
Molecular Pathways and Genomic Landscape of Glioblastoma Stem Cells: Opportunities for Targeted Therapy

 , ,
, ,  , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
2. Features of Glioblastoma Stem Cells
3. Interactions with Tumor Microenvironment
3.1. Stem Cell Niches
3.2. Hypoxia Signaling
3.3. Metabolic Environment
4. Signal Transduction Pathways
4.1. Notch
4.2. Epidermal Growth Factor Receptor
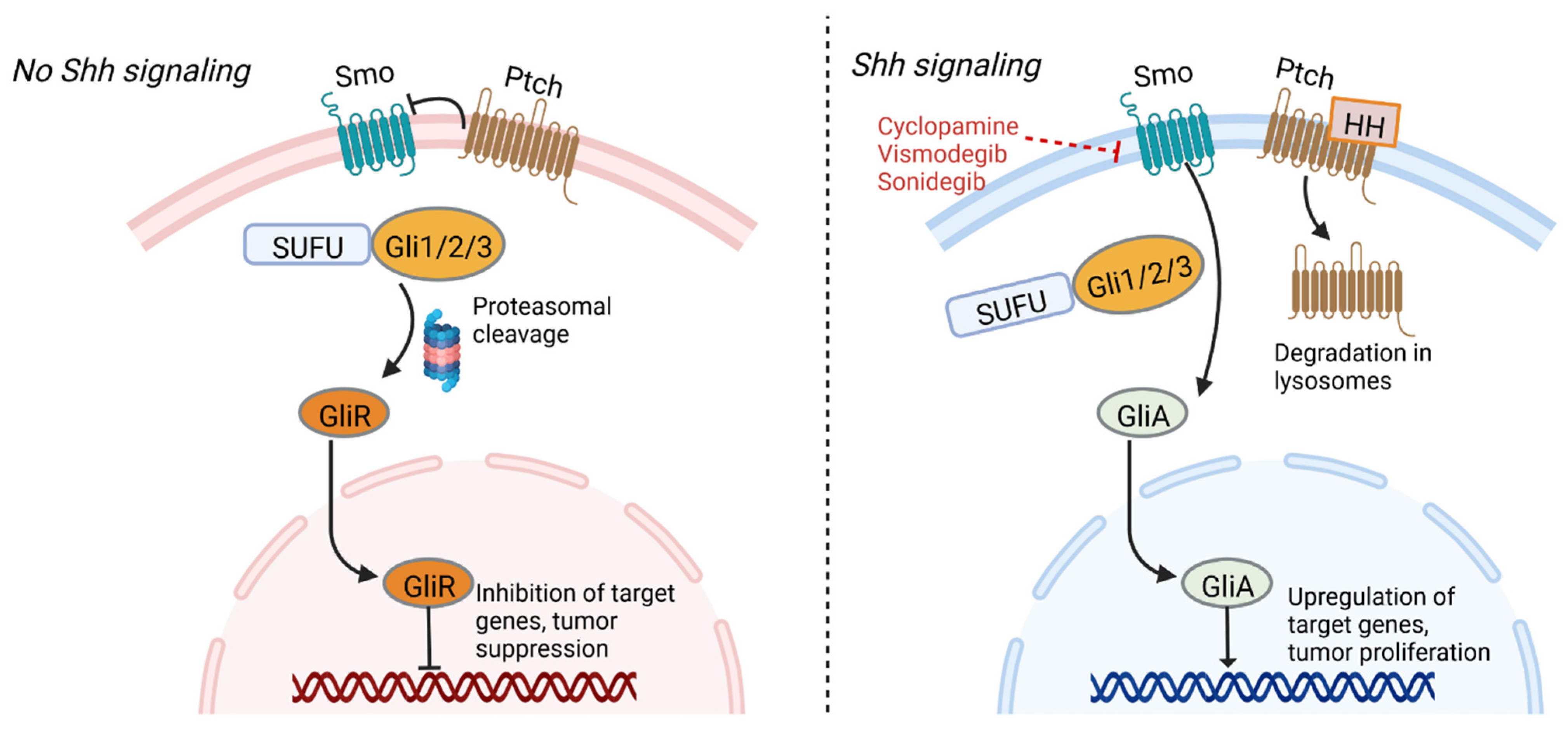
4.3. Sonic Hedgehog
4.4. Transforming Growth Factor Beta
4.5. Wnt
4.6. Signal Transducer and Activator of Transcription 3
4.7. Inhibitors of Differentiation
5. Epigenetic Regulation
5.1. DNA Methylation
5.2. Histone Post-translational Modifications
5.3. MicroRNA
6. Resistance Mechanisms to Therapy
6.1. Chemoresistance
6.2. Radioresistance
7. Targeted Therapy
7.1. Targeting Signaling Pathways
7.2. Targeting the Epigenome
7.3. Targeting the Tumor Microenvironment
7.4. Challenges
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APC | Adenomatous polyposis coli |
| CNS | Central nervous system |
| CSC | Cancer stem cell |
| EGFR | Epidermal growth factor receptor |
| EZH2 | Enhancer of Zeste Homolog 2 |
| FDA | Food and Drug Administration |
| GBM | Glioblastoma multiforme |
| GSC | Glioblastoma stem-like cell |
| HDAC | Histone deacetylase |
| HIF | Hypoxia inducible factor |
| JAK | Janus family kinase |
| MGMT | O6-Methylguanine-DNA methyltransferase |
| MiRNA | MicroRNA |
| MTOR | Mammalian target of rapamycin |
| NICD | Notch intracellular domain |
| NSC | Neural stem cell |
| PI3K | Phosphatidylinositol 3-kinase |
| ROS | Reactive oxygen species |
| RT | Radiation therapy |
| SHH | Sonic hedgehog |
| STAT3 | Signal transducer and activator of transcription 3 |
| TGF-β | Transforming growth factor beta |
| TMZ | Temozolomide |
References
- Omuro, A.; DeAngelis, L.M. Glioblastoma and Other Malignant Gliomas: A Clinical Review. JAMA 2013, 310, 1842–1850. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011–2015. Neuro-Oncology 2018, 20, iv1–iv86. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, C.; Nandhabalan, M.; Murray, S.A.; Plaha, P. Glioblastoma: Clinical Presentation, Diagnosis, and Management. BMJ 2021, 374, n1560. [Google Scholar] [CrossRef]
- Delgado-Lopez, P.D.; Corrales-Garcia, E.M. Survival in Glioblastoma: A Review on the Impact of Treatment Modalities. Clin. Transl. Oncol. 2016, 18, 1062–1071. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Komori, T. Grading of Adult Diffuse Gliomas According to the 2021 WHO Classification of Tumors of the Central Nervous System. Lab. Investig. 2021, 102, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-Cell RNA-Seq Highlights Intratumoral Heterogeneity in Primary Glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Matchett, K.B.; Lappin, T.R. Concise Reviews: Cancer Stem Cells: From Concept to Cure. Stem Cells 2014, 32, 2563–2570. [Google Scholar] [CrossRef]
- Li, X.; Wu, C.; Chen, N.; Gu, H.; Yen, A.; Cao, L.; Wang, E.; Wang, L. PI3K/Akt/MTOR Signaling Pathway and Targeted Therapy for Glioblastoma. Oncotarget 2016, 7, 33440. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wu, Q.; Guryanova, O.A.; Huang, Z.; Huang, Q.; Rich, J.N.; Bao, S. Elevated Invasive Potential of Glioblastoma Stem Cells. Biochem. Biophys. Res. Commun. 2011, 406, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Jhanwar-Uniyal, M.; Labagnara, M.; Friedman, M.; Kwasnicki, A.; Murali, R. Glioblastoma: Molecular Pathways, Stem Cells and Therapeutic Targets. Cancers 2015, 7, 538–555. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Grigore, F.; Chen, C.C.; Li, M. Selfrenewal Signaling Pathways and Differentiation Therapies of Glioblastoma Stem Cells (Review). Int. J. Oncol. 2021, 59, 45. [Google Scholar] [CrossRef] [PubMed]
- Schonberg, D.L.; Lubelski, D.; Miller, T.E.; Rich, J.N. Brain Tumor Stem Cells: Molecular Characteristics and Their Impact on Therapy. Mol. Asp. Med. 2014, 39, 82–101. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer Stem Cells in Glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Wallenborn, M.; Xu, L.X.; Kirsten, H.; Rohani, L.; Rudolf, D.; Ahnert, P.; Schmidt, C.; Schulz, R.M.; Richter, M.; Krupp, W.; et al. Molecular Analyses of Glioblastoma Stem-like Cells and Glioblastoma Tissue. PLoS ONE 2020, 15, e0234986. [Google Scholar] [CrossRef]
- Biserova, K.; Jakovlevs, A.; Uljanovs, R.; Strumfa, I. Cancer Stem Cells: Significance in Origin, Pathogenesis and Treatment of Glioblastoma. Cells 2021, 10, 621. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of Human Brain Tumour Initiating Cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma Stem Cells Promote Radioresistance by Preferential Activation of the DNA Damage Response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Becher, O.J.; Rosenblum, M.K.; Pandolfi, P.P.; Manova-Todorova, K.; Holland, E.C. PI3K Pathway Regulates Survival of Cancer Stem Cells Residing in the Perivascular Niche Following Radiation in Medulloblastoma in Vivo. Genes Dev. 2008, 22, 436–448. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A Restricted Cell Population Propagates Glioblastoma Growth after Chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of Gene Expression and Chemoresistance of CD133+ Cancer Stem Cells in Glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular Subclasses of High-Grade Glioma Predict Prognosis, Delineate a Pattern of Disease Progression, and Resemble Stages in Neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Song, T.; Yang, L.; Chen, R.; Wu, L.; Yang, Z.; Fang, J. Nestin and CD133: Valuable Stem Cell-Specific Markers for Determining Clinical Outcome of Glioma Patients. J. Exp. Clin. Cancer Res. 2008, 27, 85. [Google Scholar] [CrossRef]
- Bexell, D.; Gunnarsson, S.; Siesjo, P.; Bengzon, J.; Darabi, A. CD133+ and Nestin+ Tumor-Initiating Cells Dominate in N29 and N32 Experimental Gliomas. Int. J. Cancer 2009, 125, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Tchoghandjian, A.; Baeza, N.; Colin, C.; Cayre, M.; Metellus, P.; Beclin, C.; Ouafik, L.; Figarella-Branger, D. A2B5 Cells from Human Glioblastoma Have Cancer Stem Cell Properties. Brain Pathol. 2010, 20, 211–221. [Google Scholar] [CrossRef]
- Ge, Y.; Zhou, F.; Chen, H.; Cui, C.; Liu, D.; Li, Q.; Yang, Z.; Wu, G.; Sun, S.; Gu, J.; et al. Sox2 Is Translationally Activated by Eukaryotic Initiation Factor 4E in Human Glioma-Initiating Cells. Biochem. Biophys. Res. Commun. 2010, 397, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Hagerstrand, D.; He, X.; Bradic Lindh, M.; Hoefs, S.; Hesselager, G.; Ostman, A.; Nister, M. Identification of a SOX2-Dependent Subset of Tumor- and Sphere-Forming Glioblastoma Cells with a Distinct Tyrosine Kinase Inhibitor Sensitivity Profile. Neuro-Oncology 2011, 13, 1178–1191. [Google Scholar] [CrossRef]
- Xie, Y.; Sundstrom, A.; Maturi, N.P.; Tan, E.J.; Marinescu, V.D.; Jarvius, M.; Tirfing, M.; Jin, C.; Chen, L.; Essand, M.; et al. LGR5 Promotes Tumorigenicity and Invasion of Glioblastoma Stem-like Cells and Is a Potential Therapeutic Target for a Subset of Glioblastoma Patients. J. Pathol. 2019, 247, 228–240. [Google Scholar] [CrossRef]
- Rusu, P.; Shao, C.; Neuerburg, A.; Acikgoz, A.A.; Wu, Y.; Zou, P.; Phapale, P.; Shankar, T.S.; Doring, K.; Dettling, S.; et al. GPD1 Specifically Marks Dormant Glioma Stem Cells with a Distinct Metabolic Profile. Cell Stem Cell 2019, 25, 241–257.e8. [Google Scholar] [CrossRef] [PubMed]
- Prager, B.C.; Bhargava, S.; Mahadev, V.; Hubert, C.G.; Rich, J.N. Glioblastoma Stem Cells: Driving Resilience through Chaos. Trends Cancer 2020, 6, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.J.; Diksin, M.; Chhaya, S.; Sairam, S.; Estevez-Cebrero, M.A.; Rahman, R. The Invasive Region of Glioblastoma Defined by 5ALA Guided Surgery Has an Altered Cancer Stem Cell Marker Profile Compared to Central Tumour. Int. J. Mol. Sci. 2017, 18, 2452. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, M.; Inoue, A.; Ohnishi, T.; Kohno, S.; Ohue, S.; Matsumoto, S.; Suehiro, S.; Yamashita, D.; Ozaki, S.; Watanabe, H.; et al. Significance of Glioma Stem-Like Cells in the Tumor Periphery That Express High Levels of CD44 in Tumor Invasion, Early Progression, and Poor Prognosis in Glioblastoma. Stem Cells Int. 2018, 2018, 5387041. [Google Scholar] [CrossRef]
