
Emerging Biomarkers and the Changing Landscape of Small Cell Lung Cancer


Abstract
:Simple Summary
Abstract
1. Introduction
2. Pathogenesis
3. Genetics in SCLC

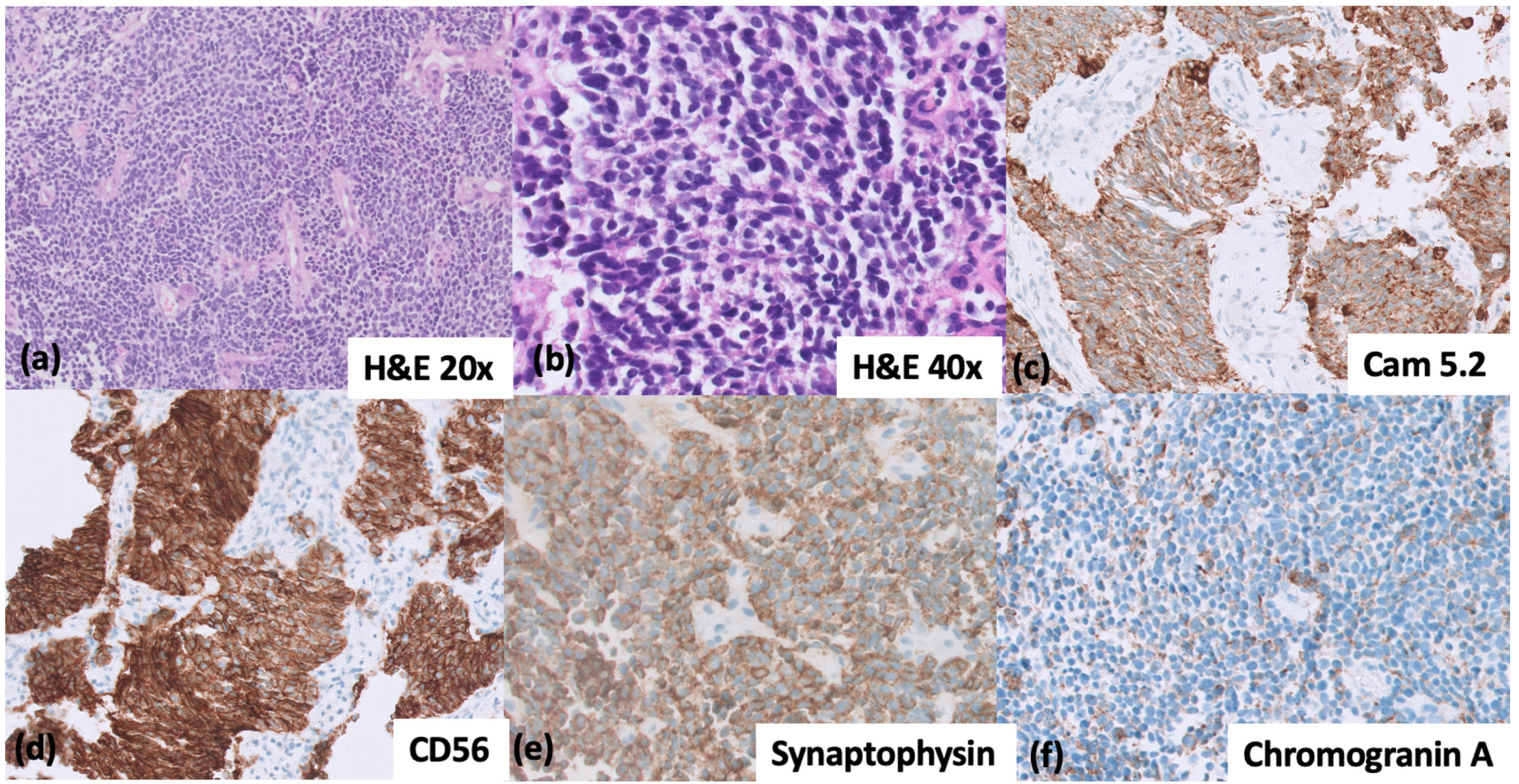
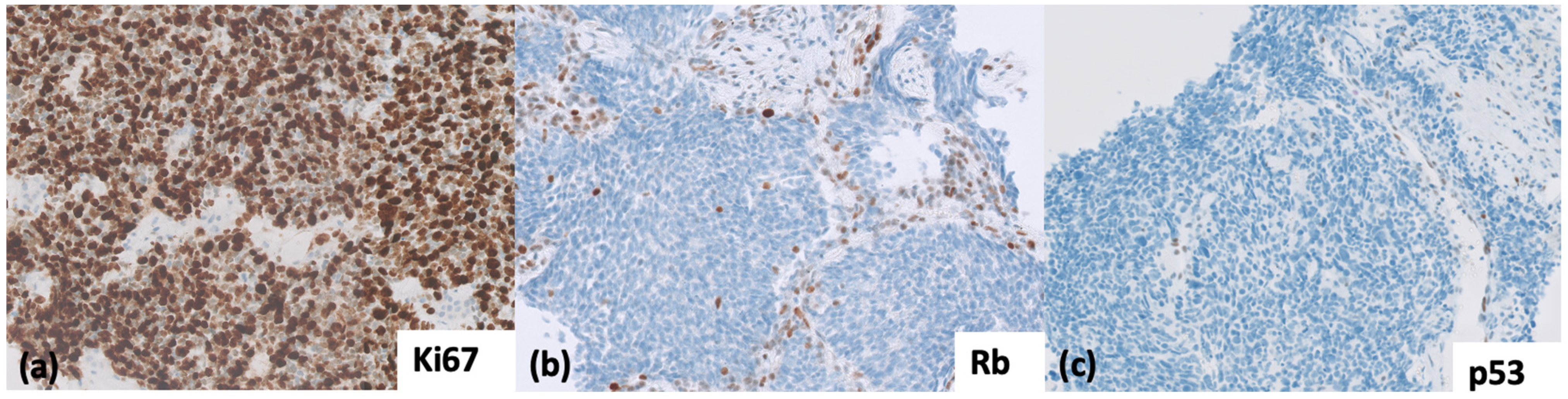
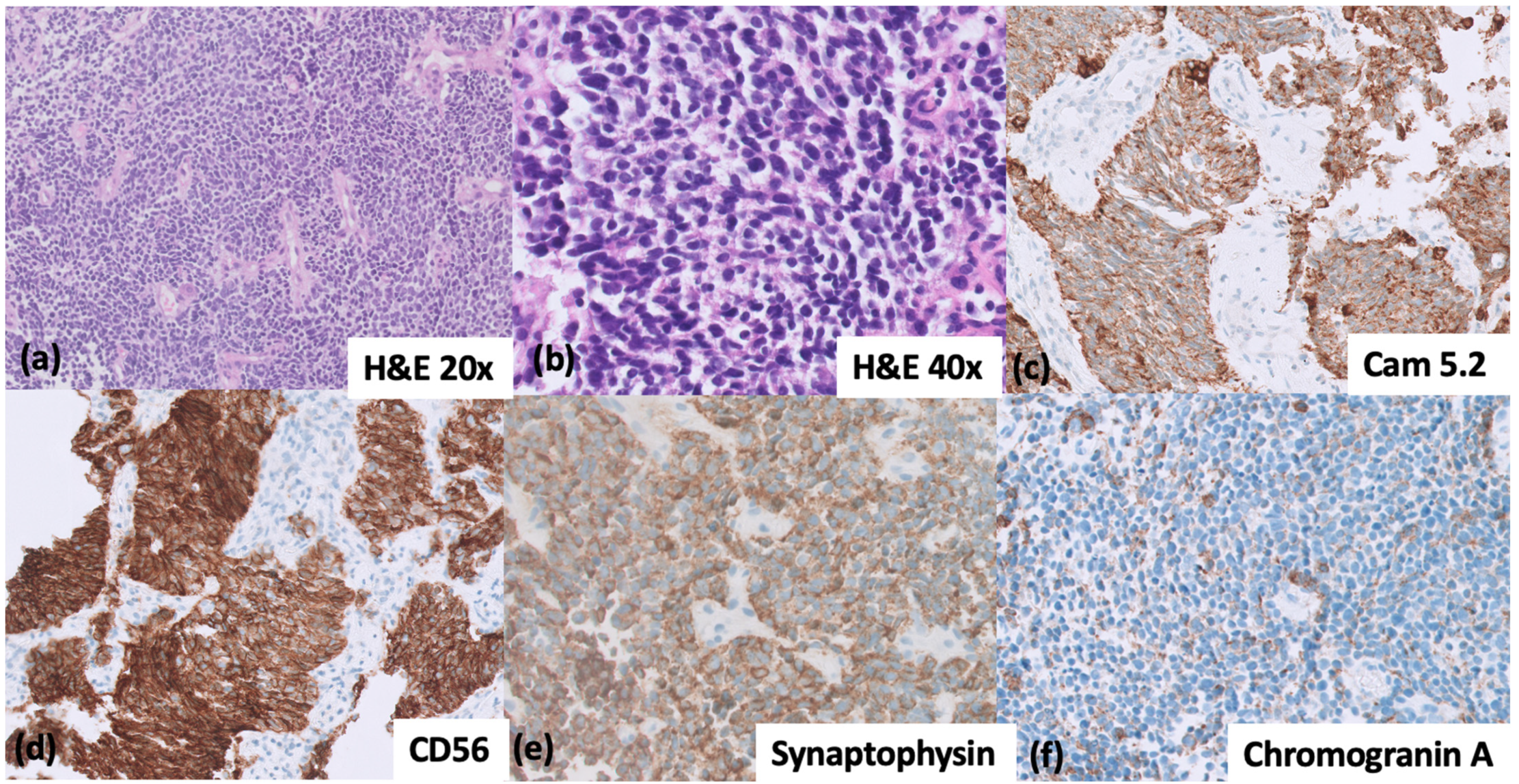{kind=link}
{kind=link}
{kind=link}
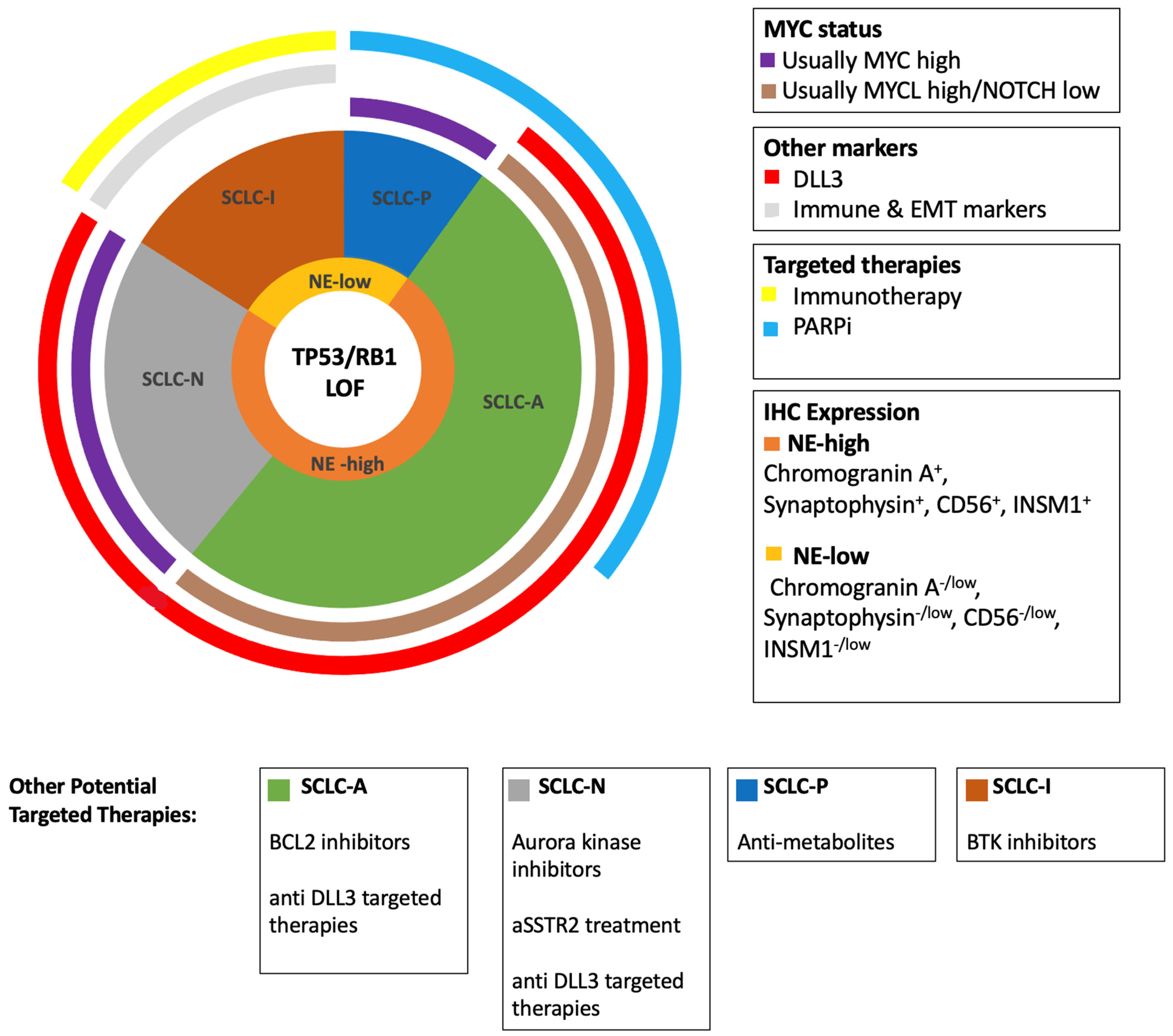{kind=link}
| Gene | Frequency in SCLC (%) | Alteration | Function | Association with SCLC Subtypes; NE Subtypes (SCLC-A, SCLC-N) Low NE Subtypes (SCLC-P, SCLC-I) |
|---|---|---|---|---|
| TP53 | 89 | LOF | TSG; cell cycle regulation; transcription regulation | SCLC-A, SCLC-N, SCLC-P, SCLC-I |
| RB1 | 64 * | LOF | TSG; cell cycle regulation; transcription regulation | SCLC-A, SCLC-N, SCLC-P, SCLC-I |
| KMT2D | 21 | LOF | TSG; epigenetic regulation | - |
| MYC family ** | 19 | Amplification | Oncogene; transcription regulation | SCLC-A, SCLC-N, SCLC-P |
| COL22A1 | 14 | LOF | Cell-cell interaction | - |
| KIAA1211 | 13 | LOF | Epithelial cell integrity | - |
| NOTCH1 | 13 | LOF | TSG; cell-cell signalling | SCLC-A [27] |
| CREBBP | 11 | LOF | TSG; epigenetic regulation | SCLC-A [27] |
| ATRX | 11 | GOF | TSG; cell-cell signalling | - |
| FAT1 | 10 | LOF | TSG; cell-cell signalling | - |
| PIK3CA | 7 | GOF | TSG; PTEN/mTOR signalling pathway | - |
| PTEN | 7 | LOF | TSG; PTEN/mTOR signalling pathway | - |
| NOTCH3 | 7 | LOF | TSG; cell-cell signalling | SCLC-A [27] |
| APC | 6 | LOF | TSG; WNT pathway | Low-NE SCLC [28] |
| AIRD1A | 6 | LOF | TSG; epigenetic regulation | - |
| PTPRD | 6 | LOF | TSG; epigenetic regulation | - |
| EP300 | 6 | LOF | TSG; epigenetic regulation | - |
| NF1 | 4 | LOF | TSG; RAS signalling pathway | - |
| TSC2 | 4 | LOF | TSG; PTEN/mTOR signalling pathway | - |
| EGFR | 4 | GOF | Oncogene; RAS signalling pathway | - |
| KRAS | 3 | GOF | Oncogene; RAS signalling pathway | - |
4. Cell of Origin of SCLC
5. Emerging Subtypes in SCLC
5.1. Neuroendocrine Subtypes
5.2. Low-Neuroendocrine Subtypes
5.3. SCLC with YAP1 Expression
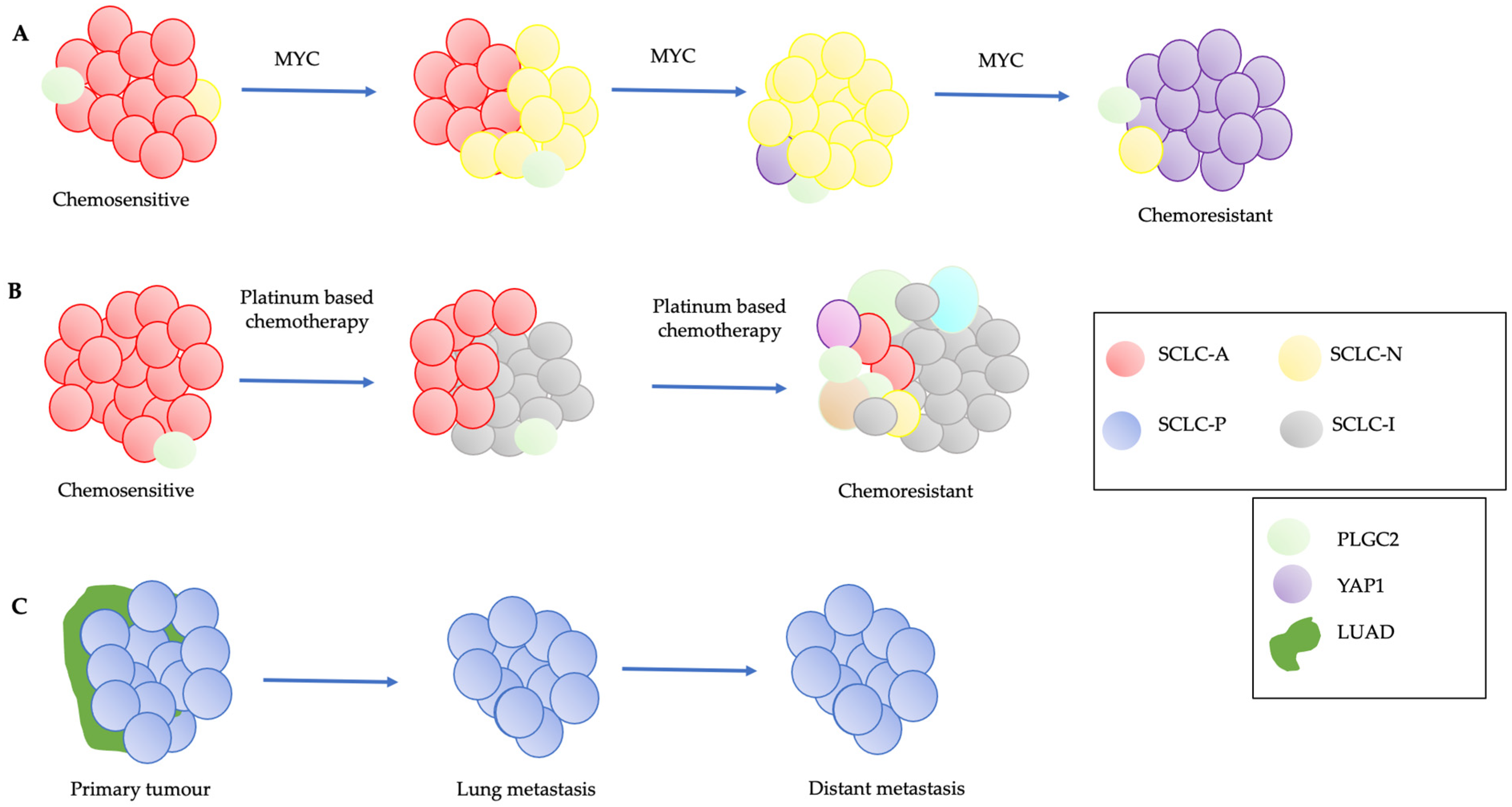
6. SCLC Heterogeneity and Plasticity
6.1. Notch and MYC Signaling Pathways
6.2. Plasticity and Chemoresistance
6.3. SCLC-P and Plasticity
6.4. PLGC2 and Intratumoral Heterogeneity
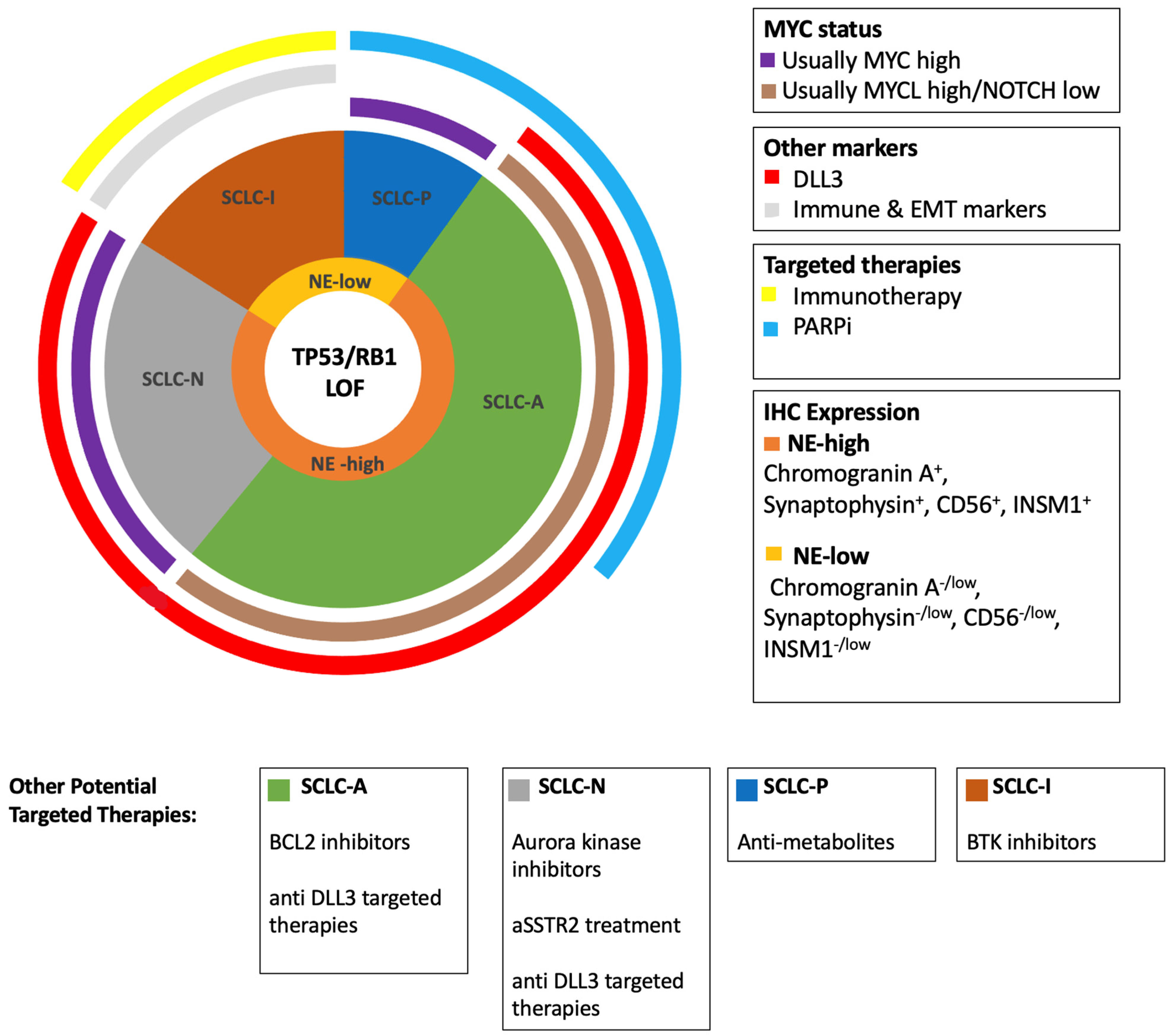
7. Potential Clinical Utility of Emerging Subtypes
7.1. Subtypes as Emerging Diagnostic Biomarkers
7.2. SCLC Subtypes as Prognostic Biomarkers
7.3. Predictive Biomarkers
7.3.1. SCLC Subtypes as Predictive Biomarkers
7.3.2. SCLC-I Predicts Response to Immunotherapy
7.3.3. SLFN11 as a Predictive Biomarker
7.3.4. DLL3 as a Predictive Biomarker
8. Potential Use of Liquid Biopsies in SCLC
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ASCL1 | Achaete–Scute Family BHLH Transcription Factor 1 |
| AT2 | Alveolar Type 2 Cells |
| CCND1 | Cyclin D1 |
| CDX | Cell Line-derived Xenograft |
| CREBBP | CREB Binding Protein |
| CTC | Circulating Tumor Cells |
| DLL3 | Delta-like Ligand 3 |
| EGFR | Epidermal Growth Factor Receptor |
| EMT | Epithelial Mesenchymal Transition |
| FGFR1 | Fibroblast Growth Factor Receptor 1 |
| FHIT | Fragile Histidine Triad Diadenosine Triphosphatase |
| GEMM | Genetically Engineered Mouse Model |
| HLA | Human Leukocyte Antigens |
| IHC | Immunohistochemistry |
| INF-y | Interferon-y |
| INSM1 | Insulinoma-associated Protein 1 |
| IRS2 | Insulin Receptor Substrate 2 |
| ITH | Intratumoral Heterogeneity |
| KMT2D | Lysine Methyltransferase 2D |
| LOH | Loss of Heterozygosity |
| NE | Neuroendocrine |
| NEUROD1 | Neurogenic Differentiation Factor 1 |
| NFIB | Nuclear Factor I B |
| NK cell | Natural Killer Cell |
| NSCLC | Non-small Cell Lung Cancer |
| ORR | Overall Response Rate |
| OS | Overall Survival |
| PARP | Poly (ADP-ribose) Polymerase |
| PARPi | Poly (ADP-ribose) Polymerase Inhibitor |
| PD-L1 | Programmed Death-ligand 1 |
| PLCG2 | Phospholipase C Gamma 2 |
| PNEC | Pulmonary Neuroendocrine Cells |
| POU2F3 | POU Class 2 Homeobox 3 |
