
Lynch Syndrome: From Carcinogenesis to Prevention Interventions


Abstract
Simple Summary
Abstract
1. Introduction
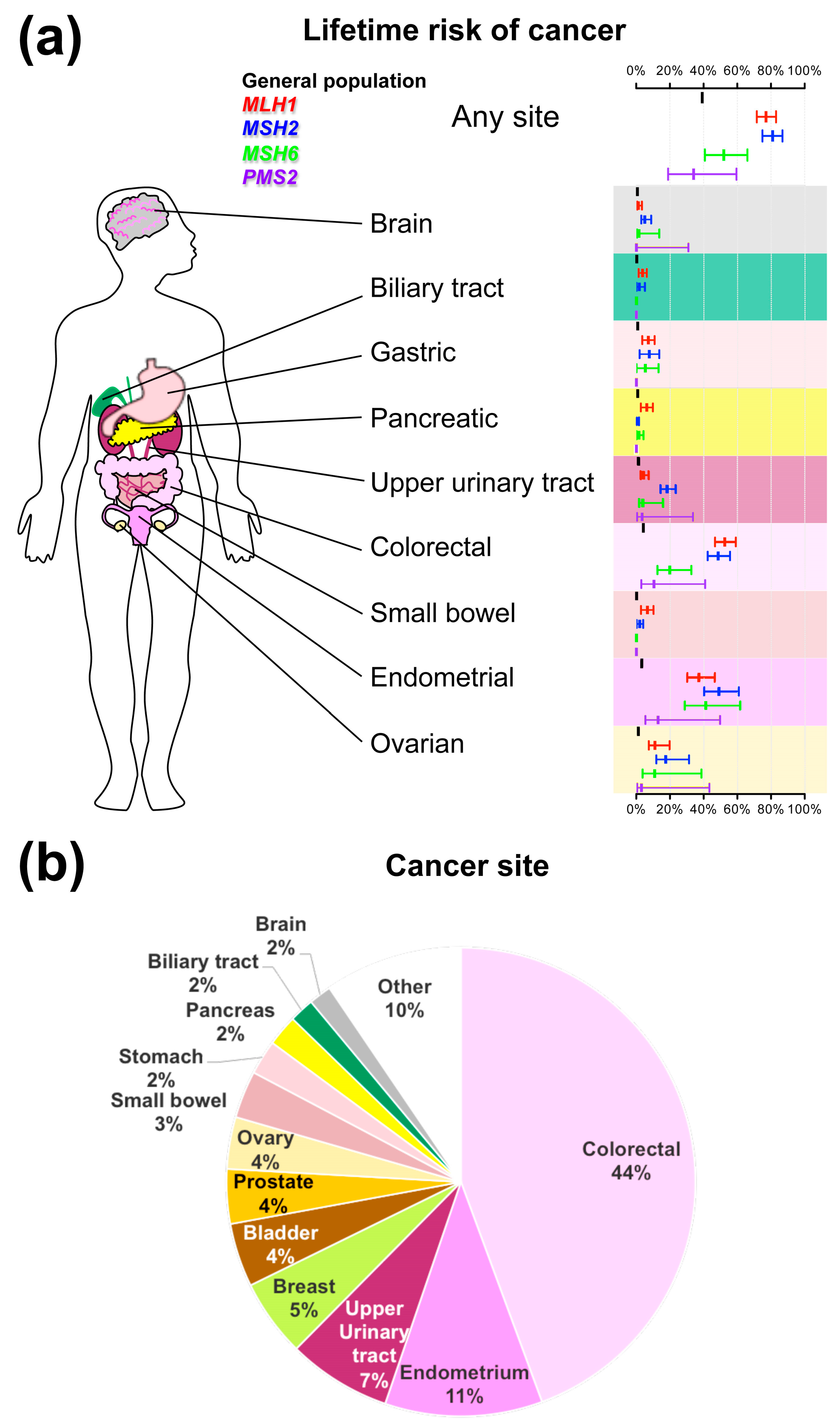
2. Clinical Features and Diagnosis
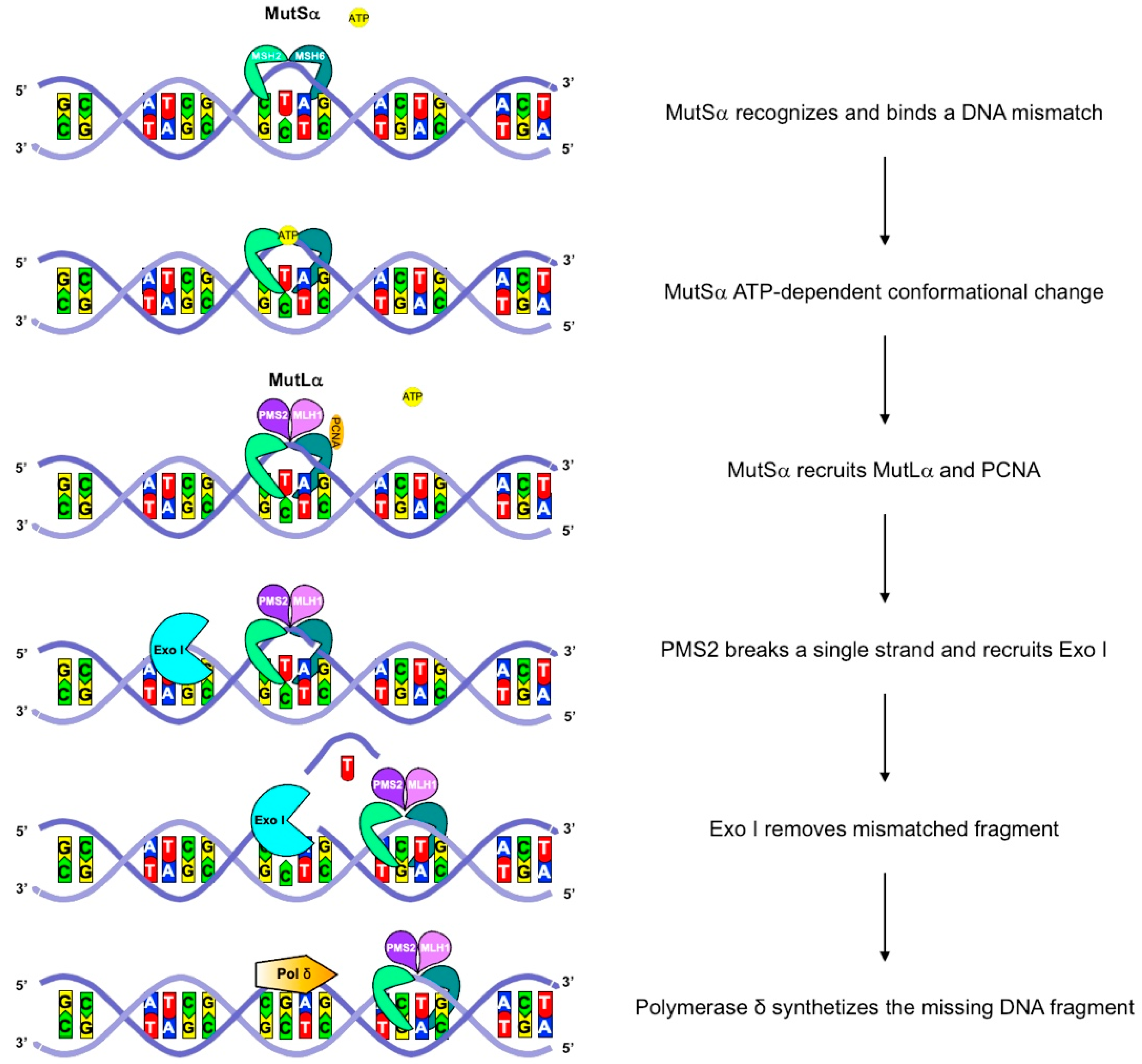
3. Biological and Genetic Features
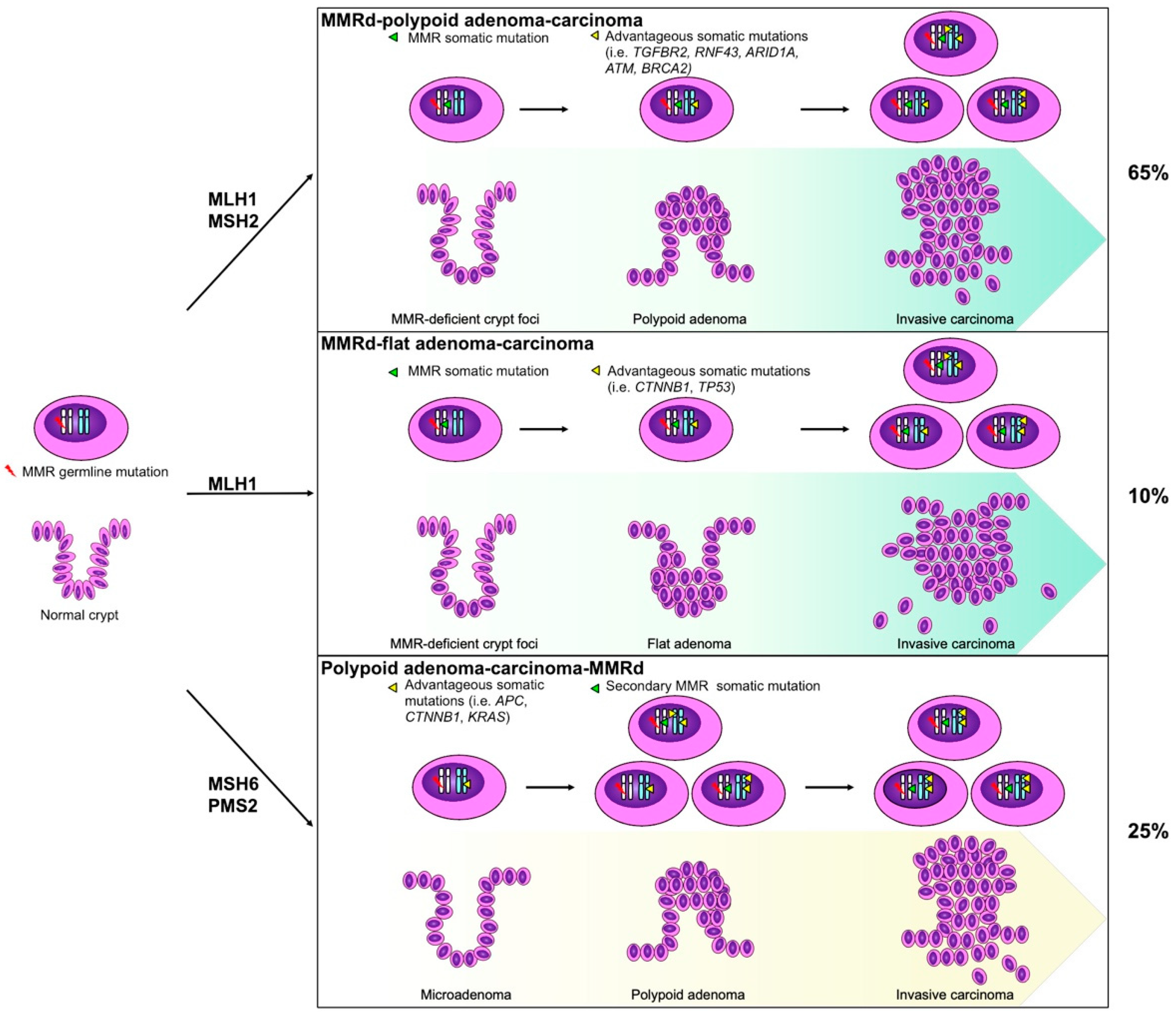
4. Pathological Features
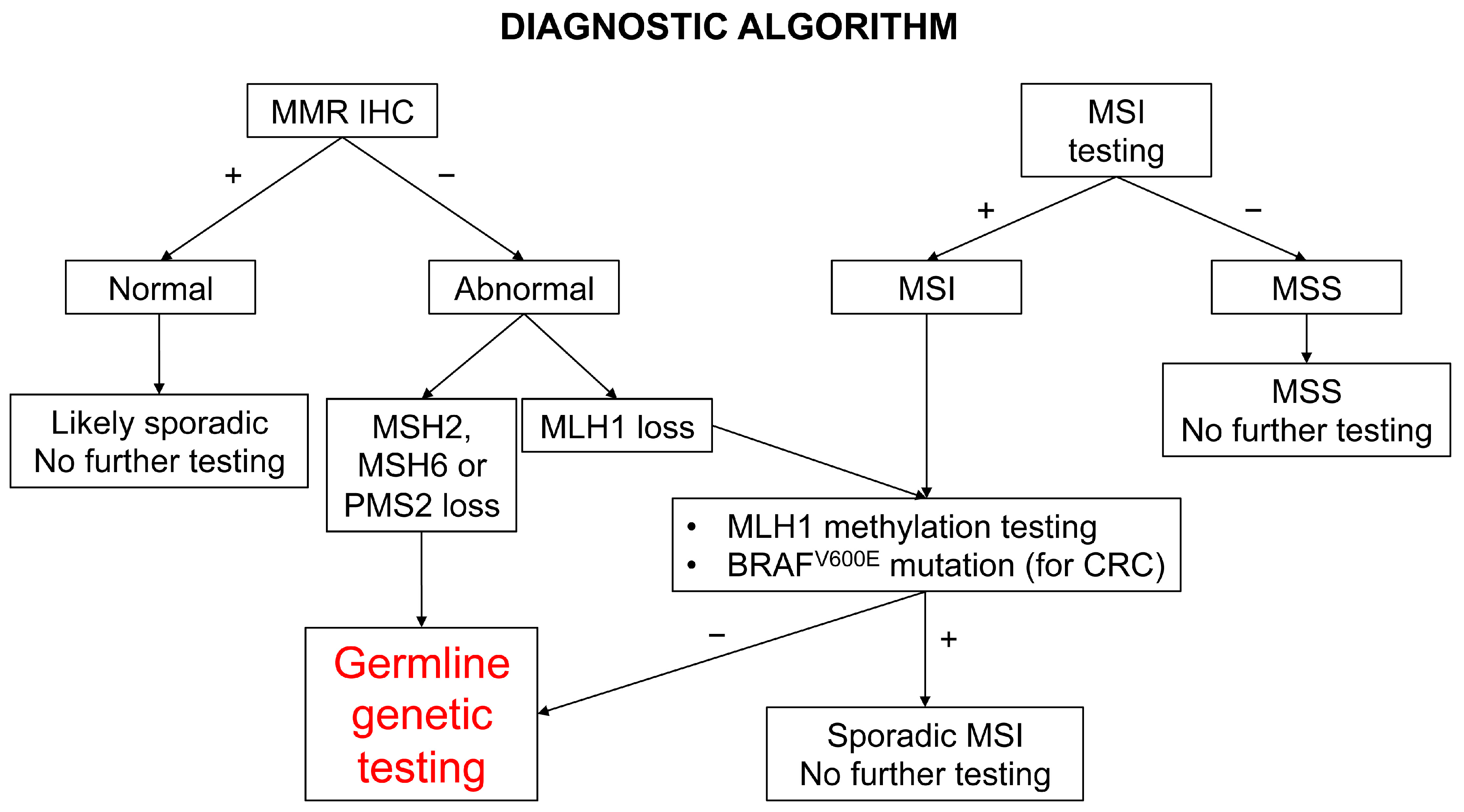
5. Diagnostic Screening
6. Immunology
7. Immunocheckpoint Inhibitors
8. Surveillance Strategies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Society Year | Colorectal Surveillance | Gynecological Surveillance | Other | Reference | |||
|---|---|---|---|---|---|---|---|
| Gene | Colonoscopy Interval | Age | Interval | Age | Procedures | ||
| AGA 2015 | |||||||
| MLH1/MSH2 | 1–2 ys | 20–25 ys * | NA | NA | NA | NA | [17] |
| MSH6/PMS2 | 1–2 ys | 20–25 ys * | |||||
| ASCO/ESMO 2015 | |||||||
| MLH1/MSH2 | 1–2 ys | 20–25 ys * | 1 y | 30–35 ys | Gynecological examination Pelvic US, Aspiration biopsy ° | HP test and subsequent eradication for gastric cancer. Upper GI endoscopy every 1 to 3 ys in some high incidence populations | [88] |
| MSH6/PMS2 | 1–2 ys | 20–25 ys * | |||||
| ACG 2015 | |||||||
| MLH1/MSH2 | 2 ys | 20–25 ys | 1 y | 30–35 ys | TV US, endometrial biopsy ° | Consider EGD with gastric biopsy and HP eradication, if found at 30–35 ys Gastric/duodenal cancer surveillance if family history | [87] |
| MSH6/PMS2 | 2 ys | 20–25 ys | |||||
| ESGE 2019 | |||||||
| MLH1/MSH2 | 2 ys | 25 ys | NA | NA | NA | Suggest HP test | [89] |
| MSH6/PMS2 | 2 ys | 35 ys | |||||
| ESMO 2019 | |||||||
| MLH1/MSH2 | 1–2 ys | 25 ys * | 1 y | 30–35 ys | TV US, CA125, End Biopsy | Consider gastric/ pancreatic/urinary tract cancer surveillance if family history | [18] |
| MSH6/PMS2 | 1–2 ys | 35 ys * | |||||
| BSG/ACPGBI/UKCGG 2020 | |||||||
| MLH1/MSH2 | 2 ys | 25 ys | NA | NA | NA | HP test and subsequent eradication for gastric cancer | [86] |
| MSH6/PMS2 | 2 ys | 35 ys | |||||
| EHTG/ESCG 2021 | |||||||
| MLH1/MSH2 | 2–3 ys | 25 ys | NA | NA | NA | No surveillance | [85] |
| MSH6 | 2–3 ys | 35 ys | |||||
| PMS2 | 5 ys | 35 ys | |||||
| NCCN 2022 | |||||||
| MLH1/MSH2/ EPCAM | 1–2 ys | 20–25 ys + | 1–2 ys | 30–35 ys | Consider Endometrial biopsy and TV US in postmenopause, serum CA125 | Consider gastric/ pancreatic/urinary tract/ prostate/ skin cancer surveillance with risk factors | [90] |
| MSH6/PMS2 | 1–3 ys | 30–35 ys + | |||||
9. Prevention Interventions
9.1. Risk Reducing Surgery
9.2. Chemoprevention
9.2.1. Aspirin
9.2.2. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) beyond Aspirin
9.2.3. Progestins
9.3. Vaccines
9.4. Exercise Intervention and Other
9.5. Ongoing Trials
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO Classification of Tumours Editorial Board. Digestive System Tumours, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2019; Volume 1. [Google Scholar]
- WHO Classification of Tumours Editorial Board. Female Genital Tumours, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2020; Volume 4. [Google Scholar]
- Ryan, N.A.J.; Glaire, M.A.; Blake, D.; Cabrera-Dandy, M.; Evans, D.G.; Crosbie, E.J. The proportion of endometrial cancers associated with Lynch syndrome: A systematic review of the literature and meta-analysis. Genet. Med. 2019, 21, 2167–2180. [Google Scholar] [CrossRef] [PubMed]
- Møller, P.; Seppälä, T.T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Gareth Evans, D.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.H.; et al. Cancer risk and survival in. Gut 2018, 67, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Valentin, M.; Sampson, J.R.; Seppälä, T.T.; Ten Broeke, S.W.; Plazzer, J.P.; Nakken, S.; Engel, C.; Aretz, S.; Jenkins, M.A.; Sunde, L.; et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: Findings from the prospective Lynch syndrome database. Genet. Med. 2020, 22, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Ryan, N.A.J.; Morris, J.; Green, K.; Lalloo, F.; Woodward, E.R.; Hill, J.; Crosbie, E.J.; Evans, D.G. Association of mismatch repair mutation with age at cancer onset in Lynch syndrome: Implications for stratified surveillance strategies. JAMA Oncol. 2017, 3, 1702–1706. [Google Scholar] [CrossRef]