- Spinelli, C.; Montermini, L.; Meehan, B.; Brisson, A.R.; Tan, S.; Choi, D.; Nakano, I.; Rak, J. Molecular Subtypes and Differentiation Programmes of Glioma Stem Cells as Determinants of Extracellular Vesicle Profiles and Endothelial Cell-Stimulating Activities. J. Extracell. Vesicles 2018, 7, 1490144. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dube, C.; Gibert, M.; Cruickshanks, N.; Wang, B.; Coughlan, M.; Yang, Y.; Setiady, I.; Deveau, C.; Saoud, K.; et al. The P53 Pathway in Glioblastoma. Cancers 2018, 10, 297. [Google Scholar] [CrossRef] [PubMed]
- Lindström, M.S.; Hede, S.M.; Nazarenko, I.; Nistér, M. Novel Perspectives on P53 Function in Neural Stem Cells and Brain Tumors. J. Oncol. 2011, 2011, 852970. [Google Scholar] [CrossRef]
- Ghatak, D.; das Ghosh, D.; Roychoudhury, S. Cancer Stemness: P53 at the Wheel. Front. Oncol. 2021, 10, 2910. [Google Scholar] [CrossRef] [PubMed]
- Crivii, C.B.; Boșca, A.B.; Melincovici, C.S.; Constantin, A.M.; Mărginean, M.; Dronca, E.; Suflețel, R.; Gonciar, D.; Bungărdean, M.; Șovrea, A. Glioblastoma Microenvironment and Cellular Interactions. Cancers 2022, 14, 1092. [Google Scholar] [CrossRef] [PubMed]
- Menna, G.; Manini, I.; Cesselli, D.; Skrap, M.; Olivi, A.; Ius, T.; Pepa, G.M.D. Immunoregulatory Effects of Glioma-Associated Stem Cells on the Glioblastoma Peritumoral Microenvironment: A Differential PD-L1 Expression from Core to Periphery? Neurosurg. Focus 2022, 52, E4. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, D.; Annovazzi, L.; Casalone, C.; Corona, C.; Mellai, M. Glioblastoma: Microenvironment and Niche Concept. Cancers 2019, 11, 5. [Google Scholar] [CrossRef] [PubMed]
- Seano, G.; Jain, R.K. Vessel Co-Option in Glioblastoma: Emerging Insights and Opportunities. Angiogenesis 2020, 23, 9–16. [Google Scholar] [CrossRef]
- Rodriguez, S.M.B.; Staicu, G.A.; Sevastre, A.S.; Baloi, C.; Ciubotaru, V.; Dricu, A.; Tataranu, L.G. Glioblastoma Stem Cells-Useful Tools in the Battle against Cancer. Int. J. Mol. Sci. 2022, 23, 4602. [Google Scholar] [CrossRef] [PubMed]
- Rath, B.H.; Fair, J.M.; Jamal, M.; Camphausen, K.; Tofilon, P.J. Astrocytes Enhance the Invasion Potential of Glioblastoma Stem-Like Cells. PLoS ONE 2013, 8, 54752. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Nguyen, H.P.T.; Jones, J.J.; Stylli, S.S.; Whitehead, C.A.; Paradiso, L.; Luwor, R.B.; Areeb, Z.; Hanssen, E.; Cho, E.; et al. Extracellular Vesicles Secreted by Glioma Stem Cells Are Involved in Radiation Resistance and Glioma Progression. Int. J. Mol. Sci. 2022, 23, 2770. [Google Scholar] [CrossRef]
- Lane, R.; Simon, T.; Vintu, M.; Solkin, B.; Koch, B.; Stewart, N.; Benstead-Hume, G.; Pearl, F.M.G.; Critchley, G.; Stebbing, J.; et al. Cell-Derived Extracellular Vesicles Can Be Used as a Biomarker Reservoir for Glioblastoma Tumor Subtyping. Commun. Biol. 2019, 2, 315. [Google Scholar] [CrossRef] [PubMed]
- Yekula, A.; Yekula, A.; Muralidharan, K.; Kang, K.; Carter, B.S.; Balaj, L. Extracellular Vesicles in Glioblastoma Tumor Microenvironment. Front. Immunol. 2019, 10, 3137. [Google Scholar] [CrossRef] [PubMed]
- Boyd, N.H.; Tran, A.N.; Bernstock, J.D.; Etminan, T.; Jones, A.B.; Gillespie, G.Y.; Friedman, G.K.; Hjelmeland, A.B. Glioma Stem Cells and Their Roles within the Hypoxic Tumor Microenvironment. Theranostics 2021, 11, 665–683. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for Cancer Therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Seidel, S.; Garvalov, B.K.; Wirta, V.; von Stechow, L.; Schanzer, A.; Meletis, K.; Wolter, M.; Sommerlad, D.; Henze, A.T.; Nister, M.; et al. A Hypoxic Niche Regulates Glioblastoma Stem Cells through Hypoxia Inducible Factor 2 Alpha. Brain 2010, 133, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-Inducible Factors and the Response to Hypoxic Stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, H.; Eyler, C.E.; Hjelmeland, A.B.; Rich, J.N. Turning Cancer Stem Cells inside out: An Exploration of Glioma Stem Cell Signaling Pathways. J. Biol. Chem. 2009, 284, 16705–16709. [Google Scholar] [CrossRef] [PubMed]
- Heddleston, J.M.; Li, Z.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. The Hypoxic Microenvironment Maintains Glioblastoma Stem Cells and Promotes Reprogramming towards a Cancer Stem Cell Phenotype. Cell Cycle 2009, 8, 3274–3284. [Google Scholar] [CrossRef]
- Bache, M.; Rot, S.; Kessler, J.; Güttler, A.; Wichmann, H.; Greither, T.; Wach, S.; Taubert, H.; Söling, A.; Bilkenroth, U.; et al. MRNA Expression Levels of Hypoxia-Induced and Stem Cell-Associated Genes in Human Glioblastoma. Oncol. Rep. 2015, 33, 3155–3161. [Google Scholar] [CrossRef]
- Soeda, A.; Park, M.; Lee, D.; Mintz, A.; Androutsellis-Theotokis, A.; McKay, R.D.; Engh, J.; Iwama, T.; Kunisada, T.; Kassam, A.B.; et al. Hypoxia Promotes Expansion of the CD133-Positive Glioma Stem Cells through Activation of HIF-1alpha. Oncogene 2009, 28, 3949–3959. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Wu, T.; Zhang, H.W.; Lu, N.; Hu, R.; Wang, Y.J.; Zhao, L.; Chen, F.H.; Wang, X.T.; You, Q.D.; et al. HIF-1alpha Is Critical for Hypoxia-Mediated Maintenance of Glioblastoma Stem Cells by Activating Notch Signaling Pathway. Cell Death Differ. 2012, 19, 284–294. [Google Scholar] [CrossRef]
- Lee, G.; Auffinger, B.; Guo, D.; Hasan, T.; Deheeger, M.; Tobias, A.L.; Kim, J.Y.; Atashi, F.; Zhang, L.; Lesniak, M.S.; et al. Dedifferentiation of Glioma Cells to Glioma Stem-like Cells By Therapeutic Stress-Induced HIF Signaling in the Recurrent GBM Model. Mol. Cancer Ther. 2016, 15, 3064–3076. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.Y.; Fu, L.A.; Li, S.Z.; Chen, Y.; Li, J.C.; Han, J.; Liang, L.; Li, L.; Ji, C.C.; Zheng, M.H.; et al. Hif-1alpha and Hif-2alpha Differentially Regulate Notch Signaling through Competitive Interaction with the Intracellular Domain of Notch Receptors in Glioma Stem Cells. Cancer Lett. 2014, 349, 67–76. [Google Scholar] [CrossRef]
- Kim, Y.H.; Yoo, K.C.; Cui, Y.H.; Uddin, N.; Lim, E.J.; Kim, M.J.; Nam, S.Y.; Kim, I.G.; Suh, Y.; Lee, S.J. Radiation Promotes Malignant Progression of Glioma Cells through HIF-1alpha Stabilization. Cancer Lett. 2014, 354, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Nusblat, L.M.; Tanna, S.; Roth, C.M. Gene Silencing of HIF-2alpha Disrupts Glioblastoma Stem Cell Phenotype. Cancer Drug Resist. 2020, 3, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Strickland, M.; Stoll, E.A. Metabolic Reprogramming in Glioma. Front. Cell Dev. Biol. 2017, 5, 43. [Google Scholar] [CrossRef] [PubMed]
- Garnier, D.; Renoult, O.; Alves-Guerra, M.C.; Paris, F.; Pecqueur, C. Glioblastoma Stem-Like Cells, Metabolic Strategy to Kill a Challenging Target. Front. Oncol. 2019, 9, 118. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.M.; Yu, J.S. Metabolic Regulation of Glioma Stem-like Cells in the Tumor Micro-Environment. Cancer Lett. 2017, 408, 174. [Google Scholar] [CrossRef] [PubMed]
- Domènech, M.; Hernández, A.; Plaja, A.; Martínez-balibrea, E.; Balañà, C. Hypoxia: The Cornerstone of Glioblastoma. Int. J. Mol. Sci. 2021, 22, 12608. [Google Scholar] [CrossRef]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic State of Glioma Stem Cells and Nontumorigenic Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar] [CrossRef] [PubMed]
- Harland, A.; Liu, X.; Ghirardello, M.; Galan, M.C.; Perks, C.M.; Kurian, K.M. Glioma Stem-Like Cells and Metabolism: Potential for Novel Therapeutic Strategies. Front. Oncol. 2021, 11, 3338. [Google Scholar] [CrossRef]
- Leão Barros, M.B.; Pinheiro, D.D.R.; Borges, B.D.N. Mitochondrial DNA Alterations in Glioblastoma (GBM). Int. J. Mol. Sci. 2021, 22, 5855. [Google Scholar] [CrossRef]