| PTEN | Phosphatase and Tensin Homolog |
| RB | Retinoblastoma Protein |
| RB1 | RB Transcriptional Corepressor 1 |
| ROBO1 | Roundabout Guidance Receptor 1 |
| SCLC | Small-cell Lung Cancer |
| SLFN11 | Schlafen Family Member 11 |
| TKI | Tyrosine Kinase Inhibitor |
| TMB | Tumor Mutational Burden |
| TSG | Tumor Suppressor Gene |
| TTF-1 | Thyroid Transcription Factor-1 |
| YAP1 | Yes1 Associated Transcriptional Regulator |
References
- Rudin, C.M.; Brambilla, E.; Faivre-Finn, C.; Sage, J. Small-cell lung cancer. Nat. Rev. Dis. Primers 2021, 7, 3. [Google Scholar] [CrossRef]
- Travis, W.D. Pathology and diagnosis of neuroendocrine tumors: Lung neuroendocrine. Thorac. Surg. Clin. 2014, 24, 257–266. [Google Scholar] [CrossRef]
- Organisation WHO. Thoracic Tumours, 5th ed.; WHO Classification of Tumours Editorial Board: Lyon, France, 2021. [Google Scholar]
- Kalemkerian, G.P.; Loo, B.W.; Akerley, W.; Attia, A.; Bassetti, M.; Boumber, Y.; Decker, R.; Dobelbower, M.C.; Dowlati, A.; Downey, R.J.; et al. NCCN Guidelines Insights: Small Cell Lung Cancer, Version 2.2018. J. Natl. Compr. Cancer Netw. 2018, 16, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Lattuca-Truc, M.; Timsit, J.-F.; Levra, M.G.; Ruckly, S.; Villa, J.; Dumas, I.; Pinsolle, J.; Ferrer, L.; Guillem, P.; Moro-Sibilot, D.; et al. Trends in response rate and survival in small-cell lung cancer patients between 1997 and 2017. Lung Cancer 2019, 131, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Forjaz, G.; Mooradian, M.J.; Meza, R.; Kong, C.Y.; Cronin, K.A.; Mariotto, A.B.; Lowy, D.R.; Feuer, E.J. The Effect of Advances in Lung-Cancer Treatment on Population Mortality. N. Engl. J. Med. 2020, 383, 640–649. [Google Scholar] [CrossRef] [PubMed]
- Horn, L.; Mansfield, A.S.; Szczęsna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Dvorkin, M.; Chen, Y.; Reinmuth, N.; Hotta, K.; Trukhin, D.; Statsenko, G.; Hochmair, M.G.; Özgüroğlu, P.M.; Ji, J.H.; et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): A randomised, controlled, open-label, phase 3 trial. Lancet 2019, 394, 1929–1939. [Google Scholar] [CrossRef]
- Rudin, C.M.; Poirier, J.T.; Byers, L.A.; Dive, C.; Dowlati, A.; George, J.; Heymach, J.V.; Johnson, J.E.; Lehman, J.M.; MacPherson, D.; et al. Molecular subtypes of small cell lung cancer: A synthesis of human and mouse model data. Nat. Rev. Cancer. 2019, 19, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Gay, C.M.; Stewart, C.A.; Park, E.M.; Diao, L.; Groves, S.M.; Heeke, S.; Nabet, B.Y.; Fujimoto, J.; Solis, L.M.; Lu, W.; et al. Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell 2021, 39, 346–360.e7. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Augert, A.; Rongione, M.; Conkrite, K.; Parazzoli, S.; Nikitin, A.Y.; Ingolia, N.; MacPherson, D. PTEN Is a Potent Suppressor of Small Cell Lung Cancer. Mol. Cancer Res. 2014, 12, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Rudin, C.M.; Durinck, S.; Stawiski, E.W.; Poirier, J.; Modrusan, Z.; Shames, D.S.; Bergbower, E.; Guan, Y.; Shin, J.; Guillory, J.; et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat. Genet. 2012, 44, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretić, L.; Kong, G.; Leenders, F.; Lu, X.; Fernández-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, E.; Gazdar, A. Pathogenesis of lung cancer signalling pathways: Roadmap for therapies. Eur. Respir. J. 2009, 33, 1485–1497. [Google Scholar] [CrossRef] [PubMed]
- Varghese, A.M.; Zakowski, M.F.; Yu, H.A.; Won, H.H.; Riely, G.J.; Krug, L.M.; Kris, M.G.; Rekhtman, N.; Ladanyi, M.; Wang, L.; et al. Small-Cell Lung Cancers in Patients Who Never Smoked Cigarettes. J. Thorac. Oncol. 2014, 9, 892–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peifer, M.; Fernandez-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- McBride, O.W.; Merry, D.; Givol, D. The gene for human p53 cellular tumor antigen is located on chromosome 17 short arm (17p13). Proc. Natl. Acad. Sci. USA 1986, 83, 130–134. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Meuwissen, R.; Linn, S.C.; Linnoila, R.; Zevenhoven, J.; Mooi, W.J.; Berns, A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 2003, 4, 181–189. [Google Scholar] [CrossRef] [Green Version]
