
Identification of Fusion Genes and Targets for Genetically Matched Therapies in a Large Cohort of Salivary Gland Cancer Patients

 , , , and
, , , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Patient Selection and Material Acquisition
2.2. DNA and RNA Extraction
2.3. Detection of RNA Gene Fusion Transcripts
2.4. DNA Next Generation Sequencing
2.5. Pan-TRK Immunohistochemistry
2.6. NTRK1, NTRK2 and NTRK3 Fluorescence In Situ Hybridization
2.7. TSO500 Data Analysis
2.8. Data Analysis
3. Results
3.1. Included Patients Comprise Diverse Subtypes of Salivary Gland Cancer
3.2. Half of SGC Cases Harbor Gene Fusions with Varying Incidence among Subtypes
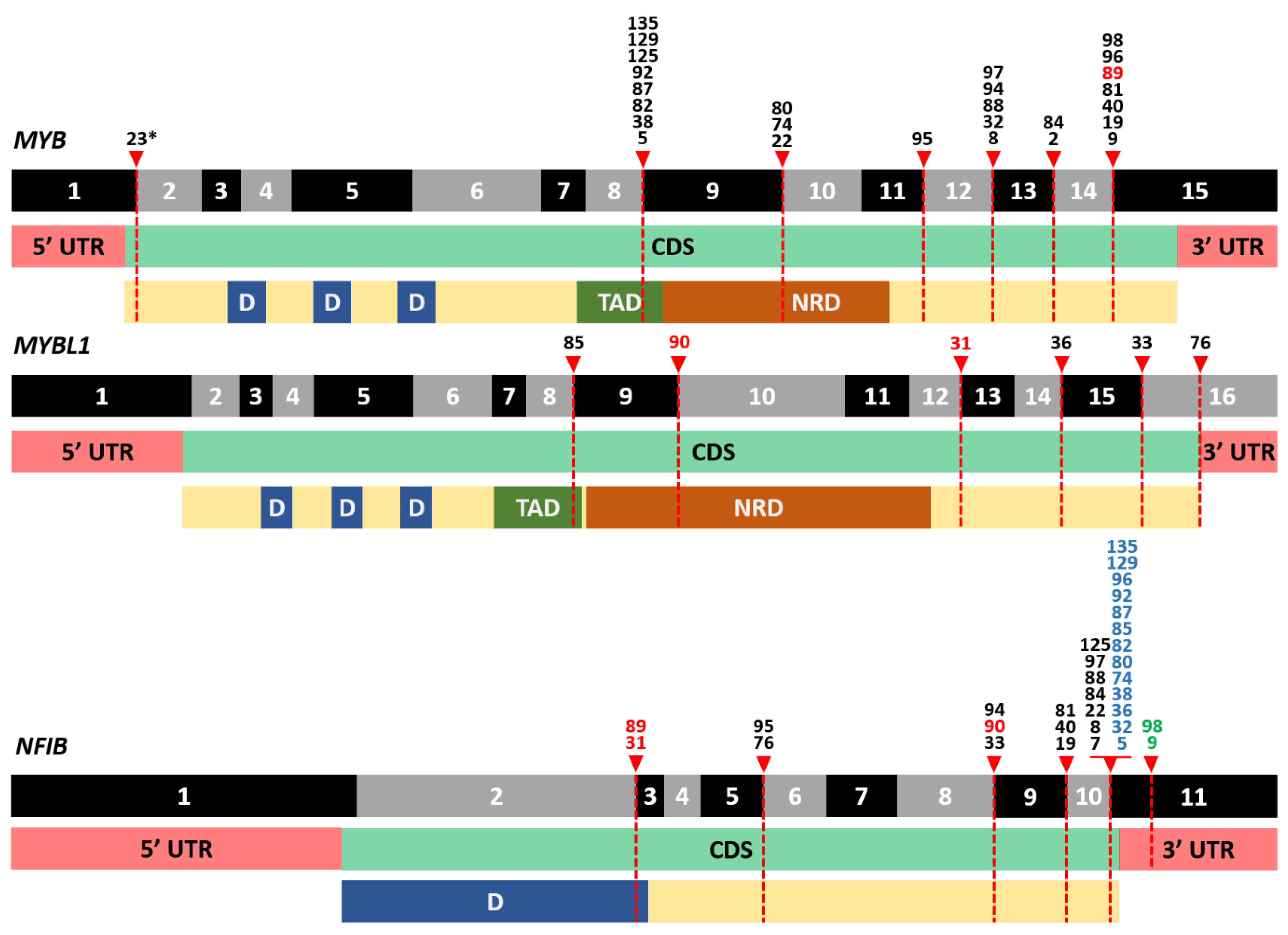
3.3. Frequent Detection of MYB- and MYBL1-NFIB Fusion Transcripts in AdCC
3.4. A Plethora of Both Known and New Fusion Transcripts was Detected in Non-AdCC Cases
3.5. Pan-TRK Immunohistochemistry Is False Positive in the Majority of SGC Cases
3.6. High TMB and MSI Are Rare in SGC
3.7. Pathogenic Small Nucleotide Variants Are Most Frequent in SDC
3.8. Copy Number Variants Are Mostly Restricted to ERBB2 Amplifications in SDC
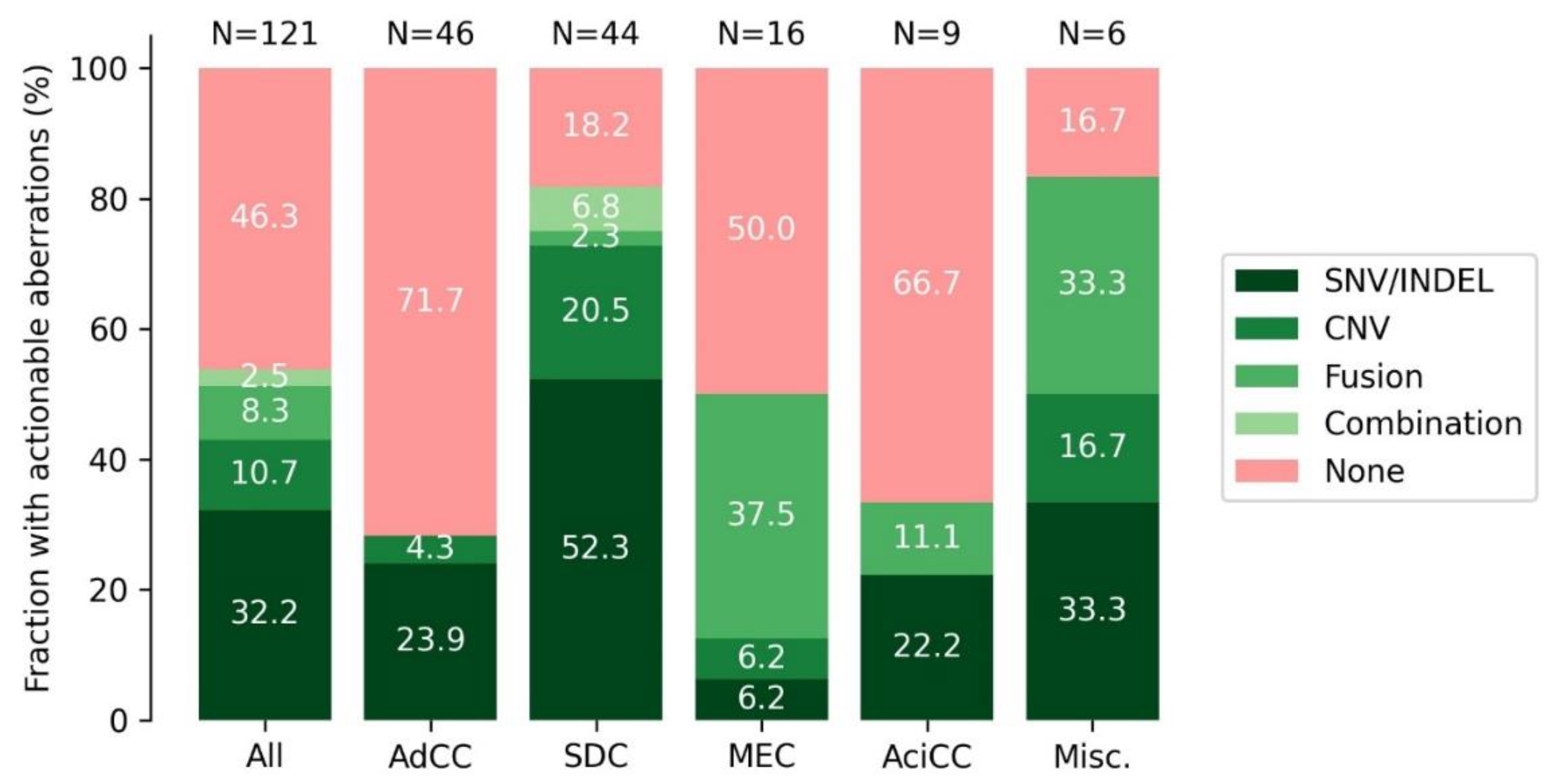
3.9. DNA and RNA Analysis Reveals Actionable Targets in the Majority of SGC Cases
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Park, J.J.H.; Hsu, G.; Siden, E.G.; Thorlund, K.; Mills, E.J. An overview of precision oncology basket and umbrella trials for clinicians. CA Cancer J. Clin. 2020, 70, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Van der Velden, D.L.; Hoes, L.R.; van der Wijngaart, H.; van Berge Henegouwen, J.M.; van Werkhoven, E.; Roepman, P.; Schilsky, R.L.; de Leng, W.W.J.; Huitema, A.D.R.; Nuijen, B.; et al. The Drug Rediscovery protocol facilitates the expanded use of existing anticancer drugs. Nature 2019, 574, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Hainsworth, J.D.; Meric-Bernstam, F.; Swanton, C.; Hurwitz, H.; Spigel, D.R.; Sweeney, C.; Burris, H.; Bose, R.; Yoo, B.; Stein, A.; et al. Targeted Therapy for Advanced Solid Tumors on the Basis of Molecular Profiles: Results From MyPathway, an Open-Label, Phase IIa Multiple Basket Study. J. Clin. Oncol. 2018, 36, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Lassche, G.; van Boxtel, W.; Ligtenberg, M.J.L.; van Engen-van Grunsven, A.C.H.; van Herpen, C.M.L. Advances and challenges in precision medicine in salivary gland cancer. Cancer Treat. Rev. 2019, 80, 101906. [Google Scholar] [CrossRef]