- Kathagen, A.; Schulte, A.; Balcke, G.; Phillips, H.S.; Martens, T.; Matschke, J.; Günther, H.S.; Soriano, R.; Modrusan, Z.; Sandmann, T.; et al. Hypoxia and Oxygenation Induce a Metabolic Switch between Pentose Phosphate Pathway and Glycolysis in Glioma Stem-like Cells. Acta Neuropathol. 2013, 126, 763–780. [Google Scholar] [CrossRef] [PubMed]
- Shafi, O.; Siddiqui, G. Tracing the Origins of Glioblastoma by Investigating the Role of Gliogenic and Related Neurogenic Genes/Signaling Pathways in GBM Development: A Systematic Review. World J. Surg. Oncol. 2022, 20, 146. [Google Scholar] [CrossRef] [PubMed]
- Kopan, R.; Ilagan, M.X. The Canonical Notch Signaling Pathway: Unfolding the Activation Mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Parmigiani, E.; Taylor, V.; Giachino, C. Oncogenic and Tumor-Suppressive Functions of NOTCH Signaling in Glioma. Cells 2020, 9, 2304. [Google Scholar] [CrossRef] [PubMed]
- Androutsellis-Theotokis, A.; Leker, R.R.; Soldner, F.; Hoeppner, D.J.; Ravin, R.; Poser, S.W.; Rueger, M.A.; Bae, S.K.; Kittappa, R.; McKay, R.D. Notch Signalling Regulates Stem Cell Numbers In Vitro and In Vivo. Nature 2006, 442, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Zhou, X.; Li, T.; Liu, P.; Hai, L.; Tong, L.; Ma, H.; Tao, Z.; Xie, Y.; Zhang, C.; et al. Notch1 Signaling Pathway Promotes Invasion, Self-Renewal and Growth of Glioma Initiating Cells via Modulating Chemokine System CXCL12/CXCR4. J. Exp. Clin. Cancer Res. 2019, 38, 339. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Rios, D.; Li, G.; Khan, D.; Tsiampali, J.; Nickel, A.C.; Aretz, P.; Hewera, M.; Suwala, A.K.; Jiang, T.; Steiger, H.J.; et al. A Computational Guided, Functional Validation of a Novel Therapeutic Antibody Proposes Notch Signaling as a Clinical Relevant and Druggable Target in Glioma. Sci. Rep. 2020, 10, 16218. [Google Scholar] [CrossRef] [PubMed]
- Kristoffersen, K.; Villingshoj, M.; Poulsen, H.S.; Stockhausen, M.T. Level of Notch Activation Determines the Effect on Growth and Stem Cell-like Features in Glioblastoma Multiforme Neurosphere Cultures. Cancer Biol. Ther. 2013, 14, 625–637. [Google Scholar] [CrossRef]
- Rajakulendran, N.; Rowland, K.J.; Selvadurai, H.J.; Ahmadi, M.; Park, N.I.; Naumenko, S.; Dolma, S.; Ward, R.J.; So, M.; Lee, L.; et al. Wnt and Notch Signaling Govern Self-Renewal and Differentiation in a Subset of Human Glioblastoma Stem Cells. Genes Dev. 2019, 33, 498–510. [Google Scholar] [CrossRef]
- Stockhausen, M.T.; Kristoffersen, K.; Stobbe, L.; Poulsen, H.S. Differentiation of Glioblastoma Multiforme Stem-like Cells Leads to Downregulation of EGFR and EGFRvIII and Decreased Tumorigenic and Stem-like Cell Potential. Cancer Biol. Ther. 2014, 15, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, R.; Ji, X.D.; Harmon, R.C.; Lazar, C.S.; Gill, G.N.; Cavenee, W.K.; Huang, H.J.S. A Mutant Epidermal Growth Factor Receptor Common in Human Glioma Confers Enhanced Tumorigenicity. Proc. Natl. Acad. Sci. USA 1994, 91, 7727–7731. [Google Scholar] [CrossRef]
- Soeda, A.; Inagaki, A.; Oka, N.; Ikegame, Y.; Aoki, H.; Yoshimura, S.; Nakashima, S.; Kunisada, T.; Iwama, T. Epidermal Growth Factor Plays a Crucial Role in Mitogenic Regulation of Human Brain Tumor Stem Cells. J. Biol. Chem. 2008, 283, 10958–10966. [Google Scholar] [CrossRef]
- Liu, X.J.; Wu, W.T.; Wu, W.H.; Yin, F.; Ma, S.H.; Qin, J.Z.; Liu, X.X.; Liu, Y.N.; Zhang, X.Y.; Li, P.; et al. A Minority Subpopulation of CD133(+)/EGFRvIII(+)/EGFR(-) Cells Acquires Stemness and Contributes to Gefitinib Resistance. CNS Neurosci. Ther. 2013, 19, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A., 3rd. Overcoming Acquired Resistance to Anticancer Therapy: Focus on the PI3K/AKT/MTOR Pathway. Cancer Chemother. Pharmacol. 2013, 71, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Akhavan, D.; Cloughesy, T.F.; Mischel, P.S. MTOR Signaling in Glioblastoma: Lessons Learned from Bench to Bedside. Neuro-Oncology 2010, 12, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Mendiburu-Elicabe, M.; Gil-Ranedo, J.; Izquierdo, M. Efficacy of Rapamycin against Glioblastoma Cancer Stem Cells. Clin. Transl. Oncol. 2014, 16, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Strobele, S.; Schneider, M.; Schneele, L.; Siegelin, M.D.; Nonnenmacher, L.; Zhou, S.; Karpel-Massler, G.; Westhoff, M.A.; Halatsch, M.E.; Debatin, K.M. A Potential Role for the Inhibition of PI3K Signaling in Glioblastoma Therapy. PLoS ONE 2015, 10, e0131670. [Google Scholar] [CrossRef]
- Man, J.; Shoemake, J.D.; Ma, T.; Rizzo, A.E.; Godley, A.R.; Wu, Q.; Mohammadi, A.M.; Bao, S.; Rich, J.N.; Yu, J.S. Hyperthermia Sensitizes Glioma Stem-like Cells to Radiation by Inhibiting AKT Signaling. Cancer Res. 2015, 75, 1760–1769. [Google Scholar] [CrossRef]
- Nanta, R.; Shrivastava, A.; Sharma, J.; Shankar, S.; Srivastava, R.K. Inhibition of Sonic Hedgehog and PI3K/Akt/MTOR Pathways Cooperate in Suppressing Survival, Self-Renewal and Tumorigenic Potential of Glioblastoma-Initiating Cells. Mol. Cell Biochem. 2019, 454, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Clark, P.A.; Iida, M.; Treisman, D.M.; Kalluri, H.; Ezhilan, S.; Zorniak, M.; Wheeler, D.L.; Kuo, J.S. Activation of Multiple ERBB Family Receptors Mediates Glioblastoma Cancer Stem-like Cell Resistance to EGFR-Targeted Inhibition. Neoplasia 2012, 14, 420–428. [Google Scholar] [CrossRef]
- Honorato, J.R.; Hauser-Davis, R.; Saggioro, E.M.; Correia, F.V.; Sales-Junior, S.F.; Soares, L.O.S.; Lima, L.D.R.; Moura-Neto, V.; Lopes, G.P.D.F.; Spohr, T.C.L.D.S. Role of Sonic Hedgehog Signaling in Cell Cycle, Oxidative Stress, and Autophagy of Temozolomide Resistant Glioblastoma. J. Cell. Physiol. 2020, 235, 3798–3814. [Google Scholar] [CrossRef]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The Role of the Hedgehog Signaling Pathway in Cancer: A Comprehensive Review. Bosn. J. Basic Med. Sci. 2018, 18, 8. [Google Scholar] [CrossRef]
- Melamed, J.R.; Morgan, J.T.; Ioele, S.A.; Gleghorn, J.P.; Sims-Mourtada, J.; Day, E.S. Investigating the Role of Hedgehog/GLI1 Signaling in Glioblastoma Cell Response to Temozolomide. Oncotarget 2018, 9, 27000. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Dontu, G.; Mantle, I.D.; Patel, S.; Ahn, N.S.; Jackson, K.W.; Suri, P.; Wicha, M.S. Hedgehog Signaling and Bmi-1 Regulate Self-Renewal of Normal and Malignant Human Mammary Stem Cells. Cancer Res. 2006, 66, 6063–6071. [Google Scholar] [CrossRef] [PubMed]
- Onishi, H.; Katano, M. Hedgehog Signaling Pathway as a Therapeutic Target in Various Types of Cancer. Cancer Sci. 2011, 102, 1756–1760. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Yuan, X.; Liu, G.; Black, K.L.; Yu, J.S. Hedgehog Signaling Regulates Brain Tumor-Initiating Cell Proliferation and Portends Shorter Survival for Patients with PTEN-Coexpressing Glioblastomas. Stem Cells 2008, 26, 3018–3026. [Google Scholar] [CrossRef] [PubMed]
- Hung, H.C.; Liu, C.C.; Chuang, J.Y.; Su, C.L.; Gean, P.W. Inhibition of Sonic Hedgehog Signaling Suppresses Glioma Stem-Like Cells Likely Through Inducing Autophagic Cell Death. Front. Oncol. 2020, 10, 1233. [Google Scholar] [CrossRef] [PubMed]
- Ferruzzi, P.; Mennillo, F.; De Rosa, A.; Giordano, C.; Rossi, M.; Benedetti, G.; Magrini, R.; Mohr, G.L.P.; Miragliotta, V.; Magnoni, L.; et al. In Vitro and in Vivo Characterization of a Novel Hedgehog Signaling Antagonist in Human Glioblastoma Cell Lines. Int. J. Cancer 2012, 131, E33–E44. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ Signalling Pathway in Disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The Roles of TGFβ in the Tumour Microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef]
- Bellomo, C.; Caja, L.; Moustakas, A. Transforming Growth Factor β as Regulator of Cancer Stemness and Metastasis. Br. J. Cancer 2016, 115, 761. [Google Scholar] [CrossRef]