- Berger, A.H.; Knudson, A.G.; Pandolfi, P.P. A continuum model for tumour suppression. Nature 2011, 476, 163–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sos, M.L.; Dietlein, F.; Peifer, M.; Schöttle, J.; Balke-Want, H.; Müller, C.; Koker, M.; Richters, A.; Heynck, S.; Malchers, F.; et al. A framework for identification of actionable cancer genome dependencies in small cell lung cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 17034–17039. [Google Scholar] [CrossRef] [Green Version]
- Mollaoglu, G.; Guthrie, M.R.; Böhm, S.; Brägelmann, J.; Can, I.; Ballieu, P.M.; Marx, A.; George, J.; Heinen, C.; Chalishazar, M.D.; et al. MYC Drives Progression of Small Cell Lung Cancer to a Variant Neuroendocrine Subtype with Vulnerability to Aurora Kinase Inhibition. Cancer Cell 2017, 31, 270–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gazzeri, S.; Brambilla, E.; Jacrot, M.; Chauvin, C.; Benabid, A.L.; Brambilla, C. Activation of myc gene family in human lung carcinomas and during heterotransplantation into nude mice. Cancer Res. 1991, 51, 2566–2571. [Google Scholar] [PubMed]
- Lim, J.S.; Ibaseta, A.; Fischer, M.M.; Cancilla, B.; O’Young, G.; Cristea, S.; Luca, V.C.; Yang, D.; Jahchan, N.S.; Hamard, C.; et al. Intratumoural heterogeneity generated by Notch signalling promotes small-cell lung cancer. Nature 2017, 545, 360–364. [Google Scholar] [CrossRef] [Green Version]
- Cheng, D.T.; Mitchell, T.N.; Zehir, A.; Shah, R.H.; Benayed, R.; Syed, A.; Chandramohan, R.; Liu, Z.Y.; Won, H.H.; Scott, H.N.; et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J. Mol. Diagn. 2015, 17, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.; Fong, C.; Luthra, A.; Smith, S.A.; DiNatale, R.G.; Nandakumar, S.; Walch, H.; Chatila, W.K.; Madupuri, R.; Kundra, R.; et al. Genomic characterization of metastatic patterns from prospective clinical sequencing of 25,000 patients. Cell 2022, 185, 563–575.e11. [Google Scholar] [CrossRef] [PubMed]
- Schwendenwein, A.; Megyesfalvi, Z.; Barany, N.; Valko, Z.; Bugyik, E.; Lang, C.; Ferencz, B.; Paku, S.; Lantos, A.; Fillinger, J.; et al. Molecular profiles of small cell lung cancer subtypes: Therapeutic implications. Mol. Ther.-Oncolytics 2021, 20, 470–483. [Google Scholar] [CrossRef]
- Wagner, A.H.; Devarakonda, S.; Skidmore, Z.L.; Krysiak, K.; Ramu, A.; Trani, L.; Kunisaki, J.; Masood, A.; Waqar, S.N.; Spies, N.C.; et al. Recurrent WNT pathway alterations are frequent in relapsed small cell lung cancer. Nat. Commun. 2018, 9, 3787. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Rho, J.Y.; Kang, S.; Yoo, K.J.; Choi, H.J. CT findings of small cell lung carcinoma: Can recognizable features be found? Medicine 2016, 95, e5426. [Google Scholar] [CrossRef] [PubMed]
- Ferone, G.; Lee, M.C.; Sage, J.; Berns, A. Cells of origin of lung cancers: Lessons from mouse studies. Genes Dev. 2020, 34, 1017–1032. [Google Scholar] [CrossRef]
- Liu, Y. Small cell lung cancer transformation from EGFR-mutated lung adenocarcinoma: A case report and literatures review. Cancer Biol. Ther. 2018, 19, 445–449. [Google Scholar] [CrossRef]
- Niederst, M.J.; Sequist, L.V.; Poirier, J.T.; Mermel, C.H.; Lockerman, E.L.; Garcia, A.R.; Katayama, R.; Costa, C.; Ross, K.N.; Moran, T.; et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat. Commun. 2015, 6, 6377. [Google Scholar] [CrossRef]
- Sutherland, K.D.; Proost, N.; Brouns, I.; Adriaensen, D.; Song, J.-Y.; Berns, A. Cell of Origin of Small Cell Lung Cancer: Inactivation of Trp53 and Rb1 in Distinct Cell Types of Adult Mouse Lung. Cancer Cell 2011, 19, 754–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Guanizo, A.; Luong, Q.; Jayasekara, W.S.N.; Jayasinghe, D.; Inampudi, C.; Szczepny, A.; Garama, D.J.; Russell, P.A.; Ganju, V.; et al. Lineage-restricted neoplasia driven by Myc defaults to small cell lung cancer when combined with loss of p53 and Rb in the airway epithelium. Oncogene 2021, 41, 138–145. [Google Scholar] [CrossRef]
