
Investigating the Retained Inhibitory Effect of Cobimetinib against p.P124L Mutated MEK1: A Combined Liquid Biopsy and in Silico Approach

 , , , , ,
, , , , ,  and
and
Abstract
:Simple Summary
Abstract

1. Introduction
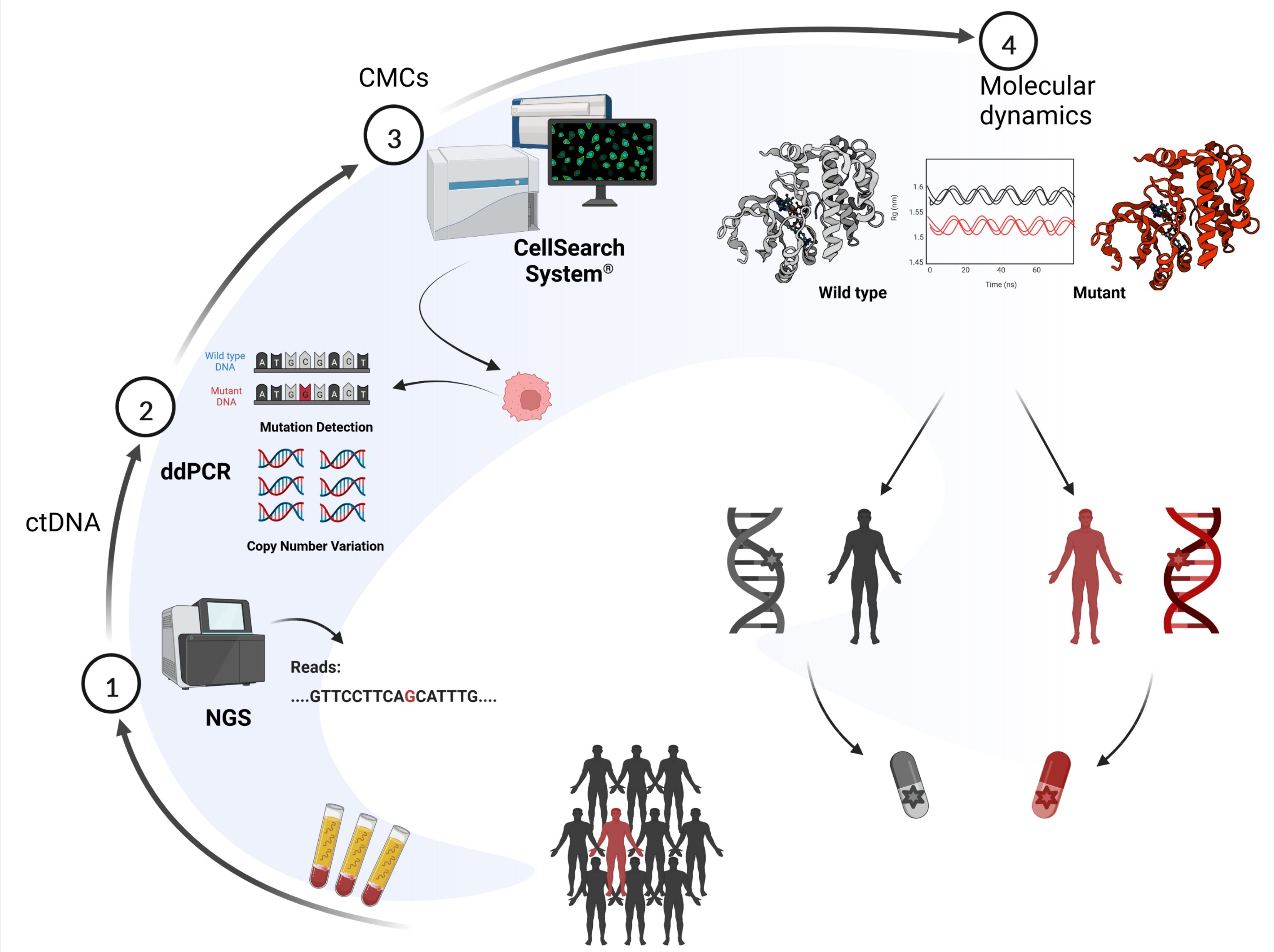
2. Materials and Methods
2.1. Patient and Samples
2.2. Molecular Dynamics Simulation
2.3. cfDNA Analysis
2.4. Circulating Melanoma Cell Enrichment, Detection and Isolation
3. Results
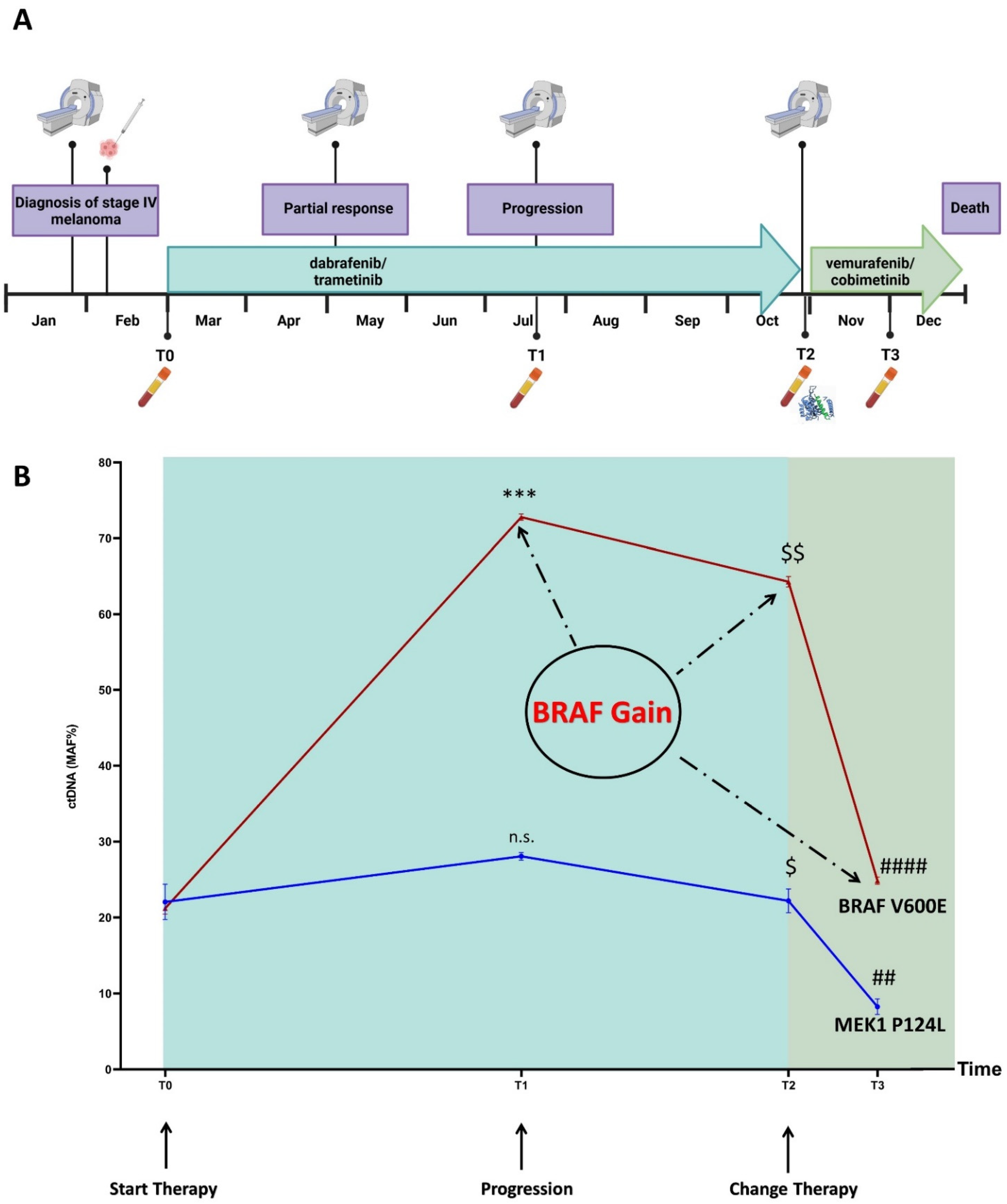
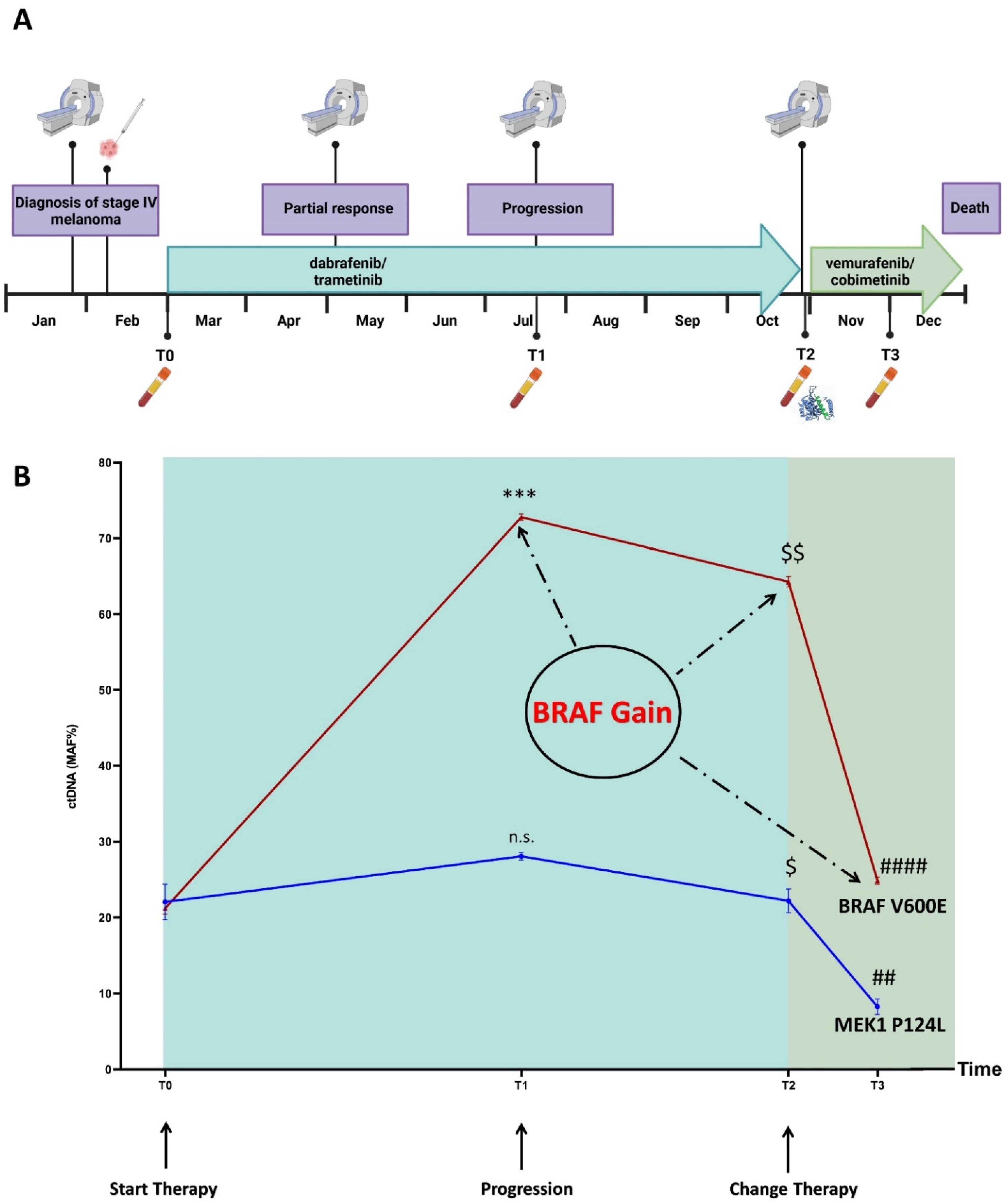
3.1. Disease and Treatment Evolution
3.2. Overview
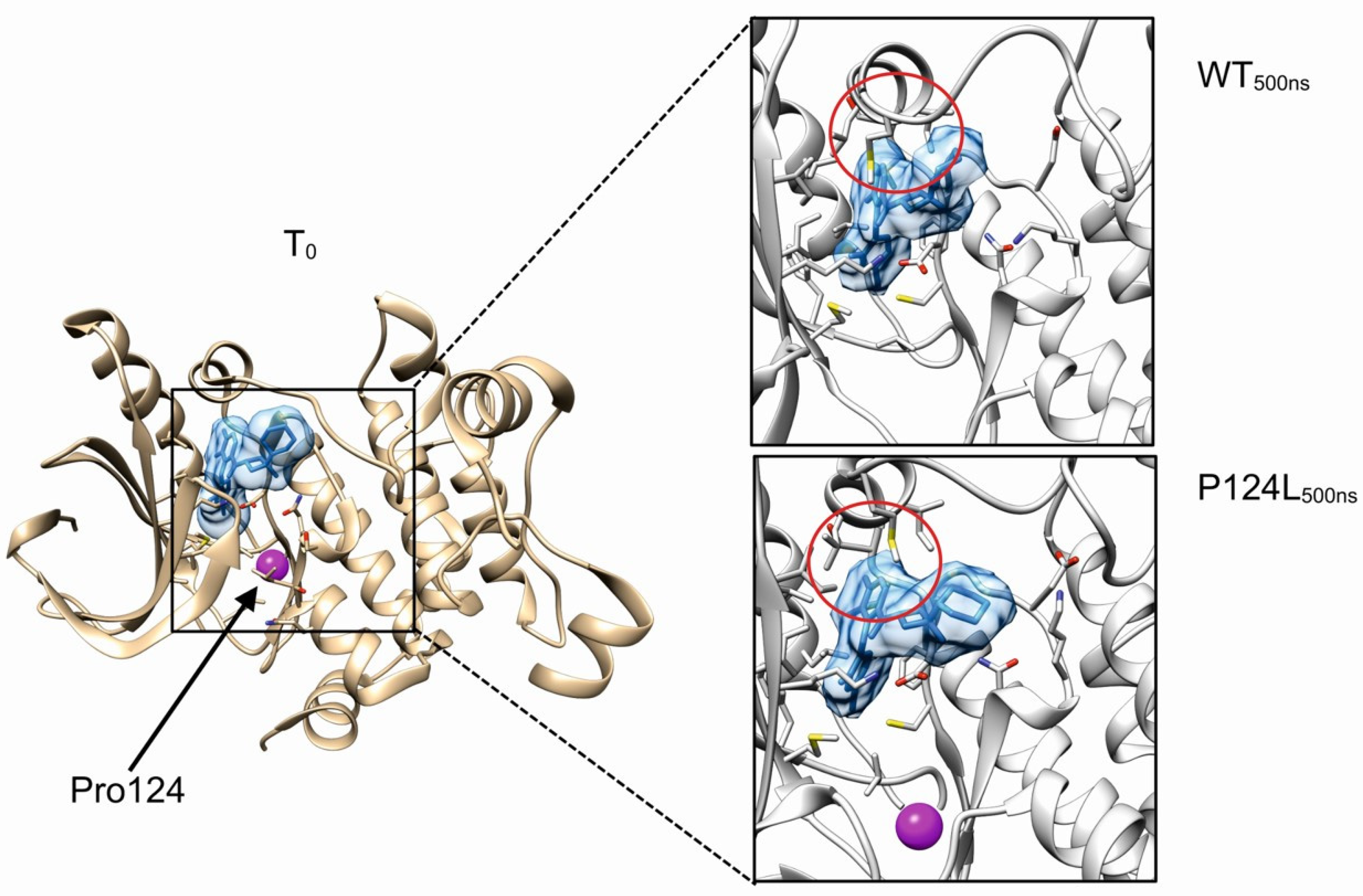
3.3. Molecular Dynamics Simulations
3.4. Mutation Profiling of ctDNA
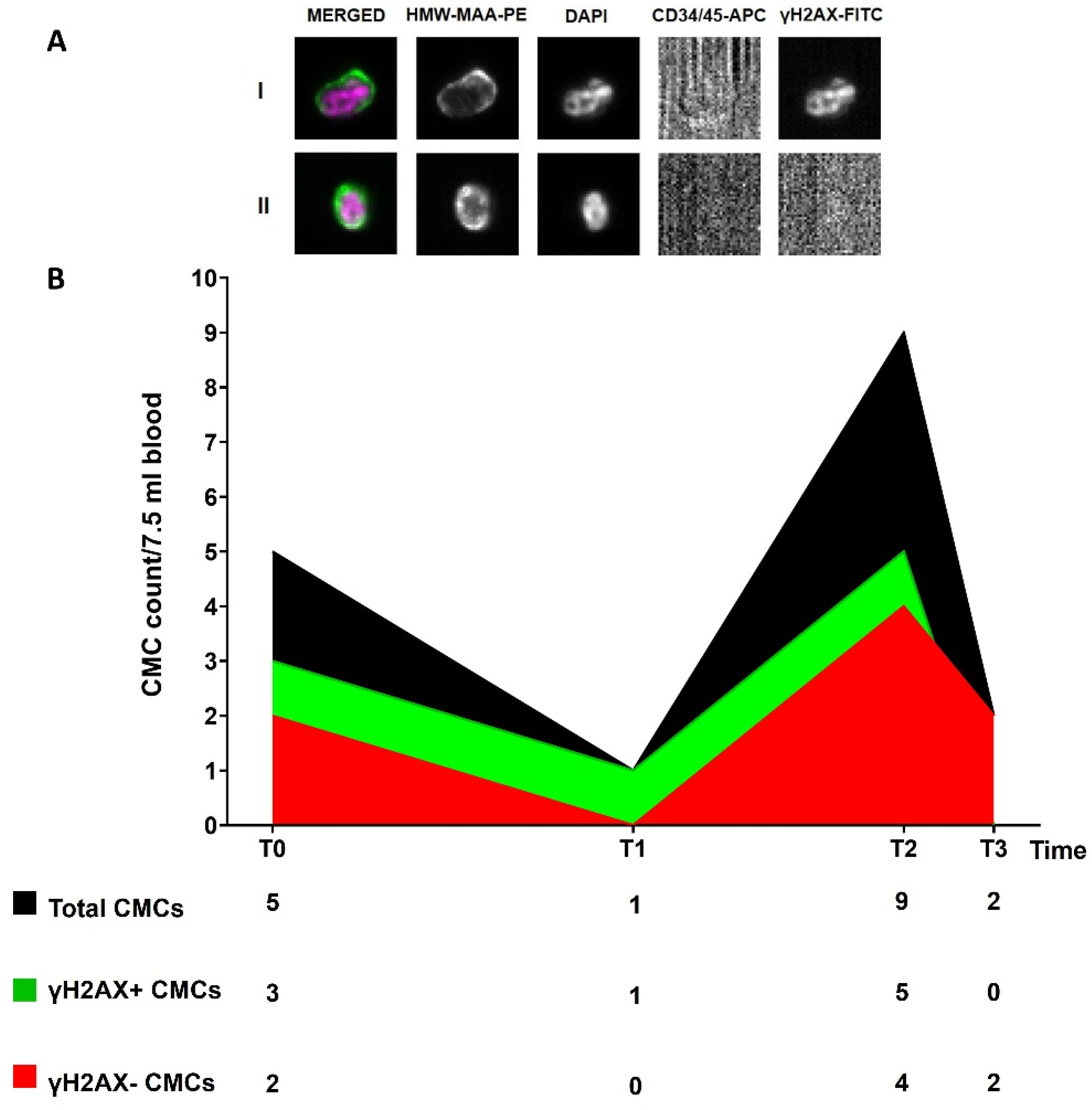
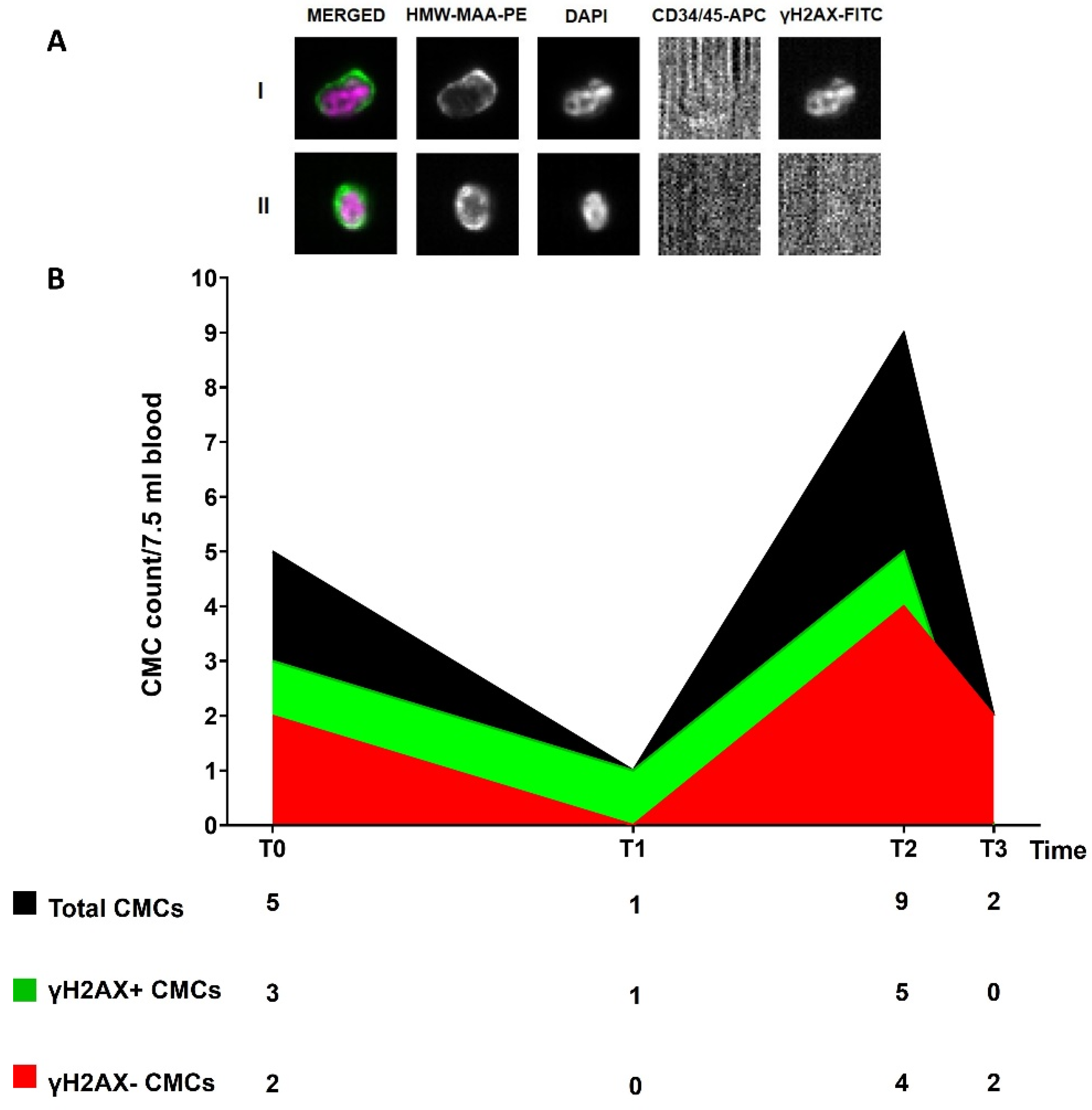
3.5. Circulating Melanoma Cell Count
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tiwari, A.; Singh, S. Computational Approaches in Drug Designing. In Bioinformatics; Singh, D.B., Pathak, R.K., Eds.; Academic Press: Cambridge, MA, USA, 2022; Chapter 13; pp. 207–217. ISBN 978-0-323-89775-4. [Google Scholar]
- Choi, G.; Kim, D.; Oh, J. AI-Based Drug Discovery of TKIs Targeting L858R/T790M/C797S-Mutant EGFR in Non-Small Cell Lung Cancer. Front. Pharmacol. 2021, 12, 313. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Hu, B. Mathematical modeling and computational prediction of cancer drug resistance. Brief. Bioinform. 2017, 19, 1382–1399. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Tsao, H.; Chin, L.; Garraway, L.A.; Fisher, D.E. Melanoma: From mutations to medicine. Genes Dev. 2012, 26, 1131–1155. [Google Scholar] [CrossRef] [PubMed]
- García-Fernández, M.; Karras, P.; Checinska, A.; Cañón, E.; Calvo, G.T.; Gómez-López, G.; Cifdaloz, M.; Colmenar, A.; Espinosa-Hevia, L.; Olmeda, D.; et al. Metastatic risk and resistance to BRAF inhibitors in melanoma defined by selective allelic loss of ATG5. Autophagy 2016, 12, 1776–1790. [Google Scholar] [CrossRef]
- Curti, B.D.; Faries, M.B. Recent Advances in the Treatment of Melanoma. N. Engl. J. Med. 2021, 384, 2229–2240. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Baik, C.; Kirkwood, J.M. Clinical Development of BRAF plus MEK Inhibitor Combinations. Trends Cancer 2020, 6, 797–810. [Google Scholar] [CrossRef]