- Golestaneh, N.; Mishra, B. TGF-β, Neuronal Stem Cells and Glioblastoma. Oncogene 2005, 24, 5722–5730. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Moses, H.L. Transforming Growth Factor β: Tumor Suppressor or Promoter? Are Host Immune Cells the Answer? Cancer Res. 2008, 68, 9107–9111. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Moustakas, A. Signaling Receptors for TGF-β Family Members. Cold Spring Harb. Perspect. Biol. 2016, 8, a022053. [Google Scholar] [CrossRef] [PubMed]
- Ten Dijke, P.; Hill, C.S. New Insights into TGF-β–Smad Signalling. Trends Biochem. Sci. 2004, 29, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Peñuelas, S.; Anido, J.; Prieto-Sánchez, R.M.; Folch, G.; Barba, I.; Cuartas, I.; García-Dorado, D.; Poca, M.A.; Sahuquillo, J.; Baselga, J.; et al. TGF-Beta Increases Glioma-Initiating Cell Self-Renewal through the Induction of LIF in Human Glioblastoma. Cancer Cell 2009, 15, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Iwadate, Y.; Matsutani, T.; Hirono, S.; Shinozaki, N.; Saeki, N. Transforming Growth Factor-β and Stem Cell Markers Are Highly Expressed around Necrotic Areas in Glioblastoma. J. Neurooncol. 2016, 129, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.; Liu, N.; Sun, Z.; Jiang, Y.; Jiang, T.; Xv, M.; Jia, L.; Tu, Y.; Wang, L. TGF-β Signaling Promotes Glioma Progression Through Stabilizing Sox9. Front. Immunol. 2021, 11, 3773. [Google Scholar] [CrossRef] [PubMed]
- Bruna, A.; Darken, R.S.; Rojo, F.; Ocaña, A.; Peñuelas, S.; Arias, A.; Paris, R.; Tortosa, A.; Mora, J.; Baselga, J.; et al. High TGFbeta-Smad Activity Confers Poor Prognosis in Glioma Patients and Promotes Cell Proliferation Depending on the Methylation of the PDGF-B Gene. Cancer Cell 2007, 11, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Ikushima, H.; Todo, T.; Ino, Y.; Takahashi, M.; Miyazawa, K.; Miyazono, K. Autocrine TGF-β Signaling Maintains Tumorigenicity of Glioma-Initiating Cells through Sry-Related HMG-Box Factors. Cell Stem Cell 2009, 5, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Guan, R.; Zhang, X.; Guo, M. Glioblastoma Stem Cells and Wnt Signaling Pathway: Molecular Mechanisms and Therapeutic Targets. Chin. Neurosurg. J. 2020, 6, S41016–S41020. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, J.K.; Ahn, S.H.; Lee, J.; Nam, D.H. WNT Signaling in Glioblastoma and Therapeutic Opportunities. Lab. Investig. 2016, 96, 137–150. [Google Scholar] [CrossRef] [PubMed]
- Parker, T.W.; Neufeld, K.L. APC Controls Wnt-Induced β-Catenin Destruction Complex Recruitment in Human Colonocytes. Sci. Rep. 2020, 10, 2957. [Google Scholar] [CrossRef]
- Yang, K.; Wang, X.; Zhang, H.; Wang, Z.; Nan, G.; Li, Y.; Zhang, F.; Mohammed, M.K.; Haydon, R.C.; Luu, H.H.; et al. The Evolving Roles of Canonical WNT Signaling in Stem Cells and Tumorigenesis: Implications in Targeted Cancer Therapies. Lab. Investig. 2016, 96, 116–136. [Google Scholar] [CrossRef] [PubMed]
- Kwong, L.N.; Dove, W.F. APC and Its Modifiers in Colon Cancer. Adv. Exp. Med. Biol. 2009, 656, 85–106. [Google Scholar] [CrossRef] [PubMed]
- Stefanski, C.D.; Prosperi, J.R. Wnt-Independent and Wnt-Dependent Effects of APC Loss on the Chemotherapeutic Response. Int. J. Mol. Sci. 2020, 21, 7844. [Google Scholar] [CrossRef]
- Tang, C.; Guo, J.; Chen, H.; Yao, C.J.; Zhuang, D.X.; Wang, Y.; Tang, W.J.; Ren, G.; Yao, Y.; Wu, J.S.; et al. Gene Mutation Profiling of Primary Glioblastoma through Multiple Tumor Biopsy Guided by 1H-Magnetic Resonance Spectroscopy. Int. J. Clin. Exp. Pathol. 2015, 8, 5327. [Google Scholar] [PubMed]
- Zhang, N.; Wei, P.; Gong, A.; Chiu, W.T.; Lee, H.T.; Colman, H.; Huang, H.; Xue, J.; Liu, M.; Wang, Y.; et al. FoxM1 Promotes β-Catenin Nuclear Localization and Controls Wnt Target-Gene Expression and Glioma Tumorigenesis. Cancer Cell 2011, 20, 427–442. [Google Scholar] [CrossRef] [PubMed]
- Behrooz, A.B.; Syahir, A. Could We Address the Interplay Between CD133, Wnt/β-Catenin, and TERT Signaling Pathways as a Potential Target for Glioblastoma Therapy? Front. Oncol. 2021, 11, 1049. [Google Scholar] [CrossRef] [PubMed]
- Yun, E.J.; Kim, S.; Hsieh, J.T.; Baek, S.T. Wnt/β-Catenin Signaling Pathway Induces Autophagy-Mediated Temozolomide-Resistance in Human Glioblastoma. Cell Death Dis. 2020, 11, 771. [Google Scholar] [CrossRef]
- Zheng, H.; Ying, H.; Wiedemeyer, R.; Yan, H.; Quayle, S.N.; Ivanova, E.V.; Paik, J.H.; Zhang, H.; Xiao, Y.; Perry, S.R.; et al. PLAGL2 Regulates Wnt Signaling to Impede Differentiation in Neural Stem Cells and Glioma. Cancer Cell 2010, 17, 497–509. [Google Scholar] [CrossRef]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 Signalling in Cancer: New and Unexpected Biological Functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef]
- Guanizo, A.C.; Fernando, C.D.; Garama, D.J.; Gough, D.J. STAT3: A Multifaceted Oncoprotein. Growth Factors 2018, 36, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Guryanova, O.A.; Zhou, W.; Liu, C.; Huang, Z.; Fang, X.; Wang, X.; Chen, C.; Wu, Q.; He, Z.; et al. Ibrutinib Inactivates BMX-STAT3 in Glioma Stem Cells to Impair Malignant Growth and Radioresistance. Sci. Transl. Med. 2018, 10, eaah6816. [Google Scholar] [CrossRef] [PubMed]
- Masliantsev, K.; Pinel, B.; Balbous, A.; Guichet, P.O.; Tachon, G.; Milin, S.; Godet, J.; Duchesne, M.; Berger, A.; Petropoulos, C.; et al. Impact of STAT3 Phosphorylation in Glioblastoma Stem Cells Radiosensitization and Patient Outcome. Oncotarget 2018, 9, 3968. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E. STATs and Gene Regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Kim, M.; Woo, D.H.; Shin, Y.; Shin, J.; Chang, N.; Oh, Y.T.; Kim, H.; Rheey, J.; Nakano, I.; et al. Phosphorylation of EZH2 Activates STAT3 Signaling via STAT3 Methylation and Promotes Tumorigenicity of Glioblastoma Stem-like Cells. Cancer Cell 2013, 23, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liao, X.; Agarwal, M.K.; Barnes, L.; Auron, P.E.; Stark, G.R. Unphosphorylated STAT3 Accumulates in Response to IL-6 and Activates Transcription by Binding to NFkappaB. Genes Dev. 2007, 21, 1396–1408. [Google Scholar] [CrossRef] [PubMed]
- West, A.J.; Tsui, V.; Stylli, S.S.; Nguyen, H.P.T.; Morokoff, A.P.; Kaye, A.H.; Luwor, R.B. The Role of Interleukin-6-STAT3 Signalling in Glioblastoma. Oncol. Lett. 2018, 16, 4095–4104. [Google Scholar] [CrossRef] [PubMed]
- Bonnin, D.A.A.; Havrda, M.C.; Lee, M.C.; Liu, H.; Zhang, Z.; Nguyen, L.N.; Harrington, L.X.; Hassanpour, S.; Cheng, C.; Israel, M.A. Secretion-Mediated STAT3 Activation Promotes Self-Renewal of Glioma Stem-like Cells during Hypoxia. Oncogene 2018, 37, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, A.; Cherryholmes, G.; Schroeder, A.; Phallen, J.; Alizadeh, D.; Xin, H.; Wang, T.; Lee, H.; Lahtz, C.; Swiderski, P.; et al. TLR9 Is Critical for Glioma Stem Cell Maintenance and Targeting. Cancer Res. 2014, 74, 5218–5228. [Google Scholar] [CrossRef] [PubMed]
- Filppu, P.; Ramanathan, J.T.; Granberg, K.J.; Gucciardo, E.; Haapasalo, H.; Lehti, K.; Nykter, M.; le Joncour, V.; Laakkonen, P. CD109-GP130 Interaction Drives Glioblastoma Stem Cell Plasticity and Chemoresistance through STAT3 Activity. JCI Insight 2021, 6, e141486. [Google Scholar] [CrossRef]
- Ouédraogo, Z.G.; Müller-Barthélémy, M.; Kemeny, J.L.; Dedieu, V.; Biau, J.; Khalil, T.; Raoelfils, L.I.; Granzotto, A.; Pereira, B.; Beaudoin, C.; et al. STAT3 Serine 727 Phosphorylation: A Relevant Target to Radiosensitize Human Glioblastoma. Brain Pathol. 2016, 26, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Sherry, M.M.; Reeves, A.; Wu, J.K.; Cochran, B.H. STAT3 Is Required for Proliferation and Maintenance of Multipotency in Glioblastoma Stem Cells. Stem Cells 2009, 27, 2383–2392. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lathia, J.D.; Wu, Q.; Wang, J.; Li, Z.; Heddleston, J.M.; Eyler, C.E.; Elderbroom, J.; Gallagher, J.; Schuschu, J.; et al. Targeting Interleukin 6 Signaling Suppresses Glioma Stem Cell Survival and Tumor Growth. Stem Cells 2009, 27, 2393–2404. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Bo, Z.; Gong, W.; Guo, Y. Inhibitor of Differentiation 1 (Id1) in Cancer and Cancer Therapy. Int. J. Med. Sci. 2020, 17, 995. [Google Scholar] [CrossRef]