- South, C.D.; Hampel, H.; Comeras, I.; Westman, J.A.; Frankel, W.L.; De La Chapelle, A. The Frequency of Muir-Torre Syndrome Among Lynch Syndrome Families. JNCI J. Natl. Cancer Inst. 2008, 100, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Hartman, D.J.; Brand, R.E.; Hu, H.; Bahary, N.; Dudley, B.; Chiosea, S.I.; Nikiforova, M.N.; Pai, R.K. Lynch syndrome–associated colorectal carcinoma: Frequent involvement of the left colon and rectum and late-onset presentation supports a universal screening approach. Hum. Pathol. 2013, 44, 2518–2528. [Google Scholar] [CrossRef] [PubMed]
- Helder-Woolderink, J.; Blok, E.; Vasen, H.; Hollema, H.; Mourits, M.; De Bock, G. Ovarian cancer in Lynch syndrome; a systematic review. Eur. J. Cancer 2016, 55, 65–73. [Google Scholar] [CrossRef]
- Ketabi, Z.; Bartuma, K.; Bernstein, I.; Malander, S.; Grönberg, H.; Björck, E.; Holck, S.; Nilbert, M. Ovarian cancer linked to lynch syndrome typically presents as early-onset, non-serous epithelial tumors. Gynecol. Oncol. 2011, 121, 462–465. [Google Scholar] [CrossRef]
- Wimmer, K.; Kratz, C.P. Constitutional mismatch repair-deficiency syndrome. Haematologica 2010, 95, 699–701. [Google Scholar] [CrossRef]
- Vasen, H.F.A.; Mecklin, J.-P.; Khan, P.M.; Lynch, H.T. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis. Colon Rectum. 1991, 34, 424–425. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Bigas, M.A.; Boland, C.R.; Hamilton, S.R.; Henson, D.E.; Jass, J.R.; Khan, P.M.; Lynch, H.; Perucho, M.; Smyrk, T.; Sobin, L.; et al. A national cancer institute workshop on hereditary nonpolyposis colorectal cancer syndrome: Meeting highlights and bethesda guidelines. J. Natl. Cancer Inst. 1997, 89, 1758–1762. [Google Scholar] [CrossRef] [PubMed]
- Vasen, H.; Watson, P.; Mecklin, J.-P.; Lynch, H.T. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative Group on HNPCC☆. Gastroenterology 1999, 116, 1453–1456. [Google Scholar] [CrossRef]
- Palomaki, G.E.; McClain, M.R.; Melillo, S.; Hampel, H.L.; Thibodeau, S.N. EGAPP supplementary evidence review: DNA testing strategies aimed at reducing morbidity and mortality from Lynch syndrome. Genet. Med. 2009, 11, 42–65. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H. Point: Justification for Lynch Syndrome Screening Among All Patients With Newly Diagnosed Colorectal Cancer. J. Natl. Compr. Cancer Netw. 2010, 8, 597–601. [Google Scholar] [CrossRef]
- Rubenstein, J.H.; Enns, R.; Heidelbaugh, J.; Barkun, A.; Adams, M.; Dorn, S.D.; Dudley-Brown, S.L.; Flamm, S.L.; Gellad, Z.F.; Gruss, C.B.; et al. American Gastroenterological Association Institute Guideline on the Diagnosis and Management of Lynch Syndrome. Gastroenterology 2015, 149, 777–782. [Google Scholar] [CrossRef]
- Stjepanovic, N.; Moreira, L.; Carneiro, F.; Balaguer, F.; Cervantes, A.; Balmaña, J.; Martinelli, E.; on behalf of the ESMO Guidelines Committee. Hereditary gastrointestinal cancers: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019, 30, 1558–1571. [Google Scholar] [CrossRef]
- Lynch, M. Rate, molecular spectrum, and consequences of human mutation. Proc. Natl. Acad. Sci. USA 2010, 107, 961–968. [Google Scholar] [CrossRef]
- Sanchez de Abajo, A.; de la Hoya, M.; van Puijenbroek, M.; Godino, J.; Díaz-Rubio, E.; Morreau, H.; Caldes, T. Dual role of loh at mmr loci in hereditary non-polyposis colorectal cancer? Oncogene 2006, 25, 2124–2130. [Google Scholar] [CrossRef][Green Version]
- Burr, K.L.-A.; van Duyn-Goedhart, A.; Hickenbotham, P.; Monger, K.; van Buul, P.P.; Dubrova, Y.E. The effects of MSH2 deficiency on spontaneous and radiation-induced mutation rates in the mouse germline. Mutat. Res. Mol. Mech. Mutagen. 2007, 617, 147–151. [Google Scholar] [CrossRef][Green Version]
- Ijsselsteijn, R.; van Hees, S.; Drost, M.; Jansen, J.G.; de Wind, N. Induction of mismatch repair deficiency, compromised DNA damage signaling and compound hypermutagenesis by a dietary mutagen in a cell-based model for Lynch syndrome. Carcinogenesis 2021, 43, 160–169. [Google Scholar] [CrossRef]