- Long, G.V.; Fung, C.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Hyman, J.; Shahheydari, H.; Tembe, V.; Thompson, J.F.; Saw, R.P.; et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat. Commun. 2014, 5, 5694. [Google Scholar] [CrossRef]
- Olbryt, M.; Pigłowski, W.; Rajczykowski, M.; Pfeifer, A.; Student, S.; Fiszer-Kierzkowska, A. Genetic Profiling of Advanced Melanoma: Candidate Mutations for Predicting Sensitivity and Resistance to Targeted Therapy. Target. Oncol. 2020, 15, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Tangella, L.P.; Clark, M.E.; Gray, E.S. Resistance mechanisms to targeted therapy in BRAF-mutant melanoma - A mini review. Biochim. Et Biophys. Acta (BBA) - Gen. Subj. 2020, 1865, 129736. [Google Scholar] [CrossRef] [PubMed]
- Gilson, P.; Merlin, J.-L.; Harlé, A. Deciphering Tumour Heterogeneity: From Tissue to Liquid Biopsy. Cancers 2022, 14, 1384. [Google Scholar] [CrossRef] [PubMed]
- Haselmann, V.; Gebhardt, C.; Brechtel, I.; Duda, A.; Czerwinski, C.; Sucker, A.; Holland-Letz, T.; Utikal, J.; Schadendorf, D.; Neumaier, M. Liquid Profiling of Circulating Tumor DNA in Plasma of Melanoma Patients for Companion Diagnostics and Monitoring of BRAF Inhibitor Therapy. Clin. Chem. 2018, 64, 830–842. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabières, C.; Pantel, K. Liquid Biopsy: From Discovery to Clinical Application. Cancer Discov. 2021, 11, 858–873. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabières, C.; Pantel, K. Clinical Applications of Circulating Tumor Cells and Circulating Tumor DNA as Liquid Biopsy. Cancer Discov. 2016, 6, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Bardelli, A.; Pantel, K. Liquid Biopsies, What We Do Not Know (Yet). Cancer Cell 2017, 31, 172–179. [Google Scholar] [CrossRef]
- Pantel, K.; Alix-Panabières, C. Liquid biopsy and minimal residual disease—Latest advances and implications for cure. Nat. Rev. Clin. Oncol. 2019, 16, 409–424. [Google Scholar] [CrossRef]
