
Genomic Landscape Alterations in Primary Tumor and Matched Lymph Node Metastasis in Hormone-Naïve Prostate Cancer Patients

 , ,
, ,  ,
,  ,
,  ,
,  , ,
, ,  ,
,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Deparaffinization Procedure and DNA Extraction
2.2. NGS Sequencing
2.3. Bioinformatics Analysis
2.4. Multiparameter Genetic Score
2.5. cBioPortal Analysis
2.6. Statistical Analysis
3. Results
3.1. DNA Amplicon Sequencing in Healty, Primary Tumor and Metastasis Samples Showed Heterogeneity in the Mutations Found
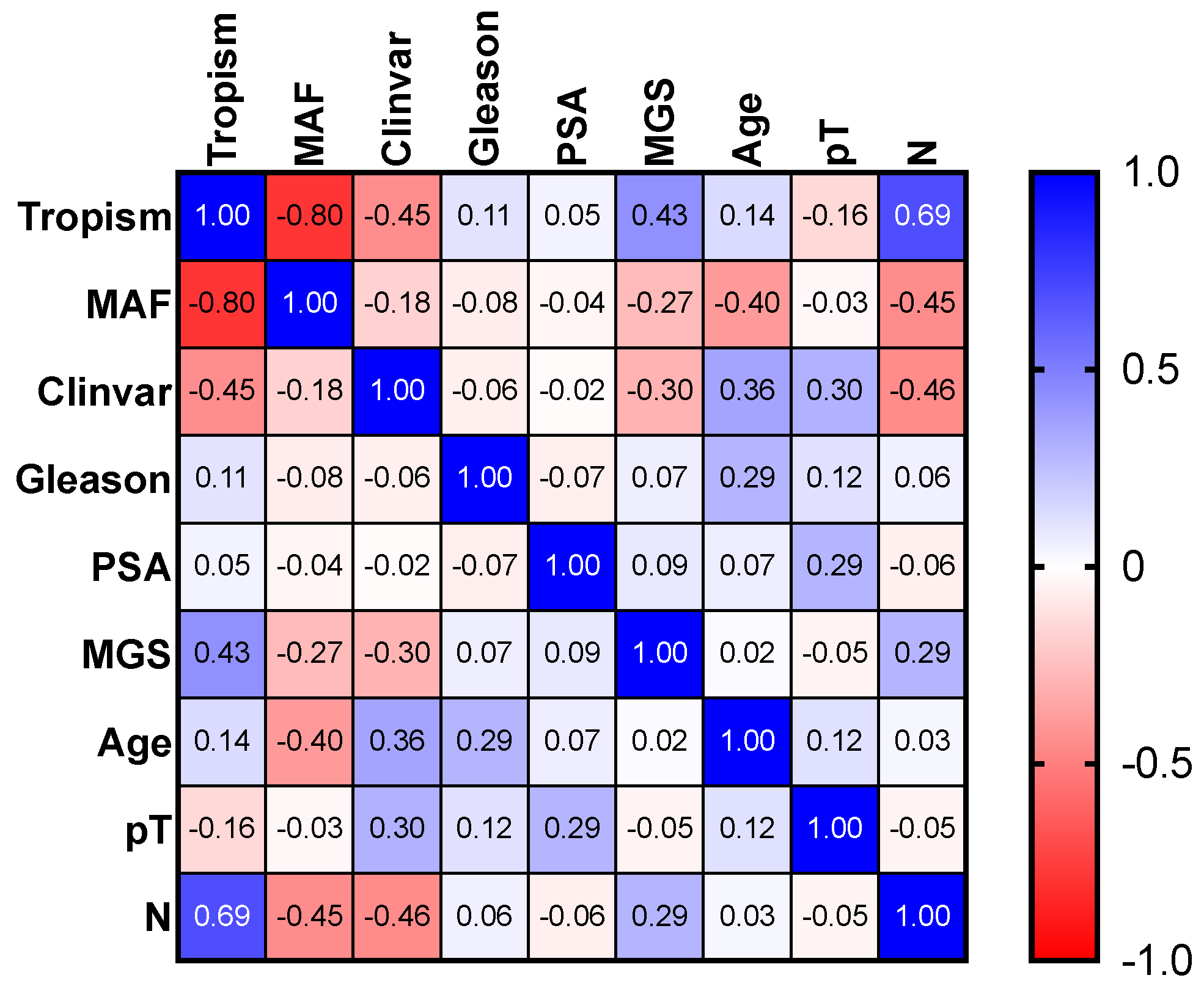
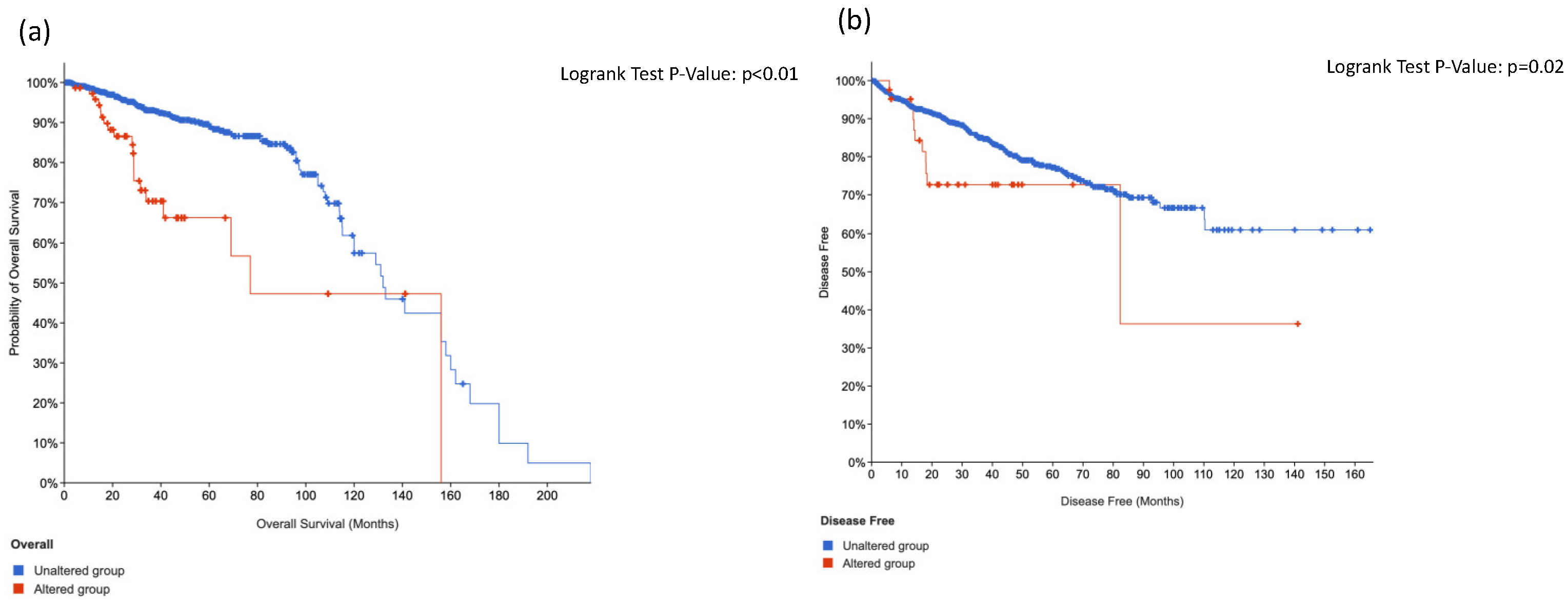
3.2. Multiparameter Genetic Score (MGS) Calculation Showed a Correlation with Lymph Node Metastasis
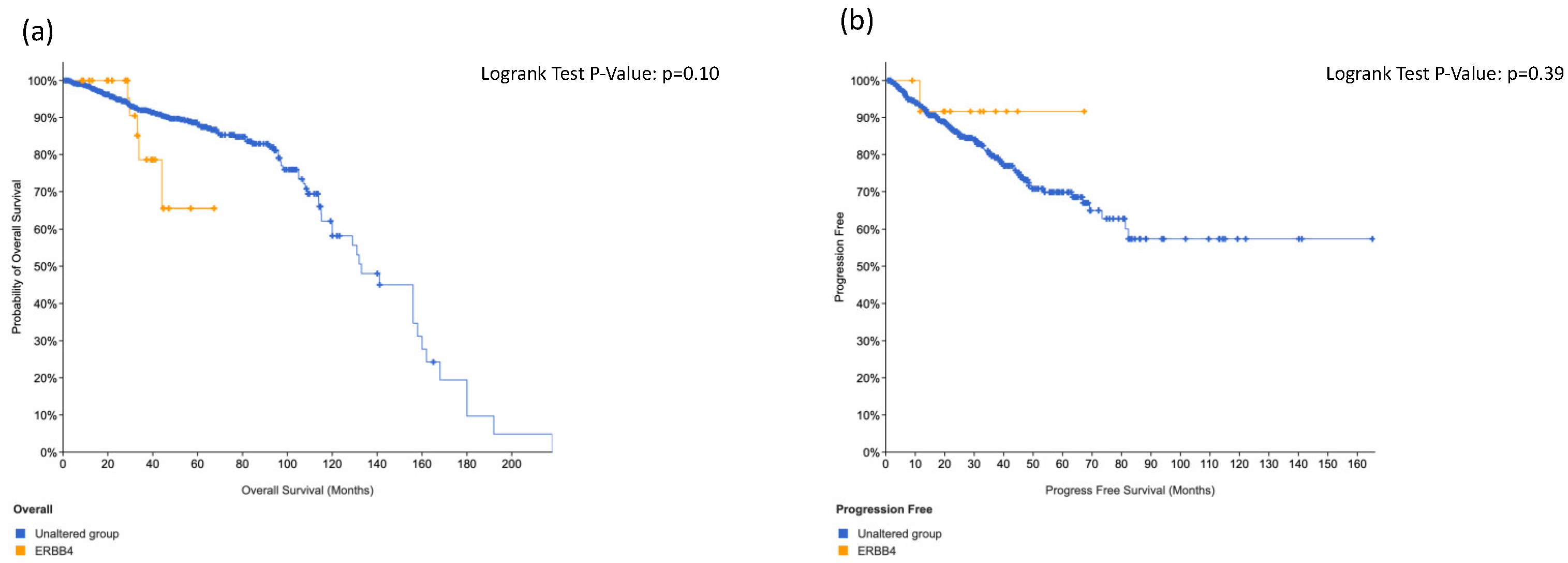
3.3. Total Impact of dbSNPs on AmpliSeq Pane Showed That ERRB4 Was the Most Frequent Mutation
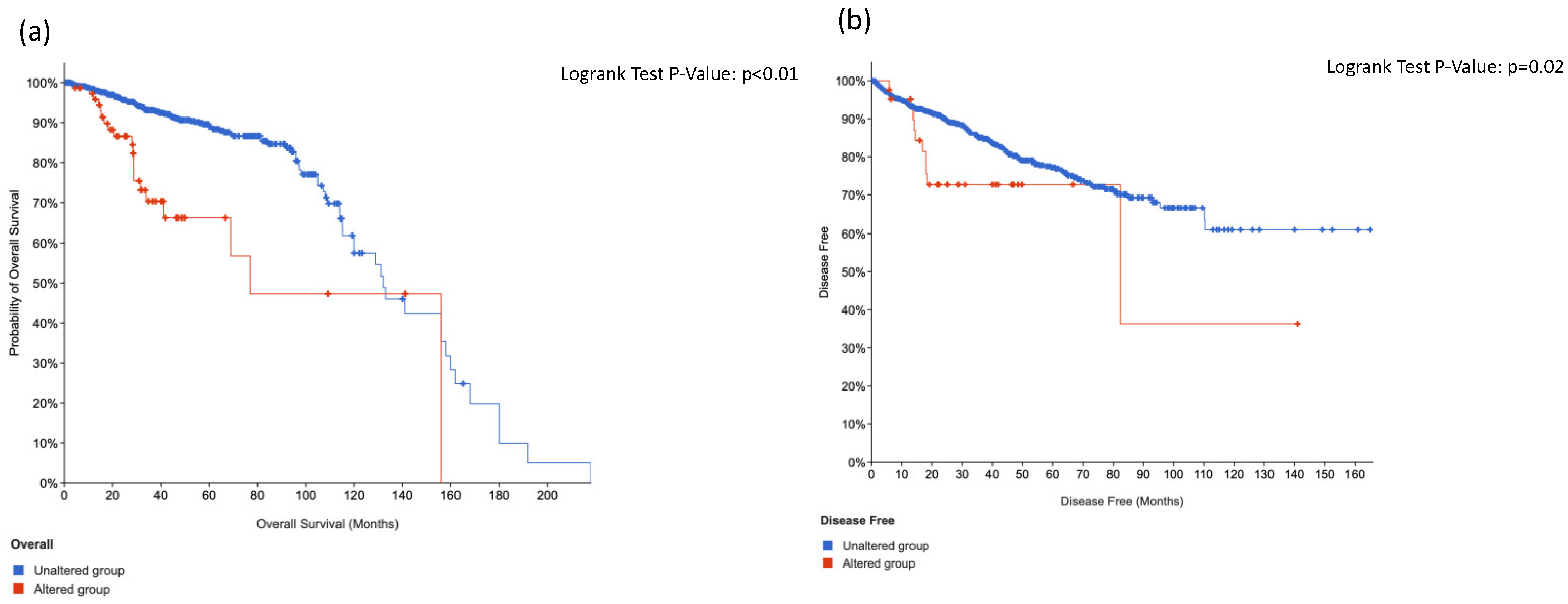
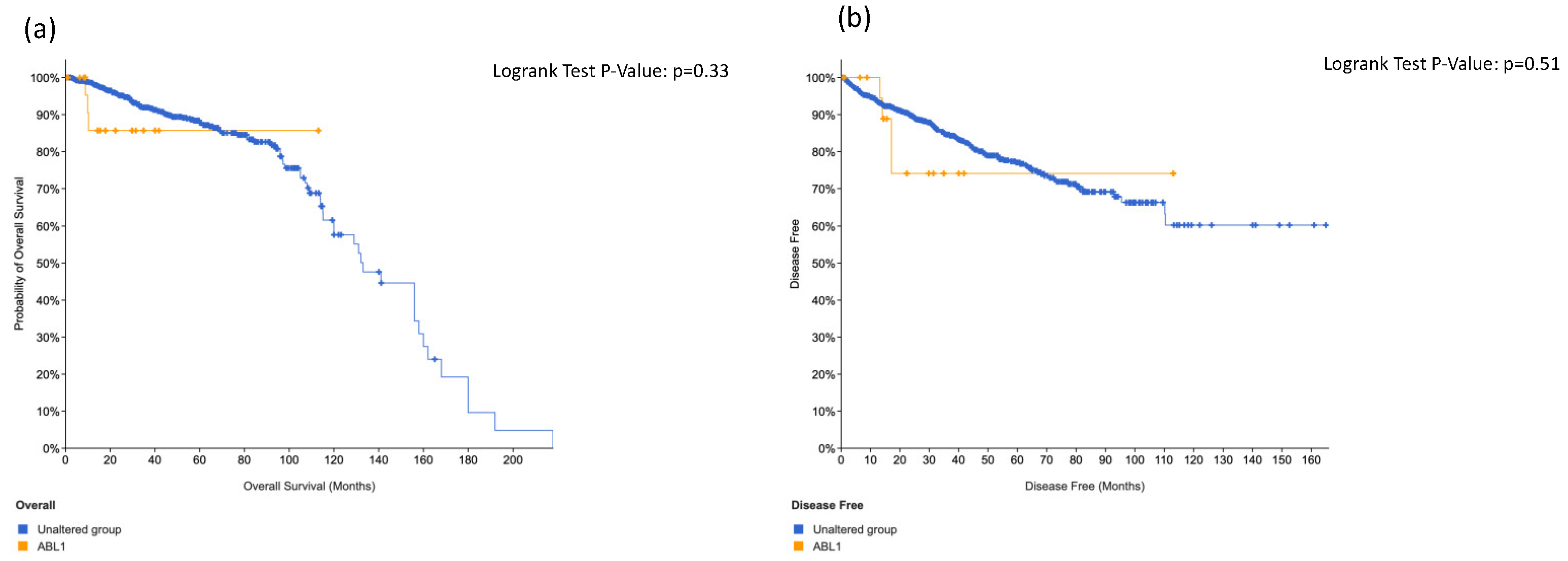
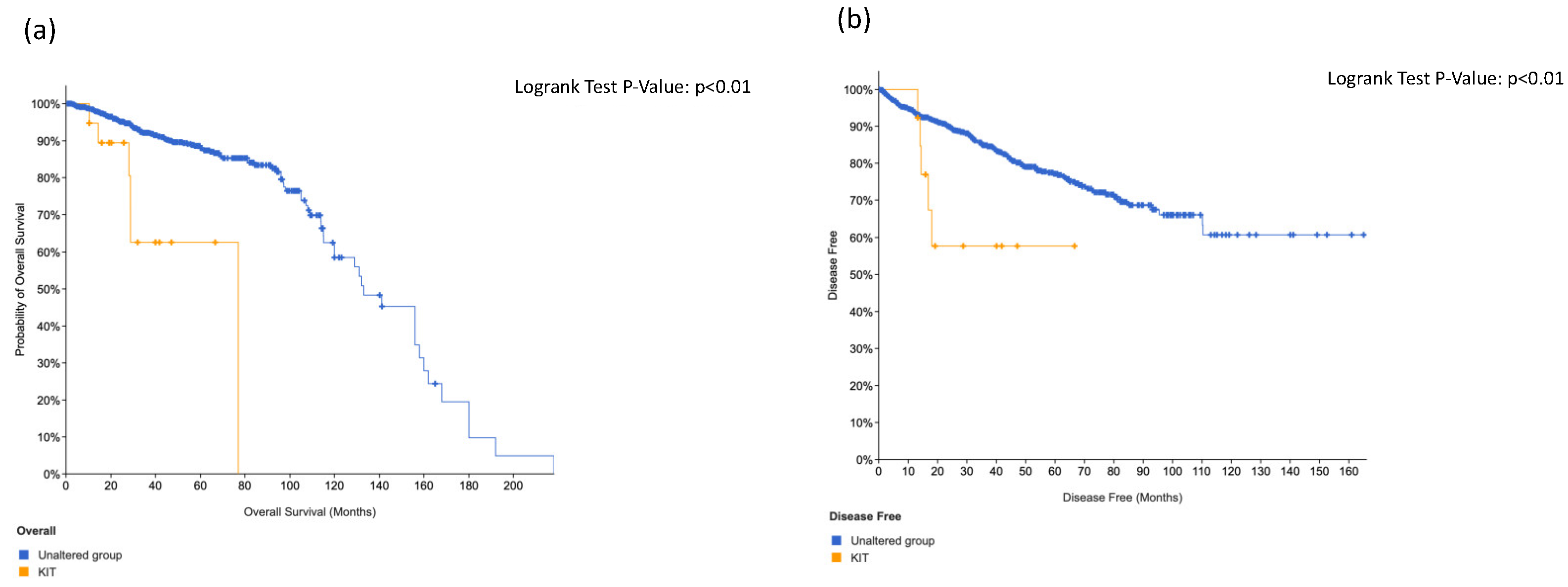
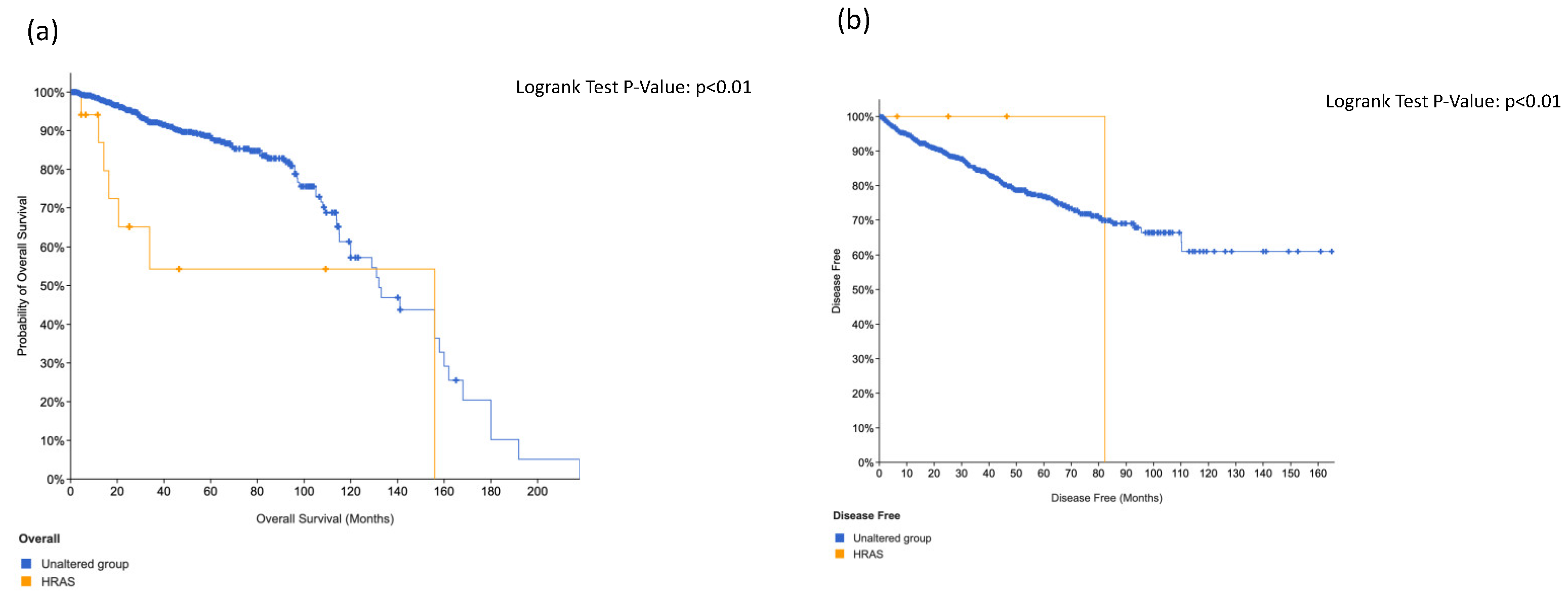
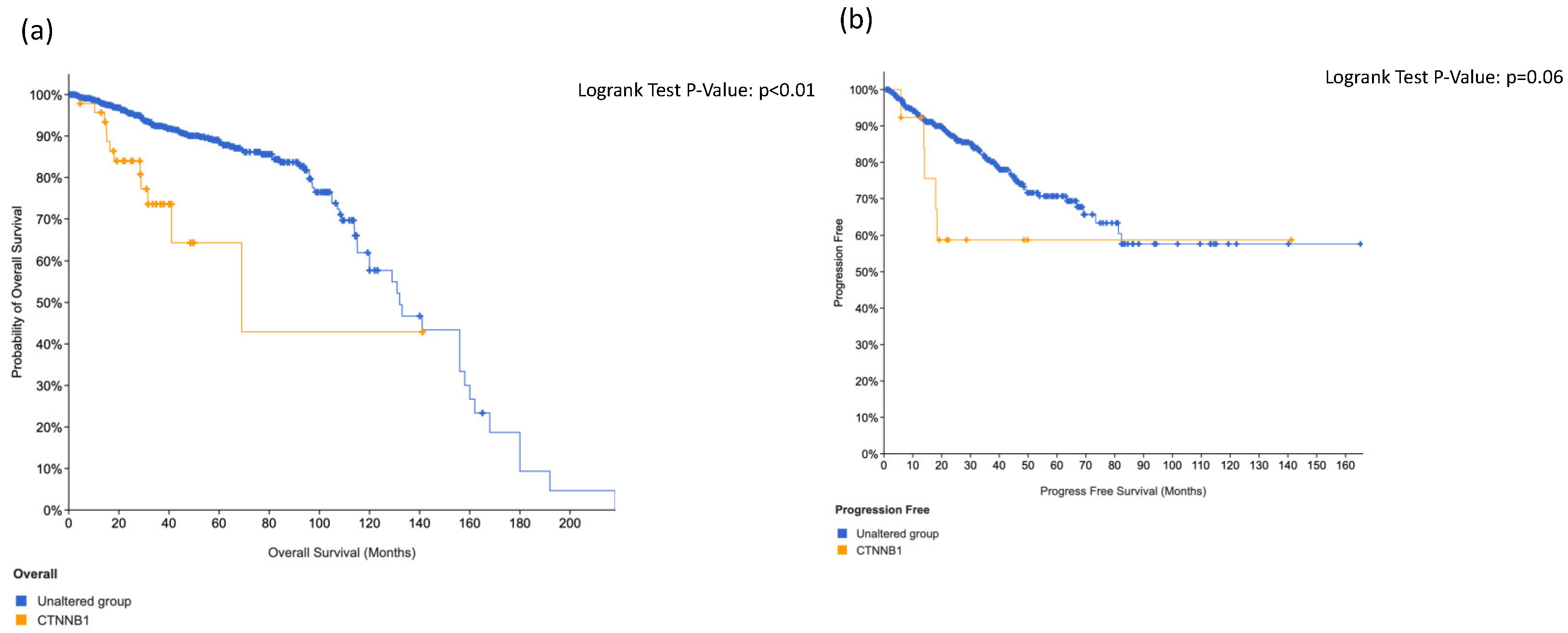
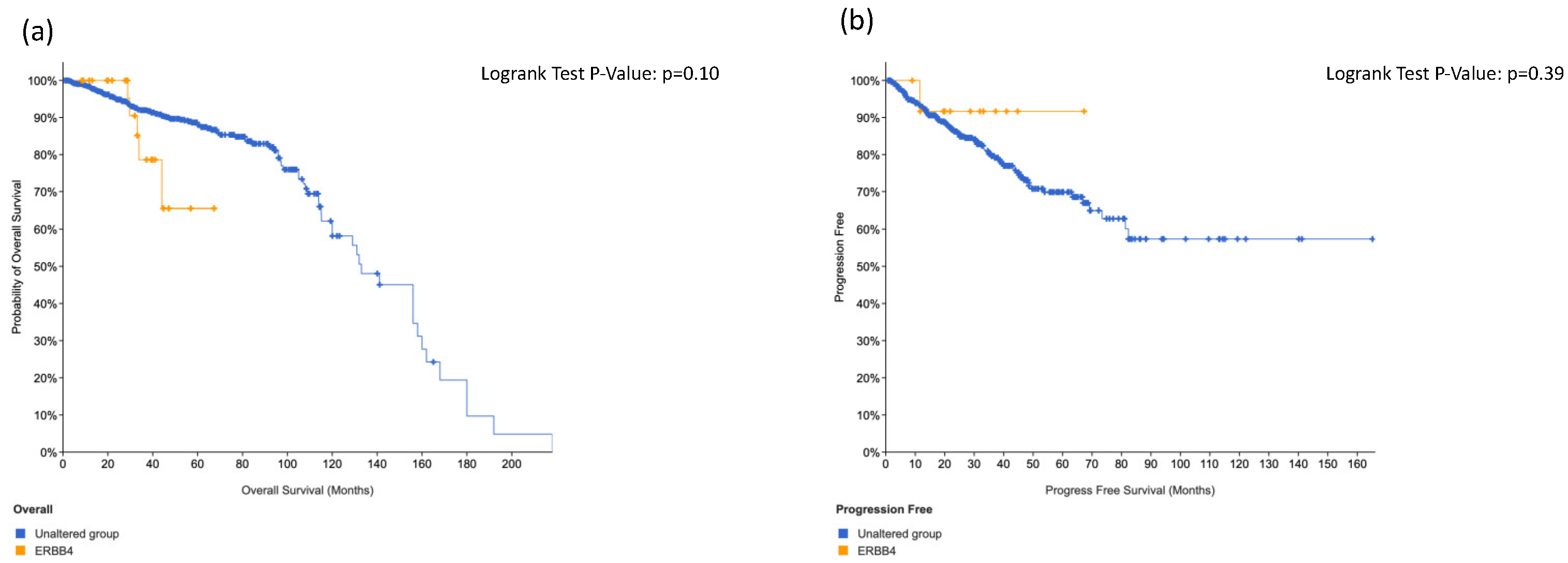
3.4. KIT, HRAS and CTNNB1 Alterations in a Combination Is Associated with Worse Surival
3.5. Tissue Micro Array Analysis and Immunohistochemistry Stainings Showed an Association between IGF1-R Expression and ERRB4 Alterations in Primary Tumor or Lymph Node Metastasis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar]
- Bernard, B.; Burnett, C.; Sweeney, C.J.; Rider, J.R.; Sridhar, S.S. Impact of age at diagnosis of de novo metastatic prostate cancer on survival. Cancer 2020, 126, 986–993. [Google Scholar] [PubMed]
- Mottet, N.; van den Bergh, R.C.N.; Briers, E.; Van den Broeck, T.; Cumberbatch, M.G.; De Santis, M.; Fanti, S.; Fossati, N.; Gandaglia, G.; Gillessen, S.; et al. EAU-EANM-ESTRO-ESUR-SIOG Guidelines on Prostate Cancer—2020 Update. Part 1: Screening, Diagnosis, and Local Treatment with Curative Intent. Eur. Urol. 2021, 79, 243–262. [Google Scholar] [PubMed]
- Touijer, K.A.; Mazzola, C.R.; Sjoberg, D.D.; Scardino, P.T.; Eastham, J.A. Long-term Outcomes of Patients with Lymph Node Metastasis Treated with Radical Prostatectomy Without Adjuvant Androgen-deprivation Therapy. Eur. Urol. 2014, 65, 20–25. [Google Scholar] [PubMed]
- Leongamornlert, D.; Mahmud, N.; Tymrakiewicz, M.; Saunders, E.; Dadaev, T.; Castro, E.; Goh, C.; Govindasami, K.; Guy, M.; O’Brien, L.; et al. Germline BRCA1 mutations increase prostate cancer risk. Br. J. Cancer 2012, 106, 1697–1701. [Google Scholar]
- Heidegger, I.; Tsaur, I.; Borgmann, H.; Surcel, C.; Kretschmer, A.; Mathieu, R.; De Visschere, P.; Valerio, M.; van den Bergh, R.C.N.; Ost, P.; et al. Hereditary prostate cancer—Primetime for genetic testing? Cancer Treat. Rev. 2019, 81, 101927. [Google Scholar]
- Kneppers, J.; Krijgsman, O.; Melis, M.; de Jong, J.; Peeper, D.S.; Bekers, E.; van der Poel, H.G.; Zwart, W.; Bergman, A.M. Frequent clonal relations between metastases and non-index prostate cancer lesions. JCI Insight 2019, 4, e124756. [Google Scholar] [CrossRef]
- Broggi, G.; Lo Giudice, A.; Di Mauro, M.; Pricoco, E.; Piombino, E.; Ferro, M.; Caltabiano, R.; Morgia, G.; Russo, G.I. Insulin signaling, androgen receptor and PSMA immunohistochemical analysis by semi-automated tissue microarray in prostate cancer with diabetes (DIAMOND study). Transl. Res. 2021, 238, 25–35. [Google Scholar]
- Broggi, G.; Lo Giudice, A.; Di Mauro, M.; Asmundo, M.G.; Pricoco, E.; Piombino, E.; Caltabiano, R.; Morgia, G.; Russo, G.I. SRSF-1 and microvessel density immunohistochemical analysis by semi-automated tissue microarray in prostate cancer patients with diabetes (DIAMOND study). Prostate 2021, 81, 882–892. [Google Scholar] [CrossRef]
- Cammarata, F.P.; Forte, G.I.; Broggi, G.; Bravatà, V.; Minafra, L.; Pisciotta, P.; Calvaruso, M.; Tringali, R.; Tomasello, B.; Torrisi, F.; et al. Molecular Investigation on a Triple Negative Breast Cancer Xenograft Model Exposed to Proton Beams. Int. J. Mol. Sci. 2020, 21, 6337. [Google Scholar]
- Broggi, G.; Filetti, V.; Ieni, A.; Rapisarda, V.; Ledda, C.; Vitale, E.; Varricchio, S.; Russo, D.; Lombardo, C.; Tuccari, G.; et al. MacroH2A1 Immunoexpression in Breast Cancer. Front. Oncol. 2020, 10, 1519. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar]
- Worthington, J.; Bertani, M.; Chan, H.-L.; Gerrits, B.; Timms, J.F. Transcriptional profiling of ErbB signalling in mammary luminal epithelial cells—interplay of ErbB and IGF1 signalling through IGFBP3 regulation. BMC Cancer 2010, 10, 490. [Google Scholar]
- Zheng, Z.; Mao, S.; Gu, Z.; Wang, R.; Guo, Y.; Zhang, W.; Yao, X. A Genomic-Clinicopathologic Nomogram for the Prediction of Lymph Node Invasion in Prostate Cancer. J. Oncol. 2021, 2021, 5554708. [Google Scholar]
- Thomas, E.; Thankan, R.S.; Purushottamachar, P.; Huang, W.; Kane, M.A.; Zhang, Y.; Ambulos, N.; Weber, D.J.; Njar, V.C. Transcriptome profiling reveals that VNPP433-3β, the lead next-generation galeterone analog inhibits prostate cancer stem cells by downregulating epithelial-mesenchymal transition and stem cell markers. Mol. Carcinog. 2022, 61, 643–654. [Google Scholar] [PubMed]
- Shamsara, E.; Shamsara, J. Bioinformatics analysis of the genes involved in the extension of prostate cancer to adjacent lymph nodes by supervised and unsupervised machine learning methods: The role of SPAG1 and PLEKHF2. Genomics 2020, 112, 3871–3882. [Google Scholar] [PubMed]
- Pudova, E.A.; Lukyanova, E.N.; Nyushko, K.M.; Mikhaylenko, D.S.; Zaretsky, A.R.; Snezhkina, A.V.; Savvateeva, M.V.; Kobelyatskaya, A.A.; Melnikova, N.V.; Volchenko, N.N.; et al. Differentially Expressed Genes Associated With Prognosis in Locally Advanced Lymph Node-Negative Prostate Cancer. Front. Genet. 2019, 10, 730. [Google Scholar] [CrossRef]
- Schmidt, L.; Møller, M.; Haldrup, C.; Strand, S.H.; Vang, S.; Hedegaard, J.; Høyer, S.; Borre, M.; Ørntoft, T.; Sørensen, K.D. Exploring the transcriptome of hormone-naive multifocal prostate cancer and matched lymph node metastases. Br. J. Cancer 2018, 119, 1527–1537. [Google Scholar] [PubMed]
- Van der Eecken, K.; Vanwelkenhuyzen, J.; Deek, M.P.; Tran, P.T.; Warner, E.; Wyatt, A.W.; Kwan, E.M.; Verbeke, S.; Van Dorpe, J.; Fonteyne, V.; et al. Tissue- and Blood-derived Genomic Biomarkers for Metastatic Hormone-sensitive Prostate Cancer: A Systematic Review. Eur. Urol. Oncol. 2021, 4, 914–923. [Google Scholar] [PubMed]
- Patel, R.; Brzezinska, E.A.; Repiscak, P.; Ahmad, I.; Mui, E.; Gao, M.; Blomme, A.; Harle, V.; Tan, E.H.; Malviya, G.; et al. Activation of β-Catenin Cooperates with Loss of Pten to Drive AR-Independent Castration-Resistant Prostate Cancer. Cancer Res. 2020, 80, 576–590. [Google Scholar] [PubMed]
- Isaacsson Velho, P.; Fu, W.; Wang, H.; Mirkheshti, N.; Qazi, F.; Lima, F.A.S.; Shaukat, F.; Carducci, M.A.; Denmeade, S.R.; Paller, C.J.; et al. Wnt-pathway Activating Mutations Are Associated with Resistance to First-line Abiraterone and Enzalutamide in Castration-resistant Prostate Cancer. Eur. Urol. 2020, 77, 14–21. [Google Scholar] [PubMed]
- El-Gamal, M.I.; Mewafi, N.H.; Abdelmotteleb, N.E.; Emara, M.A.; Tarazi, H.; Sbenati, R.M.; Madkour, M.M.; Zaraei, S.-O.; Shahin, A.I.; Anbar, H.S. A Review of HER4 (ErbB4) Kinase, Its Impact on Cancer, and Its Inhibitors. Molecules 2021, 26, 7376. [Google Scholar]
- Bian, X.; Cao, F.; Wang, X.; Hou, Y.; Zhao, H.; Liu, Y. Establishment and characterization of a new human colon cancer cell line, PUMC-CRC1. Sci. Rep. 2021, 11, 13122. [Google Scholar]
- Kassem, N.; Kassem, H.; Kassem, L.; Hassan, M. Detection of activating mutations in liquid biopsy of Egyptian breast cancer patients using targeted next-generation sequencing: A pilot study. J. Egypt Natl. Cancer Inst. 2021, 33, 10. [Google Scholar]
- Lo Iacono, M.; Buttigliero, C.; Monica, V.; Bollito, E.; Garrou, D.; Cappia, S.; Rapa, I.; Vignani, F.; Bertaglia, V.; Fiori, C.; et al. Retrospective study testing next generation sequencing of selected cancer-associated genes in resected prostate cancer. Oncotarget 2016, 7, 14394–14404. [Google Scholar]
- Mayrhofer, M.; De Laere, B.; Whitington, T.; Van Oyen, P.; Ghysel, C.; Ampe, J.; Ost, P.; Demey, W.; Hoekx, L.; Schrijvers, D.; et al. Cell-free DNA profiling of metastatic prostate cancer reveals microsatellite instability, structural rearrangements and clonal hematopoiesis. Genome Med. 2018, 10, 85. [Google Scholar]
- De Laere, B.; van Dam, P.-J.; Whitington, T.; Mayrhofer, M.; Diaz, E.H.; Van den Eynden, G.; Vandebroek, J.; Del-Favero, J.; Van Laere, S.; Dirix, L.; et al. Comprehensive Profiling of the Androgen Receptor in Liquid Biopsies from Castration-resistant Prostate Cancer Reveals Novel Intra-AR Structural Variation and Splice Variant Expression Patterns. Eur. Urol. 2017, 72, 192–200. [Google Scholar]
- Russo, G.I.; Musso, N.; Romano, A.; Caruso, G.; Petralia, S.; Lanzanò, L.; Broggi, G.; Camarda, M. The Role of Dielectrophoresis for Cancer Diagnosis and Prognosis. Cancers 2021, 14, 198. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age (years), median (IQR) | 65.0 (62.0–68.0) |
| PSA (ng/mL), median (IQR) | 15.9 (9.37–40.0) |
| Clinical stage, n (%) | |
| T1–T2 | 14 (82.35) |
| T3 | 3 (17.65) |
| Biopsy ISUP grade group, n (%) | |
| 1 | 1 (5.88) |
| 2 | 6 (35.29) |
| 3 | 2 (11.76) |
| 4 | 6 (35.29) |
| 5 | 2 (11.76) |
| Pathological stage, n (%) | |
| T2 | 1 (5.88) |
| T3a | 5 (29.41) |
| T3b | 11 (64.71) |
| Pathological ISUP grade group, n (%) | |
| 2 | 1 (5.88) |
| 3 | 4 (23.53) |
| 4 | 2 (11.76) |
| 5 | 10 (58.82) |
| Pathological lymph node metastasis, n (%) | |
| N0 | 6 (35.3) |
| N1 | 11 (64.7) |
| Patients | ||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | MAF | |||||||||||||||||||||||||||||
| Gene | dbSNP | H | T | M | H | T | M | H | T | M | H | T | H | T | M | H | T | M | H | T | M | H | T | M | H | T | M | H | T | M | H | T | M | H | T | H | T | H | T | H | T | H | T | H | T | |
| ERBB2 | rs1419499014 | x | 0.000004 | |||||||||||||||||||||||||||||||||||||||||||
| ERBB4 | rs67894136 | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | 0.24 | ||||||||||||||||
| p.Q264 * | x | - | ||||||||||||||||||||||||||||||||||||||||||||
| rs839541 | x | 0.3 | ||||||||||||||||||||||||||||||||||||||||||||
| FGFR1 | p.A268V | x | - | |||||||||||||||||||||||||||||||||||||||||||
| CSF1R | rs386693509 | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | - | ||||||||||
| TP53 | rs1042522 | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | 0.33 | ||||
| rs28934578 | x | 0.000004 | ||||||||||||||||||||||||||||||||||||||||||||
| rs587782006 | x | 0.000004 | ||||||||||||||||||||||||||||||||||||||||||||
| PIK3CA | rs780807237 | x | 0.000005 | |||||||||||||||||||||||||||||||||||||||||||
| rs2230461 | x | x | x | x | x | x | x | 0.06 | ||||||||||||||||||||||||||||||||||||||
| KDR | rs1870377 | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | 0.22 | |||||||||||||||||||||||||
| rs397772062 | x | 0.22 | ||||||||||||||||||||||||||||||||||||||||||||
| JAK3 | rs3213409 | x | x | x | x | x | 0.008 | |||||||||||||||||||||||||||||||||||||||
| SMARC1/DERL3 | rs5030613 | x | x | x | x | x | x | x | 0.144 | |||||||||||||||||||||||||||||||||||||
| KIT | rs3822214 | x | x | x | x | x | x | x | x | 0.09 | ||||||||||||||||||||||||||||||||||||
| NPM1 | rs34323200 | x | 0.000004 | |||||||||||||||||||||||||||||||||||||||||||
| APC | COSM5990839 | x | - | |||||||||||||||||||||||||||||||||||||||||||
| Frameshift Deletion | x | - | ||||||||||||||||||||||||||||||||||||||||||||
| rs121913329 | x | - | ||||||||||||||||||||||||||||||||||||||||||||
| FGF3 | rs17881656 | x | x | x | - | |||||||||||||||||||||||||||||||||||||||||
| rs149119664 | x | - | ||||||||||||||||||||||||||||||||||||||||||||
| MLH1 | rs876659657 | x | - | |||||||||||||||||||||||||||||||||||||||||||
| FLT3 | rs752729752 | x | 0.0001 | |||||||||||||||||||||||||||||||||||||||||||
| rs2491231 | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | x | 0.4 | ||||||||||
| rs199906407 | x | - | ||||||||||||||||||||||||||||||||||||||||||||
| rs75580865 | x | 0.05 | ||||||||||||||||||||||||||||||||||||||||||||
| AKT1 | rs990046031 | x | - | |||||||||||||||||||||||||||||||||||||||||||
| STK11 | rs587781474 | x | - | |||||||||||||||||||||||||||||||||||||||||||
| rs59912467 | x | x | 0.006 | |||||||||||||||||||||||||||||||||||||||||||
| RB1 | p.E137K | x | - | |||||||||||||||||||||||||||||||||||||||||||
| RET | rs1800863 | x | 0.17 | |||||||||||||||||||||||||||||||||||||||||||
| ABL1 | rs34549764 | x | x | x | 0.002 | |||||||||||||||||||||||||||||||||||||||||
| ATM | p.L2868F | x | - | |||||||||||||||||||||||||||||||||||||||||||
| rs1800056 | x | x | x | 0.01 | ||||||||||||||||||||||||||||||||||||||||||
| HRAS/LRRC56 | rs28933406 | x | x | - | ||||||||||||||||||||||||||||||||||||||||||
| CTNNB1 | rs121913403 | x | x | - | ||||||||||||||||||||||||||||||||||||||||||
| MTTP | rs748883732 | x | x | |||||||||||||||||||||||||||||||||||||||||||
| EGFR | rs766533982 | x | x | 0.000008 | ||||||||||||||||||||||||||||||||||||||||||
| p-Value | Tropism | MAF | ClinVar | Gleason | PSA | MGS | Age | pT | N |
|---|---|---|---|---|---|---|---|---|---|
| Tropism | 0.00013 | 0.07011 | 0.67739 | 0.86292 | 0.08165 | 0.59258 | 0.54789 | 0.00199 | |
| MAF | 0.00013 | 0.48214 | 0.77437 | 0.87733 | 0.28901 | 0.11517 | 0.90036 | 0.06808 | |
| ClinVar | 0.07011 | 0.48214 | 0.8074 | 0.95044 | 0.24062 | 0.16091 | 0.2357 | 0.0649 | |
| Gleason | 0.67739 | 0.77437 | 0.8074 | 0.78992 | 0.80144 | 0.25762 | 0.65732 | 0.82564 | |
| PSA | 0.86292 | 0.87733 | 0.95044 | 0.78992 | 0.7361 | 0.79574 | 0.25965 | 0.80498 | |
| MGS | 0.08165 | 0.28901 | 0.24062 | 0.80144 | 0.7361 | 0.94487 | 0.85406 | 0.26163 | |
| Age | 0.59258 | 0.11517 | 0.16091 | 0.25762 | 0.79574 | 0.94487 | 0.64361 | 0.90357 | |
| pT | 0.54789 | 0.90036 | 0.2357 | 0.65732 | 0.25965 | 0.85406 | 0.64361 | 0.86292 | |
| N | 0.00199 | 0.06808 | 0.0649 | 0.82564 | 0.80498 | 0.26163 | 0.90357 | 0.86292 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russo, G.I.; Bonacci, P.; Bivona, D.; Privitera, G.F.; Broggi, G.; Caltabiano, R.; Vella, J.; Lo Giudice, A.; Asmundo, M.G.; Cimino, S.; et al. Genomic Landscape Alterations in Primary Tumor and Matched Lymph Node Metastasis in Hormone-Naïve Prostate Cancer Patients. Cancers 2022, 14, 4212. https://doi.org/10.3390/cancers14174212
Russo GI, Bonacci P, Bivona D, Privitera GF, Broggi G, Caltabiano R, Vella J, Lo Giudice A, Asmundo MG, Cimino S, et al. Genomic Landscape Alterations in Primary Tumor and Matched Lymph Node Metastasis in Hormone-Naïve Prostate Cancer Patients. Cancers. 2022; 14(17):4212. https://doi.org/10.3390/cancers14174212
Chicago/Turabian StyleRusso, Giorgio Ivan, Paolo Bonacci, Dalida Bivona, Grete Francesca Privitera, Giuseppe Broggi, Rosario Caltabiano, Jessica Vella, Arturo Lo Giudice, Maria Giovanna Asmundo, Sebastiano Cimino, and et al. 2022. "Genomic Landscape Alterations in Primary Tumor and Matched Lymph Node Metastasis in Hormone-Naïve Prostate Cancer Patients" Cancers 14, no. 17: 4212. https://doi.org/10.3390/cancers14174212
APA StyleRusso, G. I., Bonacci, P., Bivona, D., Privitera, G. F., Broggi, G., Caltabiano, R., Vella, J., Lo Giudice, A., Asmundo, M. G., Cimino, S., Morgia, G., Stefani, S., & Musso, N. (2022). Genomic Landscape Alterations in Primary Tumor and Matched Lymph Node Metastasis in Hormone-Naïve Prostate Cancer Patients. Cancers, 14(17), 4212. https://doi.org/10.3390/cancers14174212