- El-Naggar, A.K.; Chan, J.K.C.; Grandis, J.R.; Takata, T.; Slootweg, P.J. WHO Classification of Head and Neck Tumours, 4th ed.; IARC: Lyon, France, 2017. [Google Scholar]
- Witte, H.M.; Gebauer, N.; Steinestel, K. Mutational and immunologic Landscape in malignant Salivary Gland Tumors harbor the potential for novel therapeutic strategies. Crit. Rev. Oncol. Hematol. 2022, 170, 103592. [Google Scholar] [CrossRef]
- Mertens, F.; Johansson, B.; Fioretos, T.; Mitelman, F. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer 2015, 15, 371–381. [Google Scholar] [CrossRef]
- Bubola, J.; MacMillan, C.M.; Demicco, E.G.; Chami, R.A.; Chung, C.T.; Leong, I.; Marrano, P.; Onkal, Z.; Swanson, D.; Veremis, B.M.; et al. Targeted RNA sequencing in the routine clinical detection of fusion genes in salivary gland tumors. Genes Chromosom. Cancer 2021, 60, 695–708. [Google Scholar] [CrossRef]
- Asahina, M.; Saito, T.; Hayashi, T.; Fukumura, Y.; Mitani, K.; Yao, T. Clinicopathological effect of PLAG1 fusion genes in pleomorphic adenoma and carcinoma ex pleomorphic adenoma with special emphasis on histological features. Histopathology 2019, 74, 514–525. [Google Scholar] [CrossRef]
- Fujii, K.; Murase, T.; Beppu, S.; Saida, K.; Takino, H.; Masaki, A.; Ijichi, K.; Kusafuka, K.; Iida, Y.; Onitsuka, T.; et al. MYB, MYBL1, MYBL2 and NFIB gene alterations and MYC overexpression in salivary gland adenoid cystic carcinoma. Histopathology 2017, 71, 823–834. [Google Scholar] [CrossRef]
- Ho, A.S.; Kannan, K.; Roy, D.M.; Morris, L.G.; Ganly, I.; Katabi, N.; Ramaswami, D.; Walsh, L.A.; Eng, S.; Huse, J.T.; et al. The mutational landscape of adenoid cystic carcinoma. Nat. Genet. 2013, 45, 791–798. [Google Scholar] [CrossRef]
- Luk, P.P.; Wykes, J.; Selinger, C.I.; Ekmejian, R.; Tay, J.; Gao, K.; Eviston, T.J.; Lum, T.; O’Toole, S.A.; Clark, J.R.; et al. Diagnostic and prognostic utility of Mastermind-like 2 (MAML2) gene rearrangement detection by fluorescent in situ hybridization (FISH) in mucoepidermoid carcinoma of the salivary glands. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2016, 121, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Seethala, R.R.; Stenman, G. Update from the 4th Edition of the World Health Organization Classification of Head and Neck Tumours: Tumors of the Salivary Gland. Head Neck Pathol. 