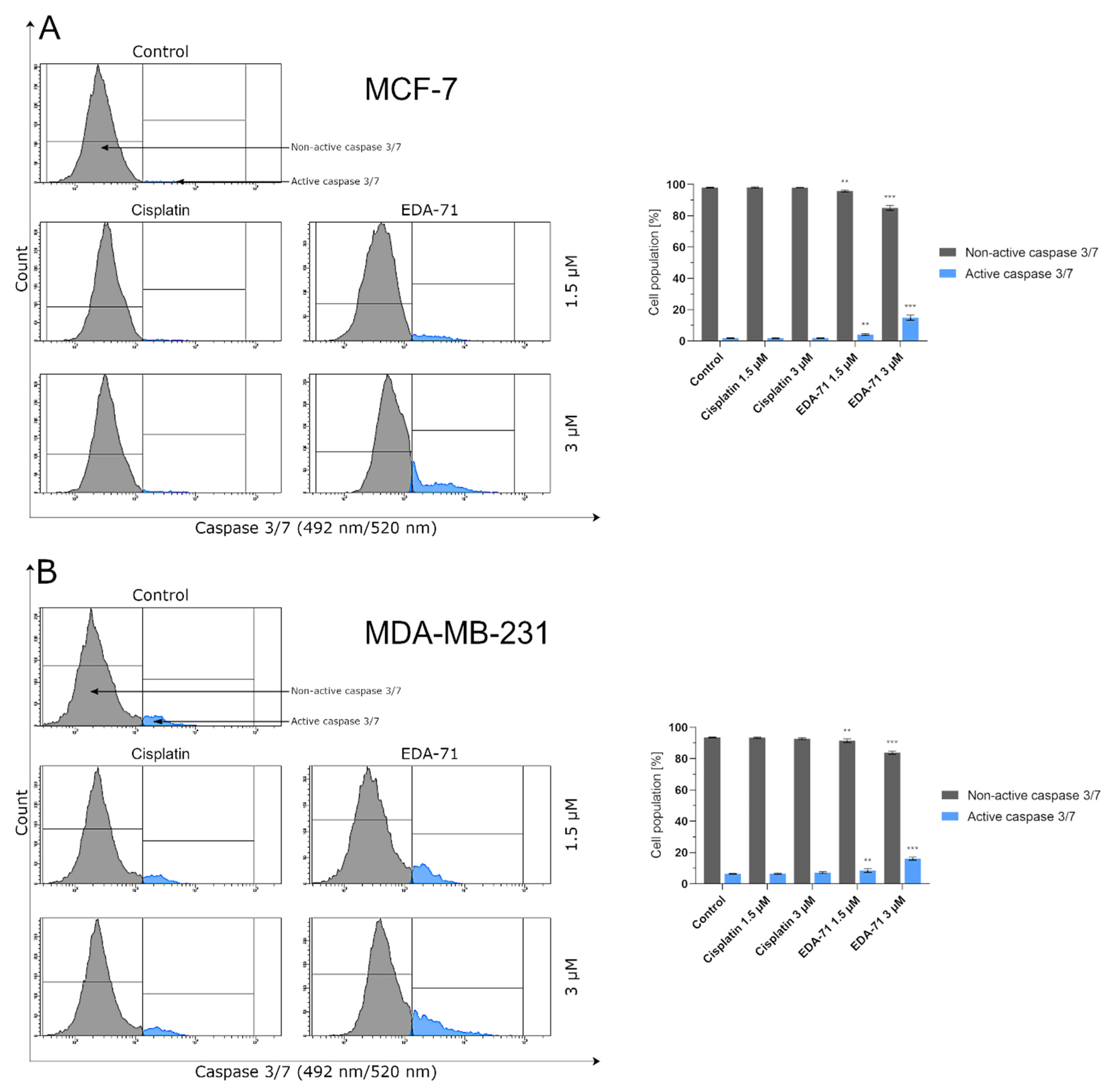
m was followed by the activation of caspases, including caspase 9 and then caspase 3, so the study of their activities was a further step to elucidate the mechanism of cell death. Our study of caspase 9 activity showed a significant increase in the activity of this protein. Along with its increase, a correlated increase in caspase 3 occurred, which is clear evidence that EDA-71 exhibits proapoptotic properties through the intrinsic pathway.