- Plasschaert, L.W.; Žilionis, R.; Choo-Wing, R.; Savova, V.; Knehr, J.; Roma, G.; Klein, A.M.; Jaffe, A.B. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 2018, 560, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Crapo, J.D.; Barry, B.E.; Gehr, P.; Bachofen, M.; Weibel, E.R. Cell number and cell characteristics of the normal human lung. Am. Rev. Respir. Dis. 1982, 126, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Denny, S.K.; Greenside, P.G.; Chaikovsky, A.C.; Brady, J.J.; Ouadah, Y.; Granja, J.M.; Jahchan, N.S.; Lim, J.S.; Kwok, S.; et al. Intertumoral Heterogeneity in SCLC Is Influenced by the Cell Type of Origin. Cancer Discov. 2018, 8, 1316–1331. [Google Scholar] [CrossRef] [Green Version]
- Burger, M.; Glodek, A.; Hartmann, T.; Schmitt-Gräff, A.; Silberstein, L.E.; Fujii, N.; Kipps, T.J.; Burger, J.A. Functional expression of CXCR4 (CD184) on small-cell lung cancer cells mediates migration, integrin activation, and adhesion to stromal cells. Oncogene 2003, 22, 8093–8101. [Google Scholar] [CrossRef] [Green Version]
- Denny, S.K.; Yang, D.; Chuang, C.-H.; Brady, J.J.; Lim, J.S.; Grüner, B.M.; Chiou, S.-H.; Schep, A.N.; Baral, J.; Hamard, C.; et al. Nfib Promotes Metastasis through a Widespread Increase in Chromatin Accessibility. Cell 2016, 166, 328–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.-H.; Klingbeil, O.; He, X.; Wu, X.S.; Arun, G.; Lu, B.; Somerville, T.D.; Milazzo, J.P.; Wilkinson, J.E.; Demerdash, O.E.; et al. POU2F3 is a master regulator of a tuft cell-like variant of small cell lung cancer. Genes Dev. 2018, 32, 915–928. [Google Scholar] [CrossRef]
- Takashima, T.; Taniyama, D.; Sakamoto, N.; Yasumoto, M.; Asai, R.; Hattori, T.; Honma, R.; Thang, P.Q.; Ukai, S.; Maruyama, R.; et al. Schlafen 11 predicts response to platinum-based chemotherapy in gastric cancers. Br. J. Cancer 2021, 125, 65–77. [Google Scholar] [CrossRef]
- McColl, K.; Wildey, G.; Sakre, N.; Lipka, M.B.; Behtaj, M.; Kresak, A.; Chen, Y.; Yang, M.; Velcheti, V.; Fu, P.; et al. Reciprocal expression of INSM1 and YAP1 defines subgroups in small cell lung cancer. Oncotarget 2017, 8, 73745–73756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baine, M.K.; Hsieh, M.-S.; Lai, W.V.; Egger, J.V.; Jungbluth, A.A.; Daneshbod, Y.; Beras, A.; Spencer, R.; Lopardo, J.; Bodd, F.; et al. SCLC Subtypes Defined by ASCL1, NEUROD1, POU2F3, and YAP1: A Comprehensive Immunohistochemical and Histopathologic Characterization. J. Thorac. Oncol. 2020, 15, 1823–1835. [Google Scholar] [CrossRef] [PubMed]
- Neptune, E.R.; Podowski, M.; Calvi, C.; Cho, J.-H.; Garcia, J.G.N.; Tuder, R.; Linnoila, R.I.; Tsai, M.-J.; Dietz, H.C. Targeted Disruption of NeuroD, a Proneural Basic Helix-Loop-Helix Factor, Impairs Distal Lung Formation and Neuroendocrine Morphology in the Neonatal Lung. J. Biol. Chem. 2008, 283, 21160–21169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borges, M.; Linnoila, R.I.; Van De Velde, H.J.K.; Chen, H.; Nelkin, B.D.; Mabry, M.; Baylin, S.B.; Ball, D.W. An achaete-scute homologue essential for neuroendocrine differentiation in the lung. Nature 1997, 386, 852–855. [Google Scholar] [CrossRef] [PubMed]
- Borromeo, M.D.; Savage, T.K.; Kollipara, R.K.; He, M.; Augustyn, A.; Osborne, J.K.; Girard, L.; Minna, J.D.; Gazdar, A.F.; Cobb, M.H.; et al. ASCL1 and NEUROD1 Reveal Heterogeneity in Pulmonary Neuroendocrine Tumors and Regulate Distinct Genetic Programs. Cell Rep. 2016, 16, 1259–1272. [Google Scholar] [CrossRef] [Green Version]
- Lantuejoul, S.; Fernandez-Cuesta, L.; Damiola, F.; Girard, N.; McLeer, A. New molecular classification of large cell neuroendocrine carcinoma and small cell lung carcinoma with potential therapeutic impacts. Transl. Lung Cancer Res. 2020, 9, 2233–2244. [Google Scholar] [CrossRef]
