
Clinical Spectrum and Tumour Risk Analysis in Patients with Beckwith-Wiedemann Syndrome Due to CDKN1C Pathogenic Variants

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gracia Bouthelier, R.; Lapunzina, P. Follow-up and risk of tumors in overgrowth syndromes. J. Pediatr. Endocrinol. Metab. 2005, 18 (Suppl. S1), 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Brioude, F.; Kalish, J.M.; Mussa, A.; Foster, A.C.; Bliek, J.; Ferrero, G.B.; Boonen, S.E.; Cole, T.; Baker, R.; Bertoletti, M.; et al. Expert consensus document: Clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: An international consensus statement. Nat. Rev. Endocrinol. 2018, 14, 229–249. [Google Scholar] [CrossRef] [PubMed]
- Brioude, F.; Hennekam, R.; Bliek, J.; Coze, C.; Eggermann, T.; Ferrero, G.B.; Kratz, C.; Bouc, Y.L.; Maas, S.M.; Mackay, D.J.G.; et al. Revisiting Wilms tumour surveillance in Beckwith-Wiedemann syndrome with IC2 methylation loss, reply. Eur. J. Hum. Genet. 2018, 26, 471–472. [Google Scholar] [CrossRef] [PubMed]
- Weksberg, R.; Shuman, C.; Beckwith, J.B. Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 2010, 18, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Tenorio, J.; Romanelli, V.; Martin-Trujillo, A.; Fernandez, G.M.; Segovia, M.; Perandones, C.; Perez Jurado, L.A.; Esteller, M.; Fraga, M.; Arias, P.; et al. Clinical and molecular analyses of Beckwith-Wiedemann syndrome: Comparison between spontaneous conception and assisted reproduction techniques. Am. J. Med. Genet. A 2016, 170, 2740–2749. [Google Scholar] [CrossRef] [Green Version]
- Mussa, A.; Molinatto, C.; Cerrato, F.; Palumbo, O.; Carella, M.; Baldassarre, G.; Carli, D.; Peris, C.; Riccio, A.; Ferrero, G.B. Assisted Reproductive Techniques and Risk of Beckwith-Wiedemann Syndrome. Pediatrics 2017, 140, e20164311. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.P.; Beischel, L.; Schwanke, C.; Styren, K.; Crunk, A.; Schoof, J.; Elias, A.F. Overrepresentation of pregnancies conceived by artificial reproductive technology in prenatally identified fetuses with Beckwith-Wiedemann syndrome. J. Assist. Reprod. Genet. 2018, 35, 985–992. [Google Scholar] [CrossRef]
- Weksberg, R.; Shuman, C.; Smith, A.C. Beckwith-Wiedemann syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2005, 137C, 12–23. [Google Scholar] [CrossRef]
- Mussa, A.; Russo, S.; Larizza, L.; Riccio, A.; Ferrero, G.B. (Epi)genotype-phenotype correlations in Beckwith-Wiedemann syndrome: A paradigm for genomic medicine. Clin. Genet. 2016, 89, 403–415. [Google Scholar] [CrossRef]