- Soroceanu, L.; Murase, R.; Limbad, C.; Singer, E.; Allison, J.; Adrados, I.; Kawamura, R.; Pakdel, A.; Fukuyo, Y.; Nguyen, D.; et al. Id-1 Is a Key Transcriptional Regulator of Glioblastoma Aggressiveness and a Novel Therapeutic Target. Cancer Res. 2013, 73, 1559–1569. [Google Scholar] [CrossRef]
- Sachdeva, R.; Wu, M.; Smiljanic, S.; Kaskun, O.; Ghannad-Zadeh, K.; Celebre, A.; Isaev, K.; Morrissy, A.S.; Guan, J.; Tong, J.; et al. ID1 Is Critical for Tumorigenesis and Regulates Chemoresistance in Glioblastoma. Cancer Res. 2019, 79, 4057–4071. [Google Scholar] [CrossRef]
- Perk, J.; Iavarone, A.; Benezra, R. Id Family of Helix-Loop-Helix Proteins in Cancer. Nat. Rev. Cancer 2005, 5, 603–614. [Google Scholar] [CrossRef]
- Ouyang, X.S.; Wang, X.; Ling, M.T.; Wong, H.L.; Tsao, S.W.; Wong, Y.C. Id-1 Stimulates Serum Independent Prostate Cancer Cell Proliferation through Inactivation of P16 INK4a/PRB Pathway. Carcinogenesis 2002, 23, 721–725. [Google Scholar] [CrossRef][Green Version]
- Jin, X.; Jeon, H.M.; Jin, X.; Kim, E.J.; Yin, J.; Jeon, H.Y.; Sohn, Y.W.; Oh, S.Y.; Kim, J.K.; Kim, S.H.; et al. The ID1-CULLIN3 Axis Regulates Intracellular SHH and WNT Signaling in Glioblastoma Stem Cells. Cell Rep. 2016, 16, 1629–1641. [Google Scholar] [CrossRef]
- Cook, P.J.; Thomas, R.; Kingsley, P.J.; Shimizu, F.; Montrose, D.C.; Marnett, L.J.; Tabar, V.S.; Dannenberg, A.J.; Benezra, R. Cox-2-Derived PGE2 Induces Id1-Dependent Radiation Resistance and Self-Renewal in Experimental Glioblastoma. Neuro-Oncology 2016, 18, 1379–1389. [Google Scholar] [CrossRef]
- Jin, X.; Jin, X.; Kim, L.J.Y.; Dixit, D.; Jeon, H.Y.; Kim, E.J.; Kim, J.K.; Lee, S.Y.; Yin, J.; Rich, J.N.; et al. Inhibition of ID1-BMPR2 Intrinsic Signaling Sensitizes Glioma Stem Cells to Differentiation Therapy. Clin. Cancer Res. 2018, 24, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K.; Kamiya, Y.; Morikawa, M. Bone Morphogenetic Protein Receptors and Signal Transduction. J. Biochem. 2010, 147, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Puerto, M.C.; Iyengar, P.V.; García de Vinuesa, A.; ten Dijke, P.; Sanchez-Duffhues, G. Bone Morphogenetic Protein Receptor Signal Transduction in Human Disease. J. Pathol. 2019, 247, 9. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Yin, J.; Kim, S.H.; Sohn, Y.W.; Beck, S.; Lim, Y.C.; Nam, D.H.; Choi, Y.J.; Kim, H. EGFR-AKT-Smad Signaling Promotes Formation of Glioma Stem-like Cells and Tumor Angiogenesis by ID3-Driven Cytokine Induction. Cancer Res. 2011, 71, 7125–7134. [Google Scholar] [CrossRef] [PubMed]
- Niola, F.; Zhao, X.; Singh, D.; Sullivan, R.; Castano, A.; Verrico, A.; Zoppoli, P.; Friedmann-Morvinski, D.; Sulman, E.; Barrett, L.; et al. Mesenchymal High-Grade Glioma Is Maintained by the ID-RAP1. J. Clin. Investig. 2013, 123, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lu, Q.; Chang, C. Epigenetics in Health and Disease. Adv. Exp. Med. Biol. 2020, 1253, 3–55. [Google Scholar] [CrossRef]
- Barnes, C.E.; English, D.M.; Cowley, S.M. Acetylation & Co: An Expanding Repertoire of Histone Acylations Regulates Chromatin and Transcription. Essays Biochem. 2019, 63, 97. [Google Scholar] [CrossRef]
- Newell-Price, J.; Clark, A.J.L.; King, P. DNA Methylation and Silencing of Gene Expression. Trends Endocrinol. Metab. 2000, 11, 142–148. [Google Scholar] [CrossRef]
- Greer, E.L.; Shi, Y. Histone Methylation: A Dynamic Mark in Health, Disease and Inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef]
- Nebbioso, A.; Tambaro, F.P.; Dell’Aversana, C.; Altucci, L. Cancer Epigenetics: Moving Forward. PLoS Genet. 2018, 14, e1007362. [Google Scholar] [CrossRef] [PubMed]
- Miranda Furtado, C.L.; Dos Santos Luciano, M.C.; Da Silva Santos, R.; Furtado, G.P.; Moraes, M.O.; Pessoa, C. Epidrugs: Targeting Epigenetic Marks in Cancer Treatment. Epigenetics 2019, 14, 1164–1176. [Google Scholar] [CrossRef] [PubMed]
- Bouyahya, A.; Mechchate, H.; Oumeslakht, L.; Zeouk, I.; Aboulaghras, S.; Balahbib, A.; Zengin, G.; Kamal, M.A.; Gallo, M.; Montesano, D.; et al. The Role of Epigenetic Modifications in Human Cancers and the Use of Natural Compounds as Epidrugs: Mechanistic Pathways and Pharmacodynamic Actions. Biomolecules 2022, 12, 367. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Alver, B.M.; Li, S.; Hlady, R.A.; Thompson, J.J.; Schroeder, M.A.; Lee, J.H.; Qiu, J.; Schwartz, P.H.; Sarkaria, J.N.; et al. Distinctive Epigenomes Characterize Glioma Stem Cells and Their Response to Differentiation Cues. Genome Biol. 2018, 19, 43. [Google Scholar] [CrossRef]
- Lu, X.; Maturi, N.P.; Jarvius, M.; Yildirim, I.; Dang, Y.; Zhao, L.; Xie, Y.; Tan, E.J.; Xing, P.; Larsson, R.; et al. Cell-Lineage Controlled Epigenetic Regulation in Glioblastoma Stem Cells Determines Functionally Distinct Subgroups and Predicts Patient Survival. Nat. Commun. 2022, 13, 2236. [Google Scholar] [CrossRef]
- Weng, J.Y.; Salazar, N. DNA Methylation Analysis Identifies Patterns in Progressive Glioma Grades to Predict Patient Survival. Int. J. Mol. Sci. 2021, 22, 1020. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Rath, P.; Liu, J.; Ryu, D.; Pei, L.; Noonepalle, S.K.; Shull, A.Y.; Feng, Q.; Litofsky, N.S.; Miller, D.C.; et al. Identification of Global DNA Methylation Signatures in Glioblastoma-Derived Cancer Stem Cells. J. Genet. Genom. 2015, 42, 355. [Google Scholar] [CrossRef][Green Version]
- Pangeni, R.P.; Zhang, Z.; Alvarez, A.A.; Wan, X.; Sastry, N.; Lu, S.; Shi, T.; Huang, T.; Lei, C.X.; James, C.D.; et al. Genome-Wide Methylomic and Transcriptomic Analyses Identify Subtype-Specific Epigenetic Signatures Commonly Dysregulated in Glioma Stem Cells and Glioblastoma. Epigenetics 2018, 13, 432–448. [Google Scholar] [CrossRef] [PubMed]
- Ninova, M.; Tóth, K.F.; Aravin, A.A. The Control of Gene Expression and Cell Identity by H3K9 Trimethylation. Development 2019, 146, dev181180. [Google Scholar] [CrossRef]
- Padeken, J.; Methot, S.P.; Gasser, S.M. Establishment of H3K9-Methylated Heterochromatin and Its Functions in Tissue Differentiation and Maintenance. Nat. Rev. Mol. Cell Biol. 2022, 2022, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.S.; Nicetto, D.; Zaret, K.S. H3K9me3-Dependent Heterochromatin: Barrier to Cell Fate Changes. Trends Genet. 2016, 32, 29. [Google Scholar] [CrossRef] [PubMed]
- Nicetto, D.; Zaret, K.S. Role of H3K9me3 Heterochromatin in Cell Identity Establishment and Maintenance. Curr. Opin. Genet. Dev. 2019, 55, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Magri, L.; Zhang, F.; Marsh, N.O.; Albrecht, S.; Huynh, J.L.; Kaur, J.; Kuhlmann, T.; Zhang, W.; Slesinger, P.A.; et al. Chromatin Landscape Defined by Repressive Histone Methylation during Oligodendrocyte Differentiation. J. Neurosci. 2015, 35, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Mallm, J.P.; Windisch, P.; Biran, A.; Gal, Z.; Schumacher, S.; Glass, R.; Herold-Mende, C.; Meshorer, E.; Barbus, M.; Rippe, K. Glioblastoma Initiating Cells Are Sensitive to Histone Demethylase Inhibition Due to Epigenetic Deregulation. Int. J. Cancer 2020, 146, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Liau, B.B.; Sievers, C.; Donohue, L.K.; Gillespie, S.M.; Flavahan, W.A.; Miller, T.E.; Venteicher, A.S.; Hebert, C.H.; Carey, C.D.; Rodig, S.J.; et al. Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem Cell 2017, 20, 233–246.e7. [Google Scholar] [CrossRef] [PubMed]
- Venneti, S.; Garimella, M.T.; Sullivan, L.M.; Martinez, D.; Huse, J.T.; Heguy, A.; Santi, M.; Thompson, C.B.; Judkins, A.R. Evaluation of Histone 3 Lysine 27 Trimethylation (H3K27me3) and Enhancer of Zest 2 (EZH2) in Pediatric Glial and Glioneuronal Tumors Shows Decreased H3K27me3 in H3F3A K27M Mutant Glioblastomas. Brain Pathol. 2013, 23, 558–564. [Google Scholar] [CrossRef]