- Khoja, L.; Lorigan, P.; Zhou, C.; Lancashire, M.; Booth, J.; Cummings, J.; Califano, R.; Clack, G.; Hughes, A.; Dive, C. Biomarker Utility of Circulating Tumor Cells in Metastatic Cutaneous Melanoma. J. Investig. Dermatol. 2013, 133, 1582–1590. [Google Scholar] [CrossRef]
- Khoja, L.; Lorigan, P.; Dive, C.; Keilholz, U.; Fusi, A. Circulating tumour cells as tumour biomarkers in melanoma: Detection methods and clinical relevance. Ann. Oncol. 2015, 26, 33–39. [Google Scholar] [CrossRef]
- Kamińska, P.; Buszka, K.; Zabel, M.; Nowicki, M.; Alix-Panabières, C.; Budna-Tukan, J. Liquid Biopsy in Melanoma: Significance in Diagnostics, Prediction and Treatment Monitoring. Int. J. Mol. Sci. 2021, 22, 9714. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Páll, S.; Zhmurov, A.; Bauer, P.; Abraham, M.; Lundborg, M.; Gray, A.; Hess, B.; Lindahl, E. Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS. J. Chem. Phys. 2020, 153, 134110. [Google Scholar] [CrossRef] [PubMed]
- Clementel, D.; Del Conte, A.; Monzon, A.M.; Camagni, G.F.; Minervini, G.; Piovesan, D.; E Tosatto, S.C. RING 3.0: Fast generation of probabilistic residue interaction networks from structural ensembles. Nucleic Acids Res. 2022, 50, W651–W656. [Google Scholar] [CrossRef]
- Bustos, B.D.U.; Estal, R.M.; Simó, G.P.; Jimenez, I.D.J.; Muñoz, B.E.; Serna, M.R.; De Miquel, V.A.; Ros, M.L.; Sánchez, R.B.; Enguídanos, E.N.; et al. Towards Personalized Medicine in Melanoma: Implementation of a Clinical Next-Generation Sequencing Panel. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Casula, M.; (Mus), M.U.O.S.; Paliogiannis, P.; Ayala, F.; De Giorgi, V.; Stanganelli, I.; Mandalà, M.; Colombino, M.; Manca, A.; Sini, M.C.; et al. Germline and somatic mutations in patients with multiple primary melanomas: A next generation sequencing study. BMC Cancer 2019, 19, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Reiman, A.; Kikuchi, H.; Scocchia, D.; Smith, P.; Tsang, Y.W.; Snead, D.; A Cree, I. Validation of an NGS mutation detection panel for melanoma. BMC Cancer 2017, 17, 150. [Google Scholar] [CrossRef]