- Leonetti, A.; Facchinetti, F.; Minari, R.; Cortellini, A.; Rolfo, C.D.; Giovannetti, E.; Tiseo, M. Notch pathway in small-cell lung cancer: From preclinical evidence to therapeutic challenges. Cell. Oncol. 2019, 42, 261–273. [Google Scholar] [CrossRef]
- Rudin, C.M.; Pietanza, M.C.; Bauer, T.M.; Ready, N.; Morgensztern, D.; Glisson, B.S.; Byers, L.A.; Johnson, M.L.; Burris, H.A., III; Robert, F.; et al. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: A first-in-human, first-in-class, open-label, phase 1 study. Lancet Oncol. 2017, 18, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Thiagalingam, A.; Chopra, H.; Borges, M.W.; Feder, J.N.; Nelkin, B.D.; Baylin, S.B.; Ball, D.W. Conservation of the Drosophila lateral inhibition pathway in human lung cancer: A hairy-related protein (HES-1) directly represses achaete-scute homolog-1 expression. Proc. Natl. Acad. Sci. USA 1997, 94, 5355–5360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geffers, I.; Serth, K.; Chapman, G.; Jaekel, R.; Schuster-Gossler, K.; Cordes, R.; Sparrow, D.; Kremmer, E.; Dunwoodie, S.; Klein, T.; et al. Divergent functions and distinct localization of the Notch ligands DLL1 and DLL3 in vivo. J. Cell Biol. 2007, 178, 465–476. [Google Scholar] [CrossRef] [Green Version]
- Saunders, L.R.; Bankovich, A.J.; Anderson, W.C.; Aujay, M.A.; Bheddah, S.; Black, K.; Desai, R.; Escarpe, P.A.; Hampl, J.; Laysang, A.; et al. A DLL3-targeted antibody-drug conjugate eradicates high-grade pulmonary neuroendocrine tumor-initiating cells in vivo. Sci. Transl. Med. 2015, 7, 302ra136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.S.; Yoo, S.; Kong, R.; Sato, T.; Sinha, A.; Karam, S.; Bao, L.; Fridrikh, M.; Emoto, K.; Nudelman, G.; et al. Prototypical oncogene family Myc defines unappreciated distinct lineage states of small cell lung cancer. Sci. Adv. 2021, 7, eabc2578. [Google Scholar] [CrossRef] [PubMed]
- Ireland, A.S.; Micinski, A.M.; Kastner, D.W.; Guo, B.; Wait, S.J.; Spainhower, K.B.; Conley, C.C.; Chen, O.S.; Guthrie, M.R.; Soltero, D.; et al. MYC Drives Temporal Evolution of Small Cell Lung Cancer Subtypes by Reprogramming Neuroendocrine Fate. Cancer Cell 2020, 38, 60–78.e12. [Google Scholar] [CrossRef]
- Stewart, C.A.; Gay, C.M.; Xi, Y.; Sivajothi, S.; Sivakamasundari, V.; Fujimoto, J.; Bolisetty, M.; Hartsfield, P.M.; Balasubramaniyan, V.; Chalishazar, M.D.; et al. Single-cell analyses reveal increased intratumoral heterogeneity after the onset of therapy resistance in small-cell lung cancer. Nat. Cancer 2020, 1, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Howitt, M.R.; Lavoie, S.; Michaud, M.; Blum, A.M.; Tran, S.V.; Weinstock, J.V.; Gallini, C.A.; Redding, K.; Margolskee, R.F.; Osborne, L.C.; et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science 2016, 351, 1329–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baine, M.K.; Febres-Aldana, C.A.; Chang, J.C.; Jungbluth, A.A.; Sethi, S.; Antonescu, C.R.; Travis, W.D.; Hsieh, M.-S.; Roh, M.S.; Homer, R.J.; et al. POU2F3 in SCLC: Clinicopathologic and genomic analysis with a focus on its diagnostic utility in neuroendocrine-low SCLC. J. Thorac. Oncol. 2022. [Google Scholar] [CrossRef]
- Caeser, R.; Egger, J.V.; Chavan, S.; Socci, N.D.; Jones, C.B.; Kombak, F.E.; Asher, M.; Roehrl, M.H.; Shah, N.S.; Allaj, V.; et al. Genomic and transcriptomic analysis of a library of small cell lung cancer patient-derived xenografts. Nat. Commun. 2022, 13, 1–12. [Google Scholar] [CrossRef]
- Owonikoko, T.K.; Dwivedi, B.; Chen, Z.; Zhang, C.; Barwick, B.; Ernani, V.; Zhang, G.; Gilbert-Ross, M.; Carlisle, J.; Khuri, F.R.; et al. YAP1 Expression in SCLC Defines a Distinct Subtype With T-cell–Inflamed Phenotype. J. Thorac. Oncol. 2020, 16, 464–476. [Google Scholar] [CrossRef]
- Song, Y.; Sun, Y.; Lei, Y.; Yang, K.; Tang, R. YAP1 promotes multidrug resistance of small cell lung cancer by CD74-related signaling pathways. Cancer Med. 2019, 9, 259–268. [Google Scholar] [CrossRef]
- Chan, J.M.; Quintanal-Villalonga, A.; Gao, V.R.; Xie, Y.; Allaj, V.; Chaudhary, O.; Masilionis, I.; Egger, J.; Chow, A.; Walle, T.; et al. Signatures of plasticity, metastasis, and immunosuppression in an atlas of human small cell lung cancer. Cancer Cell 2021, 39, 1479–1496.e18. [Google Scholar] [CrossRef]
- Rhee, S.G.; Bae, Y.S. Regulation of Phosphoinositide-specific Phospholipase C Isozymes. J. Biol. Chem. 1997, 272, 15045–15048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Zhao, R.; Yang, W.; Li, C.; Huang, J.; Wen, Z.; Du, G.; Jiang, L. PLCG2 as a potential indicator of tumor microenvironment remodeling in soft tissue sarcoma. Medicine 2021, 100, e25008. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Qin, C.; Gan, H.; Guo, X.; Zhang, L. Construction of an Immunogenomic Risk Score for Prognostication in Colon Cancer. Front. Genet. 2020, 11, 499. [Google Scholar] [CrossRef] [PubMed]
- Rekhtman, N. Lung neuroendocrine neoplasms: Recent progress and persistent challenges. Mod. Pathol. 2021, 35, 36–50. [Google Scholar] [CrossRef]
- Yuan, J.; Knorr, J.; Altmannsberger, M.; Goeckenjan, G.; Ahr, A.; Scharl, A.; Strebhardt, K. Expression of p16 and lack of pRB in primary small cell lung cancer. J Pathol. 1999, 189, 358–362. [Google Scholar] [CrossRef]
- Ding, X.-L.; Su, Y.-G.; Yu, L.; Bai, Z.-L.; Bai, X.-H.; Chen, X.-Z.; Yang, X.; Zhao, R.; He, J.-X.; Wang, Y.-Y. Clinical characteristics and patient outcomes of molecular subtypes of small cell lung cancer (SCLC). World J. Surg. Oncol. 2022, 20, 1–8. [Google Scholar] [CrossRef]
- Hanna, N.H.; Temin, S.; Masters, G. Therapy for Stage IV Non–Small-Cell Lung Cancer Without Driver Alterations: ASCO and OH (CCO) Joint Guideline Update Summary. JCO Oncol. Pract. 2020, 16, e844–e848. [Google Scholar] [CrossRef]
- Pérol, M.; Felip, E.; Dafni, U.; Polito, L.; Pal, N.; Tsourti, Z.; Ton, T.; Merritt, D.; Morris, S.; Stahel, R.; et al. Effectiveness of PD-(L)1 inhibitors alone or in combination with platinum-doublet chemotherapy in first-line (1L) non-squamous non-small-cell lung cancer (Nsq-NSCLC) with PD-L1-high expression using real-world data. Ann. Oncol. 2022, 33, 511–521. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Baas, P.; Kim, D.-W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.-Y.; Molina, J.; Kim, J.-H.; Arvis, C.D.; Ahn, M.-J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2015, 387, 1540–1550. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Nathanson, T.; Rizvi, H.; Creelan, B.C.; Sanchez-Vega, F.; Ahuja, A.; Ni, A.; Novik, J.B.; Mangarin, L.M.; Abu-Akeel, M.; et al. Genomic Features of Response to Combination Immunotherapy in Patients with Advanced Non-Small-Cell Lung Cancer. Cancer Cell 2018, 33, 843–852.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricciuti, B.; Wang, X.; Alessi, J.V.; Rizvi, H.; Mahadevan, N.R.; Li, Y.Y.; Polio, A.; Lindsay, J.; Umeton, R.; Sinha, R.; et al. Association of High Tumor Mutation Burden in Non–Small Cell Lung Cancers with Increased Immune Infiltration and Improved Clinical Outcomes of PD-L1 Blockade Across PD-L1 Expression Levels. JAMA Oncol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.C.; Piha-Paul, S.A.; Lopez-Martin, J.; Schellens, J.H.M.; Kao, S.; Miller, W.H., Jr.; Delord, J.-P.; Gao, B.; Planchard, D.; Gottfried, M.; et al. Pembrolizumab After Two or More Lines of Previous Therapy in Patients with Recurrent or Metastatic SCLC: Results From the KEYNOTE-028 and KEYNOTE-158 Studies. J. Thorac. Oncol. 2020, 15, 618–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.V.; Reck, M.; Mansfield, A.S.; Mok, T.; Scherpereel, A.; Reinmuth, N.; Garassino, M.C.; De Castro Carpeno, J.; Califano, R.; Nishio, M.; et al. Updated Overall Survival and PD-L1 Subgroup Analysis of Patients with Extensive-Stage Small-Cell Lung Cancer Treated with Atezolizumab, Carboplatin, and Etoposide (IMpower133). J. Clin. Oncol. 2021, 39, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Emi, M.; Tanabe, K. Cancer immunoediting from immune surveillance to immune escape. Immunology 2007, 121, 1–14. [Google Scholar] [CrossRef]
- Chen, M.; Chen, R.; Jin, Y.; Li, J.; Hu, X.; Zhang, J.; Fujimoto, J.; Hubert, S.M.; Gay, C.M.; Zhu, B.; et al. Cold and heterogeneous T cell repertoire is associated with copy number aberrations and loss of immune genes in small-cell lung cancer. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Hamilton, G.; Rath, B. Immunotherapy for small cell lung cancer: Mechanisms of resistance. Expert Opin. Biol. Ther. 2019, 19, 423–432. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Keung, M.; Wu, Y.; Vadgama, J. PARP Inhibitors as a Therapeutic Agent for Homologous Recombination Deficiency in Breast Cancers. J. Clin. Med. 2019, 8, 435. [Google Scholar] [CrossRef] [Green Version]
- Allison Stewart, C.; Tong, P.; Cardnell, R.J.; Sen, T.; Li, L.; Gay, C.M.; Masrorpour, F.; Fan, Y.; Bara, R.O.; Feng, Y.; et al. Dynamic variations in epithelial-to-mesenchymal transition (EMT), ATM, and SLFN11 govern response to PARP inhibitors and cisplatin in small cell lung cancer. Oncotarget 2017, 8, 28575–28587. [Google Scholar] [CrossRef] [Green Version]
- Coussy, F.; El-Botty, R.; Château-Joubert, S.; Dahmani, A.; Montaudon, E.; Leboucher, S.; Morisset, L.; Painsec, P.; Sourd, L.; Huguet, L.; et al. BRCAness, SLFN11, and RB1 loss predict response to topoisomerase I inhibitors in triple-negative breast cancers. Sci. Transl. Med. 2020, 12, eaax2625. [Google Scholar] [CrossRef] [Green Version]
- Zoppoli, G.; Regairaz, M.; Leo, E.; Reinhold, W.C.; Varma, S.; Ballestrero, A.; Doroshow, J.H.; Pommier, Y. Putative DNA/RNA helicase Schlafen-11 (SLFN11) sensitizes cancer cells to DNA-damaging agents. Proc. Natl. Acad. Sci. USA 2012, 109, 15030–15035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, L.; Song, S.; Liu, X.; Wang, Y.; Xu, X.; Hu, Y.; Xu, J. Schlafen-11 sensitizes colorectal carcinoma cells to irinotecan. Anti-Cancer Drugs 2014, 25, 1175–1181. [Google Scholar] [CrossRef] [PubMed]
- Rathkey, D.; Khanal, M.; Murai, J.; Zhang, J.; Sengupta, M.; Jiang, Q.; Morrow, B.; Evans, C.N.; Chari, R.; Fetsch, P.; et al. Sensitivity of Mesothelioma Cells to PARP Inhibitors Is Not Dependent on BAP1 but Is Enhanced by Temozolomide in Cells with High-Schlafen 11 and Low-O6-methylguanine-DNA Methyltransferase Expression. J. Thorac. Oncol. 2020, 15, 843–859. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Ramkumar, K.; Cardnell, R.J.; Gay, C.M.; Stewart, C.A.; Wang, W.-L.; Fujimoto, J.; Wistuba, I.I.; Byers, L.A. A wake-up call for cancer DNA damage: The role of Schlafen 11 (SLFN11) across multiple cancers. Br. J. Cancer 2021, 125, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.-Y.N.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP Trapping by BMN 673 and Comparison with Olaparib and Rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Branzei, D.; Foiani, M. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 2010, 11, 208–219. [Google Scholar] [CrossRef]
- Murai, J.; Feng, Y.; Yu, G.K.; Ru, Y.; Tang, S.-W.; Shen, Y.; Pommier, Y. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget 2016, 7, 76534–76550. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Huang, S.-Y.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps17. [Google Scholar] [CrossRef] [PubMed]
- Pietanza, M.C.; Waqar, S.N.; Krug, L.M.; Dowlati, A.; Hann, C.L.; Chiappori, A.; Owonikoko, T.K.; Woo, K.M.; Cardnell, R.J.; Fujimoto, J.; et al. Randomized, Double-Blind, Phase II Study of Temozolomide in Combination with Either Veliparib or Placebo in Patients with Relapsed-Sensitive or Refractory Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 2386–2394. [Google Scholar] [CrossRef]
- Trigo, J.; Subbiah, V.; Besse, B.; Moreno, V.; López, R.; Sala, M.A.; Peters, S.; Ponce, S.; Fernández, C.; Alfaro, V.; et al. Lurbinectedin as second-line treatment for patients with small-cell lung cancer: A single-arm, open-label, phase 2 basket trial. Lancet Oncol. 2020, 21, 645–654. [Google Scholar] [CrossRef]