- Laugesen, A.; Højfeldt, J.W.; Helin, K. Role of the Polycomb Repressive Complex 2 (PRC2) in Transcriptional Regulation and Cancer. Cold Spring Harb. Perspect. Med. 2016, 6, a026575. [Google Scholar] [CrossRef]
- Li, T.; Yu, C.; Zhuang, S. Histone Methyltransferase EZH2: A Potential Therapeutic Target for Kidney Diseases. Front. Physiol. 2021, 12, 45. [Google Scholar] [CrossRef]
- Tan, J.Z.; Yan, Y.; Wang, X.X.; Jiang, Y.; Xu, H.E. EZH2: Biology, Disease, and Structure-Based Drug Discovery. Acta Pharmacol. Sin. 2014, 35, 161–174. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, X.; Chen, L.; Zhang, Z.; Feng, S. EZH2 Overexpression Is Associated with Poor Prognosis in Patients with Glioma. Oncotarget 2017, 8, 565. [Google Scholar] [CrossRef]
- Chen, Y.; Hou, S.; Jiang, R.; Sun, J.; Cheng, C.; Qian, Z.-r. EZH2 Is a Potential Prognostic Predictor of Glioma. J. Cell Mol. Med. 2021, 25, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Stazi, G.; Taglieri, L.; Nicolai, A.; Romanelli, A.; Fioravanti, R.; Morrone, S.; Sabatino, M.; Ragno, R.; Taurone, S.; Nebbioso, M.; et al. Dissecting the Role of Novel EZH2 Inhibitors in Primary Glioblastoma Cell Cultures: Effects on Proliferation, Epithelial-Mesenchymal Transition, Migration, and on the pro-Inflammatory Phenotype. Clin. Epigenetics 2019, 11, 173. [Google Scholar] [CrossRef] [PubMed]
- Paskeh, M.D.A.; Mehrabi, A.; Gholami, M.H.; Zabolian, A.; Ranjbar, E.; Saleki, H.; Ranjbar, A.; Hashemi, M.; Ertas, Y.N.; Hushmandi, K.; et al. EZH2 as a New Therapeutic Target in Brain Tumors: Molecular Landscape, Therapeutic Targeting and Future Prospects. Biomed. Pharmacother. 2022, 146, 112532. [Google Scholar] [CrossRef] [PubMed]
- Suvà, M.L.; Riggi, N.; Janiszewska, M.; Radovanovic, I.; Provero, P.; Stehle, J.C.; Baumer, K.; Le Bitoux, M.A.; Marino, D.; Cironi, L.; et al. EZH2 Is Essential for Glioblastoma Cancer Stem Cell Maintenance. Cancer Res. 2009, 69, 9211–9218. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Son, M.J.; Woolard, K.; Donin, N.M.; Li, A.; Cheng, C.H.; Kotliarova, S.; Kotliarov, Y.; Walling, J.; Ahn, S.; et al. Epigenetic-Mediated Dysfunction of the Bone Morphogenetic Protein Pathway Inhibits Differentiation of Glioblastoma-Initiating Cells. Cancer Cell 2008, 13, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Valor, L.M.; Hervás-Corpión, I. The Epigenetics of Glioma Stem Cells: A Brief Overview. Front. Oncol. 2020, 10, 2656. [Google Scholar] [CrossRef] [PubMed]
- Verdone, L.; Agricola, E.; Caserta, M.; Di Mauro, E. Histone Acetylation in Gene Regulation. Brief. Funct. Genom. 2006, 5, 209–221. [Google Scholar] [CrossRef]
- Kim, Y.Z. Altered Histone Modifications in Gliomas. Brain Tumor Res. Treat. 2014, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Was, H.; Krol, S.K.; Rotili, D.; Mai, A.; Wojtas, B.; Kaminska, B.; Maleszewska, M. Histone Deacetylase Inhibitors Exert Anti-Tumor Effects on Human Adherent and Stem-like Glioma Cells. Clin. Epigenetics 2019, 11, 11. [Google Scholar] [CrossRef]
- Reddy, R.G.; Bhat, U.A.; Chakravarty, S.; Kumar, A. Advances in Histone Deacetylase Inhibitors in Targeting Glioblastoma Stem Cells. Cancer Chemother. Pharmacol. 2020, 86, 165–179. [Google Scholar] [CrossRef]
- Yang, W.; Liu, Y.; Gao, R.; Yu, H.; Sun, T. HDAC6 Inhibition Induces Glioma Stem Cells Differentiation and Enhances Cellular Radiation Sensitivity through the SHH/Gli1 Signaling Pathway. Cancer Lett. 2018, 415, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Marampon, F.; Megiorni, F.; Camero, S.; Crescioli, C.; McDowell, H.P.; Sferra, R.; Vetuschi, A.; Pompili, S.; Ventura, L.; De Felice, F.; et al. HDAC4 and HDAC6 Sustain DNA Double Strand Break Repair and Stem-like Phenotype by Promoting Radioresistance in Glioblastoma Cells. Cancer Lett. 2017, 397, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Riva, G.; Butta, V.; Cilibrasi, C.; Baronchelli, S.; Redaelli, S.; Dalprà, L.; Lavitrano, M.; Bentivegna, A. Epigenetic Targeting of Glioma Stem Cells: Short-Term and Long-Term Treatments with Valproic Acid Modulate DNA Methylation and Differentiation Behavior, but Not Temozolomide Sensitivity. Oncol. Rep. 2016, 35, 2811–2824. [Google Scholar] [CrossRef] [PubMed]
- MacFarlane, L.-A.; Murphy, P.R. MicroRNA: Biogenesis, Function and Role in Cancer. Curr. Genom. 2010, 11, 537. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Yamamoto, Y.; Ochiya, T. MiRNA Signaling Networks in Cancer Stem Cells. Regen. Ther. 2021, 17, 1. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.Q.; Ahmed, E.I.; Elareer, N.R.; Junejo, K.; Steinhoff, M.; Uddin, S. Role of MiRNA-Regulated Cancer Stem Cells in the Pathogenesis of Human Malignancies. Cells 2019, 8, 840. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.-U.; Miyazaki, H.; Ochiya, T. The Role of MicroRNAs in the Regulation of Cancer Stem Cells. Front. Genet. 2013, 4, 295. [Google Scholar] [CrossRef]
- Sun, X.; Jiao, X.; Pestell, T.G.; Fan, C.; Qin, S.; Mirabelli, E.; Ren, H.; Pestell, R.G. MicroRNAs and Cancer Stem Cells: The Sword and the Shield. Oncogene 2013, 33, 4967–4977. [Google Scholar] [CrossRef] [PubMed]
- Tomei, S.; Volontè, A.; Ravindran, S.; Mazzoleni, S.; Wang, E.; Galli, R.; Maccalli, C. Microrna Expression Profile Distinguishes Glioblastoma Stem Cells from Differentiated Tumor Cells. J. Pers. Med. 2021, 11, 264. [Google Scholar] [CrossRef]
- Sana, J.; Busek, P.; Fadrus, P.; Besse, A.; Radova, L.; Vecera, M.; Reguli, S.; Stollinova Sromova, L.; Hilser, M.; Lipina, R.; et al. Identification of MicroRNAs Differentially Expressed in Glioblastoma Stem-like Cells and Their Association with Patient Survival. Sci. Rep. 2018, 8, 2836. [Google Scholar] [CrossRef]
- Macharia, L.W.; Muriithi, W.; Nyaga, D.K.; de Mattos Coelho-Aguiar, J.; de Sampaio Spohr, T.C.L.; Moura-Neto, V. Evaluation of MiRNA Expression in Glioblastoma Stem-Like Cells: A Comparison between Normoxia and Hypoxia Microenvironment. Onco 2022, 2, 113–128. [Google Scholar] [CrossRef]
- Macharia, L.W.; Muriithi, W.; Heming, C.P.; Nyaga, D.K.; Aran, V.; Mureithi, M.W.; Ferrer, V.P.; Pane, A.; Filho, P.N.; Moura-Neto, V. The Genotypic and Phenotypic Impact of Hypoxia Microenvironment on Glioblastoma Cell Lines. BMC Cancer 2021, 21, 1248. [Google Scholar] [CrossRef] [PubMed]
- Guessous, F.; Zhang, Y.; Kofman, A.; Catania, A.; Li, Y.; Schiff, D.; Purow, B.; Abounader, R. MicroRNA-34a Is Tumor Suppressive in Brain Tumors and Glioma Stem Cells. Cell Cycle 2010, 9, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Yunqing, L.; Guessous, F.; Ying, Z.; DiPierro, C.; Kefas, B.; Johnson, E.; Marcinkiewicz, L.; Jinmai, J.; Yanzhi, Y.; Schmittgen, T.D.; et al. MicroRNA-34a Inhibits Glioblastoma Growth by Targeting Multiple Oncogenes. Cancer Res. 2009, 69, 7569–7576. [Google Scholar] [CrossRef]
- Zhang, X.; Ai, F.; Li, X.; Tian, L.; Wang, X.; Shen, S.; Liu, F. MicroRNA-34a Suppresses Colorectal Cancer Metastasis by Regulating Notch Signaling. Oncol. Lett. 2017, 14, 2325. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.X.; Zhao, Z.Y.; Weng, G.H.; He, X.Y.; Wu, C.J.; Fu, C.Y.; Sui, Z.Y.; Ma, Y.S.; Liu, T. Upregulation of MiR-181a Suppresses the Formation of Glioblastoma Stem Cells by Targeting the Notch2 Oncogene and Correlates with Good Prognosis in Patients with Glioblastoma Multiforme. Biochem. Biophys. Res. Commun. 2017, 486, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Xi, Z.; Wang, P.; Xue, Y.; Shang, C.; Liu, X.; Ma, J.; Li, Z.; Li, Z.; Bao, M.; Liu, Y. Overexpression of MiR-29a Reduces the Oncogenic Properties of Glioblastoma Stem Cells by Downregulating Quaking Gene Isoform 6. Oncotarget 2017, 8, 24949–24963. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Dodbele, S.; Park, T.; Glass, R.; Bhat, K.; Sulman, E.P.; Zhang, Y.; Abounader, R. MicroRNA-29a Inhibits Glioblastoma Stem Cells and Tumor Growth by Regulating the PDGF Pathway. J. Neurooncol. 2019, 145, 23–34. [Google Scholar] [CrossRef]