- Loeb, L.A. A mutator phenotype in cancer. Cancer Res. 2001, 61, 3230–3239. [Google Scholar] [PubMed]
- Hendriks, Y.M.; Jagmohan–Changur, S.; van der Klift, H.M.; Morreau, H.; van Puijenbroek, M.; Tops, C.; van Os, T.; Wagner, A.; Ausems, M.G.; Gomez-Garcia, E.; et al. Heterozygous Mutations in PMS2 Cause Hereditary Nonpolyposis Colorectal Carcinoma (Lynch Syndrome). Gastroenterology 2006, 130, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Berends, M.J.; Mensink, R.G.; Kempinga, C.; Sijmons, R.H.; van der Zee, A.G.; Hollema, H.; Kleibeuker, J.H.; Buys, C.H.; Hofstra, R.M. Association of Hereditary Nonpolyposis Colorectal Cancer–Related Tumors Displaying Low Microsatellite Instability with MSH6 Germline Mutations. Am. J. Hum. Genet. 1999, 65, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- Vasen, H.; Nagengast, F.; Khan, P.M. Interval cancers in hereditary non-polyposis colorectal cancer (Lynch syndrome). Lancet 1995, 345, 1183–1184. [Google Scholar] [CrossRef]
- Mori, Y.; Yin, J.; Rashid, A.; Leggett, B.A.; Young, J.; Simms, L.; Kuehl, P.M.; Langenberg, P.; Meltzer, S.J.; Stine, O.C. Instabilotyping: Comprehensive identification of frameshift mutations caused by coding region microsatellite instability. Cancer Res. 2001, 61, 6046–6049. [Google Scholar]
- Ahadova, A.; Gallon, R.; Gebert, J.; Ballhausen, A.; Endris, V.; Kirchner, M.; Stenzinger, A.; Burn, J.; von Knebel Doeberitz, M.; Bläker, H.; et al. Three molecular pathways model colorectal carcinogenesis in Lynch syndrome. Int. J. Cancer 2018, 143, 139–150. [Google Scholar] [CrossRef]
- Ahadova, A.; Doeberitz, M.V.K.; Bläker, H.; Kloor, M. CTNNB1-mutant colorectal carcinomas with immediate invasive growth: A model of interval cancers in Lynch syndrome. Fam. Cancer 2016, 15, 579–586. [Google Scholar] [CrossRef]
- Engel, C.; Ahadova, A.; Seppälä, T.T.; Aretz, S.; Bigirwamungu-Bargeman, M.; Bläker, H.; Bucksch, K.; Büttner, R.; de Vos Tot Nederveen Cappel, W.T.; Endris, V.; et al. Associations of pathogenic variants in mlh1, msh2, and msh6 with risk of colorectal adenomas and tumors and with somatic mutations in patients with Lynch syndrome. Gastroenterology 2020, 158, 1326–1333. [Google Scholar] [CrossRef] [PubMed]
- Broeke, S.W.T.; van Bavel, T.C.; Jansen, A.M.; Gomez-Garcia, E.; Hes, F.J.; van Hest, L.P.; Letteboer, T.G.; Olderode-Berends, M.J.; Ruano, D.; Spruijt, L.; et al. Molecular Background of Colorectal Tumors From Patients With Lynch Syndrome Associated With Germline Variants in PMS2. Gastroenterology 2018, 155, 844–851. [Google Scholar] [CrossRef]
- Peltomäki, P. Update on Lynch syndrome genomics. Fam. Cancer 2016, 15, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Auclair, J.; Vaissière, T.; Desseigne, F.; Lasset, C.; Bonadona, V.; Giraud, S.; Saurin, J.-C.; Joly, M.-O.; Leroux, D.; Faivre, L.; et al. Intensity-dependent constitutional MLH1 promoter methylation leads to early onset of colorectal cancer by affecting both alleles. Genes Chromosom. Cancer 2010, 50, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Hitchins, M.P. Constitutional epimutation as a mechanism for cancer causality and heritability? Nat. Rev. Cancer 2015, 15, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Yurgelun, M.B.; Allen, B.; Kaldate, R.R.; Bowles, K.R.; Judkins, T.; Kaushik, P.; Roa, B.B.; Wenstrup, R.J.; Hartman, A.R.; Syngal, S. Identification of a variety of mutations in cancer predisposition genes in patients with suspected Lynch syndrome. Gastroenterology 2015, 149, 604–613.e620. [Google Scholar] [CrossRef] [PubMed]
- Consortium, I.M.R. Variation in the risk of colorectal cancer in families with Lynch syndrome: A retrospective cohort study. Lancet Oncol. 2021, 22, 1014–1022. [Google Scholar]
- Win, A.K.; Jenkins, M.A.; Dowty, J.G.; Antoniou, A.C.; Lee, A.; Giles, G.G.; Buchanan, D.D.; Clendenning, M.; Rosty, C.; Ahnen, D.J.; et al. Prevalence and Penetrance of Major Genes and Polygenes for Colorectal Cancer. Cancer Epidemiol. Biomark. Prev. 2017, 26, 404–412. [Google Scholar] [CrossRef]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. Arid1a mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef]