- Mussa, A.; Di Candia, S.; Russo, S.; Catania, S.; De Pellegrin, M.; Di Luzio, L.; Ferrari, M.; Tortora, C.; Meazzini, M.C.; Brusati, R.; et al. Recommendations of the Scientific Committee of the Italian Beckwith-Wiedemann Syndrome Association on the diagnosis, management and follow-up of the syndrome. Eur. J. Med. Genet. 2016, 59, 52–64. [Google Scholar] [CrossRef]
- Li, M.; Squire, J.A.; Weksberg, R. Molecular genetics of Wiedemann-Beckwith syndrome. Am. J. Med. Genet. 1998, 79, 253–259. [Google Scholar] [CrossRef]
- Cooper, W.N.; Luharia, A.; Evans, G.A.; Raza, H.; Haire, A.C.; Grundy, R.; Bowdin, S.C.; Riccio, A.; Sebastio, G.; Bliek, J.; et al. Molecular subtypes and phenotypic expression of Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 2005, 13, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.P.; DeBaun, M.R.; Mitsuya, K.; Galonek, H.L.; Brandenburg, S.; Oshimura, M.; Feinberg, A.P. Loss of imprinting of a paternally expressed transcript, with antisense orientation to KVLQT1, occurs frequently in Beckwith-Wiedemann syndrome and is independent of insulin-like growth factor II imprinting. Proc. Natl. Acad. Sci. USA 1999, 96, 5203–5208. [Google Scholar] [CrossRef] [Green Version]
- Bliek, J.; Maas, S.M.; Ruijter, J.M.; Hennekam, R.C.; Alders, M.; Westerveld, A.; Mannens, M.M. Increased tumour risk for BWS patients correlates with aberrant H19 and not KCNQ1OT1 methylation: Occurrence of KCNQ1OT1 hypomethylation in familial cases of BWS. Hum. Mol. Genet. 2001, 10, 467–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weksberg, R.; Nishikawa, J.; Caluseriu, O.; Fei, Y.L.; Shuman, C.; Wei, C.; Steele, L.; Cameron, J.; Smith, A.; Ambus, I.; et al. Tumor development in the Beckwith-Wiedemann syndrome is associated with a variety of constitutional molecular 11p15 alterations including imprinting defects of KCNQ1OT1. Hum. Mol. Genet. 2001, 10, 2989–3000. [Google Scholar] [CrossRef] [Green Version]
- Slavotinek, A.M.; Biesecker, L.G. Unfolding the role of chaperones and chaperonins in human disease. Trends Genet. 2001, 17, 528–535. [Google Scholar] [CrossRef]
- Lee, M.P.; DeBaun, M.; Randhawa, G.; Reichard, B.A.; Elledge, S.J.; Feinberg, A.P. Low frequency of p57KIP2 mutation in Beckwith-Wiedemann syndrome. Am. J. Hum. Genet. 1997, 61, 304–309. [Google Scholar] [CrossRef] [Green Version]