- Garman, B.; Anastopoulos, I.N.; Krepler, C.; Brafford, P.; Sproesser, K.; Jiang, Y.; Wubbenhorst, B.; Amaravadi, R.; Bennett, J.; Beqiri, M.; et al. Genetic and Genomic Characterization of 462 Melanoma Patient-Derived Xenografts, Tumor Biopsies, and Cell Lines. Cell Rep. 2017, 21, 1936–1952. [Google Scholar] [CrossRef]
- Siroy, A.E.; Boland, G.M.; Milton, D.R.; Roszik, J.; Frankian, S.; Malke, J.; Haydu, L.; Prieto, V.G.; Tetzlaff, M.; Ivan, D.; et al. Beyond BRAF V600: Clinical Mutation Panel Testing by Next-Generation Sequencing in Advanced Melanoma. J. Investig. Dermatol. 2015, 135, 508–515. [Google Scholar] [CrossRef]
- Afshar, A.R.; Damato, B.E.; Stewart, J.M.; Zablotska, L.B.; Roy, R.; Olshen, A.B.; Joseph, N.M.; Bastian, B.C. Next-Generation Sequencing of Uveal Melanoma for Detection of Genetic Alterations Predicting Metastasis. Transl. Vis. Sci. Technol. 2019, 8, 18. [Google Scholar] [CrossRef]
- Versluis, M.; De Lange, M.J.; Van Pelt, S.I.; Ruivenkamp, C.A.L.; Kroes, W.G.M.; Cao, J.; Jager, M.J.; Luyten, G.P.M.; Van Der Velden, P.A. Digital PCR Validates 8q Dosage as Prognostic Tool in Uveal Melanoma. PLoS ONE 2015, 10, e0116371. [Google Scholar] [CrossRef] [Green Version]
- de Lange, M.J.; van Pelt, S.I.; Versluis, M.; Jordanova, E.S.; Kroes, W.G.; Ruivenkamp, C.; van der Burg, S.H.; Luyten, G.P.; van Hall, T.; Jager, M.J.; et al. Heterogeneity revealed by integrated genomic analysis uncovers a molecular switch in malignant uveal melanoma. Oncotarget 2015, 6, 37824–37835. [Google Scholar] [CrossRef] [PubMed]
- Shoda, K.; Ichikawa, D.; Fujita, Y.; Masuda, K.; Hiramoto, H.; Hamada, J.; Arita, T.; Konishi, H.; Komatsu, S.; Shiozaki, A.; et al. Monitoring the HER2 copy number status in circulating tumor DNA by droplet digital PCR in patients with gastric cancer. Gastric Cancer 2016, 20, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Rao, C.; Bui, T.; Connelly, M.; Doyle, G.; Karydis, I.; Middleton, M.R.; Clack, G.; Malone, M.; Coumans, F.A.; Terstappen, L.W. Circulating melanoma cells and survival in metastatic melanoma. Int. J. Oncol. 