- Rasmussen, M.; Lim, K.; Rambech, E.; Andersen, M.H.; Svane, I.M.; Andersen, O.; Jensen, L.H.; Nilbert, M.; Therkildsen, C. Lynch syndrome-associated epithelial ovarian cancer and its immunological profile. Gynecol. Oncol. 2021, 162, 686–693. [Google Scholar] [CrossRef]
- Kloor, M.; Huth, C.; Voigt, A.Y.; Benner, A.; Schirmacher, P.; von Knebel Doeberitz, M.; Bläker, H. Prevalence of mismatch repair-deficient crypt foci in Lynch syndrome: A pathological study. Lancet Oncol. 2012, 13, 598–606. [Google Scholar] [CrossRef]
- Staffa, L.; Echterdiek, F.; Nelius, N.; Benner, A.; Werft, W.; Lahrmann, B.; Grabe, N.; Schneider, M.; Tariverdian, M.; von Knebel Doeberitz, M.; et al. Mismatch Repair-Deficient Crypt Foci in Lynch Syndrome–Molecular Alterations and Association with Clinical Parameters. PLoS ONE 2015, 10, e0121980. [Google Scholar] [CrossRef]
- Brand, R.E.; Dudley, B.; Karloski, E.; Das, R.; Fuhrer, K.; Pai, R.K.; Pai, R.K. Detection of DNA mismatch repair deficient crypts in random colonoscopic biopsies identifies Lynch syndrome patients. Fam. Cancer 2020, 19, 169–175. [Google Scholar] [CrossRef]
- Niskakoski, A.; Pasanen, A.; Lassus, H.; Renkonen-Sinisalo, L.; Kaur, S.; Mecklin, J.-P.; Bützow, R.; Peltomäki, P. Molecular changes preceding endometrial and ovarian cancer: A study of consecutive endometrial specimens from Lynch syndrome surveillance. Mod. Pathol. 2018, 31, 1291–1301. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.; Hui, P.; Buza, N. Frequent loss of mutation-specific mismatch repair protein expression in nonneoplastic endometrium of Lynch syndrome patients. Mod. Pathol. 2020, 33, 1172–1181. [Google Scholar] [CrossRef] [PubMed]
- Hegazy, S.; Brand, R.E.; Dudley, B.; Karloski, E.; Bhargava, R.; Elishaev, E.; Pai, R.K. DNA mismatch repair-deficient non-neoplastic endometrial glands are common in Lynch syndrome patients and are present at a higher density than in the colon. Histopathology 2021, 79, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Young, J.; Simms, L.A.; Biden, K.G.; Wynter, C.; Whitehall, V.; Karamatic, R.; George, J.; Goldblatt, J.; Walpole, I.; Robin, S.-A.; et al. Features of Colorectal Cancers with High-Level Microsatellite Instability Occurring in Familial and Sporadic Settings: Parallel Pathways of Tumorigenesis. Am. J. Pathol. 2001, 159, 2107–2116. [Google Scholar] [CrossRef]
- Jass, J.R.; Walsh, M.D.; Barker, M.; Simms, L.A.; Young, J.; Leggett, B.A. Distinction between familial and sporadic forms of colorectal cancer showing DNA microsatellite instability. Eur. J. Cancer 2002, 38, 858–866. [Google Scholar] [CrossRef]
- Takemoto, N.; Konishi, F.; Yamashita, K.; Kojima, M.; Furukawa, T.; Miyakura, Y.; Shitoh, K.; Nagai, H. The correlation of microsatellite instability and tumor-infiltrating lymphocytes in hereditary non-polyposis colorectal cancer (HNPCC) and sporadic colorectal cancers: The significance of different types of lymphocyte infiltration. Jpn. J. Clin. Oncol. 2004, 34, 90–98. [Google Scholar] [CrossRef]
- Wang, T.; Lee, L.H.; Vyas, M.; Zhang, L.; Ganesh, K.; Firat, C.; Segal, N.H.; Desai, A.; Hechtman, J.F.; Ntiamoah, P.; et al. Colorectal carcinoma with double somatic mismatch repair gene inactivation: Clinical and pathological characteristics and response to immune checkpoint blockade. Mod. Pathol. 2019, 32, 1551–1562. [Google Scholar] [CrossRef]
- Surmann, E.-M.; Voigt, A.Y.; Michel, S.; Bauer, K.; Reuschenbach, M.; Ferrone, S.; von Knebel Doeberitz, M.; Kloor, M. Association of high CD4-positive T cell infiltration with mutations in HLA class II-regulatory genes in microsatellite-unstable colorectal cancer. Cancer Immunol. Immunother. 2015, 64, 357–366. [Google Scholar] [CrossRef]
- Tougeron, D.; Maby, P.; Elie, N.; Fauquembergue, E.; Le Pessot, F.; Cornic, M.; Sabourin, J.-C.; Michel, P.; Frébourg, T.; Latouche, J.-B. Regulatory T Lymphocytes Are Associated with Less Aggressive Histologic Features in Microsatellite-Unstable Colorectal Cancers. PLoS ONE 2013, 8, e61001. [Google Scholar] [CrossRef]
