
Immunization Combined with Ferroptosis Related Genes to Construct a New Prognostic Model for Head and Neck Squamous Cell Carcinoma

Abstract
:Simple Summary
Abstract
1. Introduction
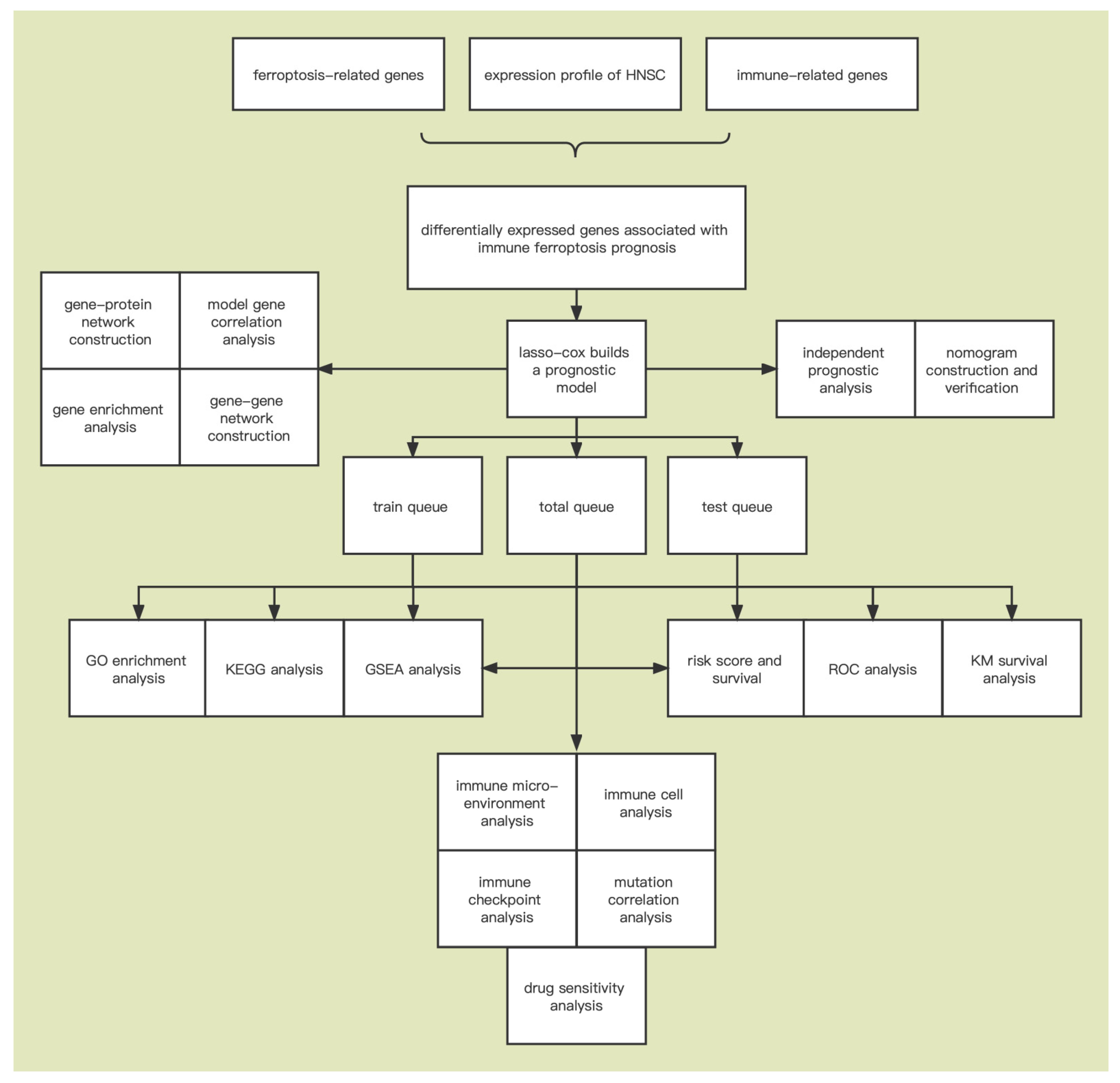
2. Materials and Methods
2.1. Data Collection
2.2. Identification of Immune-Ferroptosis-Related mRNA
2.3. Differential Expression Analysis
2.4. Construction of Immune-Ferroptosis-Related Prognostic Signature
2.5. Gene Set Enrichment Analysis (GSEA)
2.6. Functional Enrichment Analysis
2.7. Immune Cell Infiltration and Immune Microenvironment Evaluation
2.8. Model Gene Alteration Analysis
2.9. Prediction of Drug Sensitivity
2.10. Construction of Gene-Gene and Gene-Protein Networks
2.11. Statistical Analysis
3. Results
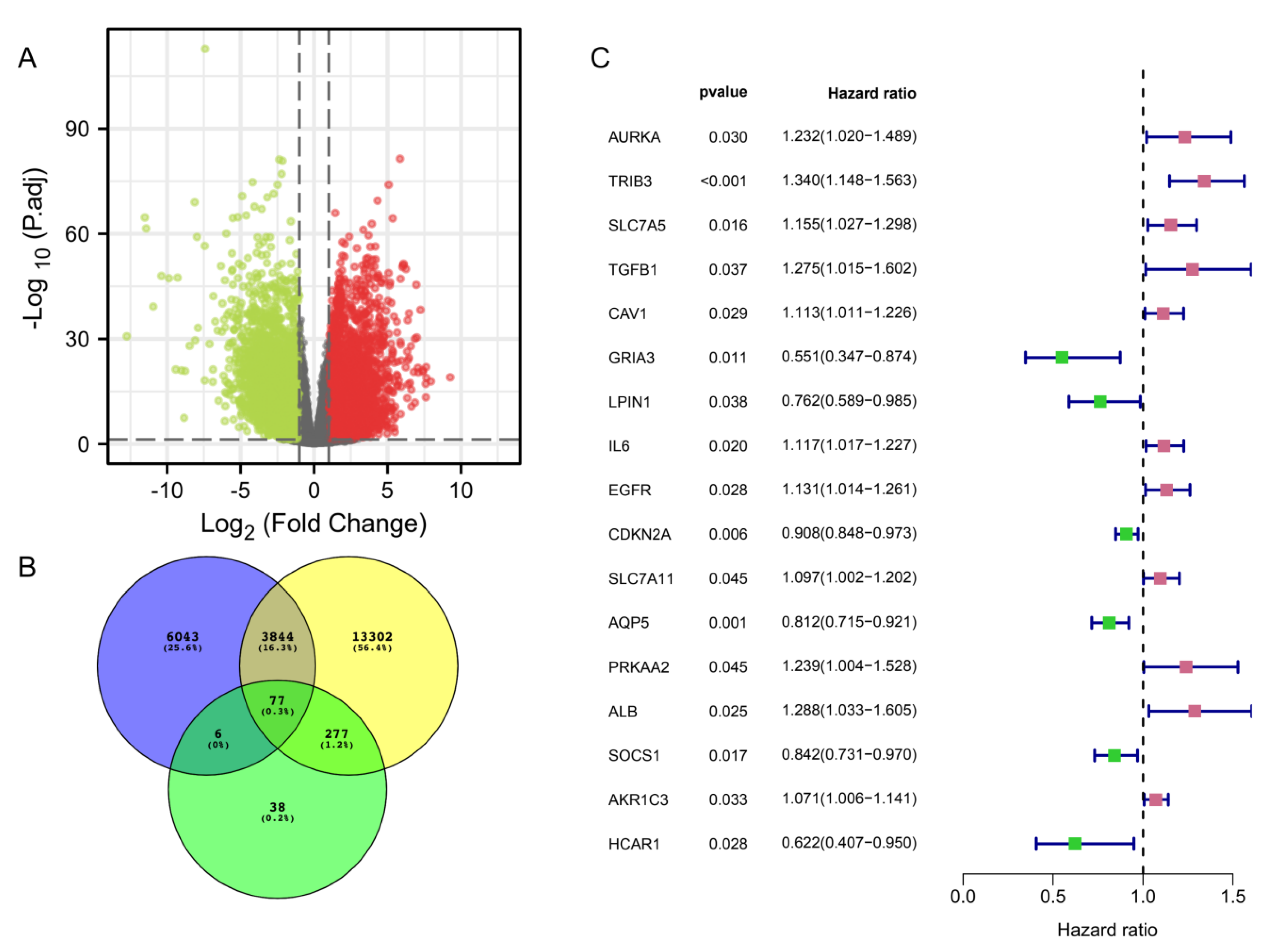
3.1. Identification of Immune-Ferroptosis-Related Differentially Expressed mRNAs in HNSC
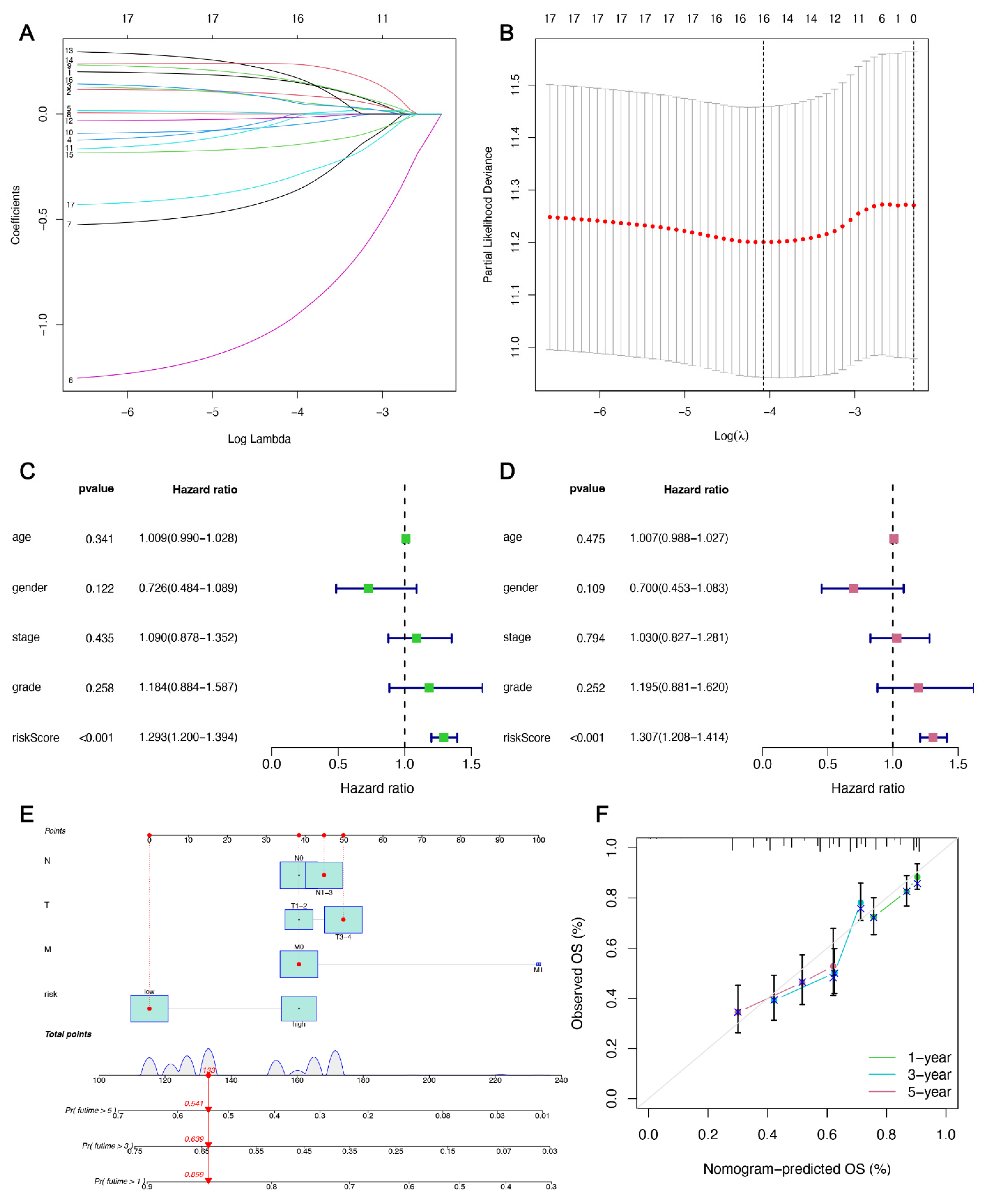
3.2. IFRMs Prognostic Model Construction and Validation
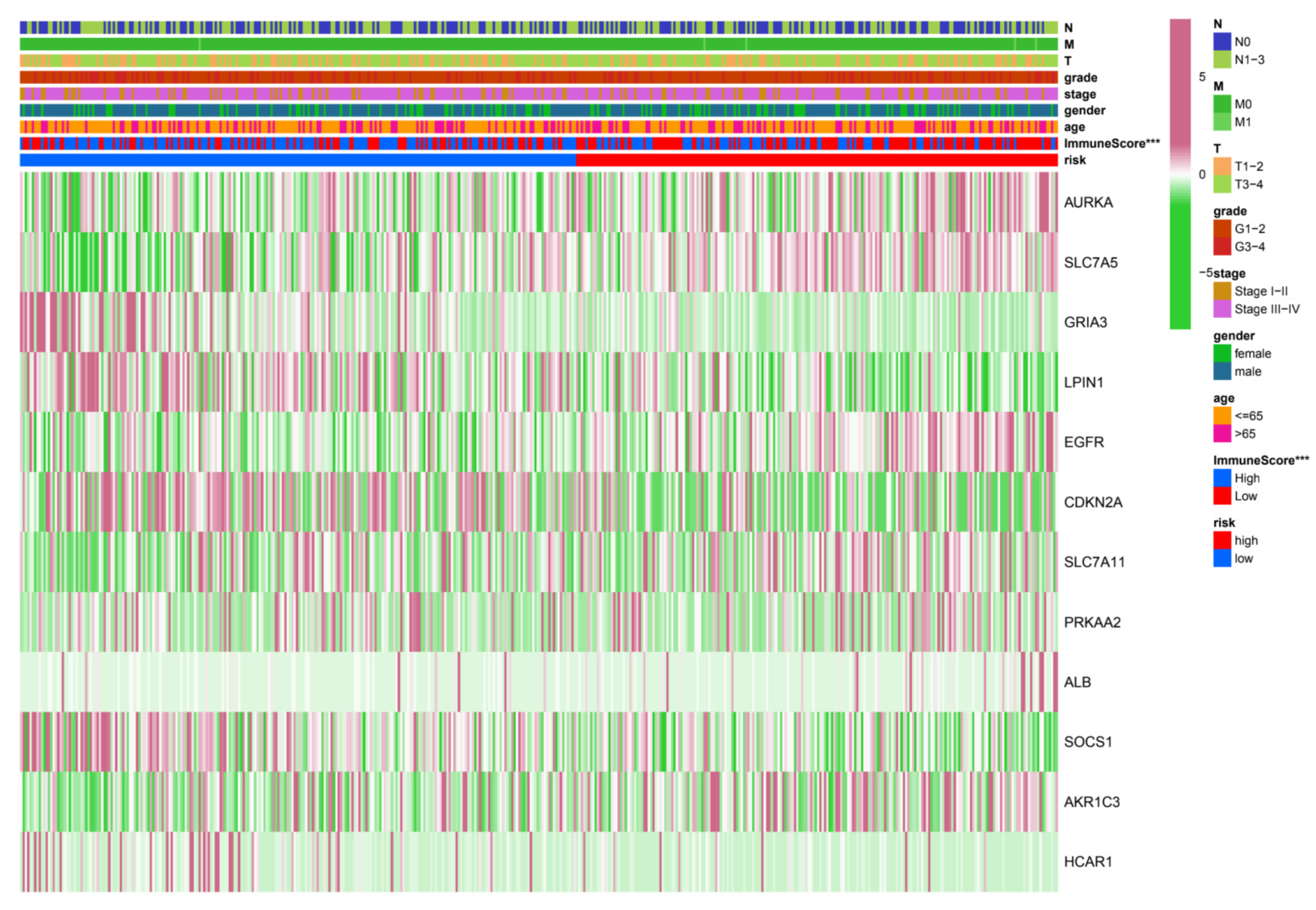
3.3. Relationship between Risk Grouping and Clinicopathological Features
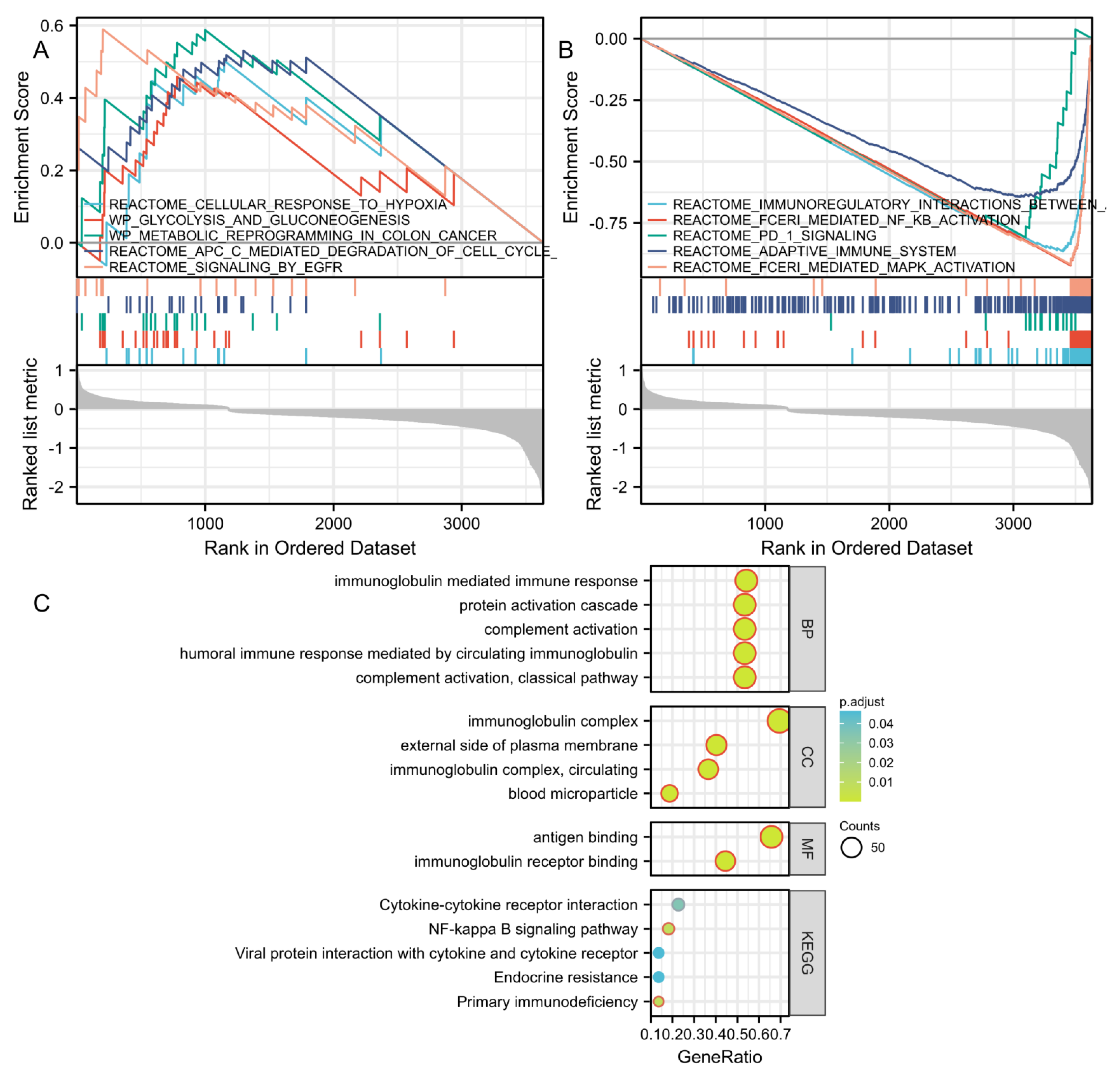
3.4. GSEA, GO, and KEGG Analysis Reveals Molecular Functions and Pathways
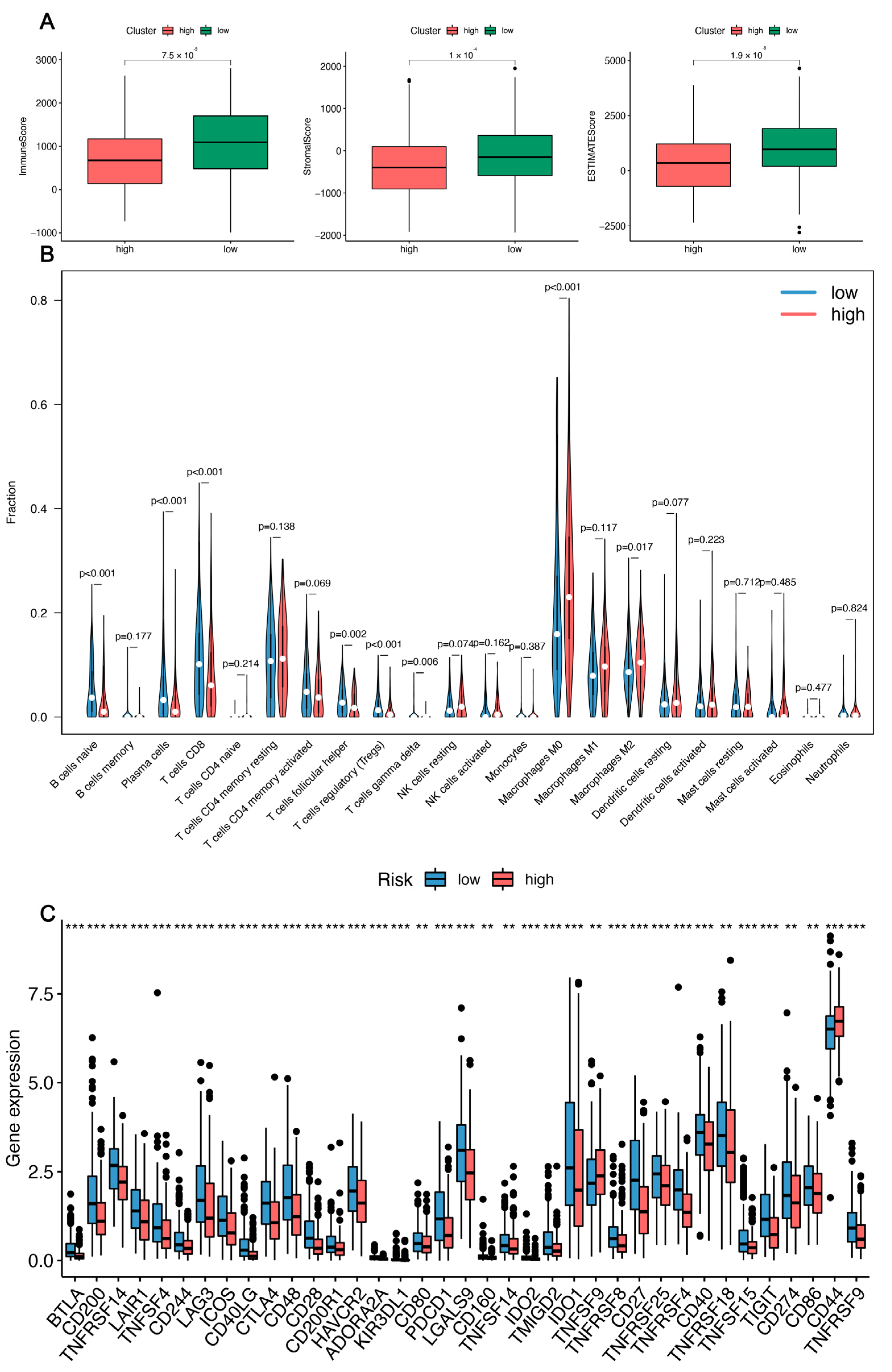
3.5. Immune-Related Analysis of HNSC Patients Using the Prognostic Signature
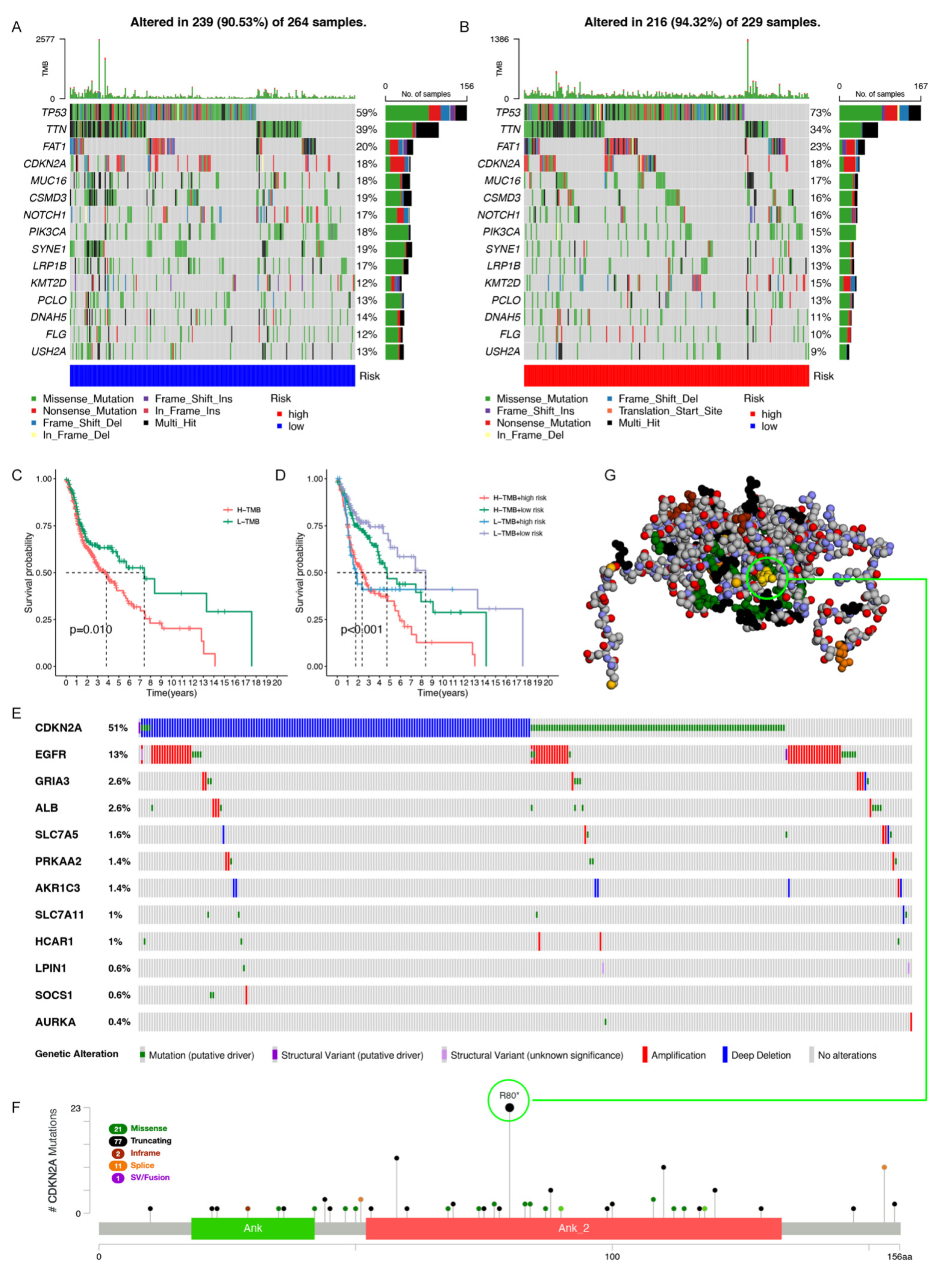
3.6. Gene Mutation Analysis in the Model
3.7. Predicting Responses to Small Drug Molecules
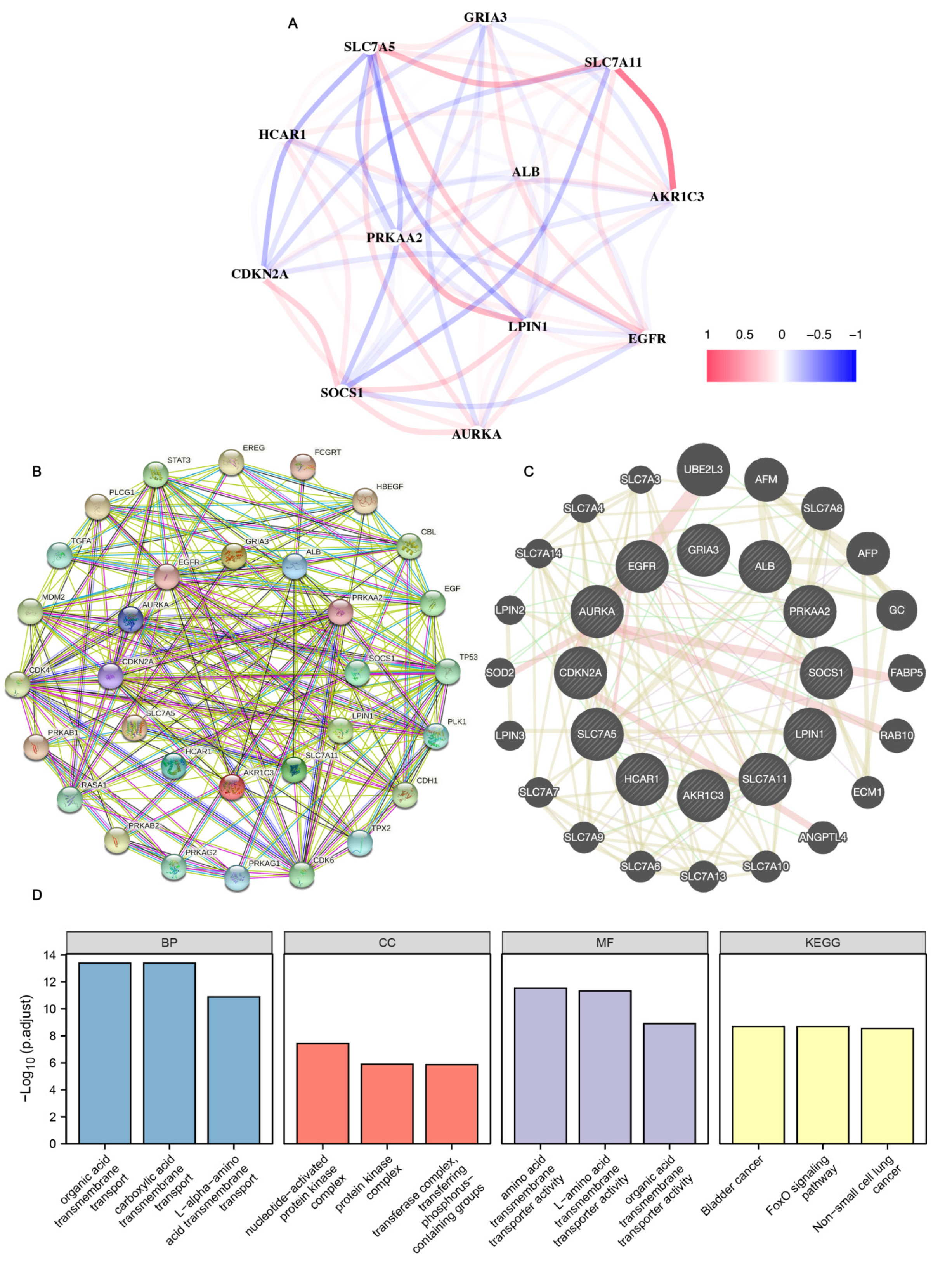
3.8. Gene Correlation Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhao, L.; Liang, J.; Zhong, W.; Han, C.; Liu, D.; Chen, X. Expression and prognostic analysis of BGN in head and neck squamous cell carcinoma. Gene 2022, 827, 146461. [Google Scholar] [CrossRef] [PubMed]