2011, 38, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Nieves-Neira, W.; Boon, C.; Pommier, Y.; Bonner, W.M. Initiation of DNA Fragmentation during Apoptosis Induces Phosphorylation of H2AX Histone at Serine 139. J. Biol. Chem. 2000, 275, 9390–9395. [Google Scholar] [CrossRef] [PubMed]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. γH2AX and cancer. Nat. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Villa, A.; Balasubramanian, P.; Miller, B.L.; Lustberg, M.B.; Ramaswamy, B.; Chalmers, J.J. Assessment of γ-H2AX levels in circulating tumor cells from patients receiving chemotherapy. Front. Oncol. 2012, 2, 128. [Google Scholar] [CrossRef]
- Wang, L.H.; Pfister, T.D.; Parchment, R.E.; Kummar, S.; Rubinstein, L.; Evrard, Y.A.; Gutierrez, M.E.; Murgo, A.J.; Tomaszewski, J.E.; Doroshow, J.H.; et al. Monitoring Drug-Induced γH2AX as a Pharmacodynamic Biomarker in Individual Circulating Tumor Cells. Clin. Cancer Res. 2010, 16, 1073–1084. [Google Scholar] [CrossRef]
- Emery, C.M.; Vijayendran, K.G.; Zipser, M.C.; Sawyer, A.M.; Niu, L.; Kim, J.J.; Hatton, C.; Chopra, R.; Oberholzer, P.A.; Karpova, M.B.; et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc. Natl. Acad. Sci. USA 2009, 106, 20411–20416. [Google Scholar] [CrossRef]
- Zhu, J.; Li, C.; Yang, H.; Guo, X.; Huang, T.; Han, W. Computational Study on the Effect of Inactivating/Activating Mutations on the Inhibition of MEK1 by Trametinib. Int. J. Mol. Sci. 2020, 21, 2167. [Google Scholar] [CrossRef]
- Khan, Z.M.; Real, A.M.; Marsiglia, W.M.; Chow, A.; Duffy, M.E.; Yerabolu, J.R.; Scopton, A.P.; Dar, A.C. Structural basis for the action of the drug trametinib at KSR-bound MEK. Nature 2020, 588, 509–514. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Aguilera, M.A.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [PubMed]
- Diamond, E.L.; Durham, B.H.; Ulaner, G.A.; Drill, E.; Buthorn, J.; Ki, M.; Bitner, L.; Cho, H.; Young, R.J.; Francis, J.H.; et al. Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature 2019, 567, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.S.; Rizos, H.; Reid, A.L.; Boyd, S.C.; Pereira, M.R.; Lo, J.; Tembe, V.; Freeman, J.; Lee, J.H.; Scolyer, R.A.; et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget 2015, 6, 42008–42018. [Google Scholar] [CrossRef] [PubMed]