- Pakish, J.B.; Zhang, Q.; Chen, Z.; Liang, H.; Chisholm, G.B.; Yuan, Y.; Mok, S.C.; Broaddus, R.R.; Lu, K.H.; Yates, M.S. Immune Microenvironment in Microsatellite-Instable Endometrial Cancers: Hereditary or Sporadic Origin Matters. Clin. Cancer Res. 2017, 23, 4473–4481. [Google Scholar] [CrossRef]
- Ramchander, N.C.; Ryan, N.A.J.; Walker, T.D.J.; Harries, L.; Bolton, J.; Bosse, T.; Evans, D.G.; Crosbie, E.J. Distinct Immunological Landscapes Characterize Inherited and Sporadic Mismatch Repair Deficient Endometrial Cancer. Front. Immunol. 2020, 10, 3023. [Google Scholar] [CrossRef] [PubMed]
- Sloan, E.A.; Ring, K.L.; Willis, B.C.; Modesitt, S.C.; Mills, A.M. Pd-l1 expression in mismatch repair-deficient endometrial carcinomas, including Lynch syndrome-associated and mlh1 promoter hypermethylated tumors. Am. J. Surg Pathol 2017, 41, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Kir, G.; Olgun, Z.C.; Soylemez, T.; Aydin, A.; Demircan, B.; Kaya, I.A.; McCluggage, W.G. Pd-l1 expression in mismatch repair-deficient endometrial carcinoma and tumor-associated immune cells: Differences between mlh1 methylated and nonmethylated subgroups. Int, J. Gynecol. Pathol. 2021, 40, 575–586. [Google Scholar] [CrossRef]
- Aarnio, M. Clinicopathological Features and Management of Cancers in Lynch Syndrome. Pathol. Res. Int. 2012, 2012, 350309. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, J.; Vermaut, C.; Buisine, M.-P. Diagnosis of Lynch Syndrome and Strategies to Distinguish Lynch-Related Tumors from Sporadic MSI/dMMR Tumors. Cancers 2021, 13, 467. [Google Scholar] [CrossRef]
- Pérez-Carbonell, L.; Ruiz-Ponte, C.; Guarinos, C.; Alenda, C.; Payá, A.; Brea-Fernández, A.; Egoavil, C.; Castillejo, A.; Barbera, V.-M.; Bessa, X.; et al. Comparison between universal molecular screening for Lynch syndrome and revised Bethesda guidelines in a large population-based cohort of patients with colorectal cancer. Gut 2011, 61, 865–872. [Google Scholar] [CrossRef] [PubMed]
- O’Kane, G.M.; Ryan, É.; McVeigh, T.; Creavin, B.; Hyland, J.M.; O’Donoghue, D.P.; Keegan, D.; Geraghty, R.; Flannery, D.; Nolan, C.; et al. Screening for mismatch repair deficiency in colorectal cancer: Data from three academic medical centers. Cancer Med. 2017, 6, 1465–1472. [Google Scholar] [CrossRef]
- Watkins, J.C.; Yang, E.J.; Muto, M.G.; Feltmate, C.M.; Berkowitz, R.S.; Horowitz, N.S.; Syngal, S.; Yurgelun, M.B.; Chittenden, A.; Hornick, J.L.; et al. Universal Screening for Mismatch-Repair Deficiency in Endometrial Cancers to Identify Patients With Lynch Syndrome and Lynch-like Syndrome. Int. J. Gynecol. Pathol. 2017, 36, 115–127. [Google Scholar] [CrossRef]
- Luchini, C.; Bibeau, F.; Ligtenberg, M.J.L.; Singh, N.; Nottegar, A.; Bosse, T.; Miller, R.; Riaz, N.; Douillard, J.Y.; Andre, F.; et al. Esmo recommendations on microsatellite instability testing for immunotherapy in cancer, and its relationship with pd-1/pd-l1 expression and tumour mutational burden: A systematic review-based approach. Ann. Oncol. 2019, 30, 1232–1243. [Google Scholar] [CrossRef]
- Goodfellow, P.J.; Billingsley, C.C.; Lankes, H.A.; Ali, S.; Cohn, D.E.; Broaddus, R.J.; Ramirez, N.; Pritchard, C.C.; Hampel, H.; Chassen, A.S.; et al. Combined microsatellite instability, mlh1 methylation analysis, and immunohistochemistry for Lynch syndrome screening in endometrial cancers from gog210: An nrg oncology and gynecologic oncology group study. J. Clin. Oncol. 2015, 33, 4301–4308. [Google Scholar] [CrossRef]
- Ryan, N.A.J.; McMahon, R.; Tobi, S.; Snowsill, T.; Esquibel, S.; Wallace, A.J.; Bunstone, S.; Bowers, N.; Mosneag, I.E.; Kitson, S.J.; et al. The proportion of endometrial tumours associated with Lynch syndrome (petals): A prospective cross-sectional study. PLoS Med. 2020, 17, e1003263. [Google Scholar] [CrossRef] [PubMed]
- National Institute for Health and Care Excellence. Testing Strategies for Lynch Syndrome in People with Endometrial Cancer (dg42). Available online: https://www.nice.org.uk/guidance/dg42 (accessed on 28 October 2020).