- Marur, S.; Forastiere, A.A. Head and Neck Squamous Cell Carcinoma: Update on Epidemiology, Diagnosis, and Treatment. Mayo Clin. Proc. 2016, 91, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ward, P.A. Opportunities and challenges of disease biomarkers: A new section in the Journal of Translational Medicine. J. Transl. Med. 2012, 10, 240. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Wang, S.; Wei, W.; Ma, N.; Qu, Y.; Liu, Q. Molecular mechanisms of ferroptosis and its role in prostate cancer therapy. Crit. Rev. Oncol. Hematol. 2022, 176, 103732. [Google Scholar] [CrossRef]
- Xu, T.; Ding, W.; Ji, X.; Ao, X.; Liu, Y.; Yu, W.; Wang, J. Molecular mechanisms of ferroptosis and its role in cancer therapy. J. Cell Mol. Med. 2019, 23, 4900–4912. [Google Scholar] [CrossRef] [PubMed]
- Fearnhead, H.O.; Vandenabeele, P.; Vanden Berghe, T. How do we fit ferroptosis in the family of regulated cell death? Cell Death Differ. 2017, 24, 1991–1998. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Yang, C.; Jian, L.; Guo, S.; Chen, R.; Li, K.; Qu, F.; Tao, K.; Fu, Y.; Luo, F.; et al. Sulfasalazineinduced ferroptosis in breast cancer cells is reduced by the inhibitory effect of estrogen receptor on the transferrin receptor. Oncol. Rep. 2019, 42, 826–838. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Louandre, C.; Marcq, I.; Bouhlal, H.; Lachaier, E.; Godin, C.; Saidak, Z.; Francois, C.; Chatelain, D.; Debuysscher, V.; Barbare, J.C.; et al. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett. 2015, 356, 971–977. [Google Scholar] [CrossRef]
- Ghoochani, A.; Hsu, E.C.; Aslan, M.; Rice, M.A.; Nguyen, H.M.; Brooks, J.D.; Corey, E.; Paulmurugan, R.; Stoyanova, T. Ferroptosis Inducers Are a Novel Therapeutic Approach for Advanced Prostate Cancer. Cancer Res. 2021, 81, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Eling, N.; Reuter, L.; Hazin, J.; Hamacher-Brady, A.; Brady, N.R. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2015, 2, 517–532. [Google Scholar] [CrossRef]