Caspase 3 is also activated by the action of caspases 8 and 10 in the extrinsic pathway of apoptosis. The activation of these caspases results from the dimerization of the FADD molecule and its interaction with the non-active forms of these proteins (procaspases 8 and 10). This leads to the formation of the DISC complex, which autocatalyzes and activates caspases 8 and 10 [
23,
24]. As a result of our research, we have shown that EDA-71 enables the expression of active caspases 8 and 10 in MCF-7 and MDA-MB 231 breast cancer cells. This may suggest that EDA-71 also induces programmed cell death by the caspase-dependent pathway of caspases 8 and caspase 10 (extrinsic pathway). In addition, these observations can be combined with the result of our study illustrating that EDA-71 affects the increase in the expression of active caspase 3.
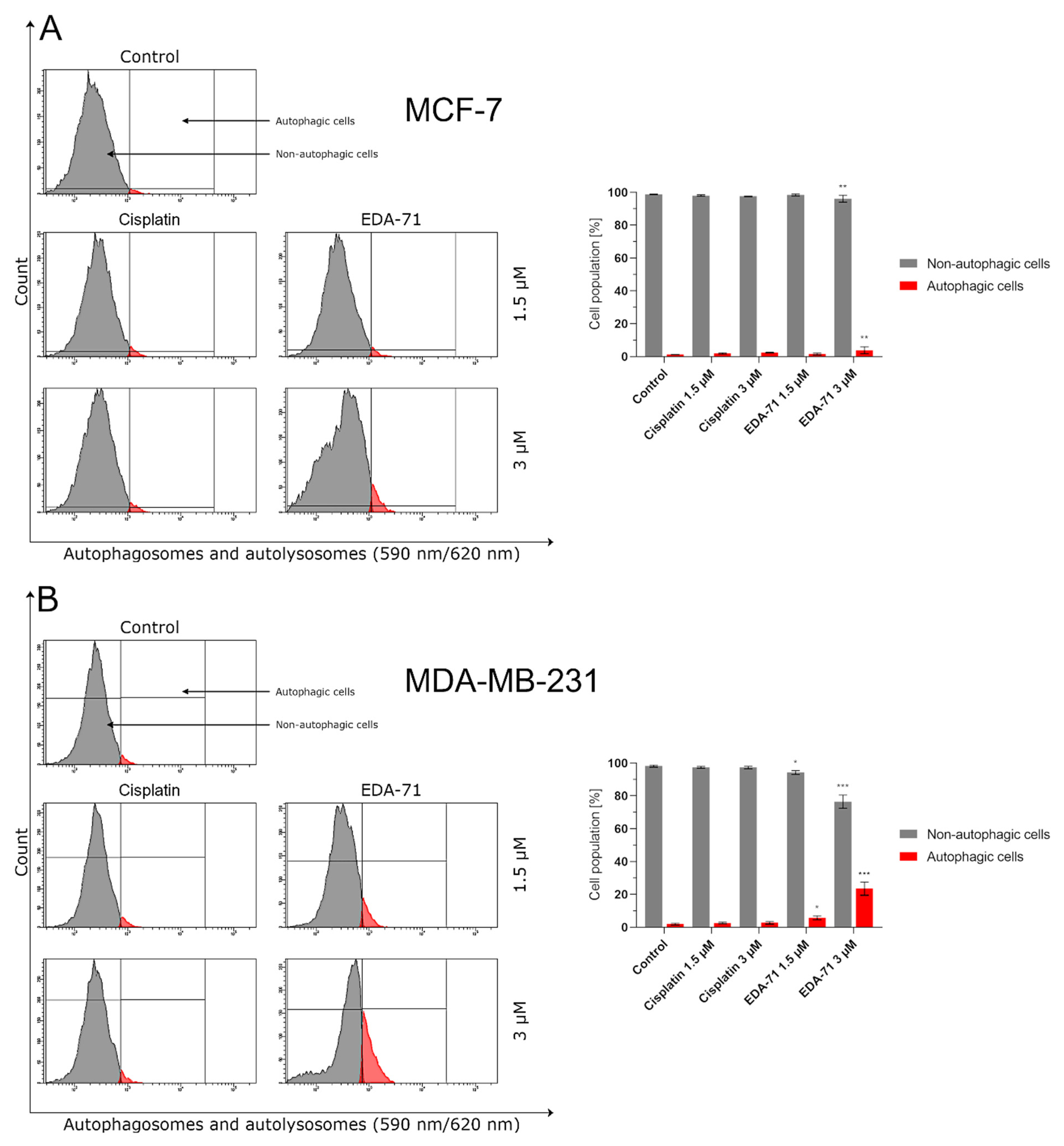
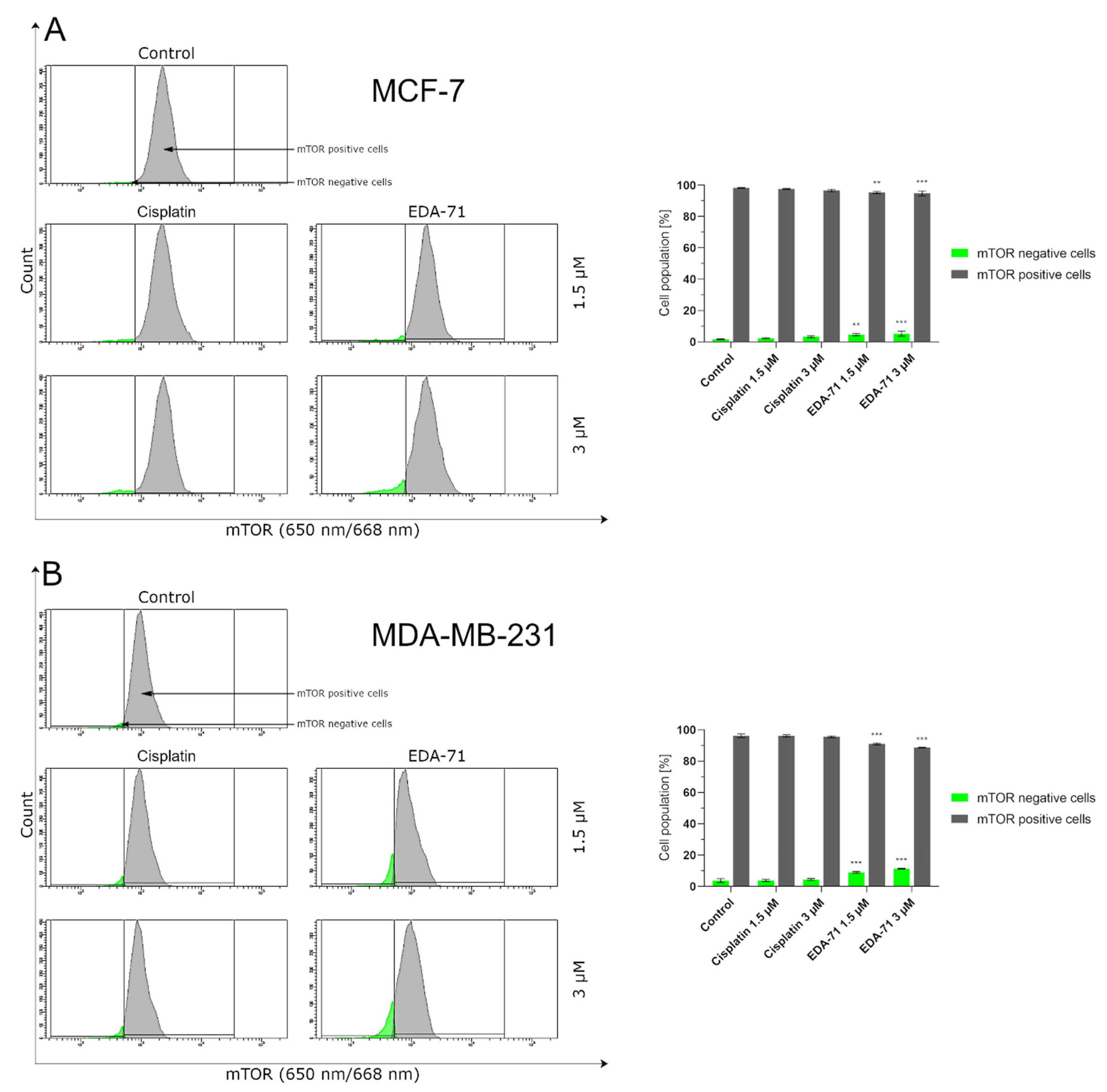
Therapy with selenium compounds can affect the activity and levels of numerous proteins, including those important in apoptosis and autophagy [
12,
42,
47]. A study conducted in 2015 by Wu et al. [