- Stelloo, E.; Jansen, A.M.L.; Osse, E.M.; Nout, R.A.; Creutzberg, C.L.; Ruano, D.; Church, D.N.; Morreau, H.; Smit, V.T.H.B.M.; van Wezel, T.; et al. Practical guidance for mismatch repair-deficiency testing in endometrial cancer. Ann. Oncol. 2016, 28, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Yurgelun, M.B.; Kulke, M.H.; Fuchs, C.S.; Allen, B.A.; Uno, H.; Hornick, J.; Ukaegbu, C.I.; Brais, L.K.; McNamara, P.G.; Mayer, R.J.; et al. Cancer Susceptibility Gene Mutations in Individuals With Colorectal Cancer. J. Clin. Oncol. 2017, 35, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Espenschied, C.R.; LaDuca, H.; Li, S.; McFarland, R.; Gau, C.-L.; Hampel, H. Multigene Panel Testing Provides a New Perspective on Lynch Syndrome. J. Clin. Oncol. 2017, 35, 2568–2575. [Google Scholar] [CrossRef]
- Pearlman, R.; Frankel, W.L.; Swanson, B.; Zhao, W.; Yilmaz, A.; Miller, K.; Bacher, J.; Bigley, C.; Nelsen, L.; Goodfellow, P.J.; et al. Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients With Early-Onset Colorectal Cancer. JAMA Oncol. 2017, 3, 464–471. [Google Scholar] [CrossRef]
- Ring, K.L.; Bruegl, A.S.; Allen, B.A.; Elkin, E.P.; Singh, N.; Hartman, A.R.; Daniels, M.S.; Broaddus, R.R. Germline multi-gene hereditary cancer panel testing in an unselected endometrial cancer cohort. Mod. Pathol. 2016, 29, 1381–1389. [Google Scholar] [CrossRef]
- Barnetson, R.A.; Tenesa, A.; Farrington, S.M.; Nicholl, I.D.; Cetnarskyj, R.; Porteous, M.E.; Campbell, H.; Dunlop, M.G. Identification and survival of carriers of mutations in DNA mismatch-repair genes in colon cancer. N. Engl. J. Med. 2006, 354, 2751–2763. [Google Scholar] [CrossRef]
- Chen, S.; Wang, W.; Lee, S.; Nafa, K.; Lee, J.; Romans, K.; Watson, P.; Gruber, S.B.; Euhus, D.; Kinzler, K.W.; et al. Prediction of Germline Mutations and Cancer Risk in the Lynch Syndrome. JAMA 2006, 296, 1479–1487. [Google Scholar] [CrossRef]
- Kastrinos, F.; Steyerberg, E.W.; Mercado, R.; Balmaña, J.; Holter, S.; Gallinger, S.; Siegmund, K.D.; Church, J.M.; Jenkins, M.A.; Lindor, N.M.; et al. The PREMM1,2,6 Model Predicts Risk of MLH1, MSH2, and MSH6 Germline Mutations Based on Cancer History. Gastroenterology 2011, 140, 73–81.e5. [Google Scholar] [CrossRef]
- Kastrinos, F.; Uno, H.; Ukaegbu, C.; Alvero, C.; McFarland, A.; Yurgelun, M.B.; Kulke, M.H.; Schrag, D.; Meyerhardt, J.A.; Fuchs, C.S.; et al. Development and validation of the premm. J. Clin. Oncol. 2017, 35, 2165–2172. [Google Scholar] [CrossRef]
- Kloor, M.; von Knebel Doeberitz, M. The immune biology of microsatellite-unstable cancer. Trends Cancer 2016, 2, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Saeterdal, I.; Bjørheim, J.; Lislerud, K.; Gjertsen, M.K.; Bukholm, I.K.; Olsen, O.C.; Nesland, J.M.; Eriksen, J.A.; Møller, M.; Lindblom, A.; et al. Frameshift-mutation-derived peptides as tumor-specific antigens in inherited and spontaneous colorectal cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 13255–13260. [Google Scholar] [CrossRef]
- Hernandez-Sanchez, A.; Grossman, M.; Yeung, K.; Sei, S.S.; Lipkin, S.; Kloor, M. Vaccines for immunoprevention of DNA mismatch repair deficient cancers. J. Immunother. Cancer 2022, 10, e004416. [Google Scholar] [CrossRef] [PubMed]
- Kloor, M.; Michel, S.; Buckowitz, B.; Rüschoff, J.; Büttner, R.; Holinski-Feder, E.; Dippold, W.; Wagner, R.; Tariverdian, M.; Benner, A.; et al. Beta2-microglobulin mutations in microsatellite unstable colorectal tumors. Int. J. Cancer 2007, 121, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Lemery, S.; Keegan, P.; Pazdur, R. First FDA Approval Agnostic of Cancer Site—When a Biomarker Defines the Indication. N. Engl. J. Med. 2017, 377, 1409–1412. [Google Scholar] [CrossRef]
- Cercek, A.; Lumish, M.; Sinopoli, J.; Weiss, J.; Shia, J.; Lamendola-Essel, M.; El Dika, I.H.; Segal, N.; Shcherba, M.; Sugarman, R.; et al. Pd-1 blockade in mismatch repair-deficient, locally advanced rectal cancer. N. Engl. J. Med. 2022, 386, 2363–2376. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Benusiglio, P.R.; Coulet, F.; Lefebvre, A.; Duval, A.; Nuel, G. Overcoming the challenges associated with universal screening for Lynch syndrome in colorectal and endometrial cancer. Genet. Med. 2020, 22, 1422–1423. [Google Scholar] [CrossRef]
- Järvinen, H.J.; Aarnio, M.; Mustonen, H.; Aktan-Collan, K.; Aaltonen, L.A.; Peltomäki, P.; De La Chapelle, A.; Mecklin, J.P. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology 2000, 118, 829–834. [Google Scholar] [CrossRef]
- Seppälä, T.T.; Latchford, A.; Negoi, I.; Soares, A.S.; Jimenez-Rodriguez, R.; Sánchez-Guillén, L.; Evans, D.G.; Ryan, N.; Crosbie, E.J.; Dominguez-Valentin, M.; et al. European guidelines from the EHTG and ESCP for Lynch syndrome: An updated third edition of the Mallorca guidelines based on gene and gender. Br. J. Surg. 2021, 108, 484–498. [Google Scholar] [CrossRef] [PubMed]
- Monahan, K.J.; Bradshaw, N.; Dolwani, S.; Desouza, B.; Dunlop, M.G.; East, J.E.; Ilyas, M.; Kaur, A.; Lalloo, F.; Latchford, A.; et al. Guidelines for the management of hereditary colorectal cancer from the british society of gastroenterology (bsg)/association of coloproctology of great britain and ireland (acpgbi)/united kingdom cancer genetics group (ukcgg). Gut 2020, 69, 411–444. [Google Scholar] [CrossRef]
- Syngal, S.; Brand, R.E.; Church, J.M.; Giardiello, F.M.; Hampel, H.L.; Burt, R.W. ACG Clinical Guideline: Genetic Testing and Management of Hereditary Gastrointestinal Cancer Syndromes. Am. J. Gastroenterol. 2015, 110, 223–262. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, E.M.; Mangu, P.B.; Gruber, S.B.; Hamilton, S.R.; Kalady, M.F.; Lau, M.W.; Lu, K.H.; Roach, N.; Limburg, P.J.; Oncology, A.S.o.C.; et al. Hereditary colorectal cancer syndromes: American society of clinical oncology clinical practice guideline endorsement of the familial risk-colorectal cancer: European society for medical oncology clinical practice guidelines. J. Clin. Oncol. 2015, 33, 209–217. [Google Scholar] [CrossRef]
- Van Leerdam, M.E.; Roos, V.H.; van Hooft, J.E.; Balaguer, F.; Dekker, E.; Kaminski, M.F.; Latchford, A.; Neumann, H.; Ricciardiello, L.; Rupińska, M.; et al. Endoscopic management of Lynch syndrome and of familial risk of colorectal cancer: European society of gastrointestinal endoscopy (esge) guideline. Endoscopy 2019, 51, 1082–1093. [Google Scholar] [CrossRef]
- NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) Genetic/Familial High-Risk Assessment: Colorectal Version 1. 2022. Available online: https://www.nccn.org/guidelines/guidelines-detail?category=2&id=1436 (accessed on 8 June 2022).
- Anele, C.C.; Adegbola, S.O.; Askari, A.; Rajendran, A.; Clark, S.K.; Latchford, A.; Faiz, O.D. Risk of metachronous colorectal cancer following colectomy in Lynch syndrome: A systematic review and meta-analysis. Colorectal Dis. 2017, 19, 528–536. [Google Scholar] [CrossRef]
- Schmeler, K.M.; Lynch, H.T.; Chen, L.M.; Munsell, M.F.; Soliman, P.T.; Clark, M.B.; Daniels, M.S.; White, K.G.; Boyd-Rogers, S.G.; Conrad, P.G.; et al. Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. N. Engl. J. Med. 2006, 354, 261–269. [Google Scholar] [CrossRef]
- Dominguez-Valentin, M.; Crosbie, E.J.; Engel, C.; Aretz, S.; Macrae, F.; Winship, I.; Capella, G.; Thomas, H.; Nakken, S.; Hovig, E.; et al. Risk-reducing hysterectomy and bilateral salpingo-oophorectomy in female heterozygotes of pathogenic mismatch repair variants: A prospective Lynch syndrome database report. Genet. Med. 2021, 23, 705–712. [Google Scholar] [CrossRef]
- Crosbie, E.J.; Ryan, N.A.J.; Arends, M.J.; Bosse, T.; Burn, J.; Cornes, J.M.; Crawford, R.; Eccles, D.; Frayling, I.M.; Ghaem-Maghami, S.; et al. The Manchester International Consensus Group recommendations for the management of gynecological cancers in Lynch syndrome. Genet. Med. 2019, 21, 2390–2400. [Google Scholar] [CrossRef]
- Rijcken, F.E.; Hollema, H.; van der Zee, A.G.; van der Sluis, T.; Boersma-van Ek, W.; Kleibeuker, J.H. Sulindac treatment in hereditary non-polyposis colorectal cancer. Eur, J. Cancer 2007, 43, 1251–1256. [Google Scholar] [CrossRef] [PubMed]
- Burn, J.; Gerdes, A.-M.; Macrae, F.; Mecklin, J.-P.; Moeslein, G.; Olschwang, S.; Eccles, D.M.; Evans, D.G.; Maher, E.R.; Bertario, L.; et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: An analysis from the CAPP2 randomised controlled trial. Lancet 2011, 378, 2081–2087. [Google Scholar] [CrossRef]
- Ricciardiello, L.; Ahnen, D.J.; Lynch, P.M. Chemoprevention of hereditary colon cancers: Time for new strategies. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Uribe, L.; Wu, W.; Gelincik, O.; Bommi, P.V.; Francisco-Cruz, A.; Solis, L.M.; Lynch, P.M.; Lim, R.; Stoffel, E.M.; Kanth, P.; et al. Naproxen chemoprevention promotes immune activation in Lynch syndrome colorectal mucosa. Gut 2020, 70, 555–566. [Google Scholar] [CrossRef]
- Kune, G.A.; Kune, S.; Watson, L.F. Colorectal cancer risk, chronic illnesses, operations, and medications: Case control results from the melbourne colorectal cancer study. Cancer Res. 1988, 48, 4399–4404. [Google Scholar] [CrossRef]
- Hybiak, J.; Broniarek, I.; Kiryczyński, G.; Los, L.D.; Rosik, J.; Machaj, F.; Sławiński, H.; Jankowska, K.; Urasińska, E. Aspirin and its pleiotropic application. Eur. J. Pharmacol 2020, 866, 172762. [Google Scholar] [CrossRef]
- Patrignani, P.; Patrono, C. Aspirin, platelet inhibition and cancer prevention. Platelets 2018, 29, 779–785. [Google Scholar] [CrossRef]
- Serrano, D.; Patrignani, P.; Stigliano, V.; Turchetti, D.; Sciallero, S.; Roviello, F.; D’Arpino, A.; Grattagliano, I.; Testa, S.; Oliani, C.; et al. Aspirin Colorectal Cancer Prevention in Lynch Syndrome: Recommendations in the Era of Precision Medicine. Genes 2022, 13, 460. [Google Scholar] [CrossRef]
- Burn, J.; Bishop, D.T.; Mecklin, J.-P.; Macrae, F.; Möslein, G.; Olschwang, S.; Bisgaard, M.-L.; Ramesar, R.; Eccles, D.M.; Maher, E.R.; et al. Effect of Aspirin or Resistant Starch on Colorectal Neoplasia in the Lynch Syndrome. N. Engl. J. Med. 2008, 359, 2567–2578. [Google Scholar] [CrossRef]
- Burn, J.; Sheth, H.; Elliott, F.; Reed, L.; Macrae, F.; Mecklin, J.-P.; Möslein, G.; McRonald, F.E.; Bertario, L.; Evans, D.G.; et al. Cancer prevention with aspirin in hereditary colorectal cancer (Lynch syndrome), 10-year follow-up and registry-based 20-year data in the CAPP2 study: A double-blind, randomised, placebo-controlled trial. Lancet 2020, 395, 1855–1863. [Google Scholar] [CrossRef]
- Soualy, A.; Deutsch, D.; Benallaoua, M.; Ait-Omar, A.; Mary, F.; Helfen, S.; Boubaya, M.; Levy, V.; Benamouzig, R.; Al-Khafaji, S.; et al. Effect of chemoprevention by low-dose aspirin of new or recurrent colorectal adenomas in patients with Lynch syndrome (AAS-Lynch): Study protocol for a multicenter, double-blind, placebo-controlled randomized controlled trial. Trials 2020, 21, 764. [Google Scholar] [CrossRef] [PubMed]
- Joharatnam-Hogan, N.; Cafferty, F.; Hubner, R.; Swinson, D.; Sothi, S.; Gupta, K.; Falk, S.; Patel, K.; Warner, N.; Kunene, V.; et al. Aspirin as an adjuvant treatment for cancer: Feasibility results from the add-aspirin randomised trial. Lancet Gastroenterol. Hepatol. 2019, 4, 854–862. [Google Scholar] [CrossRef]
- Ait Ouakrim, D.; Dashti, S.G.; Chau, R.; Buchanan, D.D.; Clendenning, M.; Rosty, C.; Winship, I.M.; Young, J.P.; Giles, G.G.; Leggett, B.; et al. Aspirin, ibuprofen, and the risk of colorectal cancer in Lynch syndrome. J. Natl. Cancer Inst. 2015, 107, djv170. [Google Scholar] [CrossRef]
- Collaborative Group on Epidemiological Studies on Endometrial Cancer. Endometrial cancer and oral contraceptives: An individual participant meta-analysis of 27 276 women with endometrial cancer from 36 epidemiological studies. Lancet Oncol. 2015, 16, 1061–1070. [Google Scholar] [CrossRef]
- Soini, T.; Hurskainen, R.; Grénman, S.; Mäenpää, J.; Paavonen, J.; Pukkala, E. Cancer risk in women using the levonorgestrel-releasing intrauterine system in finland. Obstet. Gynecol. 2014, 124, 292–299. [Google Scholar] [CrossRef]
- Jareid, M.; Thalabard, J.C.; Aarflot, M.; Bøvelstad, H.M.; Lund, E.; Braaten, T. Levonorgestrel-releasing intrauterine system use is associated with a decreased risk of ovarian and endometrial cancer, without increased risk of breast cancer. Results from the nowac study. Gynecol. Oncol. 2018, 149, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Iversen, L.; Sivasubramaniam, S.; Lee, A.J.; Fielding, S.; Hannaford, P.C. Lifetime cancer risk and combined oral contraceptives: The Royal College of General Practitioners’ Oral Contraception Study. Am. J. Obstet. Gynecol. 2017, 216, 580.e1–580.e9. [Google Scholar] [CrossRef]
- Michels, K.A.; Pfeiffer, R.M.; Brinton, L.A.; Trabert, B. Modification of the associations between duration of oral contraceptive use and ovarian, endometrial, breast, and colorectal cancers. JAMA Oncol. 2018, 4, 516–521. [Google Scholar] [CrossRef]