- Liu, X.Y.; Wei, D.G.; Li, R.S. Capsaicin induces ferroptosis of NSCLC by regulating SLC7A11/GPX4 signaling in vitro. Sci. Rep. 2022, 12, 11996. [Google Scholar] [CrossRef] [PubMed]
- Shan, K.; Feng, N.; Zhu, D.; Qu, H.; Fu, G.; Li, J.; Cui, J.; Chen, H.; Wang, R.; Qi, Y.; et al. Free docosahexaenoic acid promotes ferroptotic cell death via lipoxygenase dependent and independent pathways in cancer cells. Eur. J. Nutr. 2022. [Google Scholar] [CrossRef]
- Huang, F.; Pang, J.; Xu, L.; Niu, W.; Zhang, Y.; Li, S.; Li, X. Hedyotis diffusa injection induces ferroptosis via the Bax/Bcl2/VDAC2/3 axis in lung adenocarcinoma. Phytomedicine 2022, 104, 154319. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Shi, J.; Wang, L.; Huang, Y.; Zhao, B.; Ding, H.; Liu, Y.; Wang, W.; Chen, Z.; Yang, J. Loss of EMP1 promotes the metastasis of human bladder cancer cells by promoting migration and conferring resistance to ferroptosis through activation of PPAR gamma signaling. Free Radic. Biol. Med. 2022, 189, 42–57. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, L.; Yao, C.; Ma, Y.; Liu, Y. Epithelial Membrane Protein 1 Promotes Sensitivity to RSL3-Induced Ferroptosis and Intensifies Gefitinib Resistance in Head and Neck Cancer. Oxid. Med. Cell Longev. 2022, 2022, 4750671. [Google Scholar] [CrossRef]
- Liu, Y.; Li, L.; Yang, Z.; Wen, D.; Hu, Z. Circular RNA circACAP2 Suppresses Ferroptosis of Cervical Cancer during Malignant Progression by miR-193a-5p/GPX4. J. Oncol. 2022, 2022, 5228874. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Wu, X.; Xu, F.; Ma, H.; Wu, M.; Xia, Y. Targeting Ferroptosis Pathway to Combat Therapy Resistance and Metastasis of Cancer. Front. Pharmacol. 2022, 13, 909821. [Google Scholar] [CrossRef]
- Wang, D.; DuBois, R.N. Immunosuppression associated with chronic inflammation in the tumor microenvironment. Carcinogenesis 2015, 36, 1085–1093. [Google Scholar] [CrossRef] [Green Version]
- Stockwell, B.R.; Jiang, X. A Physiological Function for Ferroptosis in Tumor Suppression by the Immune System. Cell Metab. 2019, 30, 14–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Green, M.; Choi, J.E.; Gijon, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-de-Angulo, A.; Bilbao-Asensio, M.; Cronin, J.; Evans, S.J.; Clift, M.J.D.; Llop, J.; Feiner, I.V.J.; Beadman, R.; Bascaran, K.Z.; Mareque-Rivas, J.C. Chemically Programmed Vaccines: Iron Catalysis in Nanoparticles Enhances Combination Immunotherapy and Immunotherapy-Promoted Tumor Ferroptosis. iScience 2020, 23, 101499. [Google Scholar] [CrossRef] [PubMed]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef]
- Yi, J.; Zhu, J.; Wu, J.; Thompson, C.B.; Jiang, X. Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 31189–31197. [Google Scholar] [CrossRef]
- Zhou, N.; Bao, J. FerrDb: A manually curated resource for regulators and markers of ferroptosis and ferroptosis-disease associations. Database 2020, 2020, baaa021. [Google Scholar] [CrossRef]
- Song, Y.; Tian, S.; Zhang, P.; Zhang, N.; Shen, Y.; Deng, J. Construction and Validation of a Novel Ferroptosis-Related Prognostic Model for Acute Myeloid Leukemia. Front. Genet. 2021, 12, 708699. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Tu, Z.; Wu, L.; Wang, P.; Hu, Q.; Tao, C.; Li, K.; Huang, K.; Zhu, X. N6-Methylandenosine-Related lncRNAs Are Potential Biomarkers for Predicting the Overall Survival of Lower-Grade Glioma Patients. Front. Cell Dev. Biol. 2020, 8, 642. [Google Scholar] [CrossRef]
- Kamarudin, A.N.; Cox, T.; Kolamunnage-Dona, R. Time-dependent ROC curve analysis in medical research: Current methods and applications. BMC Med. Res. Methodol. 2017, 17, 53. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- The Gene Ontology, C. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Geeleher, P.; Cox, N.; Huang, R.S. pRRophetic: An R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS ONE 2014, 9, e107468. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef] [PubMed]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef]
- Wu, Z.; Lu, Z.; Li, L.; Ma, M.; Long, F.; Wu, R.; Huang, L.; Chou, J.; Yang, K.; Zhang, Y.; et al. Identification and Validation of Ferroptosis-Related LncRNA Signatures as a Novel Prognostic Model for Colon Cancer. Front. Immunol. 2021, 12, 783362. [Google Scholar] [CrossRef]
- Yue, Z.; Sun, J.; Shi, L. Construction and Validation of a 6-Ferroptosis Related Gene Signature for Prognosis and Immune Landscape Prediction in Melanoma. Front. Genet. 2022, 13, 887542. [Google Scholar] [CrossRef]
- Tan, L.; Shen, X.; He, Z.; Lu, Y. The Role of Photodynamic Therapy in Triggering Cell Death and Facilitating Antitumor Immunology. Front. Oncol. 2022, 12, 863107. [Google Scholar] [CrossRef]
- Liu, P.; Lin, C.; Liu, Z.; Zhu, C.; Lin, Z.; Xu, D.; Chen, J.; Huang, Q.; Li, C.Y.; Hou, L.; et al. Inhibition of ALG3 stimulates cancer cell immunogenic ferroptosis to potentiate immunotherapy. Cell Mol. Life Sci. 2022, 79, 352. [Google Scholar] [CrossRef]