2017, 11, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Skalova, A.; Vanecek, T.; Sima, R.; Laco, J.; Weinreb, I.; Perez-Ordonez, B.; Starek, I.; Geierova, M.; Simpson, R.H.; Passador-Santos, F.; et al. Mammary analogue secretory carcinoma of salivary glands, containing the ETV6-NTRK3 fusion gene: A hitherto undescribed salivary gland tumor entity. Am. J. Surg. Pathol. 2010, 34, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.E.; Farago, A.F.; Tan, D.S.-W.; Kummar, S.; McDermott, R.S.; Berlin, J.; Patel, J.D.; Brose, M.S.; Leyvraz, S.; Tahara, M.; et al. Activity and safety of larotrectinib in adult patients with TRK fusion cancer: An expanded data set. J. Clin. Oncol. 2020, 38, 3610. [Google Scholar] [CrossRef]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Casparie, M.; Tiebosch, A.T.; Burger, G.; Blauwgeers, H.; van de Pol, A.; van Krieken, J.H.; Meijer, G.A. Pathology databanking and biobanking in The Netherlands, a central role for PALGA, the nationwide histopathology and cytopathology data network and archive. Cell Oncol. 2007, 29, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Eijkelenboom, A.; Kamping, E.J.; Kastner-van Raaij, A.W.; Hendriks-Cornelissen, S.J.; Neveling, K.; Kuiper, R.P.; Hoischen, A.; Nelen, M.R.; Ligtenberg, M.J.; Tops, B.B. Reliable Next-Generation Sequencing of Formalin-Fixed, Paraffin-Embedded Tissue Using Single Molecule Tags. J. Mol. Diagn. 2016, 18, 851–863. [Google Scholar] [CrossRef]
- Kroeze, L.I.; de Voer, R.M.; Kamping, E.J.; von Rhein, D.; Jansen, E.A.M.; Hermsen, M.J.W.; Barberis, M.C.P.; Botling, J.; Garrido-Martin, E.M.; Haller, F.; et al. Evaluation of a Hybrid Capture-Based Pan-Cancer Panel for Analysis of Treatment Stratifying Oncogenic Aberrations and Processes. J. Mol. Diagn. 2020, 22, 757–769. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Eijkelenboom, A.; Tops, B.B.J.; van den Berg, A.; van den Brule, A.J.C.; Dinjens, W.N.M.; Dubbink, H.J.; Ter Elst, A.; Geurts-Giele, W.R.R.; Groenen, P.; Groenendijk, F.H.; et al. Recommendations for the clinical interpretation and reporting of copy number gains using gene panel NGS analysis in routine diagnostics. Virchows Arch. 2019, 474, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Pierini, T.; Di Giacomo, D.; Pierini, V.; Gorello, P.; Barba, G.; Lema Fernandez, A.G.; Pellanera, F.; Iannotti, T.; Falzetti, F.; La Starza, R.; et al. MYB deregulation from a EWSR1-MYB fusion at leukemic evolution of a JAK2 (V617F) positive primary myelofibrosis. Mol. Cytogenet. 2016, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- Haller, F.; Bieg, M.; Will, R.; Körner, C.; Weichenhan, D.; Bott, A.; Ishaque, N.; Lutsik, P.; Moskalev, E.A.; Mueller, S.K.; et al. Enhancer hijacking activates oncogenic transcription factor NR4A3 in acinic cell carcinomas of the salivary glands. Nat. Commun. 2019, 10, 368. [Google Scholar] [CrossRef] [PubMed]