- Girotti, M.R.; Gremel, G.; Lee, R.; Galvani, E.; Rothwell, D.; Viros, A.; Mandal, A.K.; Lim, K.H.J.; Saturno, G.; Furney, S.J.; et al. Application of Sequencing, Liquid Biopsies, and Patient-Derived Xenografts for Personalized Medicine in Melanoma. Cancer Discov. 2016, 6, 286–299. [Google Scholar] [CrossRef]
- Berger, M.; Thueringer, A.; Franz, D.; Dandachi, N.; Talakić, E.; Richtig, G.; Richtig, E.; Rohrer, P.; Koch, L.; Wolf, I.; et al. Circulating Tumor DNA as a Marker for Treatment Response in Metastatic Melanoma Patients Using Next-Generation Sequencing—A Prospective Feasibility Study. Cancers 2021, 13, 3101. [Google Scholar] [CrossRef]
- Hall, C.S.; Ross, M.; Bauldry, J.B.B.; Upshaw, J.; Karhade, M.G.; Royal, R.; Patel, S.; Lucci, A. Circulating Tumor Cells in Stage IV Melanoma Patients. J. Am. Coll. Surg. 2018, 227, 116–124. [Google Scholar] [CrossRef]
- Hanssen, A.; Riebensahm, C.; Mohme, M.; Joosse, S.A.; Velthaus, J.-L.; Berger, L.A.; Bernreuther, C.; Glatzel, M.; Loges, S.; Lamszus, K.; et al. Frequency of Circulating Tumor Cells (CTC) in Patients with Brain Metastases: Implications as a Risk Assessment Marker in Oligo-Metastatic Disease. Cancers 2018, 10, 527. [Google Scholar] [CrossRef]
- Sacco, A.; Forgione, L.; Carotenuto, M.; De Luca, A.; Ascierto, P.A.; Botti, G.; Normanno, N. Circulating Tumor DNA Testing Opens New Perspectives in Melanoma Management. Cancers 2020, 12, 2914. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224. [Google Scholar] [CrossRef] [Green Version]
- Calapre, L.; Warburton, L.; Millward, M.; Ziman, M.; Gray, E. Circulating tumour DNA (ctDNA) as a liquid biopsy for melanoma. Cancer Lett. 2017, 404, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Calapre, L.; Giardina, T.; Robinson, C.; Reid, A.; Al-Ogaili, Z.; Pereira, M.R.; McEvoy, A.; Warburton, L.; Hayward, N.; Khattak, M.A.; et al. Locus-specific concordance of genomic alterations between tissue and plasma circulating tumor DNA in metastatic melanoma. Mol. Oncol. 2018, 13, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Valpione, S.; Gremel, G.; Mundra, P.; Middlehurst, P.; Galvani, E.; Girotti, M.; Lee, R.; Garner, G.; Dhomen, N.; Lorigan, P.; et al. Plasma total cell-free DNA (cfDNA) is a surrogate biomarker for tumour burden and a prognostic biomarker for survival in metastatic melanoma patients. Eur. J. Cancer 2017, 88, 1–9. [Google Scholar] [CrossRef]
- de Kock, R.; Borne, B.V.D.; Soud, M.Y.-E.; Belderbos, H.; Brunsveld, L.; Scharnhorst, V.; Deiman, B. Therapy Monitoring of EGFR-Positive Non–Small-Cell Lung Cancer Patients Using ddPCR Multiplex Assays. J. Mol. Diagn. 2021, 23, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Lyskjær, I.; Kronborg, C.S.; Rasmussen, M.; Sorensen, B.; DeMuth, C.; Rosenkilde, M.; Johansen, A.F.B.; Knudsen, M.; Vang, S.; Krag, S.R.P.; et al. Correlation between early dynamics in circulating tumour DNA and outcome from FOLFIRI treatment in metastatic colorectal cancer. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Aleotti, V.; Catoni, C.; Poggiana, C.; Rosato, A.; Facchinetti, A.; Scaini, M.C. Methylation Markers in Cutaneous Melanoma: Unravelling the Potential Utility of Their Tracking by Liquid Biopsy. Cancers 2021, 13, 6217. [Google Scholar] [CrossRef] [PubMed]
- Bozic, I.; Antal, T.; Ohtsuki, H.; Carter, H.; Kim, D.; Chen, S.; Karchin, R.; Kinzler, K.W.; Vogelstein, B.; Nowak, M.A. Accumulation of driver and passenger mutations during tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 18545–18550. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Position | Coding Change | Amino Acid Change | COSMIC ID | ACMG Classification $ | T0 (MAF%) | T1 (MAF%) | T2 (MAF%) | T3 (MAF%) |
|---|---|---|---|---|---|---|---|---|---|
| ALK | 2:29,228,936 | c.2763C > G | p.F921L | COSM9118654 | Uncertain significance | ND | ND | ND | 2.5 |
| ATM | 11:108,330,374 | c.7468C > T | p.L2490F | COSM327924 | Uncertain significance | 26.1 | 35.5 | 27.6 | 7.8 |
| BRAF * | 7:140,753,336 | c.1799T > A | p.V600E | COSM476 | Pathogenic | 20.6 | 73.1 | 63.9 | 24.7 |
| CDKN2A | 9:21,971,138 | c.221A > C | p.D74A | COSM4163709 | Uncertain significance | ND | ND | 2.1 | ND |
| HOXD8 | 2:176,130,574 | c.208G > C | p.A70P | COSM3391142 | Uncertain Significance | 3.8 | 3.8 | 4.6 | 4.7 |
| MEK1 * | 15:66,436,825 | c.371C > T | p.P124L | COSM1315861 | Pathogenic | 23.7 | 27.7 | 24 | 9.1 |
| Time Points | ddPCR Output | Copy Number Assessment | |
|---|---|---|---|
| BRAF/TTC5 | BRAF/VOPP1 | ||
| T0 | 2.0 | 1.9 | Diploid |
| T1 | 6.5 | 5.7 | Gain |
| T2 | 4.8 | 4.5 | Gain |
| T3 | 2.7 | 2.6 | Gain |
| Time Points | BRAF p.V600E (Copies/mL) | MEK1 p.P124L (Copies/mL) |
|---|---|---|
| T0 | 10,432 | 14,766 |
| T1 | 8469 | 1054 |
| T2 | 11,715 | 2326 |
| T3 | 18,080 | 6578 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Catoni, C.; Poggiana, C.; Facchinetti, A.; Pigozzo, J.; Piccin, L.; Chiarion-Sileni, V.; Rosato, A.; Minervini, G.; Scaini, M.C. Investigating the Retained Inhibitory Effect of Cobimetinib against p.P124L Mutated MEK1: A Combined Liquid Biopsy and in Silico Approach. Cancers 2022, 14, 4153. https://doi.org/10.3390/cancers14174153
Catoni C, Poggiana C, Facchinetti A, Pigozzo J, Piccin L, Chiarion-Sileni V, Rosato A, Minervini G, Scaini MC. Investigating the Retained Inhibitory Effect of Cobimetinib against p.P124L Mutated MEK1: A Combined Liquid Biopsy and in Silico Approach. Cancers. 2022; 14(17):4153. https://doi.org/10.3390/cancers14174153
Chicago/Turabian StyleCatoni, Cristina, Cristina Poggiana, Antonella Facchinetti, Jacopo Pigozzo, Luisa Piccin, Vanna Chiarion-Sileni, Antonio Rosato, Giovanni Minervini, and Maria Chiara Scaini. 2022. "Investigating the Retained Inhibitory Effect of Cobimetinib against p.P124L Mutated MEK1: A Combined Liquid Biopsy and in Silico Approach" Cancers 14, no. 17: 4153. https://doi.org/10.3390/cancers14174153
APA StyleCatoni, C., Poggiana, C., Facchinetti, A., Pigozzo, J., Piccin, L., Chiarion-Sileni, V., Rosato, A., Minervini, G., & Scaini, M. C. (2022). Investigating the Retained Inhibitory Effect of Cobimetinib against p.P124L Mutated MEK1: A Combined Liquid Biopsy and in Silico Approach. Cancers, 14(17), 4153. https://doi.org/10.3390/cancers14174153