- Brioude, F.; Netchine, I.; Praz, F.; Le Jule, M.; Calmel, C.; Lacombe, D.; Edery, P.; Catala, M.; Odent, S.; Isidor, B.; et al. Mutations of the Imprinted CDKN1C Gene as a Cause of the Overgrowth Beckwith-Wiedemann Syndrome: Clinical Spectrum and Functional Characterization. Hum. Mutat. 2015, 36, 894–902. [Google Scholar] [CrossRef]
- Romanelli, V.; Belinchon, A.; Campos-Barros, A.; Heath, K.E.; Garcia-Minaur, S.; Martinez-Glez, V.; Palomo, R.; Mercado, G.; Gracia, R.; Lapunzina, P. CDKN1C mutations in HELLP/preeclamptic mothers of Beckwith-Wiedemann Syndrome (BWS) patients. Placenta 2009, 30, 551–554. [Google Scholar] [CrossRef]
- Choufani, S.; Shuman, C.; Weksberg, R. Beckwith-Wiedemann syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154C, 343–354. [Google Scholar] [CrossRef]
- Maas, S.M.; Vansenne, F.; Kadouch, D.J.; Ibrahim, A.; Bliek, J.; Hopman, S.; Mannens, M.M.; Merks, J.H.; Maher, E.R.; Hennekam, R.C. Phenotype, cancer risk, and surveillance in Beckwith-Wiedemann syndrome depending on molecular genetic subgroups. Am. J. Med. Genet. A 2016, 170, 2248–2260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bliek, J.; Gicquel, C.; Maas, S.; Gaston, V.; Le Bouc, Y.; Mannens, M. Epigenotyping as a tool for the prediction of tumor risk and tumor type in patients with Beckwith-Wiedemann syndrome (BWS). J. Pediatr. 2004, 145, 796–799. [Google Scholar] [CrossRef] [PubMed]
- Romanelli, V.; Belinchon, A.; Benito-Sanz, S.; Martinez-Glez, V.; Gracia-Bouthelier, R.; Heath, K.E.; Campos-Barros, A.; Garcia-Minaur, S.; Fernandez, L.; Meneses, H.; et al. CDKN1C (p57Kip2) analysis in Beckwith-Wiedemann syndrome (BWS) patients: Genotype-phenotype correlations, novel mutations, and polymorphisms. Am. J. Med. Genet. A 2010, 152A, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Kantaputra, P.N.; Sittiwangkul, R.; Sonsuwan, N.; Romanelli, V.; Tenorio, J.; Lapunzina, P. A novel mutation in CDKN1C in sibs with Beckwith-Wiedemann syndrome and cleft palate, sensorineural hearing loss, and supernumerary flexion creases. Am. J. Med. Genet. A 2013, 161A, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Eggermann, T.; Algar, E.; Lapunzina, P.; Mackay, D.; Maher, E.R.; Mannens, M.; Netchine, I.; Prawitt, D.; Riccio, A.; Temple, I.K.; et al. Clinical utility gene card for: Beckwith-Wiedemann Syndrome. Eur. J. Hum. Genet. 2014, 22, 435. [Google Scholar] [CrossRef]
- Lam, W.W.; Hatada, I.; Ohishi, S.; Mukai, T.; Joyce, J.A.; Cole, T.R.; Donnai, D.; Reik, W.; Schofield, P.N.; Maher, E.R. Analysis of germline CDKN1C (p57KIP2) mutations in familial and sporadic Beckwith-Wiedemann syndrome (BWS) provides a novel genotype-phenotype correlation. J. Med. Genet. 1999, 36, 518–523. [Google Scholar] [CrossRef]