54] showed that mTOR protein levels may be downregulated during Se-allylselenocysteine therapy, possibly due to AMPK protein inhibition. Thus, the authors of this article suggest that the decrease in mTOR protein levels can lead to autophagy resulting in cell death. Similar results were obtained by Lee and Facompre with their co-workers, but they also demonstrated an additional role in the inhibition of the PI3K/Akt/mTOR signaling pathway by Se-containing compounds [
55,
56]. The results obtained in our work indicate that EDA-71 can lead to a reduction in mTOR protein levels, a factor that may promote cell death by autophagy. Knowledge of the involvement of this process in the elimination of EDA-71-treated cells seems to be of utmost importance, since modulation of signal transduction pathways involved in autophagy, on the one hand, enables the additional sensitization of cells to EDA-71 and, on the other hand, may facilitate the elimination of the resistance phenomenon. Interestingly, while the autophagy process was already observed at a concentration of 1.5 μM in the case of MDA-MB-231 cells, this process was activated in the case of MCF-7 cells over a concentration of 3.0 μM. The obtained results suggest that one of the possible cell death pathways under the influence of EDA-71 may be the process of autophagy, which significantly depends on the dose used.
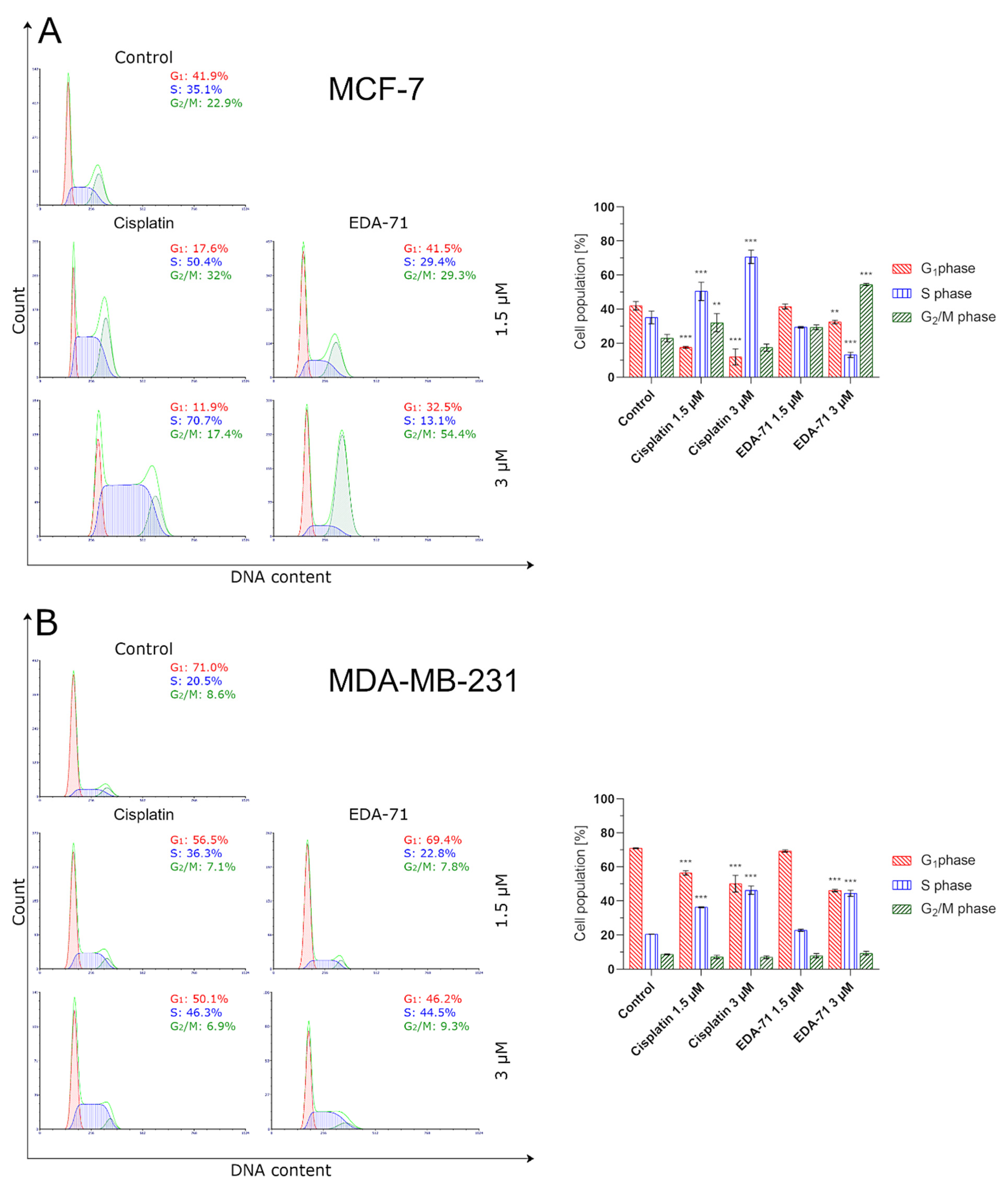
Considering the results obtained in MTT and apoptosis assays, we decided to determine the effect of compound EDA-71 on the cell cycle. A study of the effects of EDA-71 on cell cycle phases showed cell cycle inhibition in the G
2/M phase in MCF-7 cells and the S phase for MDA-MB-231 cells. Similar results were obtained with selenocystine treatment of the same cell line [
57]. The results obtained for the compound EDA-71 suggest that it affects specific phases of the cell cycle. Analysis of cell cycle phases showed an increase in the number of cells stained with propidium iodide with increasing dose.
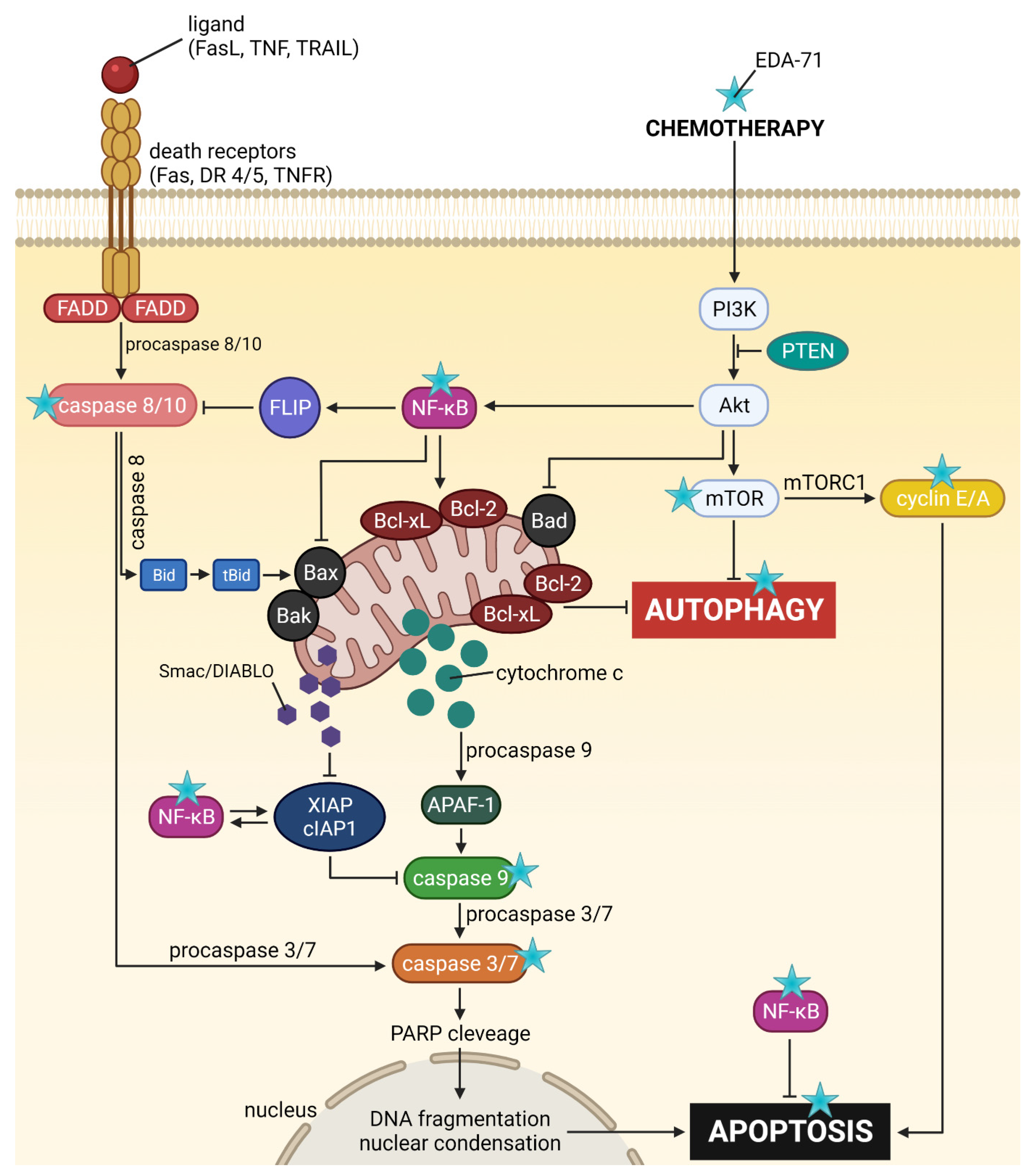
The studies conducted so far in this work suggest that EDA-71 is a compound with very interesting anticancer activity. Due to the fact that it is a completely new compound with biological activity that is difficult to predict, the experiments conducted within the framework of this work covered many aspects related to its activity (the potential molecular mechanism of its anticancer activity is summarized in
Figure 16), so that it is possible to delineate the most important directions in which further research should be directed. First of all, an unfavorable fact in the potential future clinical application of this agent is that it has high toxicity to normal cells, so as a first step it would be appropriate to undertake to reduce this undesirable phenomenon. The development of a targeted drug delivery system based, for example, on lipid nanocarriers, could help overcome the cytotoxicity of this compound against normal cells with simultaneous preservation of high anticancer activity. In the next stage,
in vitro studies evaluating the developed formulation of the drug and the associated reduction of toxic side effects should be planned, and if the results are satisfactory,
in vivo studies should be conducted. In addition, further detailed studies should be carried out to elucidate the mechanism of cytotoxic activity in cancer cells, as well as to allow for evaluation of the application of the tested compound with the developed drug formulation as an effective anticancer drug candidate.
5. Conclusions
In the present study, we aimed to evaluate the cytotoxic activity and antiproliferation effects of two selenoesters differing in the end fragment against MCF-7 and MDA-MB-231 breast cancer cells. The compound containing the ketone end fragment (EDA-71) was more cytotoxic and exhibited a stronger inhibitory effect on the cell proliferation of both breast cancer lines than the selenoester containing the nitrile end fragment (E-NS-4), but, importantly, both were very active at low micromolar concentrations (<5 µM). Similar results were observed in normal human breast epithelial cells (MCF-10A). In further experiments performed with the more active compound (EDA-71), a strong induction of apoptosis was observed, especially in MDA-MB-231 triple-negative breast cancer cells and normal MCF-10A cells. This process follows both an extrinsic pathway with activation of caspases 8 and 10 and an intrinsic pathway, as evidenced by a decrease in transcription factor NF-κB activity and mitochondrial potential and an increase in caspase 9 activity. Additionally, increased activity of caspases 3 and 7, which are common executioner caspases for both apoptotic pathways, was observed.
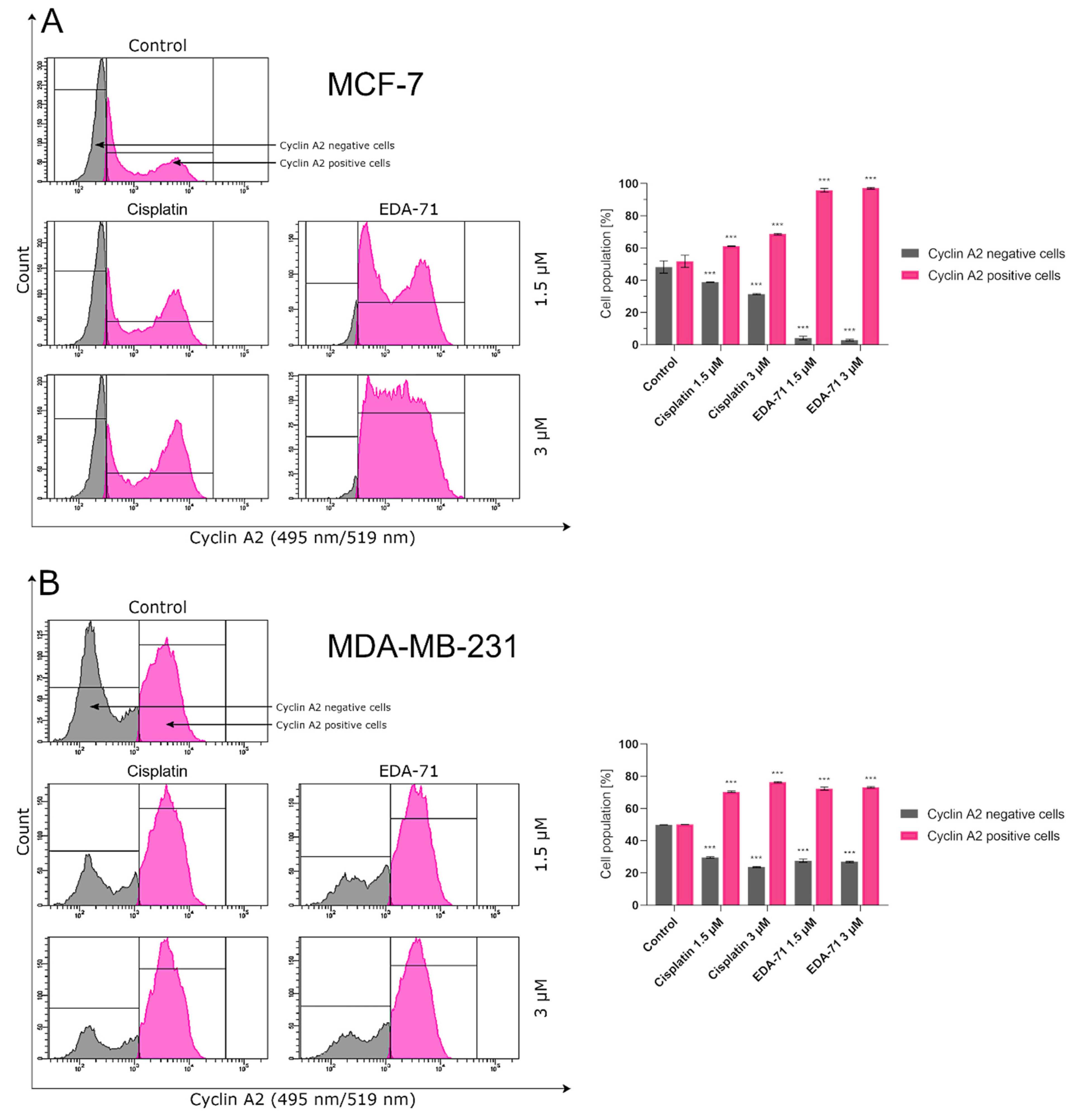
Furthermore, another investigation of the molecular mechanism of action of EDA-71 revealed that its autophagy activity is dose-dependent, and one possible target is the inhibition of mTOR protein. Apart from the above, our study also showed that the novel selenoorganic compound—EDA-71—can exhibit its anticancer activity through cell cycle arrest, and its effect on specific phases of the cell cycle was dependent on the type of breast cancer line (MCF-7, G2/M phase; MDA-MB-231, S phase). Confirmation of cell arrest in specific phases of the cell cycle was observed by a decrease in cyclin E1 levels with a concomitant increase in cyclin A2 levels.
To conclude, the tested novel selenoester EDA-71 is a highly interesting compound whose probably broad, molecular mechanism of action has not yet been fully understood, prompting further research in this field. The above results, as well as the very low doses of the agent used, suggest that EDA-71 seems to be a really promising candidate as a future potential anticancer drug for breast cancer therapy. However, this requires extensive research involving this substance.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}