

Mutant p53 Depletion by Novel Inhibitors for HSP40/J-Domain Proteins Derived from the Natural Compound Plumbagin
 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Molecular Docking Studies
2.3. Chemicals and Compounds
2.4. Plasmids
2.5. Generation of DNAJA1 and p53 Knockout or Knockdown Cell Lines
2.6. Antibodies
2.7. Western Blotting
2.8. Immunofluorescence
2.9. F-Actin Staining
2.10. Transwell Migration Assay
2.11. Cellular Thermal Shift Assay (CETSA)
2.12. Rac1/Cdc42 Pull-Down Activation Assay
2.13. Statistical Analysis
3. Results
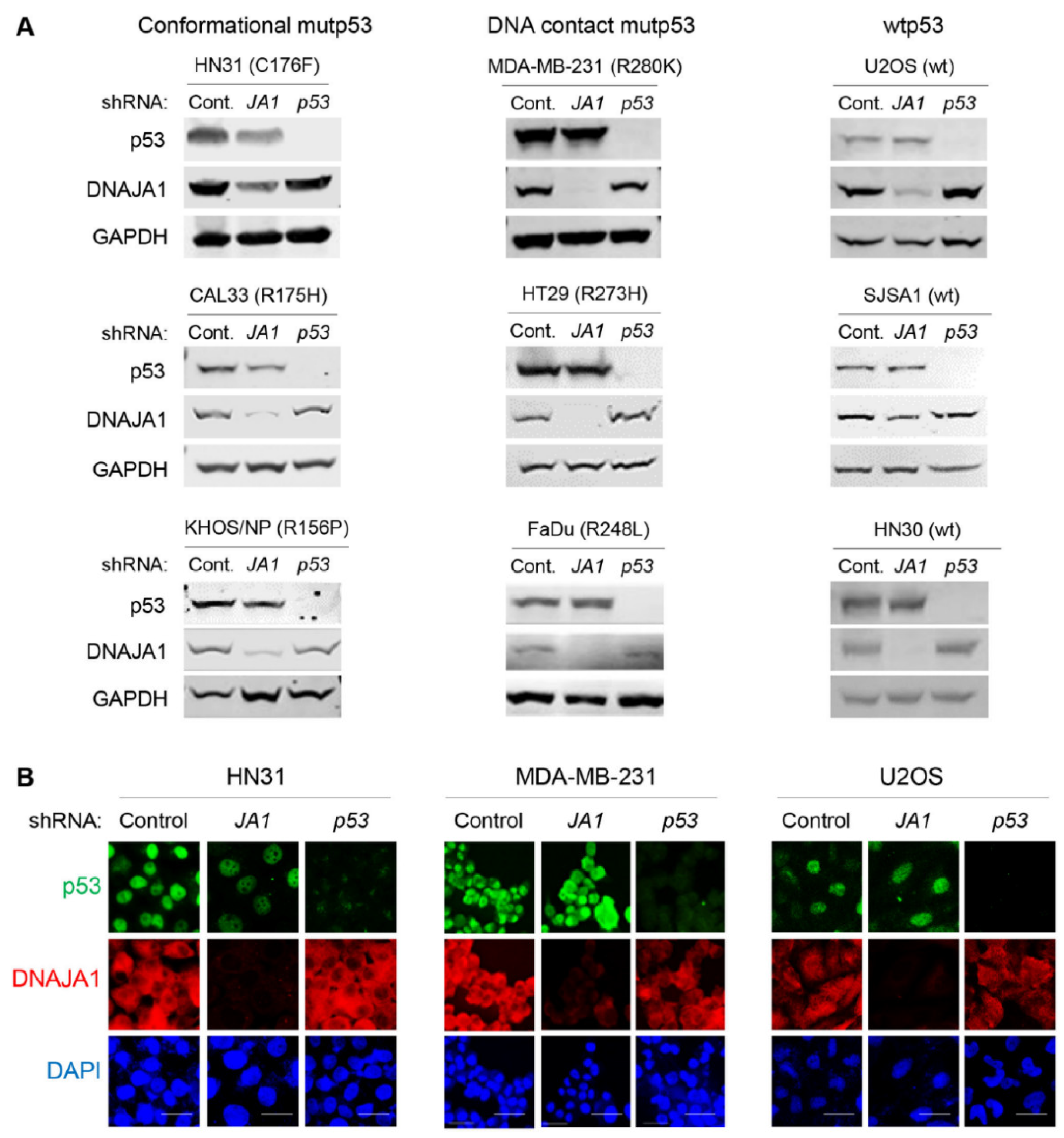
3.1. Knockdown of DNAJA1 Specifically Reduces Protein Levels of Conformational mutp53, but Not DNA Contact mutp53 and wtp53
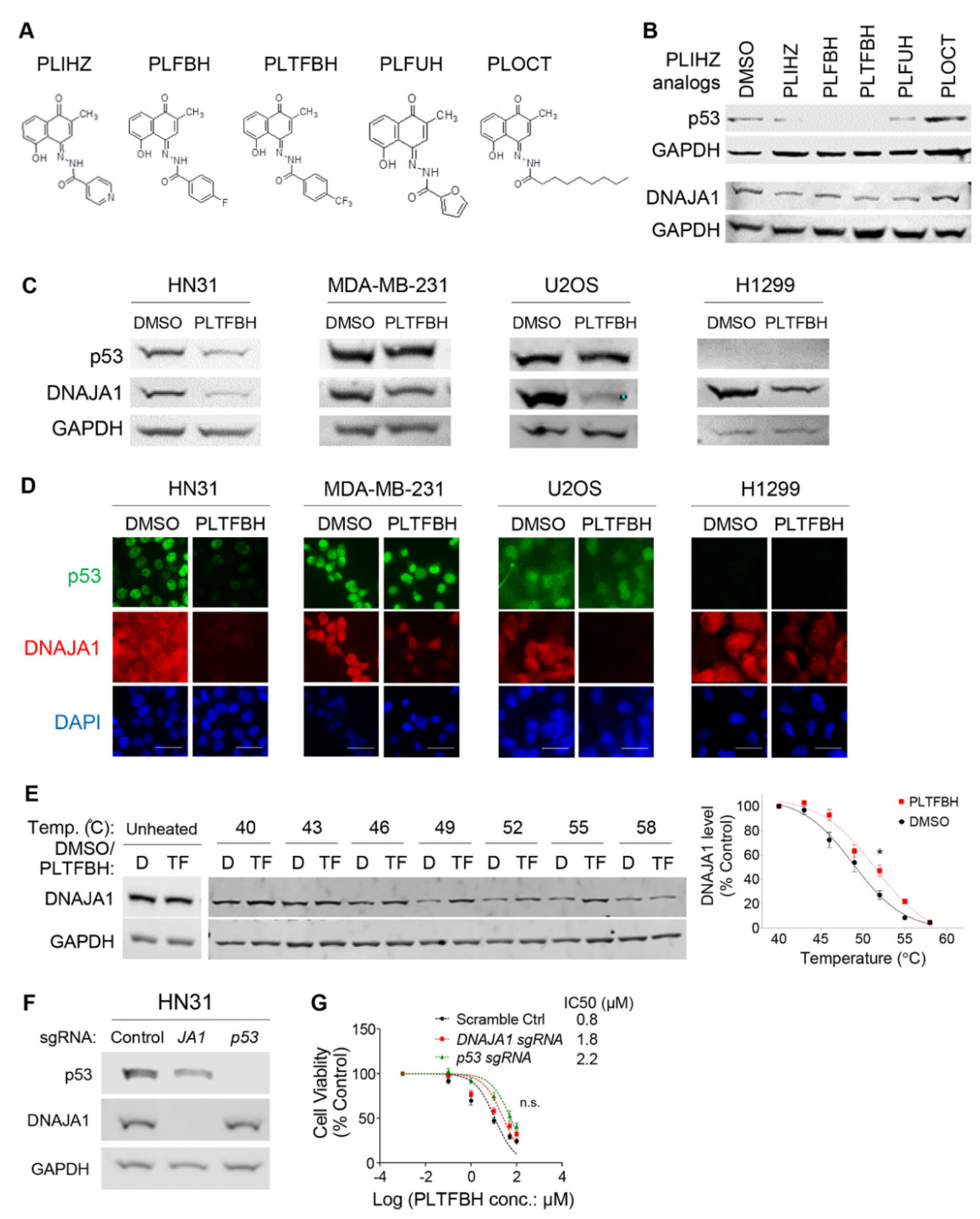
3.2. Identification of a Compound That Binds DNAJA1 to Specifically Reduce Conformational mutp53
3.3. PLTFBH, An Analog of PLIHZ, Specifically Reduces Conformational mutp53 Levels Similar to PLIHZ
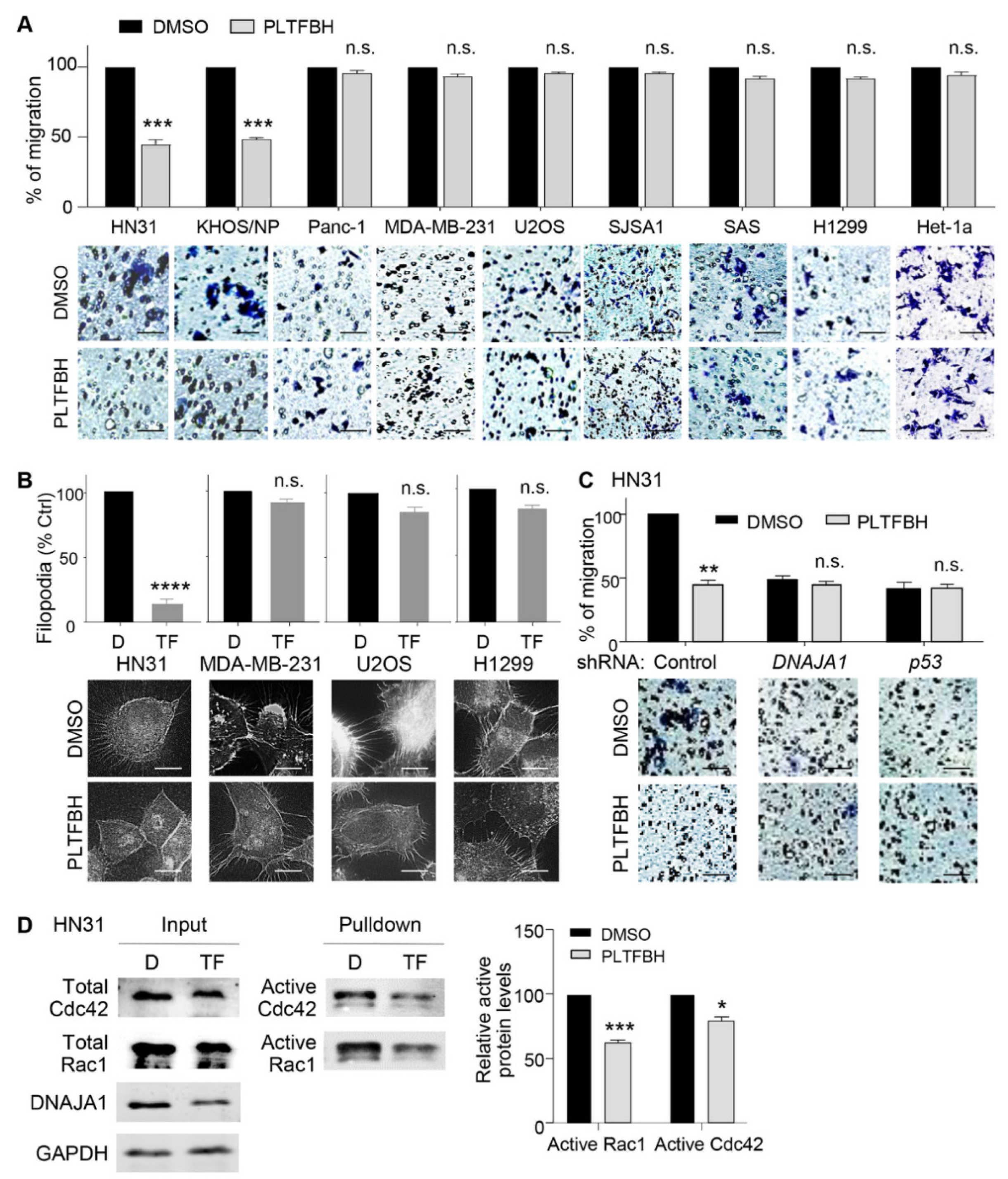
3.4. PLTFBH Inhibits Migratory Potential of Cancer Cells in a Manner Dependent on DNAJA1 and Conformational mutp53
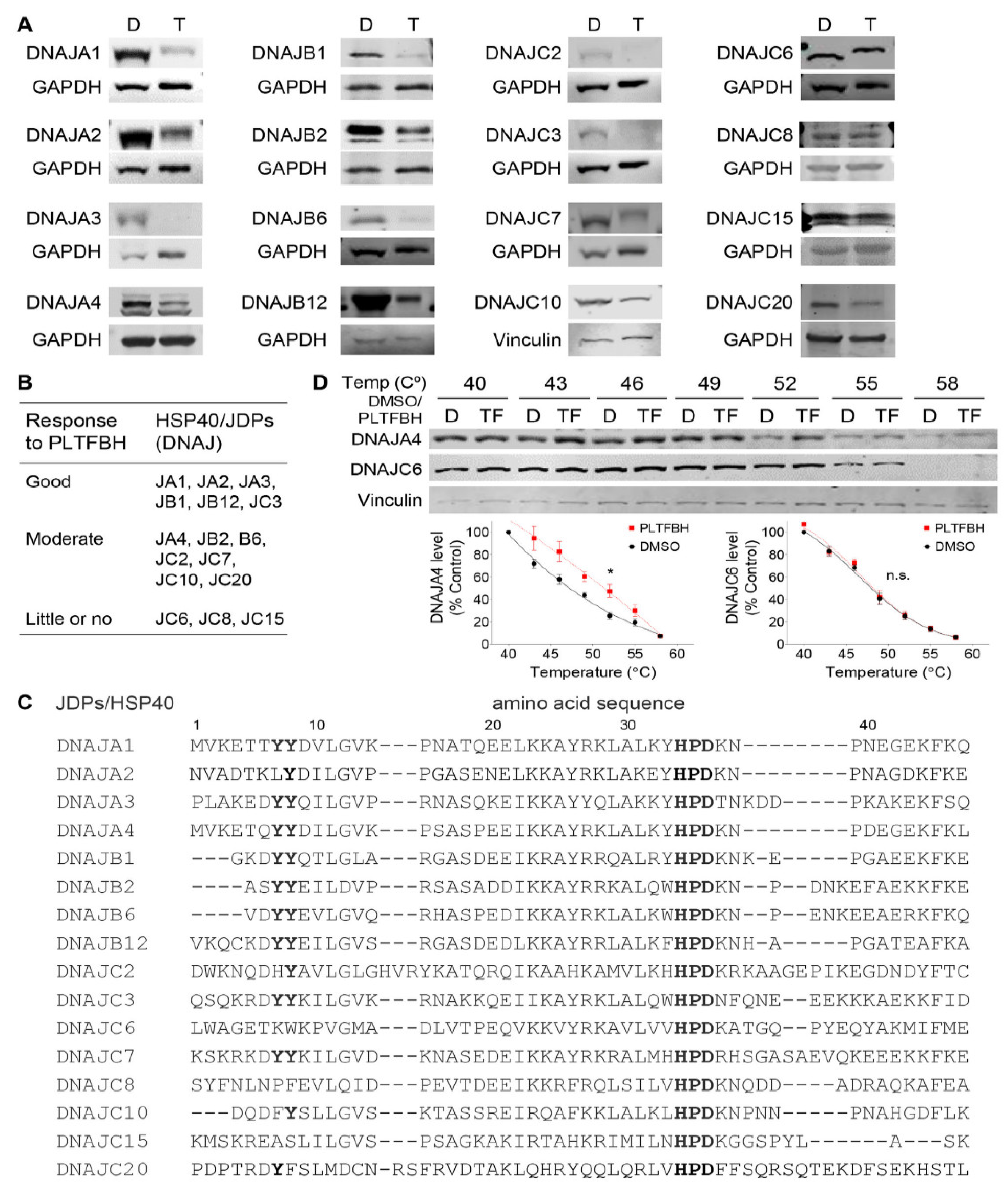
3.5. PLTFBH Selectively Decreases Protein Levels of Certain Members of HSP40/JDPs
3.6. Mutations at Y7 and Y8 Residues in DNAJA1 Abrogate the Ability of PLTFBH to Deplete DNAJA1 and Conformational mutp53
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HSP | heat shock protein |
| JDP | J-domain protein |
| IC50 | half maximal inhibitory concentration |
| GOF | gain of function |