- Lu, K.H.; Loose, D.S.; Yates, M.S.; Nogueras-Gonzalez, G.M.; Munsell, M.F.; Chen, L.-M.; Lynch, H.; Cornelison, T.; Boyd-Rogers, S.; Rubin, M.; et al. Prospective Multicenter Randomized Intermediate Biomarker Study of Oral Contraceptive versus Depo-Provera for Prevention of Endometrial Cancer in Women with Lynch Syndrome. Cancer Prev. Res. 2013, 6, 774–781. [Google Scholar] [CrossRef]
- Dashti, S.G.; Chau, R.; Ouakrim, D.A.; Buchanan, D.D.; Clendenning, M.; Young, J.P.; Winship, I.M.; Arnold, J.; Ahnen, D.J.; Haile, R.W.; et al. Female hormonal factors and the risk of endometrial cancer in Lynch syndrome. JAMA 2015, 314, 61–71. [Google Scholar] [CrossRef]
- Pastor, D.M.; Schlom, J. Immunology of Lynch Syndrome. Curr. Oncol. Rep. 2021, 23, 96. [Google Scholar] [CrossRef] [PubMed]
- Doeberitz, M.V.K.; Kloor, M. Towards a vaccine to prevent cancer in Lynch syndrome patients. Fam. Cancer 2013, 12, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Kloor, M.; Reuschenbach, M.; Pauligk, C.; Karbach, J.; Rafiyan, M.R.; Al-Batran, S.E.; Tariverdian, M.; Jäger, E.; von Knebel Doeberitz, M. A frameshift peptide neoantigen-based vaccine for mismatch repair-deficient cancers: A phase i/iia clinical trial. Clin. Cancer Res. 2020, 26, 4503–4510. [Google Scholar] [CrossRef] [PubMed]
- Gebert, J.; Gelincik, O.; Oezcan-Wahlbrink, M.; Marshall, J.D.; Hernandez-Sanchez, A.; Urban, K.; Long, M.; Cortes, E.; Tosti, E.; Katzenmaier, E.M.; et al. Recurrent frameshift neoantigen vaccine elicits protective immunity with reduced tumor burden and improved overall survival in a Lynch syndrome mouse model. Gastroenterology 2021, 161, 1288–1302.e13. [Google Scholar] [CrossRef]
- Overman, M.; Leoni, G.; D’Alise, A.; Cotugno, G.; Langone, F.; Capone, S.; Del Sorbo, M.; Fakih, M.; Le, D.; Shields, A.; et al. 1004P Initial results from a phase I study of Nous-209, an off-the-shelf viral vectored immunotherapy encoding 209 shared frame shift peptide neoantigens, with pembrolizumab, for the treatment of tumors with a deficiency in mismatch repair/microsatellite instability. Ann. Oncol. 2021, 32, S850. [Google Scholar] [CrossRef]
- Greiner, J.W.; Zeytin, H.; Anver, M.R.; Schlom, J. Vaccine-based therapy directed against carcinoembryonic antigen demonstrates antitumor activity on spontaneous intestinal tumors in the absence of autoimmunity. Cancer Res. 2002, 62, 6944–6951. [Google Scholar]
- Zeytin, H.E.; Patel, A.C.; Rogers, C.J.; Canter, D.; Hursting, S.D.; Schlom, J.; Greiner, J.W. Combination of a Poxvirus-Based Vaccine with a Cyclooxygenase-2 Inhibitor (Celecoxib) Elicits Antitumor Immunity and Long-Term Survival in CEA.Tg/MIN Mice. Cancer Res. 2004, 64, 3668–3678. [Google Scholar] [CrossRef]
- Moore, S.C.; Lee, I.-M.; Weiderpass, E.; Campbell, P.T.; Sampson, J.N.; Kitahara, C.M.; Keadle, S.K.; Arem, H.; de Gonzalez, A.B.; Hartge, P.; et al. Association of Leisure-Time Physical Activity With Risk of 26 Types of Cancer in 1.44 Million Adults. JAMA Intern. Med. 2016, 176, 816–825. [Google Scholar] [CrossRef]
- Matthews, C.E.; Moore, S.C.; Arem, H.; Cook, M.B.; Trabert, B.; Håkansson, N.; Larsson, S.C.; Wolk, A.; Gapstur, S.M.; Lynch, B.M.; et al. Amount and Intensity of Leisure-Time Physical Activity and Lower Cancer Risk. J. Clin. Oncol. 2020, 38, 686–697. [Google Scholar] [CrossRef]
- Friedenreich, C.M.; Orenstein, M.R. Physical activity and cancer prevention: Etiologic evidence and biological mechanisms. J. Nutr. 2002, 132, 3456S–3464S. [Google Scholar] [CrossRef]
- Dashti, S.G.; Win, A.K.; Hardikar, S.S.; Glombicki, S.E.; Mallenahalli, S.; Thirumurthi, S.; Peterson, S.K.; You, Y.N.; Buchanan, D.D.; Figueiredo, J.C.; et al. Physical activity and the risk of colorectal cancer in Lynch syndrome. Int. J. Cancer 2018, 143, 2250–2260. [Google Scholar] [CrossRef] [PubMed]
- Mathers, J.C.; Elliott, F.; Macrae, F.; Mecklin, J.-P.; Möslein, G.; McRonald, F.E.; Bertario, L.; Evans, D.G.; Gerdes, A.-M.; Ho, J.W.; et al. Cancer Prevention with Resistant Starch in Lynch Syndrome Patients in the CAPP2-Randomized Placebo Controlled Trial: Planned 10-Year Follow-Up. Cancer Prev. Res. 2022, OF1–OF12. [Google Scholar] [CrossRef] [PubMed]
- Panebianco, C.; Adamberg, K.; Adamberg, S.; Saracino, C.; Jaagura, M.; Kolk, K.; Di Chio, A.G.; Graziano, P.; Vilu, R.; Pazienza, V. Engineered Resistant-Starch (ERS) Diet Shapes Colon Microbiota Profile in Parallel with the Retardation of Tumor Growth in In Vitro and In Vivo Pancreatic Cancer Models. Nutrients 2017, 9, 331. [Google Scholar] [CrossRef] [PubMed]




| Amsterdam II Revised Criteria |
|---|
| All of the following criteria should be met: |
| Three or more blood relatives with a Lynch-related cancer (CRC, endometrial, small bowel, ureter, or renal pelvis) |
| One relative must be a first-degree relative of the other two |
| One or more cancer cases diagnosed before the age of 50 years |
| Two or more successive generations affected |
| Familial adenomatous polyposis excluded in CRC |
| Tumor diagnosis confirmed by histopathologic examination |
| Revised Bethesda Guidelines |
| At least one of the following criteria should be met: |
| CRC or endometrial cancer diagnosed before the age if 50 years |
| Synchronous or metachronous CRC or other LS-associated tumors, regardless of age |
| Colorectal cancer with MSI-high-associated morphologic features (Crohn-like lymphocytic reaction, mu-cinous/signet cell differentiation, or medullary growth pattern) in a patient younger than 60 years |
| CRC in a patient with one or more first-degree relatives with an LS-associated tumor, with one of the can-cers being diagnosed before the age of 50 years |
| CRC diagnosed in two or more first- or second-degree relatives with LS-associated tumors, regardless of age |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gambini, D.; Ferrero, S.; Kuhn, E. Lynch Syndrome: From Carcinogenesis to Prevention Interventions. Cancers 2022, 14, 4102. https://doi.org/10.3390/cancers14174102
Gambini D, Ferrero S, Kuhn E. Lynch Syndrome: From Carcinogenesis to Prevention Interventions. Cancers. 2022; 14(17):4102. https://doi.org/10.3390/cancers14174102
Chicago/Turabian StyleGambini, Donatella, Stefano Ferrero, and Elisabetta Kuhn. 2022. "Lynch Syndrome: From Carcinogenesis to Prevention Interventions" Cancers 14, no. 17: 4102. https://doi.org/10.3390/cancers14174102
APA StyleGambini, D., Ferrero, S., & Kuhn, E. (2022). Lynch Syndrome: From Carcinogenesis to Prevention Interventions. Cancers, 14(17), 4102. https://doi.org/10.3390/cancers14174102