- Bilgin, B.; Kabacam, S.; Taskiran, E.; Simsek-Kiper, P.O.; Alanay, Y.; Boduroglu, K.; Utine, G.E. Epigenotype and phenotype correlations in patients with Beckwith-Wiedemann syndrome. Turk. J. Pediatr. 2018, 60, 506–513. [Google Scholar] [CrossRef]
- Wang, K.H.; Kupa, J.; Duffy, K.A.; Kalish, J.M. Diagnosis and Management of Beckwith-Wiedemann Syndrome. Front. Pediatr. 2019, 7, 562. [Google Scholar] [CrossRef]
- Kalish, J.M.; Biesecker, L.G.; Brioude, F.; Deardorff, M.A.; Di Cesare-Merlone, A.; Druley, T.; Ferrero, G.B.; Lapunzina, P.; Larizza, L.; Maas, S.; et al. Nomenclature and definition in asymmetric regional body overgrowth. Am. J. Med. Genet. A 2017, 173, 1735–1738. [Google Scholar] [CrossRef]
- Brioude, F.; Nicolas, C.; Marey, I.; Gaillard, S.; Bernier, M.; Das Neves, C.; Le Bouc, Y.; Touraine, P.; Netchine, I. Hypercortisolism due to a Pituitary Adenoma Associated with Beckwith-Wiedemann Syndrome. Horm. Res. Paediatr. 2016, 86, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Jurkiewicz, D.; Skorka, A.; Ciara, E.; Kugaudo, M.; Pelc, M.; Chrzanowska, K.; Krajewska-Walasek, M. Rare clinical findings in three sporadic cases of Beckwith-Wiedemann syndrome due to novel mutations in the CDKN1C gene. Clin. Dysmorphol. 2020, 29, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Abadie, C.; Bernard, F.; Netchine, I.; Sanlaville, D.; Roque, A.; Rossignol, S.; Coupier, I. Acute lymphocytic leukaemia in a child with Beckwith-Wiedemann syndrome harbouring a CDKN1C mutation. Eur. J. Med. Genet. 2010, 53, 400–403. [Google Scholar] [CrossRef]
- Brioude, F.; Oliver-Petit, I.; Blaise, A.; Praz, F.; Rossignol, S.; Le Jule, M.; Thibaud, N.; Faussat, A.M.; Tauber, M.; Le Bouc, Y.; et al. CDKN1C mutation affecting the PCNA-binding domain as a cause of familial Russell Silver syndrome. J. Med. Genet. 2013, 50, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Gazzin, A.; Carli, D.; Sirchia, F.; Molinatto, C.; Cardaropoli, S.; Palumbo, G.; Zampino, G.; Ferrero, G.B.; Mussa, A. Phenotype evolution and health issues of adults with Beckwith-Wiedemann syndrome. Am. J. Med. Genet. A 2019, 179, 1691–1702. [Google Scholar] [CrossRef]
- Piersigilli, F.; Auriti, C.; Mondi, V.; Francalanci, P.; Salvatori, G.; Danhaive, O. Decreased CDKN1C Expression in Congenital Alveolar Rhabdomyosarcoma Associated with Beckwith-Wiedemann Syndrome. Indian J. Pediatr. 2016, 83, 1476–1478. [Google Scholar] [CrossRef]