- Bier, A.; Giladi, N.; Kronfeld, N.; Lee, H.K.; Cazacu, S.; Finniss, S.; Xiang, C.; Poisson, L.; deCarvalho, A.C.; Slavin, S.; et al. MicroRNA-137 Is Downregulated in Glioblastoma and Inhibits the Stemness of Glioma Stem Cells by Targeting RTVP-1. Oncotarget 2013, 4, 665–676. [Google Scholar] [CrossRef]
- Silber, J.; Lim, D.A.; Petritsch, C.; Persson, A.I.; Maunakea, A.K.; Yu, M.; Vandenberg, S.R.; Ginzinger, D.G.; James, C.D.; Costello, J.F.; et al. MiR-124 and MiR-137 Inhibit Proliferation of Glioblastoma Multiforme Cells and Induce Differentiation of Brain Tumor Stem Cells. BMC Med. 2008, 6, 14. [Google Scholar] [CrossRef]
- Godlewski, J.; Nowicki, M.O.; Bronisz, A.; Williams, S.; Otsuki, A.; Nuovo, G.; RayChaudhury, A.; Newton, H.B.; Chiocca, E.A.; Lawler, S. Targeting of the Bmi-1 Oncogene/Stem Cell Renewal Factor by MicroRNA-128 Inhibits Glioma Proliferation and Self-Renewal. Cancer Res. 2008, 68, 9125–9130. [Google Scholar] [CrossRef]
- Li, H.; Yang, B.B.; Li, H.; Yang, B.B. Stress Response of Glioblastoma Cells Mediated by MiR-17-5p Targeting PTEN and the Passenger Strand MiR-17-3p Targeting MDM2. Oncotarget 2012, 3, 1653–1668. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Costa, A.; Osório, L.; Lago, R.C.; Linhares, P.; Carvalho, B.; Caeiro, C. Current Standards of Care in Glioblastoma Therapy. In Glioblastoma; Elsevier Inc.: Amsterdam, The Netherlands, 2017; pp. 197–241. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y. Temozolomide Resistance in Glioblastoma Multiforme. Genes Dis. 2016, 3, 198. [Google Scholar] [CrossRef] [PubMed]
- Rivera, M.; Sukhdeo, K.; Yu, J. Ionizing Radiation in Glioblastoma Initiating Cells. Front. Oncol. 2013, 3, 74. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Lv, L.; Yang, K. Chemotherapy Targeting Cancer Stem Cells. Am. J. Cancer Res. 2015, 5, 880. [Google Scholar] [PubMed]
- Olivier, J.C. Drug Transport to Brain with Targeted Nanoparticles. NeuroRx 2005, 2, 108. [Google Scholar] [CrossRef]
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of Temozolomide Resistance in Glioblastoma—A Comprehensive Review. Cancer Drug Resist. 2021, 4, 17. [Google Scholar] [CrossRef]
- Prieto-Vila, M.; Takahashi, R.U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef]
- Auffinger, B.; Spencer, D.; Pytel, P.; Ahmed, A.U.; Lesniak, M.S. The Role of Glioma Stem Cells in Chemotherapy Resistance and Glioblastoma Multiforme Recurrence. Expert Rev. Neurother. 2015, 15, 741. [Google Scholar] [CrossRef]
- Yu, W.; Zhang, L.; Wei, Q.; Shao, A. O6-Methylguanine-DNA Methyltransferase (MGMT): Challenges and New Opportunities in Glioma Chemotherapy. Front. Oncol. 2020, 9, 1547. [Google Scholar] [CrossRef] [PubMed]
- Smiley, S.B.; Zarrinmayeh, H.; Das, S.K.; Pollok, K.E.; Vannier, M.W.; Veronesi, M.C. Novel Therapeutics and Drug-Delivery Approaches in the Modulation of Glioblastoma Stem Cell Resistance. Ther. Deliv. 2022, 13, 249–273. [Google Scholar] [CrossRef] [PubMed]
- Beier, D.; Röhrl, S.; Pillai, D.R.; Schwarz, S.; Kunz-Schughart, L.A.; Leukel, P.; Proescholdt, M.; Brawanski, A.; Bogdahn, U.; Trampe-Kieslich, A.; et al. Temozolomide Preferentially Depletes Cancer Stem Cells in Glioblastoma. Cancer Res. 2008, 68, 5706–5715. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, G.; Bozek, D.A.; Rajakulendran, N.; Monteiro, V.; Ahmadi, M.; Steinhart, Z.; Kushida, M.M.; Yu, H.; Coutinho, F.J.; Cavalli, F.M.G.; et al. Genome-Wide CRISPR-Cas9 Screens Expose Genetic Vulnerabilities and Mechanisms of Temozolomide Sensitivity in Glioblastoma Stem Cells. Cell Rep. 2019, 27, 971–986.e9. [Google Scholar] [CrossRef] [PubMed]
- Olivier, C.; Oliver, L.; Lalier, L.; Vallette, F.M. Drug Resistance in Glioblastoma: The Two Faces of Oxidative Stress. Front. Mol. Biosci. 2021, 7, 468. [Google Scholar] [CrossRef] [PubMed]
- Tentori, L.; Ricci-Vitiani, L.; Muzi, A.; Ciccarone, F.; Pelacchi, F.; Calabrese, R.; Runci, D.; Pallini, R.; Caiafa, P.; Graziani, G. Pharmacological Inhibition of Poly(ADP-Ribose) Polymerase-1 Modulates Resistance of Human Glioblastoma Stem Cells to Temozolomide. BMC Cancer 2014, 14, 151. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wan, Y.; Pan, T.; Gu, X.; Qian, C.; Sun, G.; Sun, L.; Xiang, Y.; Wang, Z.; Shi, L. MicroRNA-21 Inhibitor Sensitizes Human Glioblastoma U251 Stem Cells to Chemotherapeutic Drug Temozolomide. J. Mol. Neurosci. 2012, 47, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.; Wang, B.; Zou, Y.; Zhang, Y.; Hu, X.; Sun, W.; Xiao, H.; Liu, H.; Shi, L. MicroRNA 145 Enhances Chemosensitivity of Glioblastoma Stem Cells to Demethoxycurcumin. Cancer Manag. Res. 2019, 11, 6829. [Google Scholar] [CrossRef] [PubMed]
- Tezcan, G.; Tunca, B.; Bekar, A.; Preusser, M.; Berghoff, A.S.; Egeli, U.; Cecener, G.; Ricken, G.; Budak, F.; Taskapılıoglu, M.O.; et al. MicroRNA Expression Pattern Modulates Temozolomide Response in GBM Tumors with Cancer Stem Cells. Cell. Mol. Neurobiol. 2014, 34, 679–692. [Google Scholar] [CrossRef]
- Chien, C.H.; Hsueh, W.T.; Chuang, J.Y.; Chang, K.Y. Dissecting the Mechanism of Temozolomide Resistance and Its Association with the Regulatory Roles of Intracellular Reactive Oxygen Species in Glioblastoma. J. Biomed. Sci. 2021, 28, 18. [Google Scholar] [CrossRef]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.G.; Lee, S.H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and Their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef] [PubMed]
- Ulasov, I.V.; Nandi, S.; Dey, M.; Sonabend, A.M.; Lesniak, M.S. Inhibition of Sonic Hedgehog and Notch Pathways Enhances Sensitivity of CD133+ Glioma Stem Cells to Temozolomide Therapy. Mol. Med. 2011, 17, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, B.; Tobias, A.L.; Han, Y.; Lee, G.; Guo, D.; Dey, M.; Lesniak, M.S.; Ahmed, A.U. Conversion of Differentiated Cancer Cells into Cancer Stem-like Cells in a Glioblastoma Model after Primary Chemotherapy. Cell Death Differ. 2014, 21, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Kolenda, J.; Jensen, S.S.; Aaberg-Jessen, C.; Christensen, K.; Andersen, C.; Brünner, N.; Kristensen, B.W. Effects of Hypoxia on Expression of a Panel of Stem Cell and Chemoresistance Markers in Glioblastoma-Derived Spheroids. J. Neurooncol. 2011, 103, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Pistollato, F.; Abbadi, S.; Rampazzo, E.; Persano, L.; della Puppa, A.; Frasson, C.; Sarto, E.; Scienza, R.; D’Avella, D.; Basso, G. Intratumoral Hypoxic Gradient Drives Stem Cells Distribution and MGMT Expression in Glioblastoma. Stem Cells 2010, 28, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Hersh, A.M.; Alomari, S.; Tyler, B.M. Crossing the Blood-Brain Barrier: Advances in Nanoparticle Technology for Drug Delivery in Neuro-Oncology. Int. J. Mol. Sci. 2022, 23, 4153. [Google Scholar] [CrossRef] [PubMed]
- Alves, A.L.V.; Gomes, I.N.F.; Carloni, A.C.; Rosa, M.N.; da Silva, L.S.; Evangelista, A.F.; Reis, R.M.; Silva, V.A.O. Role of Glioblastoma Stem Cells in Cancer Therapeutic Resistance: A Perspective on Antineoplastic Agents from Natural Sources and Chemical Derivatives. Stem Cell Res. Ther. 2021, 12, 206. [Google Scholar] [CrossRef] [PubMed]
- Lötsch, D.; Steiner, E.; Holzmann, K.; Spiegl-Kreinecker, S.; Pirker, C.; Hlavaty, J.; Petznek, H.; Hegedus, B.; Garay, T.; Mohr, T.; et al. Major Vault Protein Supports Glioblastoma Survival and Migration by Upregulating the EGFR/PI3K Signalling Axis. Oncotarget 2013, 4, 1904. [Google Scholar] [CrossRef] [PubMed]
- Noh, K.H.; Lee, S.H.; Lee, H.; Jeong, A.J.; Kim, K.O.; Shin, H.M.; Kim, H.R.; Park, M.J.; Park, J.B.; Lee, J.; et al. Novel Cancer Stem Cell Marker MVP Enhances Temozolomide-Resistance in Glioblastoma. Transl. Oncol. 2022, 15, 101255. [Google Scholar] [CrossRef]