| FBS | fetal bovine serum |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| RIPA | Radioimmunoprecipitation assay |
| PVDF | polyvinylidene fluoride |
| DMSO | dimethyl sulfoxide |
| PBS | phosphate-buffered saline |
| ATCC | American Type Culture Collection |
| shRNA | short hairpin RNA |
| NCBI | National Center for Biotechnology Information |
| ICLAC | International Cell Line Authentication Committee |
References
- Tsimberidou, A.M. Targeted therapy in cancer. Cancer Chemother. Pharmacol. 2015, 76, 1113–1132. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer 2011, 2, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Pfister, N.T.; Prives, C. Transcriptional Regulation by Wild-Type and Cancer-Related Mutant Forms of p53. Cold Spring Harb. Perspect. Med. 2017, 7, a026054. [Google Scholar] [CrossRef]
- Terzian, T.; Suh, Y.A.; Iwakuma, T.; Post, S.M.; Neumann, M.; Lang, G.A.; Van Pelt, C.S.; Lozano, G. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 2008, 22, 1337–1344. [Google Scholar] [CrossRef]
- Boeckler, F.M.; Joerger, A.C.; Jaggi, G.; Rutherford, T.J.; Veprintsev, D.B.; Fersht, A.R. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl. Acad. Sci. USA 2008, 105, 10360–10365. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Zhao, Y.; Xu, Y.; Zheng, M.; Feng, Z.; Hu, W. Mutant p53 in Cancer: Accumulation, Gain-of-Function, and Therapy. J. Mol. Biol. 2017, 429, 1595–1606. [Google Scholar] [CrossRef]