- Bolomiti, M.; Batnes-Pedersen, E.; Telman, G.; Januszkiewicz-Lewandowska, D. A Case report: Co-occurrence of IMAGe syndrome and Rhabdomyosarcoma. Cancer Genet. 2021, 256–257, 100–105. [Google Scholar] [CrossRef]
- Li, M.; Squire, J.; Shuman, C.; Fei, Y.L.; Atkin, J.; Pauli, R.; Smith, A.; Nishikawa, J.; Chitayat, D.; Weksberg, R. Imprinting status of 11p15 genes in Beckwith-Wiedemann syndrome patients with CDKN1C mutations. Genomics 2001, 74, 370–376. [Google Scholar] [CrossRef]
- Sirinelli, D.; Silberman, B.; Baudon, J.J.; Sinnassamy, P.; Gruner, M.; Montagne, J.P. Beckwith-wiedemann syndrome and neural crest tumors. A report of two cases. Pediatr. Radiol. 1989, 19, 242–245. [Google Scholar] [CrossRef]
- Sotelo-Avila, C.; Gonzalez-Crussi, F.; Starling, K.A. Wilms’ tumor in a patient with an incomplete form of Beckwith-Wiedemann syndrome. Pediatrics 1980, 66, 121–123. [Google Scholar] [CrossRef]
- Thornburg, C.D.; Shulkin, B.L.; Castle, V.P.; McAllister-Lucas, L.M. Thoracic neural crest tumors in Beckwith-Wiedemann syndrome. Med. Pediatr. Oncol. 2003, 41, 468–469. [Google Scholar] [CrossRef] [PubMed]
- Kamihara, J.; Bourdeaut, F.; Foulkes, W.D.; Molenaar, J.J.; Mosse, Y.P.; Nakagawara, A.; Parareda, A.; Scollon, S.R.; Schneider, K.W.; Skalet, A.H.; et al. Retinoblastoma and Neuroblastoma Predisposition and Surveillance. Clin. Cancer Res. 2017, 23, e98–e106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
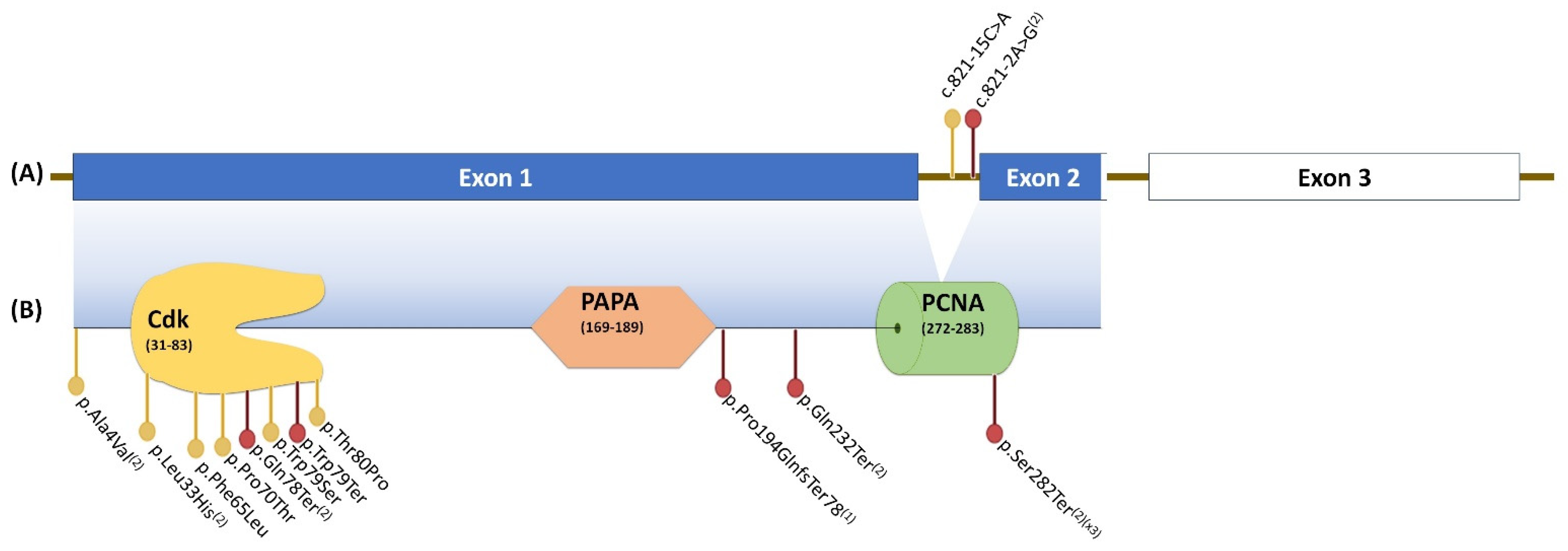

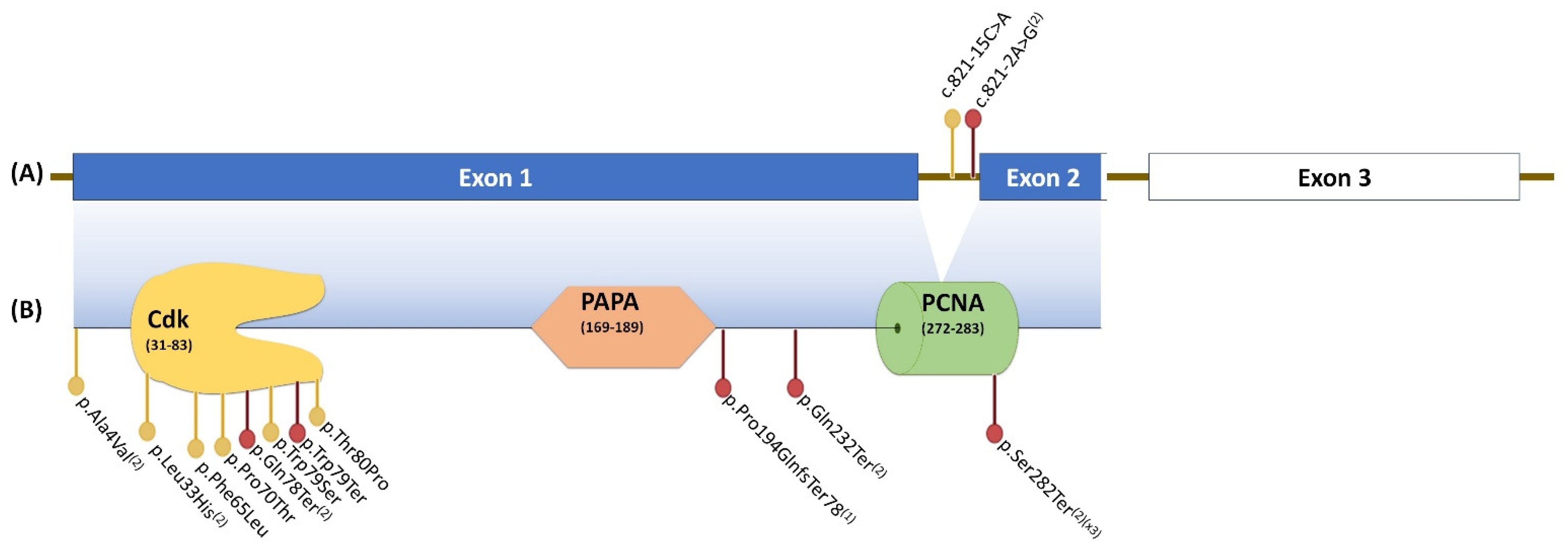{kind=link}
| Patient | Age * | Family | Relative | Genomic Coordinate (hg19) | Variant | Exon/ Intron | Variant Type | Inheritance | Allele Frequency † | Pathogenicity Predictors ‡ | CADD | ACMG Prediction § | Detection Method | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OGS1360 | 13 | 1 | Pr | chr11:2906484 | CDKN1C(NM_000076.2):c.236G>C,p.(Trp79Ser) | 1 | missense | maternal | - | 24/26 | 28.39 | VUS | SS | This study |
| OGS1810 | 10 | 1 | S | chr11:2906484 | CDKN1C(NM_000076.2):c.236G>C,p.(Trp79Ser) | 1 | missense | maternal | - | 24/26 | 28.39 | VUS | SS | This study |
| OGS1380 | 12 | 2 | Pr | chr11:2906527 | CDKN1C(NM_000076.2):c.193T>C,p.(Phe65Leu) | 1 | missense | maternal | - | 23/26 | 28.5 | VUS | SS | This study |
| OGS1774 | 9 | 2 | B | chr11:2906527 | CDKN1C(NM_000076.2):c.193T>C,p.(Phe65Leu) | 1 | missense | maternal | - | 23/26 | 28.5 | VUS | SS | This study |