- Kundu, K.; Cardnell, R.J.; Zhang, B.; Shen, L.; Stewart, C.A.; Ramkumar, K.; Cargill, K.R.; Wang, J.; Gay, C.M.; Byers, L.A. SLFN11 biomarker status predicts response to lurbinectedin as a single agent and in combination with ATR inhibition in small cell lung cancer. Transl. Lung Cancer Res. 2021, 10, 4095–4105. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Stewart, C.A.; Wang, Q.; Cardnell, R.J.; Rocha, P.; Fujimoto, J.; Soto, L.M.S.; Wang, R.; Novegil, V.; Ansell, P.; et al. Dynamic expression of Schlafen 11 (SLFN11) in circulating tumour cells as a liquid biomarker in small cell lung cancer. Br. J. Cancer 2022, 127, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Isse, K.; Fujihira, T.; Takenoyama, M.; Saunders, L.; Bheddah, S.; Nakanishi, Y.; Okamoto, I. Prevalence of Delta-like protein 3 expression in patients with small cell lung cancer. Lung Cancer 2017, 115, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Taniguchi, K.; Hamamoto, H.; Inomata, Y.; Komura, K.; Tanaka, T.; Lee, S.; Uchiyama, K. Delta-like canonical Notch ligand 3 as a potential therapeutic target in malignancies: A brief overview. Cancer Sci. 2021, 112, 2984–2992. [Google Scholar] [CrossRef] [PubMed]
- Hipp, S.; Voynov, V.; Drobits-Handl, B.; Giragossian, C.; Trapani, F.; Nixon, A.E.; Scheer, J.M.; Adam, P.J. A Bispecific DLL3/CD3 IgG-Like T-Cell Engaging Antibody Induces Antitumor Responses in Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 5258–5268. [Google Scholar] [CrossRef] [PubMed]
- Tully, K.M.; Tendler, S.; Carter, L.M.; Sharma, S.K.; Samuels, Z.V.; Mandleywala, K.; Korsen, J.A.; Reyes, A.M.D.; Piersigilli, A.; Travis, W.D.; et al. Radioimmunotherapy Targeting Delta-like Ligand 3 in Small Cell Lung Cancer Exhibits Antitumor Efficacy with Low Toxicity. Clin. Cancer Res. 2022, 28, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.-M.; Greystoke, A.; Lancashire, L.; Cummings, J.; Ward, T.; Board, R.; Amir, E.; Hughes, S.; Krebs, M.; Hughes, A.; et al. Evaluation of Circulating Tumor Cells and Serological Cell Death Biomarkers in Small Cell Lung Cancer Patients Undergoing Chemotherapy. Am. J. Pathol. 2009, 175, 808–816. [Google Scholar] [CrossRef] [Green Version]
- Pantel, K.; Speicher, M. The biology of circulating tumor cells. Oncogene 2015, 35, 1216–1224. [Google Scholar] [CrossRef]
- Hou, J.-M.; Krebs, M.G.; Lancashire, L.; Sloane, R.; Backen, A.; Swain, R.K.; Priest, L.J.C.; Greystoke, A.; Zhou, C.; Morris, K.; et al. Clinical Significance and Molecular Characteristics of Circulating Tumor Cells and Circulating Tumor Microemboli in Patients with Small-Cell Lung Cancer. J. Clin. Oncol. 2012, 30, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Tay, R.; Fernández-Gutiérrez, F.; Foy, V.; Burns, K.; Pierce, J.; Morris, K.; Priest, L.; Tugwood, J.; Ashcroft, L.; Lindsay, C.; et al. Prognostic value of circulating tumour cells in limited-stage small-cell lung cancer: Analysis of the concurrent once-daily versus twice-daily radiotherapy (CONVERT) randomised controlled trial. Ann. Oncol. 2019, 30, 1114–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keogh, A.; Finn, S.; Radonic, T. Emerging Biomarkers and the Changing Landscape of Small Cell Lung Cancer. Cancers 2022, 14, 3772. https://doi.org/10.3390/cancers14153772
Keogh A, Finn S, Radonic T. Emerging Biomarkers and the Changing Landscape of Small Cell Lung Cancer. Cancers. 2022; 14(15):3772. https://doi.org/10.3390/cancers14153772
Chicago/Turabian StyleKeogh, Anna, Stephen Finn, and Teodora Radonic. 2022. "Emerging Biomarkers and the Changing Landscape of Small Cell Lung Cancer" Cancers 14, no. 15: 3772. https://doi.org/10.3390/cancers14153772
APA StyleKeogh, A., Finn, S., & Radonic, T. (2022). Emerging Biomarkers and the Changing Landscape of Small Cell Lung Cancer. Cancers, 14(15), 3772. https://doi.org/10.3390/cancers14153772