- Schulz-Heddergott, R.; Moll, U.M. Gain-of-Function (GOF) Mutant p53 as Actionable Therapeutic Target. Cancers 2018, 10, 188. [Google Scholar] [CrossRef]
- Parrales, A.; Iwakuma, T. Targeting Oncogenic Mutant p53 for Cancer Therapy. Front. Oncol. 2015, 5, 288. [Google Scholar] [CrossRef]
- Oren, M.; Rotter, V. Mutant p53 gain-of-function in cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a001107. [Google Scholar] [CrossRef]
- Joerger, A.C.; Fersht, A.R. Structural biology of the tumor suppressor p53 and cancer-associated mutants. Adv. Cancer Res. 2007, 97, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, C.A.; Singh, S.; Subler, M.A.; Windle, J.J.; Inoue, K.; Fry, E.A.; Pillappa, R.; Grossman, S.R.; Windle, B.; Andrew Yeudall, W.; et al. The oncogenicity of tumor-derived mutant p53 is enhanced by the recruitment of PLK3. Nat. Commun. 2021, 12, 704. [Google Scholar] [CrossRef]
- Alexandrova, E.M.; Yallowitz, A.R.; Li, D.; Xu, S.; Schulz, R.; Proia, D.A.; Lozano, G.; Dobbelstein, M.; Moll, U.M. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 2015, 523, 352–356. [Google Scholar] [CrossRef]
- Lukashchuk, N.; Vousden, K.H. Ubiquitination and degradation of mutant p53. Mol. Cell Biol. 2007, 27, 8284–8295. [Google Scholar] [CrossRef]
- Parrales, A.; Ranjan, A.; Iyer, S.V.; Padhye, S.; Weir, S.J.; Roy, A.; Iwakuma, T. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 2016, 18, 1233–1243. [Google Scholar] [CrossRef]
- Qiu, X.B.; Shao, Y.M.; Miao, S.; Wang, L. The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell Mol. Life Sci. 2006, 63, 2560–2570. [Google Scholar] [CrossRef]