| OGS1449 | 12 | 3 | Pr | chr11:2906512 | CDKN1C(NM_000076.2):c.208C>A,p.(Pro70Thr) | 1 | missense | maternal | - | 24/26 | 25.6 | VUS | SS | This study |
| OGS1584 | 20 | 4 | S | chr11:2905379 | CDKN1C(NM_000076.2):c.821-15C>A | intron 1 | Non-coding | maternal | - | 1/2 | 19.38 | VUS | SS | This study |
| OGS1775 | 16 | 4 | Pr | chr11:2905379 | CDKN1C(NM_000076.2):c.821-15C>A | intron 1 | Non-coding | maternal | - | 1/2 | 19.38 | VUS | SS | This study |
| OGS2051 | 18 | 5 | Pr | chr11:2906483 | CDKN1C(NM_000076.2):c.237G>A,p.(Trp79Ter) | 1 | nonsense | maternal | - | 8/9 | 37 | P | SS | This study |
| OGS2052 | 12 | 6 | Pr | chr11:2906482 | CDKN1C(NM_000076.2):c.238A>C,p.(Thr80Pro) | 1 | missense | maternal | - | ago-26 | 23.7 | VUS | GP | This study |
| OGS2166 | 17 | 6 | S | chr11:2906482 | CDKN1C(NM_000076.2):c.238A>C,p.(Thr80Pro) | 1 | missense | maternal | - | 8/26 | 23.7 | VUS | GP | This study |
| OGS1492 | 32 | 7 | Pr | chr11:2906141 | CDKN1C(NM_000076.2):c.579del,p.(Pro194GlnfsTer78) | 1 | frameshift | maternal | - | - | - | LP | SS | [24] |
| OGS1493 | 23 | 7 | B | chr11:2906141 | CDKN1C(NM_000076.2):c.579del,p.(Pro194GlnfsTer78) | 1 | frameshift | maternal | - | - | - | LP | SS | [24] |
| OGS1494 | 28 | 7 | S | chr11:2906141 | CDKN1C(NM_000076.2):c.579del,p.(Pro194GlnfsTer78) | 1 | frameshift | maternal | - | - | - | LP | SS | [24] |
| OGS1074 | 14 | 8 | Pr | chr11:2906709 | CDKN1C(NM_000076.2):c.11C>T,p.(Ala4Val) | 1 | missense | maternal | 1/36.514 | 5/24 | 14.67 | VUS | GP | [23] |
| OGS542 | 16 | 9 | Pr | chr11:2906622 | CDKN1C(NM_000076.2):c.98T>A,p.(Leu33His) | 1 | missense | maternal | - | 8/10 | 26.4 | VUS | SS | [23] |
| OGS4 | 24 | 10 | Pr | chr11:2906488 | CDKN1C(NM_000076.2):c.232C>T,p.(Gln78Ter) | 1 | nonsense | de novo | - | 7/9 | 36 | P | SS | [23] |
| OGS851 | 15 | 11 | Pr | chr11:2906026 | CDKN1C(NM_000076.2):c.694C>T,p.(Gln232Ter) | 1 | nonsense | maternal | - | 5/9 | 37 | LP | SS | [23] |
| OGS145 | 19 | 12 | Pr | chr11:2905366 | CDKN1C(NM_000076.2):c.821-2A>G | intron 1 | splicing | maternal | - | 7/9 | 33 | P | SS | [23] |
| OGS1234 | 14 | 13 | Pr | chr11:2905340 | CDKN1C(NM_000076.2):c.845delC,p.(Ser282Ter) | 2 | nonsense | maternal | - | - | - | LP | SS | [23] |
| OGS491 | 20 | 14 | Pr | chr11:2905340 | CDKN1C(NM_000076.2):c.845C>A,p.(Ser282Ter) | 2 | nonsense | maternal | - | 7/23 | 37 | P | SS | [23] |
| OGS1356 | 45 | 15 | Pr | chr11:2905340 | CDKN1C(NM_000076.2):c.845C>G,p.(Ser282Ter) | 2 | nonsense | N/E | - | 7/23 | 36 | LP | SS | [23] |