- Date, O.; Katsura, M.; Ishida, M.; Yoshihara, T.; Kinomura, A.; Sueda, T.; Miyagawa, K. Haploinsufficiency of RAD51B causes centrosome fragmentation and aneuploidy in human cells. Cancer Res. 2006, 66, 6018–6024. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Isaacson, A.L.; Guseva, N.V.; Bossler, A.D.; Ma, D. Urothelial carcinoma with an NRF1-BRAF rearrangement and response to targeted therapy. Cold Spring Harb. Mol. Case Stud. 2019, 5, a003848. [Google Scholar] [CrossRef] [PubMed]
- Ferrarotto, R.; Mitani, Y.; Diao, L.; Guijarro, I.; Wang, J.; Zweidler-McKay, P.; Bell, D.; William, W.N., Jr.; Glisson, B.S.; Wick, M.J.; et al. Activating NOTCH1 Mutations Define a Distinct Subgroup of Patients With Adenoid Cystic Carcinoma Who Have Poor Prognosis, Propensity to Bone and Liver Metastasis, and Potential Responsiveness to Notch1 Inhibitors. J. Clin. Oncol. 2017, 35, 352–360. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, Q.; Li, D.; Ching, K.; Zhang, C.; Zheng, X.; Ozeck, M.; Shi, S.; Li, X.; Wang, H.; et al. PEST domain mutations in Notch receptors comprise an oncogenic driver segment in triple-negative breast cancer sensitive to a γ-secretase inhibitor. Clin. Cancer Res. 2015, 21, 1487–1496. [Google Scholar] [CrossRef]
- Lobry, C.; Oh, P.; Aifantis, I. Oncogenic and tumor suppressor functions of Notch in cancer: It’s NOTCH what you think. J. Exp. Med. 2011, 208, 1931–1935. [Google Scholar] [CrossRef]
- Rosen, E.Y.; Goldman, D.A.; Hechtman, J.F.; Benayed, R.; Schram, A.M.; Cocco, E.; Shifman, S.; Gong, Y.; Kundra, R.; Solomon, J.P.; et al. TRK Fusions Are Enriched in Cancers with Uncommon Histologies and the Absence of Canonical Driver Mutations. Clin. Cancer Res. 2020, 26, 1624–1632. [Google Scholar] [CrossRef]
- Solomon, J.P.; Linkov, I.; Rosado, A.; Mullaney, K.; Rosen, E.Y.; Frosina, D.; Jungbluth, A.A.; Zehir, A.; Benayed, R.; Drilon, A.; et al. NTRK fusion detection across multiple assays and 33,997 cases: Diagnostic implications and pitfalls. Mod. Pathol. 2020, 33, 38–46. [Google Scholar] [CrossRef]
- Hechtman, J.F.; Benayed, R.; Hyman, D.M.; Drilon, A.; Zehir, A.; Frosina, D.; Arcila, M.E.; Dogan, S.; Klimstra, D.S.; Ladanyi, M.; et al. Pan-Trk Immunohistochemistry Is an Efficient and Reliable Screen for the Detection of NTRK Fusions. Am. J. Surg. Pathol. 2017, 41, 1547–1551. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, S.V.; Panaccione, A.; Brown, B.; Guo, Y.; Moskaluk, C.A.; Wick, M.J.; Brown, J.L.; Ivanova, A.V.; Issaeva, N.; El-Naggar, A.K.; et al. TrkC signaling is activated in adenoid cystic carcinoma and requires NT-3 to stimulate invasive behavior. Oncogene 2013, 32, 3698–3710. [Google Scholar] [CrossRef]
- Csanyi-Bastien, M.; Lanic, M.D.; Beaussire, L.; Ferric, S.; François, A.; Meseure, D.; Jardin, F.; Wassef, M.; Ruminy, P.; Laé, M. Pan-TRK Immunohistochemistry Is Highly Correlated with NTRK3 Gene Rearrangements in Salivary Gland Tumors. Am. J. Surg. Pathol. 2021, 45, 1487–1498. [Google Scholar] [CrossRef]
- Togashi, Y.; Dobashi, A.; Sakata, S.; Sato, Y.; Baba, S.; Seto, A.; Mitani, H.; Kawabata, K.; Takeuchi, K. MYB and MYBL1 in adenoid cystic carcinoma: Diversity in the mode of genomic rearrangement and transcripts. Mod. Pathol. 2018, 31, 934–946. [Google Scholar] [CrossRef]
- Brayer, K.J.; Frerich, C.A.; Kang, H.; Ness, S.A. Recurrent Fusions in MYB and MYBL1 Define a Common, Transcription Factor-Driven Oncogenic Pathway in Salivary Gland Adenoid Cystic Carcinoma. Cancer Discov. 2016, 6, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Drier, Y.; Cotton, M.J.; Williamson, K.E.; Gillespie, S.M.; Ryan, R.J.; Kluk, M.J.; Carey, C.D.; Rodig, S.J.; Sholl, L.M.; Afrogheh, A.H.; et al. An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nat. Genet. 2016, 48, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Hoes, L.R.; van Berge Henegouwen, J.M.; van der Wijngaart, H.; Zeverijn, L.J.; van der Velden, D.L.; van de Haar, J.; Roepman, P.; de Leng, W.J.; Jansen, A.M.L.; van Werkhoven, E.; et al. Patients With Rare Cancers in the Drug Rediscovery Protocol (DRUP) benefit from Genomics-Guided Treatment. Clin. Cancer Res. 2022. [Google Scholar] [CrossRef]
- Rack, S.; Feeney, L.; Hapuarachi, B.; Adderley, H.; Woodhouse, L.; Betts, G.; Burghel, G.J.; Harrington, K.J.; Metcalf, R. Evaluation of the Clinical Utility of Genomic Profiling to Inform Selection of Clinical Trial Therapy in Salivary Gland Cancer. Cancers 2022, 14, 1133. [Google Scholar] [CrossRef]