- Uribe, D.; Torres, Á.; Rocha, J.D.; Niechi, I.; Oyarzún, C.; Sobrevia, L.; San Martín, R.; Quezada, C. Multidrug Resistance in Glioblastoma Stem-like Cells: Role of the Hypoxic Microenvironment and Adenosine Signaling. Mol. Asp. Med. 2017, 55, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Aoyagi, M.; Wakimoto, H.; Ando, N.; Nariai, T.; Yamamoto, M.; Ohno, K. Accumulation of CD133-Positive Glioma Cells after High-Dose Irradiation by Gamma Knife Surgery plus External Beam Radiation: Clinical Article. J. Neurosurg. 2010, 113, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Arnold, C.R.; Mangesius, J.; Skvortsova, I.I.; Ganswindt, U. The Role of Cancer Stem Cells in Radiation Resistance. Front. Oncol. 2020, 10, 164. [Google Scholar] [CrossRef] [PubMed]
- Mannino, M.; Chalmers, A.J. Radioresistance of Glioma Stem Cells: Intrinsic Characteristic or Property of the ‘Microenvironment-stem Cell Unit’? Mol. Oncol. 2011, 5, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhou, K.; Gao, L.; Zhang, B.; Li, W.; Yan, W.; Song, X.; Yu, H.; Wang, S.; Yu, N.; et al. Radiation Induces the Generation of Cancer Stem Cells: A Novel Mechanism for Cancer Radioresistance. Oncol. Lett. 2016, 12, 3059–3065. [Google Scholar] [CrossRef]
- Tsai, C.Y.; Ko, H.J.; Huang, C.Y.F.; Lin, C.Y.; Chiou, S.J.; Su, Y.F.; Lieu, A.S.; Loh, J.K.; Kwan, A.L.; Chuang, T.H.; et al. Ionizing Radiation Induces Resistant Glioblastoma Stem-Like Cells by Promoting Autophagy via the Wnt/β-Catenin Pathway. Life 2021, 11, 451. [Google Scholar] [CrossRef] [PubMed]
- King, H.O.; Brend, T.; Payne, H.L.; Wright, A.; Ward, T.A.; Patel, K.; Egnuni, T.; Stead, L.F.; Patel, A.; Wurdak, H.; et al. RAD51 Is a Selective DNA Repair Target to Radiosensitize Glioma Stem Cells. Stem Cell Rep. 2017, 8, 125. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma Stem-like Cells Give Rise to Tumour Endothelium. Nature 2010, 468, 829–835. [Google Scholar] [CrossRef]
- Danielsson, A.; Barreau, K.; Kling, T.; Tisell, M.; Carén, H. Accumulation of DNA Methylation Alterations in Paediatric Glioma Stem Cells Following Fractionated Dose Irradiation. Clin. Epigenetics 2020, 12, 26. [Google Scholar] [CrossRef]
- Fan, X.; Khaki, L.; Zhu, T.S.; Soules, M.E.; Talsma, C.E.; Gul, N.; Koh, C.; Zhang, J.; Li, Y.M.; Maciaczyk, J.; et al. NOTCH Pathway Blockade Depletes CD133-Positive Glioblastoma Cells and Inhibits Growth of Tumor Neurospheres and Xenografts. Stem Cells 2010, 28, 5. [Google Scholar] [CrossRef]
- Vengoji, R.; Ponnusamy, M.P.; Rachagani, S.; Mahapatra, S.; Batra, S.K.; Shonka, N.; Macha, M.A. Novel Therapies Hijack the Blood–Brain Barrier to Eradicate Glioblastoma Cancer Stem Cells. Carcinogenesis 2019, 40, 2. [Google Scholar] [CrossRef]
- Jiang, W.; Finniss, S.; Cazacu, S.; Xiang, C.; Brodie, Z.; Mikkelsen, T.; Poisson, L.; Shackelford, D.B.; Brodie, C. Repurposing Phenformin for the Targeting of Glioma Stem Cells and the Treatment of Glioblastoma. Oncotarget 2016, 7, 56456–56470. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Sunayama, J.; Okada, M.; Watanabe, E.; Seino, S.; Shibuya, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Glioma-Initiating Cell Elimination by Metformin Activation of FOXO3 via AMPK. Stem Cells Transl. Med. 2012, 1, 811. [Google Scholar] [CrossRef] [PubMed]
- Raizer, J.J.; Abrey, L.E.; Lassman, A.B.; Chang, S.M.; Lamborn, K.R.; Kuhn, J.G.; Yung, W.K.A.; Gilbert, M.R.; Aldape, K.A.; Wen, P.Y.; et al. A Phase II Trial of Erlotinib in Patients with Recurrent Malignant Gliomas and Nonprogressive Glioblastoma Multiforme Postradiation Therapy. Neuro-Oncology 2010, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.K.; Taipale, J.; Cooper, M.K.; Beachy, P.A. Inhibition of Hedgehog Signaling by Direct Binding of Cyclopamine to Smoothened. Genes Dev. 2002, 16, 2743–2748. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Bustos, C.; Mella, J.; Soto-Delgado, J.; Salas, C.O. State of the Art of Smo Antagonists for Cancer Therapy: Advances in the Target Receptor and New Ligand Structures. Future Med. Chem. 2019, 11, 617–638. [Google Scholar] [CrossRef] [PubMed]
- Jeng, K.S.; Sheen, I.S.; Leu, C.M.; Tseng, P.H.; Chang, C.F. The Role of Smoothened in Cancer. Int. J. Mol. Sci. 2020, 21, 6863. [Google Scholar] [CrossRef] [PubMed]
- Esposito, C.L.; Nuzzo, S.; Ibba, M.L.; Ricci-Vitiani, L.; Pallini, R.; Condorelli, G.; Catuogno, S.; de Franciscis, V. Combined Targeting of Glioblastoma Stem-Like Cells by Neutralizing RNA-Bio-Drugs for STAT3. Cancers 2020, 12, 1434. [Google Scholar] [CrossRef]
- Horiguchi, A.; Asano, T.; Kuroda, K.; Sato, A.; Asakuma, J.; Ito, K.; Hayakawa, M.; Sumitomo, M.; Asano, T. STAT3 Inhibitor WP1066 as a Novel Therapeutic Agent for Renal Cell Carcinoma. Br. J. Cancer 2010, 102, 1592–1599. [Google Scholar] [CrossRef]
- Stechishin, O.D.; Luchman, H.A.; Ruan, Y.; Blough, M.D.; Nguyen, S.A.; Kelly, J.J.; Cairncross, J.G.; Weiss, S. On-Target JAK2/STAT3 Inhibition Slows Disease Progression in Orthotopic Xenografts of Human Glioblastoma Brain Tumor Stem Cells. Neuro-Oncology 2013, 15, 198–207. [Google Scholar] [CrossRef] [PubMed]
- de Groot, J.; Ott, M.; Wei, J.; Kassab, C.; Fang, D.; Najem, H.; O’Brien, B.; Weathers, S.-P.; Matsouka, C.K.; Majd, N.K.; et al. A First-in-Human Phase I Trial of the Oral p-STAT3 Inhibitor WP1066 in Patients with Recurrent Malignant Glioma. CNS Oncol. 2022, 11, CNS87. [Google Scholar] [CrossRef] [PubMed]
- Wightman, S.M.; Alban, T.J.; Chen, X.; Lathia, J.D.; Wang, Y.; Stark, G.R. Bazedoxifene Inhibits Sustained STAT3 Activation and Increases Survival in GBM. Transl. Oncol. 2021, 14, 101192. [Google Scholar] [CrossRef] [PubMed]
- Xiao, A.; Brenneman, B.; Floyd, D.; Comeau, L.; Spurio, K.; Olmez, I.; Lee, J.; Nakano, I.; Godlewski, J.; Bronisz, A.; et al. Statins Affect Human Glioblastoma and Other Cancers through TGF-β Inhibition. Oncotarget 2019, 10, 1716–1728. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Wu, C.L.; Lin, M.X.; Sze, C.I.; Gean, P.W. Disulfiram Sensitizes a Therapeutic-Resistant Glioblastoma to the TGF-β Receptor Inhibitor. Int. J. Mol. Sci. 2021, 22, 10496. [Google Scholar] [CrossRef]
- Anido, J.; Sáez-Borderías, A.; Gonzàlez-Juncà, A.; Rodón, L.; Folch, G.; Carmona, M.A.; Prieto-Sánchez, R.M.; Barba, I.; Martínez-Sáez, E.; Prudkin, L.; et al. TGF-β Receptor Inhibitors Target the CD44(High)/Id1(High) Glioma-Initiating Cell Population in Human Glioblastoma. Cancer Cell 2010, 18, 655–668. [Google Scholar] [CrossRef]
- Kuang, X.; Xiong, J.; Lu, T.; Wang, W.; Zhang, Z.; Wang, J. Inhibition of USP1 Induces Apoptosis via ID1/AKT Pathway in B-Cell Acute Lymphoblastic Leukemia Cells. Int. J. Med. Sci. 2021, 18, 245–255. [Google Scholar] [CrossRef]
- Mistry, H.; Hsieh, G.; Buhrlage, S.J.; Huang, M.; Park, E.; Cuny, G.D.; Galinsky, I.; Stone, R.M.; Gray, N.S.; D’Andrea, A.D.; et al. Small-Molecule Inhibitors of USP1 Target ID1 Degradation in Leukemic Cells. Mol. Cancer Ther. 2013, 12, 2651–2662. [Google Scholar] [CrossRef] [PubMed]
- McCord, M.; Mukouyama, Y.S.; Gilbert, M.R.; Jackson, S. Targeting WNT Signaling for Multifaceted Glioblastoma Therapy. Front. Cell. Neurosci. 2017, 11, 318. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Yue, X.; Han, L.; Yuan, X.; Shi, Z.; Huang, K.; Yang, Y.; Zou, J.; Zhang, J.; Jiang, T.; et al. Antitumor Effect of Aspirin in Glioblastoma Cells by Modulation of β-Catenin/T-Cell Factor–Mediated Transcriptional Activity: Laboratory Investigation. J. Neurosurg. 2011, 115, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Stockhammer, F.; Misch, M.; Koch, A.; Czabanka, M.; Plotkin, M.; Blechschmidt, C.; Tuettenberg, J.; Vajkoczy, P. Continuous Low-Dose Temozolomide and Celecoxib in Recurrent Glioblastoma. J. Neurooncol. 2010, 100, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Carra, E.; Barbieri, F.; Marubbi, D.; Pattarozzi, A.; Favoni, R.E.; Florio, T.; Daga, A. Sorafenib Selectively Depletes Human Glioblastoma Tumor-Initiating Cells from Primary Cultures. Cell Cycle 2013, 12, 491. [Google Scholar] [CrossRef]
- Hainsworth, J.D.; Ervin, T.; Friedman, E.; Priego, V.; Murphy, P.B.; Clark, B.L.; Lamar, R.E. Concurrent Radiotherapy and Temozolomide Followed by Temozolomide and Sorafenib in the First-Line Treatment of Patients with Glioblastoma Multiforme. