
Targeted Sequencing of Plasma-Derived vs. Urinary cfDNA from Patients with Triple-Negative Breast Cancer


Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Characteristics
2.2. Acquisition and Processing of Blood and Urine Samples
2.3. Isolation and Quantification of Cell-Free DNA (cfDNA)
2.4. Library Preparation and Targeted Sequencing of Breast-Cancer-Related Genes
2.5. Sequencing Parameters
2.6. Bioinformatical Analysis and Filtering of Somatic Variants in ctDNA
2.7. Statistical Analysis
3. Results
3.1. Cell-Free DNA (cfDNA) Concentration in Matched Plasma and Urine Samples
3.2. Investigation of Somatic Variants in ctDNA Derived from Matched Plasma and Urine Samples
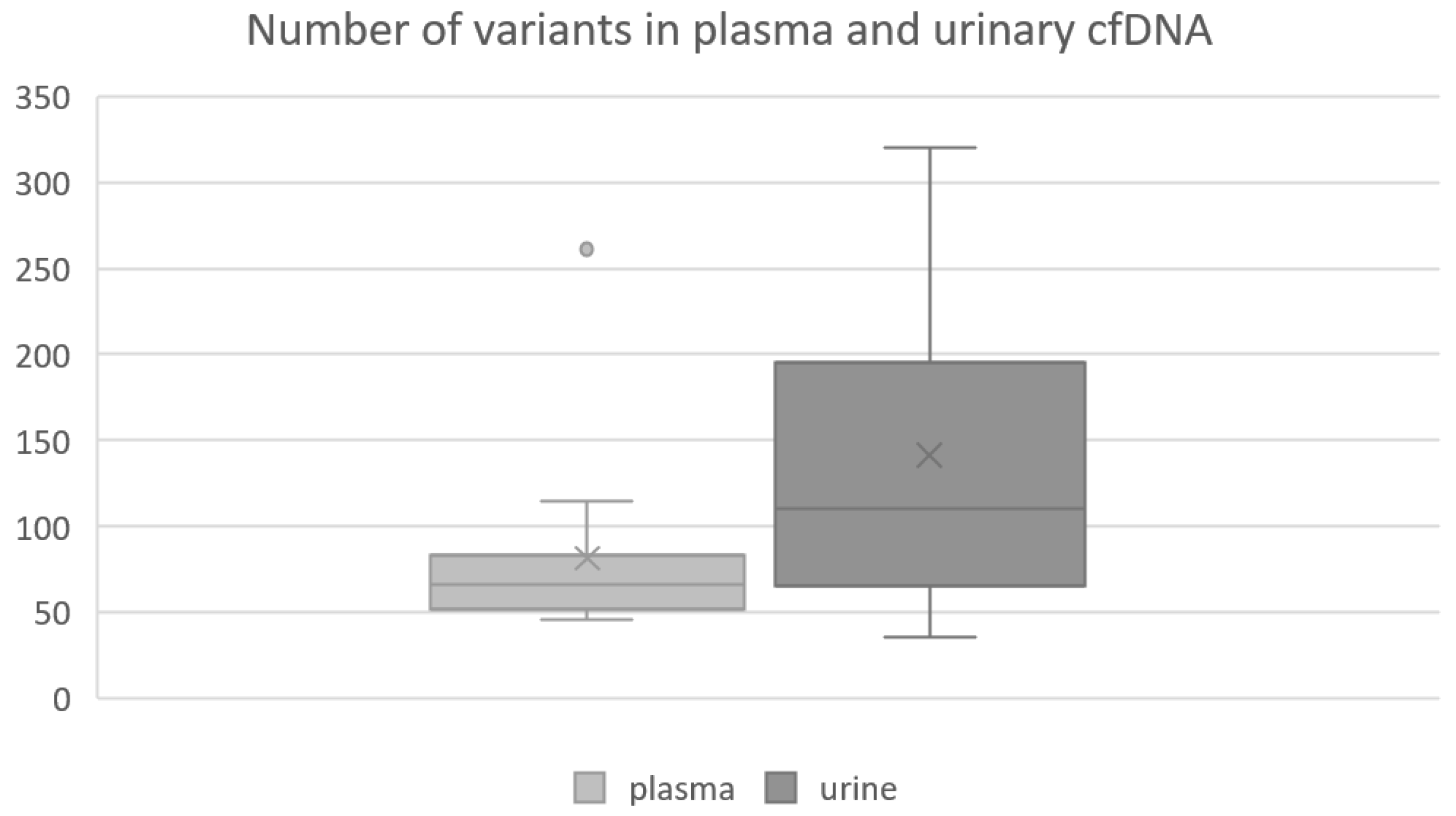
3.3. Abundance of Detected Variants in Plasma and Urinary ctDNA
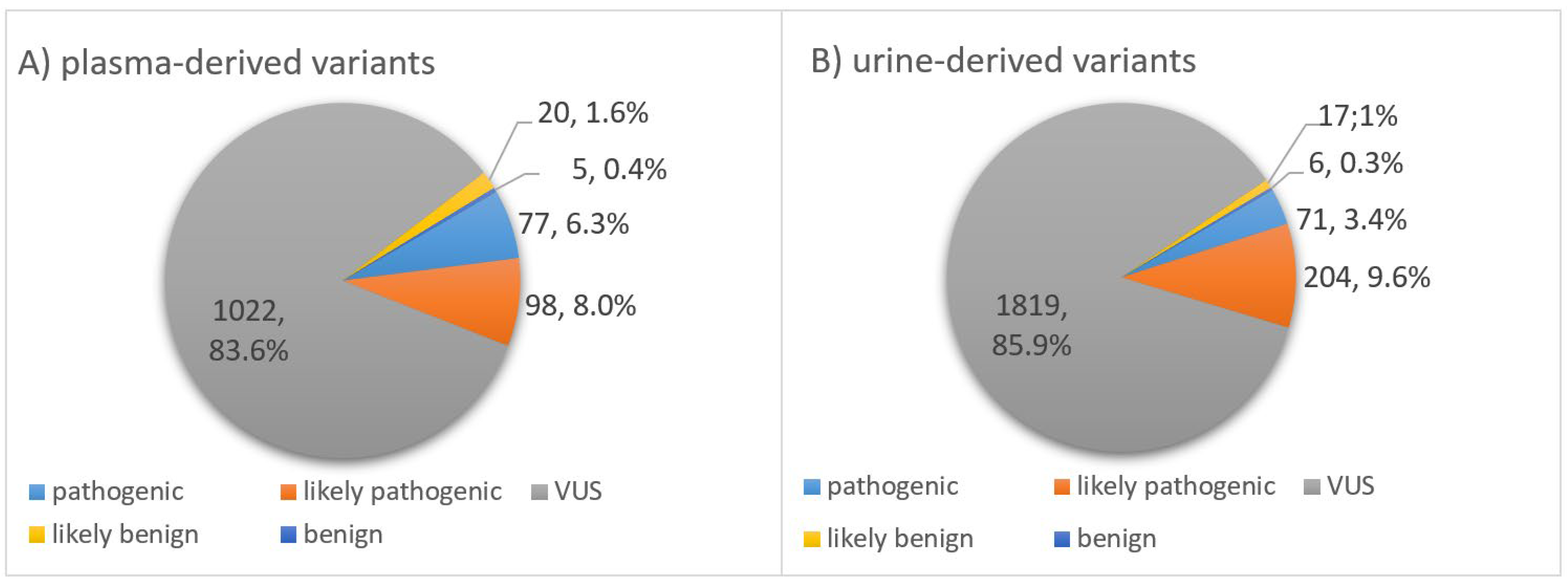
3.4. Classification of Somatic Variants According to Biological Impact
3.5. Classification of Somatic Variants According to Clinical Significance
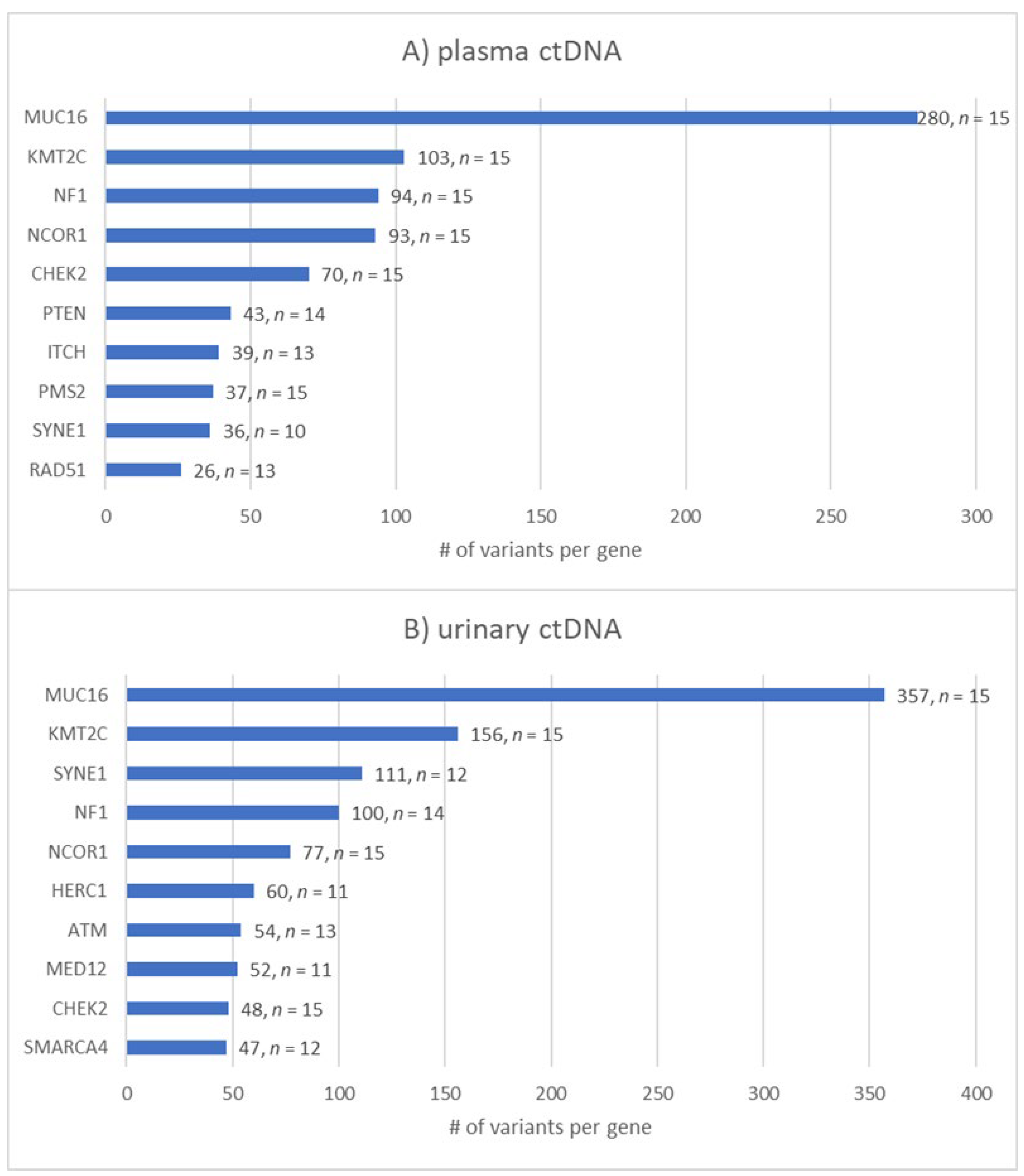
3.6. Analysis of Most Frequently Mutated Breast-Cancer-Related Genes in Plasma vs. Urinary ctDNA
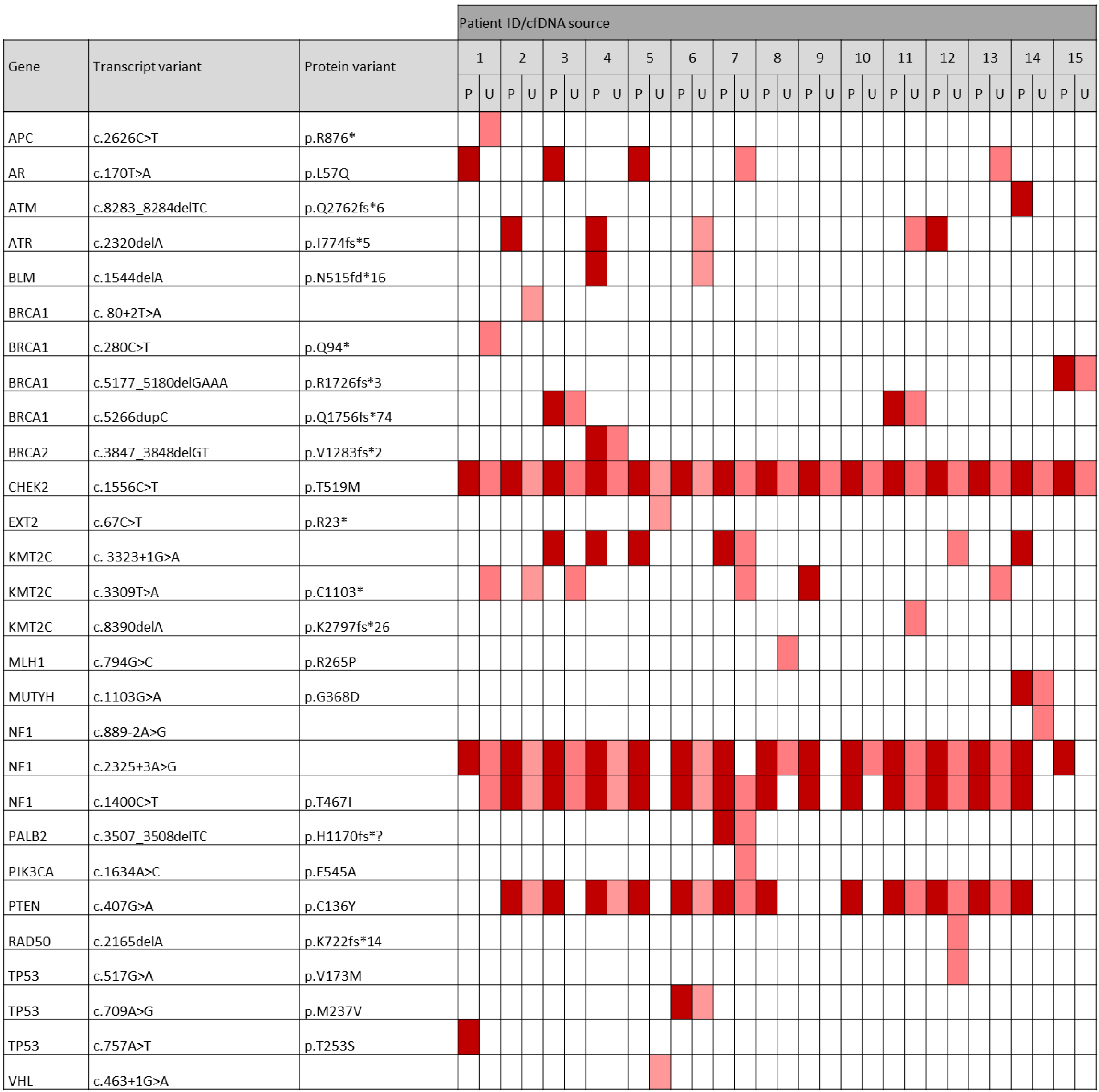
3.7. Occurrence of Pathogenic and Likely Pathogenic Variants among the Most Frequently Altered Genes in the TNBC Cohort
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- Craig, D.W.; O’Shaughnessy, J.A.; Kiefer, J.A.; Aldrich, J.; Sinari, S.; Moses, T.M.; Wong, S.; Dinh, J.; Christoforides, A.; Blum, J.L.; et al. Genome and transcriptome sequencing in prospective metastatic triple-negative breast cancer uncovers therapeutic vulnerabilities. Mol. Cancer Ther. 2013, 12, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Cortes, J.; Dent, R.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; et al. Event-free Survival with Pembrolizumab in Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2022, 386, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef]
- Radovich, M.; Jiang, G.; Hancock, B.A.; Chitambar, C.; Nanda, R.; Falkson, C.; Lynce, F.C.; Gallagher, C.; Isaacs, C.; Blaya, M.; et al. Association of Circulating Tumor DNA and Circulating Tumor Cells After Neoadjuvant Chemotherapy with Disease Recurrence in Patients with Triple-Negative Breast Cancer: Preplanned Secondary Analysis of the BRE12-158 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1410–1415. [Google Scholar] [CrossRef]
- Marotti, J.D.; de Abreu, F.B.; Wells, W.A.; Tsongalis, G.J. Triple-Negative Breast Cancer: Next-Generation Sequencing for Target Identification. Am. J. Pathol. 2017, 187, 2133–2138. [Google Scholar] [CrossRef]
- Gupta, G.K.; Collier, A.L.; Lee, D.; Hoefer, R.A.; Zheleva, V.; van Siewertsz Reesema, L.L.; Tang-Tan, A.M.; Guye, M.L.; Chang, D.Z.; Winston, J.S.; et al. Perspectives on Triple-Negative Breast Cancer: Current Treatment Strategies, Unmet Needs, and Potential Targets for Future Therapies. Cancers 2020, 12, 2392. [Google Scholar] [CrossRef]
- Pelizzari, G.; Gerratana, L.; Basile, D.; Fanotto, V.; Bartoletti, M.; Liguori, A.; Fontanella, C.; Spazzapan, S.; Puglisi, F. Post-neoadjuvant strategies in breast cancer: From risk assessment to treatment escalation. Cancer Treat. Rev. 2019, 72, 7–14. [Google Scholar] [CrossRef]
- Gass, P.; Lux, M.P.; Rauh, C.; Hein, A.; Bani, M.R.; Fiessler, C.; Hartmann, A.; Häberle, L.; Pretscher, J.; Erber, R.; et al. Prediction of pathological complete response and prognosis in patients with neoadjuvant treatment for triple-negative breast cancer. BMC Cancer 2018, 18, 1051. [Google Scholar] [CrossRef]
- Denkert, C.; Liedtke, C.; Tutt, A.; Minckwitz von, G. Molecular alterations in triple-negative breast cancer—The road to new treatment strategies. Lancet 2017, 389, 2430–2442. [Google Scholar] [CrossRef]
- Li, Y.; Zhan, Z.; Yin, X.; Fu, S.; Deng, X. Targeted Therapeutic Strategies for Triple-Negative Breast Cancer. Front. Oncol. 2021, 11, 731535. [Google Scholar] [CrossRef]
- Cao, L.; Niu, Y. Triple negative breast cancer: Special histological types and emerging therapeutic methods. Cancer Biol. Med. 2020, 17, 293–306. [Google Scholar] [CrossRef]
- Keenan, T.E.; Tolaney, S.M. Role of Immunotherapy in Triple-Negative Breast Cancer. J. Natl. Compr. Cancer Netw. JNCCN 2020, 18, 479–489. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Howlader, N.; Altekruse, S.F.; Li, C.I.; Chen, V.W.; Clarke, C.A.; Ries, L.A.G.; Cronin, K.A. US incidence of breast cancer subtypes defined by joint hormone receptor and HER2 status. J. Natl. Cancer Inst. 2014, 106, dju055. [Google Scholar] [CrossRef]
- Brackstone, M.; Townson, J.L.; Chambers, A.F. Tumour dormancy in breast cancer: An update. Breast Cancer Res. 2007, 9, 208. [Google Scholar] [CrossRef]
- Diaz, L.A.; Bardelli, A. Liquid biopsies: Genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Kustanovich, A.; Schwartz, R.; Peretz, T.; Grinshpun, A. Life and death of circulating cell-free DNA. Cancer Biol. Ther. 2019, 20, 1057–1067. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Diamandis, E.P.; Crown, J. Circulating tumor DNA (ctDNA) as a pan-cancer screening test: Is it finally on the horizon? Clin. Chem. Lab. Med. 2021, 59, 1353–1361. [Google Scholar] [CrossRef]
- Chung, J.H.; Pavlick, D.; Hartmaier, R.; Schrock, A.B.; Young, L.; Forcier, B.; Ye, P.; Levin, M.K.; Goldberg, M.; Burris, H.; et al. Hybrid capture-based genomic profiling of circulating tumor DNA from patients with estrogen receptor-positive metastatic breast cancer. Ann. Oncol. 2017, 28, 2866–2873. [Google Scholar] [CrossRef]
- Ivonne, N. The Potential Use of Urinary CtDNA Profiling in the Treatment of Breast Cancer. WJGWH 2020, 4. [Google Scholar] [CrossRef]
- Greytak, S.R.; Engel, K.B.; Parpart-Li, S.; Murtaza, M.; Bronkhorst, A.J.; Pertile, M.D.; Moore, H.M. Harmonizing Cell-Free DNA Collection and Processing Practices through Evidence-Based Guidance. Clin. Cancer Res. 2020, 26, 3104–3109. [Google Scholar] [CrossRef] [PubMed]
- Nel, I.; Herzog, H.; Aktas, B. Combined Analysis of Disseminated Tumor Cells (DTCs) and Circulating Tumor DNA (ctDNA) in a Patient Suffering from Triple Negative Breast Cancer Revealed Elevated Risk. Front. Biosci. 2022, 27, 208. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- Keup, C.; Hahn, P.; Hauch, S.; Sprenger-Haussels, M.; Tewes, M.; Mach, P.; Bittner, A.-K.; Kimmig, R.; Kasimir-Bauer, S.; Benyaa, K. Protocols Io 2018. Available online: https://www.protocols.io/view/targeted-pcr-based-deep-sequencing-of-cfdna-with-u-q26g7r8w9vwz/v1 (accessed on 21 August 2022).
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Reva, B.; Antipin, Y.; Sander, C. Determinants of protein function revealed by combinatorial entropy optimization. Genome Biol. 2007, 8, R232. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef]
- Zelli, V.; Compagnoni, C.; Cannita, K.; Capelli, R.; Capalbo, C.; Di Vito Nolfi, M.; Alesse, E.; Zazzeroni, F.; Tessitore, A. Applications of Next Generation Sequencing to the Analysis of Familial Breast/Ovarian Cancer. High Throughput 2020, 9, 1. [Google Scholar] [CrossRef]
- Bewicke-Copley, F.; Arjun Kumar, E.; Palladino, G.; Korfi, K.; Wang, J. Applications and analysis of targeted genomic sequencing in cancer studies. Comput. Struct. Biotechnol. J. 2019, 17, 1348–1359. [Google Scholar] [CrossRef]
- Bos, M.K.; Nasserinejad, K.; Jansen, M.P.H.M.; Angus, L.; Atmodimedjo, P.N.; de Jonge, E.; Dinjens, W.N.M.; van Schaik, R.H.N.; Del Re, M.; Dubbink, H.J.; et al. Comparison of variant allele frequency and number of mutant molecules as units of measurement for circulating tumor DNA. Mol. Oncol. 2021, 15, 57–66. [Google Scholar] [CrossRef]
- Strom, S.P. Current practices and guidelines for clinical next-generation sequencing oncology testing. Cancer Biol. Med. 2016, 13, 3–11. [Google Scholar] [CrossRef]
- He, M.M.; Li, Q.; Yan, M.; Cao, H.; Hu, Y.; He, K.Y.; Cao, K.; Li, M.M.; Wang, K. Variant Interpretation for Cancer (VIC): A computational tool for assessing clinical impacts of somatic variants. Genome Med. 2019, 11, 53. [Google Scholar] [CrossRef]
- Yan, Y.H.; Chen, S.X.; Cheng, L.Y.; Rodriguez, A.Y.; Tang, R.; Cabrera, K.; Zhang, D.Y. Confirming putative variants at ≤ 5% allele frequency using allele enrichment and Sanger sequencing. Sci Rep. 2021, 11, 11640. [Google Scholar] [CrossRef]
- Tan, H.; Bao, J.; Zhou, X. Genome-wide mutational spectra analysis reveals significant cancer-specific heterogeneity. Sci. Rep. 2015, 5, 12566. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, W. Association of urinary and plasma DNA in early breast cancer patients and its links to disease relapse. Clin. Transl. Oncol. 2018, 20, 1053–1060. [Google Scholar] [CrossRef]
- Zuo, Z.; Tang, J.; Cai, X.; Ke, F.; Shi, Z. Probing of breast cancer using a combination of plasma and urinary circulating cell-free DNA. Biosci. Rep. 2020, 40, BSR20194306. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Hoon, D.S.B.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef]
- Swaminathan, R.; Butt, A.N. Circulating nucleic acids in plasma and serum: Recent developments. Ann. N. Y. Acad. Sci. 2006, 1075, 1–9. [Google Scholar] [CrossRef]
- Jung, K.; Fleischhacker, M.; Rabien, A. Cell-free DNA in the blood as a solid tumor biomarker-A critical appraisal of the literature. Clin. Chim. Acta 2010, 411, 1611–1624. [Google Scholar] [CrossRef]
- Guan, G.; Wang, Y.; Sun, Q.; Wang, L.; Xie, F.; Yan, J.; Huang, H.; Liu, H. Utility of urinary ctDNA to monitoring minimal residual disease in early breast cancer patients. Cancer Biomark. 2020, 28, 111–119. [Google Scholar] [CrossRef]
- Thierry, A.R.; El Messaoudi, S.; Gahan, P.B.; Anker, P.; Stroun, M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016, 35, 347–376. [Google Scholar] [CrossRef]
- Thierry, A.R.; Mouliere, F.; El Messaoudi, S.; Mollevi, C.; Lopez-Crapez, E.; Rolet, F.; Gillet, B.; Gongora, C.; Dechelotte, P.; Robert, B.; et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat. Med. 2014, 20, 430–435. [Google Scholar] [CrossRef]
- Pascual, T.; Gonzalez-Farre, B.; Teixidó, C.; Oleaga, L.; Oses, G.; Ganau, S.; Chic, N.; Riu, G.; Adamo, B.; Galván, P.; et al. Significant Clinical Activity of Olaparib in a Somatic BRCA1-Mutated Triple-Negative Breast Cancer with Brain Metastasis. JCO Precis. Oncol. 2019, 3, 1–6. [Google Scholar] [CrossRef]
- den Brok, W.D.; Schrader, K.A.; Sun, S.; Tinker, A.V.; Zhao, E.Y.; Aparicio, S.; Gelmon, K.A. Homologous Recombination Deficiency in Breast Cancer: A Clinical Review. JCO Precis. Oncol. 2017, 1, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat. Rev. Cancer 2004, 4, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.-T.; Choi, Y.-L.; Yun, J.W.; Kim, N.K.D.; Kim, S.-Y.; Jeon, H.J.; Nam, J.-Y.; Lee, C.; Ryu, D.; Kim, S.C.; et al. Prevalence and detection of low-allele-fraction variants in clinical cancer samples. Nat. Commun. 2017, 8, 1377. [Google Scholar] [CrossRef]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Cavallone, L.; Aguilar-Mahecha, A.; Lafleur, J.; Brousse, S.; Aldamry, M.; Roseshter, T.; Lan, C.; Alirezaie, N.; Bareke, E.; Majewski, J.; et al. Prognostic and predictive value of circulating tumor DNA during neoadjuvant chemotherapy for triple negative breast cancer. Sci. Rep. 2020, 10, 14704. [Google Scholar] [CrossRef]
- Lodish, M.B.; Stratakis, C.A. Endocrine tumours in neurofibromatosis type 1, tuberous sclerosis and related syndromes. Best Pract. Res. Clin. Endocrinol. Metab. 2010, 24, 439–449. [Google Scholar] [CrossRef]
- Courtney, K.D.; Corcoran, R.B.; Engelman, J.A. The PI3K pathway as drug target in human cancer. J. Clin. Oncol. 2010, 28, 1075–1083. [Google Scholar] [CrossRef]
- Wu, R.; Hu, T.C.; Rehemtulla, A.; Fearon, E.R.; Cho, K.R. Preclinical testing of PI3K/AKT/mTOR signaling inhibitors in a mouse model of ovarian endometrioid adenocarcinoma. Clin. Cancer Res. 2011, 17, 7359–7372. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; Oudard, S.; Hutson, T.E.; Porta, C.; Bracarda, S.; Grünwald, V.; Thompson, J.A.; Figlin, R.A.; Hollaender, N.; et al. Phase 3 trial of everolimus for metastatic renal cell carcinoma: Final results and analysis of prognostic factors. Cancer 2010, 116, 4256–4265. [Google Scholar] [CrossRef]
- Hudes, G.; Carducci, M.; Tomczak, P.; Dutcher, J.; Figlin, R.; Kapoor, A.; Staroslawska, E.; Sosman, J.; McDermott, D.; Bodrogi, I.; et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 2271–2281. [Google Scholar] [CrossRef]
- Saal, L.H.; Holm, K.; Maurer, M.; Memeo, L.; Su, T.; Wang, X.; Yu, J.S.; Malmström, P.-O.; Mansukhani, M.; Enoksson, J.; et al. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res. 2005, 65, 2554–2559. [Google Scholar] [CrossRef]
- Tomlinson, I.P.M.; Houlston, R.S.; Montgomery, G.W.; Sieber, O.M.; Dunlop, M.G. Investigation of the effects of DNA repair gene polymorphisms on the risk of colorectal cancer. Mutagenesis 2012, 27, 219–223. [Google Scholar] [CrossRef][Green Version]
- Apostolou, P.; Papasotiriou, I. Current perspectives on CHEK2 mutations in breast cancer. Breast Cancer 2017, 9, 331–335. [Google Scholar] [CrossRef] [PubMed]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Bialkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef] [PubMed]
- Cortesi, L.; Rugo, H.S.; Jackisch, C. An Overview of PARP Inhibitors for the Treatment of Breast Cancer. Target. Oncol. 2021, 16, 255–282. [Google Scholar] [CrossRef] [PubMed]
- Ansari, K.I.; Mandal, S.S. Mixed lineage leukemia: Roles in gene expression, hormone signaling and mRNA processing. FEBS J. 2010, 277, 1790–1804. [Google Scholar] [CrossRef] [PubMed]
- Stumpel, D.J.P.M.; Schneider, P.; Seslija, L.; Osaki, H.; Williams, O.; Pieters, R.; Stam, R.W. Connectivity mapping identifies HDAC inhibitors for the treatment of t(4;11)-positive infant acute lymphoblastic leukemia. Leukemia 2012, 26, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, Y.; Rappaport, A.R.; Kitzing, T.; Schultz, N.; Zhao, Z.; Shroff, A.S.; Dickins, R.A.; Vakoc, C.R.; Bradner, J.E.; et al. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell 2014, 25, 652–665. [Google Scholar] [CrossRef] [PubMed]
- Niyomnaitham, S.; Parinyanitikul, N.; Roothumnong, E.; Jinda, W.; Samarnthai, N.; Atikankul, T.; Suktitipat, B.; Thongnoppakhun, W.; Limwongse, C.; Pithukpakorn, M. Tumor mutational profile of triple negative breast cancer patients in Thailand revealed distinctive genetic alteration in chromatin remodeling gene. PeerJ 2019, 7, e6501. [Google Scholar] [CrossRef] [PubMed]
- Serio, P.A.d.M.P.; de Lima, P.G.F.; Katayama, M.L.H.; Roela, R.A.; Maistro, S.; Folgueira, M.A.A.K. Somatic Mutational Profile of High-Grade Serous Ovarian Carcinoma and Triple-Negative Breast Carcinoma in Young and Elderly Patients: Similarities and Divergences. Cells 2021, 10, 3586. [Google Scholar] [CrossRef]
- Li, Y.-Z.; Chen, B.; Lin, X.-Y.; Zhang, G.-C.; Lai, J.-G.; Li, C.; Lin, J.-L.; Guo, L.-P.; Xiao, W.-K.; Mok, H.; et al. Clinicopathologic and Genomic Features in Triple-Negative Breast Cancer between Special and No-Special Morphologic Pattern. Front. Oncol. 2022, 12, 830124. [Google Scholar] [CrossRef]
- Garcia-Murillas, I.; Chopra, N.; Comino-Méndez, I.; Beaney, M.; Tovey, H.; Cutts, R.J.; Swift, C.; Kriplani, D.; Afentakis, M.; Hrebien, S.; et al. Assessment of Molecular Relapse Detection in Early-Stage Breast Cancer. JAMA Oncol. 2019, 5, 1473–1478. [Google Scholar] [CrossRef]
- Garcia-Murillas, I.; Schiavon, G.; Weigelt, B.; Ng, C.; Hrebien, S.; Cutts, R.J.; Cheang, M.; Osin, P.; Nerurkar, A.; Kozarewa, I.; et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci. Transl. Med. 2015, 7, 302ra133. [Google Scholar] [CrossRef]
- Hai, L.; Li, L.; Liu, Z.; Tong, Z.; Sun, Y. Whole-genome circulating tumor DNA methylation landscape reveals sensitive biomarkers of breast cancer. Med. Comm. 2022, 3, e134. [Google Scholar] [CrossRef]
- Chen, M.; Zhao, H. Next-generation sequencing in liquid biopsy: Cancer screening and early detection. Hum. Genom. 2019, 13, 34. [Google Scholar] [CrossRef]
- Tangvarasittichai, O.; Jaiwang, W.; Tangvarasittichai, S. The plasma DNA concentration as a potential breast cancer screening marker. Indian J. Clin. Biochem. 2015, 30, 55–58. [Google Scholar] [CrossRef]
- Shang, M.; Chang, C.; Pei, Y.; Guan, Y.; Chang, J.; Li, H. Potential Management of Circulating Tumor DNA as a Biomarker in Triple-Negative Breast Cancer. J. Cancer 2018, 9, 4627–4634. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | n = 15 | % |
|---|---|---|
| Age at diagnosis | ||
| Median (years) | 48 | |
| Range (years) | 26–69 | |
| KI-67 proliferation rate | ||
| Median (%) | 75 | |
| Range (%) | 10–90 | |
| Histology | ||
| Ductal | 14 | 93 |
| Medullary | 1 | 7 |
| Grade | ||
| G2 | 4 | 27 |
| G3 | 11 | 73 |
| Tumor size | ||
| T1 | 7 | 47 |
| T2 | 7 | 47 |
| T3 | 0 | 0 |
| T4 | 1 | 7 |
| Nodal status | ||
| cN0 | 12 | 80 |
| ≥pN1 | 3 | 20 |
| BRCA1/2 germline mutation | ||
| yes | 9 | 60 |
| no | 5 | 33 |
| unknown | 1 | 7 |
| Neoadjuvant systemic therapy | ||
| yes | 13 | 87 |
| EC * + Paclitacel/Carboplatin | 9 | 60 |
| AC * + Paclitacel/Carboplatin | 2 | 13 |
| Docetaxel + Carboplatin | 2 | 13 |
| no | 2 | 13 |
| Pathologic complete response after NACT (n = 13) | ||
| yes | 12 | 92 |
| no | 1 | 8 |
| Patient ID # | cfDNA Concentration (ng/mL) | |
|---|---|---|
| Plasma | Urine | |
| 1 | 37 | 206 |
| 2 | 218 | 160 |
| 3 | 80 | 180 |
| 4 | 356 | 412 |
| 5 | 252 | 0 |
| 6 | 172 | 1820 |
| 7 | 244 | 196 |
| 8 | 420 | 0 |
| 9 | 69 | 650 |
| 10 | 212 | 192 |
| 11 | 118 | 1730 |
| 12 | 442 | 1410 |
| 13 | 112 | 180 |
| 14 | 158 | 110 |
| 15 | 80 | 254 |
| median | 172 | 196 |
| range | 37–442 | 0–1820 |
| Gene | Variants in Plasma (n) | Variants in Urine (n) |
|---|---|---|
| NF1 | 49 | 54 |
| KMT2C | 25 | 32 |
| CHEK2 | 25 | 19 |
| PTEN | 25 | 13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herzog, H.; Dogan, S.; Aktas, B.; Nel, I. Targeted Sequencing of Plasma-Derived vs. Urinary cfDNA from Patients with Triple-Negative Breast Cancer. Cancers 2022, 14, 4101. https://doi.org/10.3390/cancers14174101
Herzog H, Dogan S, Aktas B, Nel I. Targeted Sequencing of Plasma-Derived vs. Urinary cfDNA from Patients with Triple-Negative Breast Cancer. Cancers. 2022; 14(17):4101. https://doi.org/10.3390/cancers14174101
Chicago/Turabian StyleHerzog, Henrike, Senol Dogan, Bahriye Aktas, and Ivonne Nel. 2022. "Targeted Sequencing of Plasma-Derived vs. Urinary cfDNA from Patients with Triple-Negative Breast Cancer" Cancers 14, no. 17: 4101. https://doi.org/10.3390/cancers14174101
APA StyleHerzog, H., Dogan, S., Aktas, B., & Nel, I. (2022). Targeted Sequencing of Plasma-Derived vs. Urinary cfDNA from Patients with Triple-Negative Breast Cancer. Cancers, 14(17), 4101. https://doi.org/10.3390/cancers14174101