KRAS Pathway Alterations in Malignant Pleural Mesothelioma: An Underestimated Player

{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Review Body
3.1. KRAS Pathway Alterations in Older Studies of MPM
3.2. Molecular Alterations in Published Next Generation Sequencing Studies of MPM
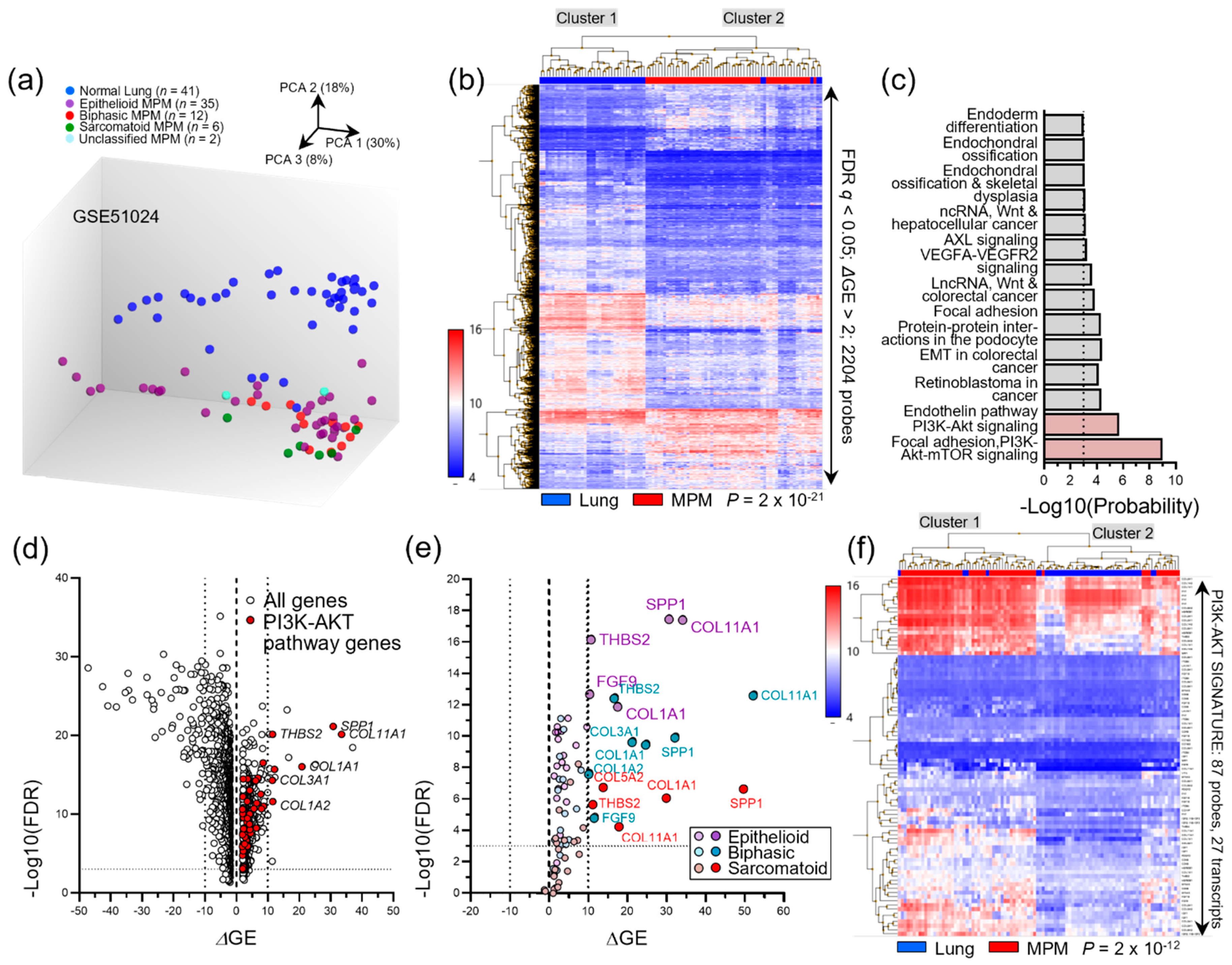
3.3. Transcriptomic MPM Studies
3.4. Molecular Alterations in MPM Cell Lines
3.5. Findings from Newer Sensitive Methods
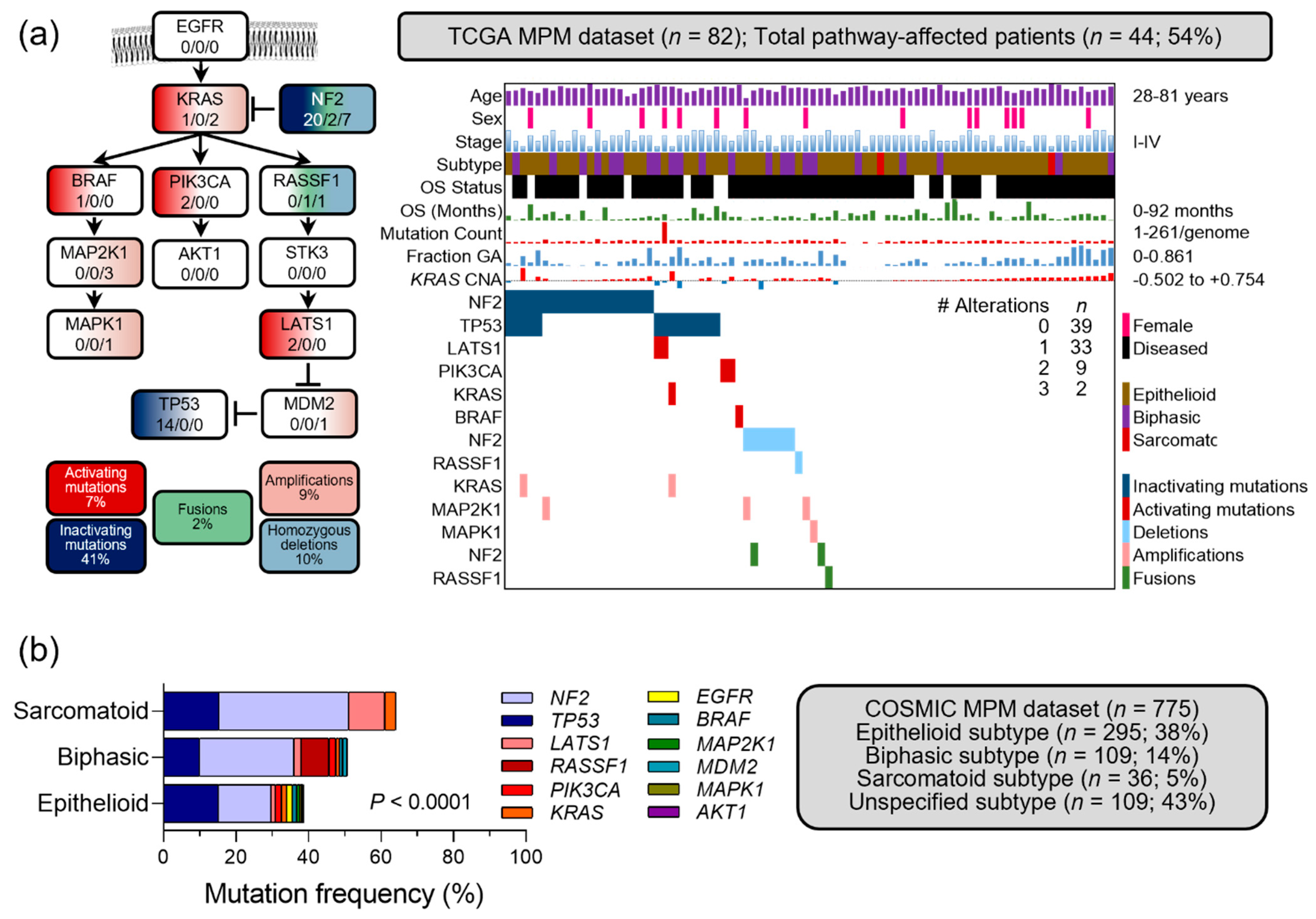
3.6. Occult KRAS Pathway Alterations in Published MPM Datasets
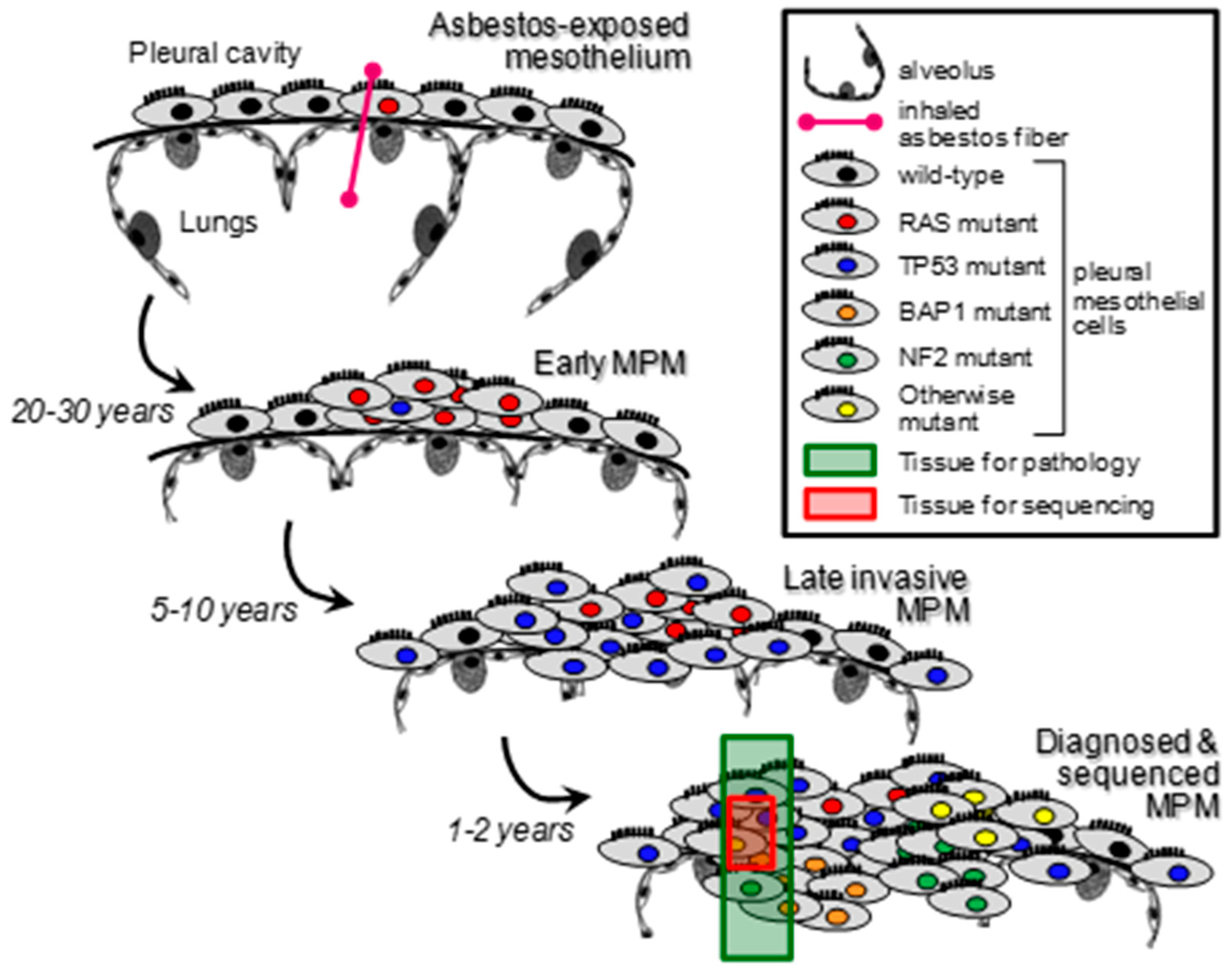
3.7. Studies on the Clonal Evolution of Mesothelioma
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bibby, A.C.; Tsim, S.; Kanellakis, N.; Ball, H.; Talbot, D.C.; Blyth, K.G.; Maskell, N.A.; Psallidas, I. Malignant Pleural Mesothelioma: An Update on Investigation, Diagnosis and Treatment. Eur. Respir. Rev. 2016, 25, 472–486. [Google Scholar] [CrossRef] [PubMed]
- Sekido, Y. Molecular Pathogenesis of Malignant Mesothelioma. Carcinogenesis 2013, 34, 1413–1419. [Google Scholar] [CrossRef]
- Park, E.-K.; Takahashi, K.; Hoshuyama, T.; Cheng, T.-J.; Delgermaa, V.; Le, G.V.; Sorahan, T. Global Magnitude of Reported and Unreported Mesothelioma. Environ. Health Perspect. 2011, 119, 514–518. [Google Scholar] [CrossRef]
- Hmeljak, J.; Sanchez-Vega, F.; Hoadley, K.A.; Shih, J.; Stewart, C.; Heiman, D.; Tarpey, P.; Danilova, L.; Drill, E.; Gibb, E.A.; et al. Integrative Molecular Characterization of Malignant Pleural Mesothelioma. Cancer Discov. 2018, 8, 1548–1565. [Google Scholar] [CrossRef] [PubMed]
- Vogelzang, N.J.; Rusthoven, J.J.; Symanowski, J.; Denham, C.; Kaukel, E.; Ruffie, P.; Gatzemeier, U.; Boyer, M.; Emri, S.; Manegold, C.; et al. Phase III Study of Pemetrexed in Combination with Cisplatin versus Cisplatin Alone in Patients with Malignant Pleural Mesothelioma. J. Clin. Oncol. 2003, 21, 2636–2644. [Google Scholar] [CrossRef]
- Zalcman, G.; Mazieres, J.; Margery, J.; Greillier, L.; Audigier-Valette, C.; Moro-Sibilot, D.; Molinier, O.; Corre, R.; Monnet, I.; Gounant, V.; et al. Bevacizumab for Newly Diagnosed Pleural Mesothelioma in the Mesothelioma Avastin Cisplatin Pemetrexed Study (MAPS): A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet 2016, 387, 1405–1414. [Google Scholar] [CrossRef]
- Galateau-Salle, F.; Churg, A.; Roggli, V.; Travis, W.D.; World Health Organization Committee for Tumors of the Pleura. The 2015 World Health Organization Classification of Tumors of the Pleura: Advances since the 2004 Classification. J. Thorac. Oncol. 2016, 11, 142–154. [Google Scholar] [CrossRef]
- Baas, P.; Scherpereel, A.; Nowak, A.K.; Fujimoto, N.; Peters, S.; Tsao, A.S.; Mansfield, A.S.; Popat, S.; Jahan, T.; Antonia, S.; et al. First-Line Nivolumab plus Ipilimumab in Unresectable Malignant Pleural Mesothelioma (CheckMate 743): A Multicentre, Randomised, Open-Label, Phase 3 Trial. Lancet 2021, 397, 375–386. [Google Scholar] [CrossRef]
- Klotz, L.V.; Lindner, M.; Eichhorn, M.E.; Grützner, U.; Koch, I.; Winter, H.; Kauke, T.; Duell, T.; Hatz, R.A. Pleurectomy/Decortication and Hyperthermic Intrathoracic Chemoperfusion Using Cisplatin and Doxorubicin for Malignant Pleural Mesothelioma. J. Thorac. Dis. 2019, 11, 1963–1972. [Google Scholar] [CrossRef]
- Zhang, M.; Luo, J.-L.; Sun, Q.; Harber, J.; Dawson, A.G.; Nakas, A.; Busacca, S.; Sharkey, A.J.; Waller, D.; Sheaff, M.T.; et al. Clonal Architecture in Mesothelioma Is Prognostic and Shapes the Tumour Microenvironment. Nat. Commun. 2021, 12, 1751. [Google Scholar] [CrossRef]
- Bueno, R.; Stawiski, E.W.; Goldstein, L.D.; Durinck, S.; De Rienzo, A.; Modrusan, Z.; Gnad, F.; Nguyen, T.T.; Jaiswal, B.S.; Chirieac, L.R.; et al. Comprehensive Genomic Analysis of Malignant Pleural Mesothelioma Identifies Recurrent Mutations, Gene Fusions and Splicing Alterations. Nat. Genet. 2016, 48, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Chmielecki, J.; Goparaju, C.; Heguy, A.; Dolgalev, I.; Carbone, M.; Seepo, S.; Meyerson, M.; Pass, H.I. Whole-Exome Sequencing Reveals Frequent Genetic Alterations in BAP1, NF2, CDKN2A, and CUL1 in Malignant Pleural Mesothelioma. Cancer Res. 2015, 75, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Bott, M.; Brevet, M.; Taylor, B.S.; Shimizu, S.; Ito, T.; Wang, L.; Creaney, J.; Lake, R.A.; Zakowski, M.F.; Reva, B.; et al. The Nuclear Deubiquitinase BAP1 is Commonly Inactivated by Somatic Mutations and 3p21.1 Losses in Malignant Pleural Mesothelioma. Nat. Genet. 2011, 43, 668–672. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Tomson, B.N.; Buys, T.P.H.; Elkin, S.K.; Carter, J.L.; Kurzrock, R. Genomic Landscape of Malignant Mesotheliomas. Mol. Cancer Ther. 2016, 15, 2498–2507. [Google Scholar] [CrossRef] [PubMed]
- Iacono, M.L.; Monica, V.; Righi, L.; Grosso, F.; Libener, R.; Vatrano, S.; Bironzo, P.; Novello, S.; Musmeci, L.; Volante, M.; et al. Targeted Next-Generation Sequencing of Cancer Genes in Advanced Stage Malignant Pleural Mesothelioma: A Retrospective Study. J. Thorac. Oncol. 2015, 10, 492–499. [Google Scholar] [CrossRef]
- Mezzapelle, R.; Miglio, U.; Rena, O.; Paganotti, A.; Allegrini, S.; Antona, J.; Molinari, F.; Frattini, M.; Monga, G.; Alabiso, O.; et al. Mutation Analysis of the EGFR Gene and Downstream Signalling Pathway in Histologic Samples of Malignant Pleural Mesothelioma. Br. J. Cancer 2013, 108, 1743–1749. [Google Scholar] [CrossRef]
- Rienzo, A.D.; Archer, M.A.; Yeap, B.Y.; Dao, N.; Sciaranghella, D.; Sideris, A.C.; Zheng, Y.; Holman, A.G.; Wang, Y.E.; Cin, P.S.D.; et al. Gender-Specific Molecular and Clinical Features Underlie Malignant Pleural Mesothelioma. Cancer Res. 2016, 76, 319–328. [Google Scholar] [CrossRef]
- Marazioti, A.; Krontira, A.C.; Behrend, S.J.; Giotopoulou, G.A.; Ntaliarda, G.; Blanquart, C.; Bayram, H.; Iliopoulou, M.; Vreka, M.; Trassl, L.; et al. KRAS Signaling in Malignant Pleural Mesothelioma. EMBO Mol. Med. 2022, 14, e13631. [Google Scholar] [CrossRef]