- Zarouchlioti, C.; Parfitt, D.A.; Li, W.; Gittings, L.M.; Cheetham, M.E. DNAJ Proteins in neurodegeneration: Essential and protective factors. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20160534. [Google Scholar] [CrossRef]
- Li, J.; Qian, X.; Sha, B. Heat shock protein 40: Structural studies and their functional implications. Protein Pept. Lett. 2009, 16, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Moses, M.A.; Kim, Y.S.; Rivera-Marquez, G.M.; Oshima, N.; Watson, M.J.; Beebe, K.E.; Wells, C.; Lee, S.; Zuehlke, A.D.; Shao, H.; et al. Targeting the Hsp40/Hsp70 Chaperone Axis as a Novel Strategy to Treat Castration-Resistant Prostate Cancer. Cancer Res. 2018, 78, 4022–4035. [Google Scholar] [CrossRef]
- Kaida, A.; Yamamoto, S.; Parrales, A.; Young, E.D.; Ranjan, A.; Alalem, M.A.; Morita, K.I.; Oikawa, Y.; Harada, H.; Ikeda, T.; et al. DNAJA1 promotes cancer metastasis through interaction with mutant p53. Oncogene 2021, 40, 5013–5025. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Dandawate, P.; Khan, E.; Padhye, S.; Gaba, H.; Sinha, S.; Deshpande, J.; Venkateswara Swamy, K.; Khetmalas, M.; Ahmad, A.; Sarkar, F.H. Synthesis, characterization, molecular docking and cytotoxic activity of novel plumbagin hydrazones against breast cancer cells. Bioorg. Med. Chem. Lett. 2012, 22, 3104–3108. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Xu, D.; Mishra, R.K.; Jones, R.D.; Sun, L.; Schiltz, G.E.; Liao, J.; Yang, G.Y. Identification of a druggable protein-protein interaction site between mutant p53 and its stabilizing chaperone DNAJA1. J. Biol. Chem. 2021, 296, 100098. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Tong, X.; Sun, L.; Li, H.; Jones, R.D.; Liao, J.; Yang, G.Y. Inhibition of mutant Kras and p53-driven pancreatic carcinogenesis by atorvastatin: Mainly via targeting of the farnesylated DNAJA1 in chaperoning mutant p53. Mol. Carcinog. 2019, 58, 2052–2064. [Google Scholar] [CrossRef]
- Dandawate, P.; Vemuri, K.; Venkateswara Swamy, K.; Khan, E.M.; Sritharan, M.; Padhye, S. Synthesis, characterization, molecular docking and anti-tubercular activity of Plumbagin–Isoniazid Analog and its β-cyclodextrin conjugate. Bioorg. Med. Chem. Lett. 2014, 24, 5070–5075. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Zhang, J.; Chen, L.; Guo, Q.; Yang, B.; Zhang, W.; Kang, W. Anticancer Effects and Mechanisms of Action of Plumbagin: Review of Research Advances. Biomed Res. Int. 2020, 2020, 6940953. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Yin, D.; Ren, Y.; Gong, C.; Chen, A.; Guo, F.-J. Plumbagin induces apoptosis via the p53 pathway and generation of reactive oxygen species in human osteosarcoma cells. Mol. Med. Rep. 2012, 5, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.H.; Dreckschmidt, N.E.; Verma, A.K. Plumbagin, a medicinal plant-derived naphthoquinone, is a novel inhibitor of the growth and invasion of hormone-refractory prostate cancer. Cancer Res. 2008, 68, 9024–9032. [Google Scholar] [CrossRef]
- Jafari, R.; Almqvist, H.; Axelsson, H.; Ignatushchenko, M.; Lundback, T.; Nordlund, P.; Martinez Molina, D. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 2014, 9, 2100–2122. [Google Scholar] [CrossRef]
- Martinez Molina, D.; Jafari, R.; Ignatushchenko, M.; Seki, T.; Larsson, E.A.; Dan, C.; Sreekumar, L.; Cao, Y.; Nordlund, P. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 2013, 341, 84–87. [Google Scholar] [CrossRef]
- Saxena, C. Identification of protein binding partners of small molecules using label-free methods. Expert Opin. Drug Discov. 2016, 11, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.V.; Parrales, A.; Begani, P.; Narkar, A.; Adhikari, A.S.; Martinez, L.A.; Iwakuma, T. Allele-specific silencing of mutant p53 attenuates dominant-negative and gain-of-function activities. Oncotarget 2016, 7, 5401–5415. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Iwakuma, T. Regulators of Oncogenic Mutant TP53 Gain of Function. Cancers 2018, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Vousden, K.H.; Norman, J.C. p53 and its mutants in tumor cell migration and invasion. J. Cell Biol. 2011, 192, 209–218. [Google Scholar] [CrossRef]
- Fukata, M.; Nakagawa, M.; Kaibuchi, K. Roles of Rho-family GTPases in cell polarisation and directional migration. Curr. Opin. Cell Biol. 2003, 15, 590–597. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Craig, E.A. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 2010, 11, 579–592. [Google Scholar] [CrossRef]
- Shukla, B.; Saxena, S.; Usmani, S.; Kushwaha, P. Phytochemistry and pharmacological studies of Plumbago zeylanica L.: A medicinal plant review. Clin. Phytoscience 2021, 7, 34. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alalem, M.; Bhosale, M.; Ranjan, A.; Yamamoto, S.; Kaida, A.; Nishikawa, S.; Parrales, A.; Farooki, S.; Anant, S.; Padhye, S.; et al. Mutant p53 Depletion by Novel Inhibitors for HSP40/J-Domain Proteins Derived from the Natural Compound Plumbagin. Cancers 2022, 14, 4187. https://doi.org/10.3390/cancers14174187
Alalem M, Bhosale M, Ranjan A, Yamamoto S, Kaida A, Nishikawa S, Parrales A, Farooki S, Anant S, Padhye S, et al. Mutant p53 Depletion by Novel Inhibitors for HSP40/J-Domain Proteins Derived from the Natural Compound Plumbagin. Cancers. 2022; 14(17):4187. https://doi.org/10.3390/cancers14174187
Chicago/Turabian StyleAlalem, Mohamed, Mrinalini Bhosale, Atul Ranjan, Satomi Yamamoto, Atsushi Kaida, Shigeto Nishikawa, Alejandro Parrales, Sana Farooki, Shrikant Anant, Subhash Padhye, and et al. 2022. "Mutant p53 Depletion by Novel Inhibitors for HSP40/J-Domain Proteins Derived from the Natural Compound Plumbagin" Cancers 14, no. 17: 4187. https://doi.org/10.3390/cancers14174187
APA StyleAlalem, M., Bhosale, M., Ranjan, A., Yamamoto, S., Kaida, A., Nishikawa, S., Parrales, A., Farooki, S., Anant, S., Padhye, S., & Iwakuma, T. (2022). Mutant p53 Depletion by Novel Inhibitors for HSP40/J-Domain Proteins Derived from the Natural Compound Plumbagin. Cancers, 14(17), 4187. https://doi.org/10.3390/cancers14174187