- Dong, Y.; Liu, D.; Zhou, H.; Gao, Y.; Nueraihemaiti, Y.; Xu, Y. A Prognostic Signature for Clear Cell Renal Cell Carcinoma Based on Ferroptosis-Related lncRNAs and Immune Checkpoints. Front. Genet. 2022, 13, 912190. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Henry, W.S.; Ricq, E.L.; Graham, E.T.; Phadnis, V.V.; Maretich, P.; Paradkar, S.; Boehnke, N.; Deik, A.A.; Reinhardt, F.; et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 2020, 585, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef]
- Veglia, F.; Tyurin, V.A.; Blasi, M.; De Leo, A.; Kossenkov, A.V.; Donthireddy, L.; To, T.K.J.; Schug, Z.; Basu, S.; Wang, F.; et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature 2019, 569, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Kalinski, P. Regulation of immune responses by prostaglandin E2. J. Immunol. 2012, 188, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Ostroumov, D.; Fekete-Drimusz, N.; Saborowski, M.; Kuhnel, F.; Woller, N. CD4 and CD8 T lymphocyte interplay in controlling tumor growth. Cell Mol. Life Sci. 2018, 75, 689–713. [Google Scholar] [CrossRef]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef]
- George, S.; Miao, D.; Demetri, G.D.; Adeegbe, D.; Rodig, S.J.; Shukla, S.; Lipschitz, M.; Amin-Mansour, A.; Raut, C.P.; Carter, S.L.; et al. Loss of PTEN Is Associated with Resistance to Anti-PD-1 Checkpoint Blockade Therapy in Metastatic Uterine Leiomyosarcoma. Immunity 2017, 46, 197–204. [Google Scholar] [CrossRef]
- Burr, M.L.; Sparbier, C.E.; Chan, Y.C.; Williamson, J.C.; Woods, K.; Beavis, P.A.; Lam, E.Y.N.; Henderson, M.A.; Bell, C.C.; Stolzenburg, S.; et al. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature 2017, 549, 101–105. [Google Scholar] [CrossRef]
- Zhong, Y.; Zhang, W.; Yu, H.; Lin, L.; Gao, X.; He, J.; Li, D.; Chen, Y.; Zeng, Z.; Xu, Y.; et al. Multi-platform-based characterization of ferroptosis in human colorectal cancer. iScience 2022, 25, 104750. [Google Scholar] [CrossRef]
- Deneka, A.Y.; Baca, Y.; Serebriiskii, I.G.; Nicolas, E.; Parker, M.I.; Nguyen, T.T.; Xiu, J.; Korn, W.M.; Demeure, M.J.; Wise-Draper, T.; et al. Association of TP53 and CDKN2A Mutation Profile with Tumor Mutation Burden in Head and Neck Cancer. Clin. Cancer Res. 2022, 28, 1925–1937. [Google Scholar] [CrossRef]
- Plath, M.; Gass, J.; Hlevnjak, M.; Li, Q.; Feng, B.; Hostench, X.P.; Bieg, M.; Schroeder, L.; Holzinger, D.; Zapatka, M.; et al. Unraveling most abundant mutational signatures in head and neck cancer. Int. J. Cancer 2021, 148, 115–127. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ontology | ID | Description | GeneRatio | BgRatio | p-Value | p. Adjust | q-Value |
|---|---|---|---|---|---|---|---|
| BP | GO:0006958 | Complement activation, classical pathway | 71/133 | 137/18,670 | 2.64 × 10123 | 3.96 × 10120 | 3.90 × 10120 |
| BP | GO:0002455 | Humoral immune response mediated by circulating immunoglobulin | 71/133 | 150/18,670 | 1.75 × 10119 | 1.31 × 10116 | 1.29 × 10116 |
| BP | GO:0006956 | Complement activation | 71/133 | 175/18,670 | 2.76 × 10113 | 1.38 × 10110 | 1.36 × 10110 |
| BP | GO:0072376 | Protein activation cascade | 71/133 | 198/18,670 | 1.54 × 10108 | 5.77 × 10106 | 5.69 × 10106 |
| BP | GO:0016064 | Immunoglobulin mediated immune response | 72/133 | 218/18,670 | 4.14 × 10107 | 1.24 × 10104 | 1.22 × 10104 |
| CC | GO:0019814 | Immunoglobulin complex | 93/134 | 159/19,717 | 1.15 × 10175 | 1.27 × 10173 | 1.27 × 10173 |
| CC | GO:0042571 | Immunoglobulin complex, circulating | 49/134 | 72/19,717 | 9.47 × 1093 | 5.26 × 1091 | 5.26 × 1091 |
| CC | GO:0009897 | External side of plasma membrane | 54/134 | 393/19,717 | 1.09 × 1056 | 4.02 × 1055 | 4.02 × 1055 |
| CC | GO:0072562 | Blood microparticle | 25/134 | 147/19,717 | 3.50 × 1028 | 9.72 × 1027 | 9.72 × 1027 |
| MF | GO:0003823 | Antigen binding | 71/108 | 160/17,697 | 7.75 × 10125 | 1.15 × 10122 | 1.12 × 10122 |
| MF | GO:0034987 | Immunoglobulin receptor binding | 48/108 | 76/17,697 | 9.71 × 1092 | 7.23 × 1090 | 7.00 × 1090 |
| KEGG | hsa05340 | Primary immunodeficiency | 3/22 | 38/8076 | 1.39 × 1004 | 0.008 | 0.006 |
| KEGG | hsa04064 | NF-kappa B signaling pathway | 4/22 | 104/8076 | 1.59 × 1004 | 0.008 | 0.006 |
| KEGG | hsa04060 | Cytokine–cytokine receptor interaction | 5/22 | 295/8076 | 9.92 × 1004 | 0.035 | 0.025 |
| KEGG | hsa01522 | Endocrine resistance | 3/22 | 98/8076 | 0.002 | 0.046 | 0.033 |
| KEGG | hsa04061 | Viral protein interaction with cytokine and cytokine receptor | 3/22 | 100/8076 | 0.002 | 0.046 | 0.033 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, L.; Chen, Z.; Liu, Y.; Wang, X.; Li, J.; Ye, Q. Immunization Combined with Ferroptosis Related Genes to Construct a New Prognostic Model for Head and Neck Squamous Cell Carcinoma. Cancers 2022, 14, 4099. https://doi.org/10.3390/cancers14174099
Yang L, Chen Z, Liu Y, Wang X, Li J, Ye Q. Immunization Combined with Ferroptosis Related Genes to Construct a New Prognostic Model for Head and Neck Squamous Cell Carcinoma. Cancers. 2022; 14(17):4099. https://doi.org/10.3390/cancers14174099
Chicago/Turabian StyleYang, Linhui, Zhiwei Chen, Yunliang Liu, Xiaoyan Wang, Jing Li, and Qing Ye. 2022. "Immunization Combined with Ferroptosis Related Genes to Construct a New Prognostic Model for Head and Neck Squamous Cell Carcinoma" Cancers 14, no. 17: 4099. https://doi.org/10.3390/cancers14174099
APA StyleYang, L., Chen, Z., Liu, Y., Wang, X., Li, J., & Ye, Q. (2022). Immunization Combined with Ferroptosis Related Genes to Construct a New Prognostic Model for Head and Neck Squamous Cell Carcinoma. Cancers, 14(17), 4099. https://doi.org/10.3390/cancers14174099