| Clinical Features | No. Patients | % Patients | ||
|---|---|---|---|---|
| Macroglossia | HP:0000158 | 21 | 100.0 | |
| Omphalocele | HP:0001539 | 13 | 61.9 | |
| Posterior helix pits | HP0008523 | 8 | 38.1 | |
| Anterior earlobe creases | HP:0009908 | 8 | 38.1 | |
| Transient hypoglycaemia | HP:0001998 | 8 | 38.1 | |
| Cleft palate | HP:0000175 | 7 | 33.3 | |
| Overgrowth | HP:0001548 | 7 | 33.3 | |
| Premature birth | HP:0001622 | 6 | 28.6 | (ranging from 29 to 36 weeks) |
| Umbilical hernia | HP:0001537 | 5 | 23.8 | |
| Nevus flammeus | HP:0001052 | 5 | 23.8 | |
| Inguinal hernia | HP:0000023 | 4 | 19.0 | |
| Hemihypertrophy | HP:0001528 | 3 | 14.3 | |
| Thin upper lip vermilion | HP:0000219 | 3 | 14.3 | |
| Coarse facial features | HP:0000280 | 3 | 14.3 | |
| Anterior open-bite malocclusion | HP:0009102 | 3 | 14.3 | |
| Capillary malformation | HP:0025104 | 2 | 9.5 | |
| Hepatomegaly | HP:0002240 | 2 | 9.5 | |
| Postaxial foot polydactyly | HP:0001830 | 2 | 9.5 | |
| Premature tooth eruption | HP:0006288 | 2 | 9.5 | |
| Sensorineural hearing impairment | HP:0000407 | 2 | 9.5 | |
| Supernumerary flexion creases of the fingers | HP:0040064 | 2 | 9.5 | |
| Neoplasm (ganglioneuroma) | HP:0002664 | 1 | 4.8 | |
| Macrocephaly, craniosynostosis, hypotonia, cryptorchidism, hypospadias, thyroglossal cyst, renal cyst, nephromegaly, splenomegaly, haemangioma, prominent upper eyelids, strabismus, apnoea, psoriasiform dermatitis, visceromegaly | 1 | 4.8 | ||
| Clinical Features | Patients and Consensus BWSp Scoring * | ||||||||||||
| OGS1360 | OGS1810 | OGS1380 | OGS1774 | OGS1449 | OGS1584 | OGS1775 | OGS2051 | OGS2052 | OGS2166 | ||||
| Score 5 | Score 5 | Score 4 | Score 3 | Score 4 | Score 8 | Score 5 | Score 6 | Score 5 | Score 5 | ||||
| Macroglossia | HP:0000158 | CF | X | X | X | X | X | X | X | X | X | X | |
| Omphalocele | HP:0001539 | CF | X | X | X | X | |||||||
| Anterior earlobe creases | HP:0009908 | CF | X | X | X | ||||||||
| Overgrowth | HP:0001548 | SF | X | X | X | X | |||||||
| Posterior helix pits | HP0008523 | SF | X | X | |||||||||
| Transient hypoglycaemia | HP:0001998 | SF | X | X | X | X | |||||||
| Umbilical hernia | HP:0001537 | SF | X | X | |||||||||
| Nevus flammeus | HP:0001052 | SF | X | X | X | ||||||||
| Hemihypertrophy | HP:0001528 | SF | X | X | |||||||||
| Hepatomegaly | HP:0002240 | SF | |||||||||||
| Nephromegaly | HP:0000105 | SF | |||||||||||
| Clinical Features | Patients and Consensus BWSp Scoring * | ||||||||||||
| OGS1492 | OGS1493 | OGS1494 | OGS1074 | OGS542 | OGS4 | OGS851 | OGS145 | OGS1234 | OGS491 | OGS1356 | |||
| Score 5 | Score 5 | Score 5 | Score 5 | Score 11 | Score 6 | Score 6 | Score 4 | Score 6 | Score 6 | Score 6 | |||
| Macroglossia | HP:0000158 | CF | X | X | X | X | X | X | X | X | X | X | X |
| Omphalocele | HP:0001539 | CF | X | X | X | X | X | X | X | X | X | ||
| Anterior earlobe creases | HP:0009908 | CF | X | X | X | X | X | ||||||
| Overgrowth | HP:0001548 | SF | X | X | X | X | |||||||
| Posterior helix pits | HP0008523 | SF | X | X | X | X | X | X | |||||
| Transient hypoglycaemia | HP:0001998 | SF | X | X | X | X | |||||||
| Umbilical hernia | HP:0001537 | SF | X | X | X | ||||||||
| Nevus flammeus | HP:0001052 | SF | X | X | |||||||||
| Hemihypertrophy | HP:0001528 | SF | X | ||||||||||
| Hepatomegaly | HP:0002240 | SF | X | X | |||||||||
| Nephromegaly | HP:0000105 | SF | X | ||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardoso, L.C.d.A.; Parra, A.; Gil, C.R.; Arias, P.; Gallego, N.; Romanelli, V.; Kantaputra, P.N.; Lima, L.; Llerena Júnior, J.C.; Arberas, C.; et al. Clinical Spectrum and Tumour Risk Analysis in Patients with Beckwith-Wiedemann Syndrome Due to CDKN1C Pathogenic Variants. Cancers 2022, 14, 3807. https://doi.org/10.3390/cancers14153807
Cardoso LCdA, Parra A, Gil CR, Arias P, Gallego N, Romanelli V, Kantaputra PN, Lima L, Llerena Júnior JC, Arberas C, et al. Clinical Spectrum and Tumour Risk Analysis in Patients with Beckwith-Wiedemann Syndrome Due to CDKN1C Pathogenic Variants. Cancers. 2022; 14(15):3807. https://doi.org/10.3390/cancers14153807
Chicago/Turabian StyleCardoso, Leila Cabral de Almeida, Alejandro Parra, Cristina Ríos Gil, Pedro Arias, Natalia Gallego, Valeria Romanelli, Piranit Nik Kantaputra, Leonardo Lima, Juan Clinton Llerena Júnior, Claudia Arberas, and et al. 2022. "Clinical Spectrum and Tumour Risk Analysis in Patients with Beckwith-Wiedemann Syndrome Due to CDKN1C Pathogenic Variants" Cancers 14, no. 15: 3807. https://doi.org/10.3390/cancers14153807
APA StyleCardoso, L. C. d. A., Parra, A., Gil, C. R., Arias, P., Gallego, N., Romanelli, V., Kantaputra, P. N., Lima, L., Llerena Júnior, J. C., Arberas, C., Guillén-Navarro, E., Nevado, J., Spanish OverGrowth Registry Initiative, Tenorio-Castano, J., & Lapunzina, P. (2022). Clinical Spectrum and Tumour Risk Analysis in Patients with Beckwith-Wiedemann Syndrome Due to CDKN1C Pathogenic Variants. Cancers, 14(15), 3807. https://doi.org/10.3390/cancers14153807