- Ho, A.S.; Ochoa, A.; Jayakumaran, G.; Zehir, A.; Valero Mayor, C.; Tepe, J.; Makarov, V.; Dalin, M.G.; He, J.; Bailey, M.; et al. Genetic hallmarks of recurrent/metastatic adenoid cystic carcinoma. J. Clin. Investig. 2019, 129, 4276–4289. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All (n = 121) | AdCC (n = 46) | SDC (n = 44) | MEC (n = 16) | AciCC (n = 9) | Misc. (n = 6) | ||
|---|---|---|---|---|---|---|---|
| n (%) | |||||||
| Age at diagnosis | |||||||
| Median (range) | 57 (17–90) | 53 (21–83) | 63 (35–90) | 53 (17–72) | 56 (45–71) | 67 (48–79) | |
| Gender | |||||||
| Male | 58 (47.9) | 15 (32.6) | 28 (63.6) | 8 (50) | 5 (55.6) | 2 (33.3) | |
| Female | 63 (52.1) | 31 (67.4) | 16 (36.4) | 8 (50) | 4 (44.4) | 4 (66.7) | |
| Location primary tumor | |||||||
| Major salivary gland | 81 (66.9) | 21 (45.7) | 42 (95.5) | 6 (37.5) | 9 (100.0) | 3 (50.0) | |
| Minor salivary gland | 28 (23.1) | 16 (34.8) | 1 (2.3) | 8 (50.0) | 0 (0.0) | 3 (50.0) | |
| Other | 12 (9.9) | 9 (19.6) | 1 (2.3) | 2 (12.5) | 0 (0.0) | 0 (0.0) | |
| T-stage at diagnosis | |||||||
| 1–2 | 39 (32.3) | 6 (13.0) | 16 (36.4) | 10 (62.5) | 4 (44.4) | 3 (50.0) | |
| 3–4 | 56 (46.3) | 27 (58.7) | 21 (47.7) | 4 (25.0) | 2 (22.2) | 2 (33.3) | |
| Tx | 26 (21.5) | 13 (28.3) | 7 (15.9) | 2 (12.5) | 3 (33.3) | 1 (16.7) | |
| N-stage at diagnosis | |||||||
| 0 | 52 (43.0) | 27 (58.7) | 8 (18.2) | 7 (43.8) | 5 (55.6) | 5 (83.3) | |
| 1–3 | 44 (36.4) | 7 (15.2) | 28 (63.6) | 7 (43.8) | 1 (11.1) | 1 (16.7) | |
| Nx | 25 (20.7) | 12 (26.1) | 8 (18.2) | 2 (12.5) | 3 (33.3) | 0 (0.0) | |
| M-stage at diagnosis | |||||||
| 0 | 100 (82.6) | 36 (78.3) | 39 (88.6) | 14 (87.5) | 7 (77.8) | 4 (66.7) | |
| 1 | 19 (15.7) | 8 (17.4) | 5 (11.4) | 2 (12.5) | 2 (22.2) | 2 (33.3) | |
| Mx | 2 (1.7) | 2 (4.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | |
| Initial treatment intent | |||||||
| Curative | 99 (83.5) | 35 (76.1) | 39 (88.6) | 14 (87.5) | 7 (77.8) | 4 (66.7) | |
| Palliative | 22 (18.2) | 11 (23.9) | 5 (11.4) | 2 (12.5) | 2 (22.2) | 2 (33.3) | |
| R/M disease after initial curative treatment (n = 99) | |||||||
| Yes | 63 (63.4) | 28 (80.0) | 24 (61.5) | 6 (42.9) | 4 (57.1) | 1 (25%) | |
| No | 36 (36.4) | 7 (20.0) | 15 (38.5) | 8 (57.1) | 3 (42.9) | 3 (75%) | |
| Underwent surgery primary tumor | |||||||
| Yes | 101 (83.5) | 36 (78.3) | 38 (86.4) | 15 (93.8) | 8 (88.9) | 4 (66.7) | |
| No | 20 (16.5) | 10 (21.7) | 6 (13.6) | 1 (6.3) | 1 (11.1) | 2 (33.3) | |
| Palliative systemic therapy | |||||||
| Yes | 57 (47.1) | 18 (39.1) | 28 (63.6) | 4 (25.0) | 5 (55.6) | 2 (33.3) | |
| No | 64 (52.9) | 28 (60.9) | 16 (36.4) | 12 (75.0) | 4 (44.4) | 4 (66.7) | |
| Lines of systemic therapy | |||||||
| Median (range) | 1 (1–7) | 1 (1–4) | 2 (1–5) | 2 (1–7) | 2 (1–2) | 1 (1) | |
| First line systemic therapy | |||||||
| Chemotherapy | 18 (14.9) | 11 (23.9) | 2 (4.5) | 2 (12.5) | 3 (33.3) | 0 (0.0) | |
| Targeted | 12 (9.9) | 6 (13.0) | 1 (2.3) | 2 (12.5) | 1 (11.1) | 2 (33.3) | |
| Hormonal | 17 (14.0) | 0 (0.0) | 16 (36.4) | 0 (0.0) | 1 (11.1) | 0 (0.0) | |
| Immunotherapy | 1 (0.8) | 0 (0.0) | 1 (2.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | |
| Combination | 8 (6.6) | 0 (0.0) | 8 (18.2) | 0 (0.0) | 0 (0.0) | 0 (0.0) | |
| Other | 1 (0.8) | 1 (2.2) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lassche, G.; van Helvert, S.; Eijkelenboom, A.; Tjan, M.J.H.; Jansen, E.A.M.; van Cleef, P.H.J.; Verhaegh, G.W.; Kamping, E.J.; Grünberg, K.; van Engen-van Grunsven, A.C.H.; et al. Identification of Fusion Genes and Targets for Genetically Matched Therapies in a Large Cohort of Salivary Gland Cancer Patients. Cancers 2022, 14, 4156. https://doi.org/10.3390/cancers14174156
Lassche G, van Helvert S, Eijkelenboom A, Tjan MJH, Jansen EAM, van Cleef PHJ, Verhaegh GW, Kamping EJ, Grünberg K, van Engen-van Grunsven ACH, et al. Identification of Fusion Genes and Targets for Genetically Matched Therapies in a Large Cohort of Salivary Gland Cancer Patients. Cancers. 2022; 14(17):4156. https://doi.org/10.3390/cancers14174156
Chicago/Turabian StyleLassche, Gerben, Sjoerd van Helvert, Astrid Eijkelenboom, Martijn J. H. Tjan, Erik A. M. Jansen, Patricia H. J. van Cleef, Gerald W. Verhaegh, Eveline J. Kamping, Katrien Grünberg, Adriana C. H. van Engen-van Grunsven, and et al. 2022. "Identification of Fusion Genes and Targets for Genetically Matched Therapies in a Large Cohort of Salivary Gland Cancer Patients" Cancers 14, no. 17: 4156. https://doi.org/10.3390/cancers14174156
APA StyleLassche, G., van Helvert, S., Eijkelenboom, A., Tjan, M. J. H., Jansen, E. A. M., van Cleef, P. H. J., Verhaegh, G. W., Kamping, E. J., Grünberg, K., van Engen-van Grunsven, A. C. H., Ligtenberg, M. J. L., & van Herpen, C. M. L. (2022). Identification of Fusion Genes and Targets for Genetically Matched Therapies in a Large Cohort of Salivary Gland Cancer Patients. Cancers, 14(17), 4156. https://doi.org/10.3390/cancers14174156