- Patel, M.R.; Jacobson, B.A.; De, A.; Frizelle, S.P.; Janne, P.; Thumma, S.C.; Whitson, B.A.; Farassati, F.; Kratzke, R.A. Ras Pathway Activation in Malignant Mesothelioma. J. Thorac. Oncol. 2007, 2, 789–795. [Google Scholar] [CrossRef]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-Targeted Therapies: Is the Undruggable Drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef]
- Tikoo, A.; Varga, M.; Ramesh, V.; Gusella, J.; Maruta, H. An Anti-Ras Function of Neurofibromatosis Type 2 Gene Product (NF2/Merlin). J. Biol. Chem. 1994, 269, 23387–23390. [Google Scholar] [CrossRef]
- Matallanas, D.; Romano, D.; Al-Mulla, F.; O’Neill, E.; Al-Ali, W.; Crespo, P.; Doyle, B.; Nixon, C.; Sansom, O.; Drosten, M.; et al. Mutant K-Ras Activation of the Proapoptotic MST2 Pathway Is Antagonized by Wild-Type K-Ras. Mol. Cell 2011, 44, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Arizti, P.; Fang, L.; Park, I.; Yin, Y.; Solomon, E.; Ouchi, T.; Aaronson, S.A.; Lee, S.W. Tumor Suppressor P53 is Required To Modulate BRCA1 Expression. Mol. Cell. Biol. 2000, 20, 7450–7459. [Google Scholar] [CrossRef] [PubMed]
- Bi, M.; Zhao, S.; Said, J.W.; Merino, M.J.; Adeniran, A.J.; Xie, Z.; Nawaf, C.B.; Choi, J.; Belldegrun, A.S.; Pantuck, A.J.; et al. Genomic Characterization of Sarcomatoid Transformation in Clear Cell Renal Cell Carcinoma. Proc. Natl. Acad. Sci. USA 2016, 113, 2170–2175. [Google Scholar] [CrossRef]
- Stott, F.J.; Bates, S.; James, M.C.; McConnell, B.B.; Starborg, M.; Brookes, S.; Palmero, I.; Ryan, K.; Hara, E.; Vousden, K.H.; et al. The Alternative Product from the Human CDKN2A Locus, P14(ARF), Participates in a Regulatory Feedback Loop with P53 and MDM2. EMBO J. 1998, 17, 5001–5014. [Google Scholar] [CrossRef]
- Agalioti, T.; Giannou, A.D.; Krontira, A.C.; Kanellakis, N.I.; Kati, D.; Vreka, M.; Pepe, M.; Spella, M.; Lilis, I.; Zazara, D.E.; et al. Mutant KRAS Promotes Malignant Pleural Effusion Formation. Nat. Commun. 2017, 8, 15205. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Metcalf, R.A.; Welsh, J.A.; Bennett, W.P.; Seddon, M.B.; Lehman, T.A.; Pelin, K.; Linnainmaa, K.; Tammilehto, L.; Mattson, K.; Gerwin, B.I.; et al. P53 and Kirsten-Ras Mutations in Human Mesothelioma Cell Lines. Cancer Res. 1992, 52, 2610–2615. [Google Scholar]
- Ni, Z.; Liu, Y.-Q.; Keshava, N.; Zhou, G.; Whong, W.; Ong, T. Analysis of K-Ras and P53 Mutations in Mesotheliomas from Humans and Rats Exposed to Asbestos. Mutat. Res. Genet. Toxicol. Environ. Mutagenesis 2000, 468, 87–92. [Google Scholar] [CrossRef]
- Suraokar, M.B.; Nunez, M.I.; Diao, L.; Chow, C.W.; Kim, D.; Behrens, C.; Lin, H.; Lee, S.; Raso, G.; Moran, C.; et al. Expression Profiling Stratifies Mesothelioma Tumors and Signifies Deregulation of Spindle Checkpoint Pathway and Microtubule Network with Therapeutic Implications. Ann. Oncol. 2014, 25, 1184–1192. [Google Scholar] [CrossRef]
- Blum, Y.; Meiller, C.; Quetel, L.; Elarouci, N.; Ayadi, M.; Tashtanbaeva, D.; Armenoult, L.; Montagne, F.; Tranchant, R.; Renier, A.; et al. Dissecting Heterogeneity in Malignant Pleural Mesothelioma through Histo-Molecular Gradients for Clinical Applications. Nat. Commun. 2019, 10, 1333. [Google Scholar] [CrossRef] [Green Version]
- de Reyniès, A.; Jaurand, M.-C.; Renier, A.; Couchy, G.; Hysi, I.; Elarouci, N.; Galateau-Sallé, F.; Copin, M.-C.; Hofman, P.; Cazes, A.; et al. Molecular Classification of Malignant Pleural Mesothelioma: Identification of a Poor Prognosis Subgroup Linked to the Epithelial-to-Mesenchymal Transition. Clin. Cancer Res. 2014, 20, 1323–1334. [Google Scholar] [CrossRef]
- Sneddon, S.; Dick, I.; Lee, Y.C.G.; Musk, A.W.B.; Patch, A.-M.; Pearson, J.V.; Waddell, N.; Allcock, R.J.N.; Holt, R.A.; Robinson, B.W.S.; et al. Malignant Cells from Pleural Fluids in Malignant Mesothelioma Patients Reveal Novel Mutations. Lung Cancer 2018, 119, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Quispel-Janssen, J.M.; Badhai, J.; Schunselaar, L.; Price, S.; Brammeld, J.; Iorio, F.; Kolluri, K.; Garnett, M.; Berns, A.; Baas, P.; et al. Comprehensive Pharmacogenomic Profiling of Malignant Pleural Mesothelioma Identifies a Subgroup Sensitive to FGFR Inhibition. Clin. Cancer Res. 2018, 24, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Cerami1, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Forbes, S.A.; Beare, D.; Gunasekaran, P.; Leung, K.; Bindal, N.; Boutselakis, H.; Ding, M.; Bamford, S.; Cole, C.; Ward, S.; et al. COSMIC: Exploring the World’s Knowledge of Somatic Mutations in Human Cancer. Nucleic Acids Res. 2015, 43, D805–D811. [Google Scholar] [CrossRef]
- Kelder, T.; van Iersel, M.P.; Hanspers, K.; Kutmon, M.; Conklin, B.R.; Evelo, C.T.; Pico, A.R. WikiPathways: Building Research Communities on Biological Pathways. Nucleic Acids Res. 2012, 40, D1301–D1307. [Google Scholar] [CrossRef]
- Psallidas, I.; Stathopoulos, G.T.; Maniatis, N.A.; Magkouta, S.; Moschos, C.; Karabela, S.P.; Kollintza, A.; Simoes, D.C.M.; Kardara, M.; Vassiliou, S.; et al. Secreted Phosphoprotein-1 Directly Provokes Vascular Leakage to Foster Malignant Pleural Effusion. Oncogene 2013, 32, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Pass, H.I.; Lott, D.; Lonardo, F.; Harbut, M.; Liu, Z.; Tang, N.; Carbone, M.; Webb, C.; Wali, A. Asbestos Exposure, Pleural Mesothelioma, and Serum Osteopontin Levels. N. Engl. J. Med. 2005, 353, 1564–1573. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef]
- Singh, A.; Pruett, N.; Pahwa, R.; Mahajan, A.P.; Schrump, D.S.; Hoang, C.D. MicroRNA-206 Suppresses Mesothelioma Progression via the Ras Signaling Axis. Mol. Ther. Nucleic Acids 2021, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Abbosh, C.; Birkbak, N.J.; Wilson, G.A.; Jamal-Hanjani, M.; Constantin, T.; Salari, R.; Le Quesne, J.; Moore, D.A.; Veeriah, S.; Rosenthal, R.; et al. Phylogenetic CtDNA Analysis Depicts Early-Stage Lung Cancer Evolution. Nature 2017, 545, 446–451. [Google Scholar] [CrossRef]
- Awad, M.M.; Liu, S.; Rybkin, I.I.; Arbour, K.C.; Dilly, J.; Zhu, V.W.; Johnson, M.L.; Heist, R.S.; Patil, T.; Riely, G.J.; et al. Acquired Resistance to KRASG12C Inhibition in Cancer. N. Engl. J. Med. 2021, 384, 2382–2393. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Goswami, D.; Adiseshaiah, P.P.; Burgan, W.; Yi, M.; Guerin, T.M.; Kozlov, S.V.; Nissley, D.V.; McCormick, F. Undermining Glutaminolysis Bolsters Chemotherapy While NRF2 Promotes Chemoresistance in KRAS-Driven Pancreatic Cancers. Cancer Res. 2020, 80, 1630–1643. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Vander Heiden, M.G.; McCormick, F. The Metabolic Landscape of RAS-Driven Cancers from Biology to Therapy. Nat. Cancer 2021, 2, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Kimmelman, A.C. Metabolic Dependencies in RAS-Driven Cancers. Clin. Cancer Res. 2015, 21, 1828–1834. [Google Scholar] [CrossRef]
- Li, S.; MacAlpine, D.M.; Counter, C.M. Capturing the Primordial Kras Mutation Initiating Urethane Carcinogenesis. Nat. Commun. 2020, 11, 1800. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trassl, L.; Stathopoulos, G.T. KRAS Pathway Alterations in Malignant Pleural Mesothelioma: An Underestimated Player. Cancers 2022, 14, 4303. https://doi.org/10.3390/cancers14174303
Trassl L, Stathopoulos GT. KRAS Pathway Alterations in Malignant Pleural Mesothelioma: An Underestimated Player. Cancers. 2022; 14(17):4303. https://doi.org/10.3390/cancers14174303
Chicago/Turabian StyleTrassl, Lilith, and Georgios T. Stathopoulos. 2022. "KRAS Pathway Alterations in Malignant Pleural Mesothelioma: An Underestimated Player" Cancers 14, no. 17: 4303. https://doi.org/10.3390/cancers14174303
APA StyleTrassl, L., & Stathopoulos, G. T. (2022). KRAS Pathway Alterations in Malignant Pleural Mesothelioma: An Underestimated Player. Cancers, 14(17), 4303. https://doi.org/10.3390/cancers14174303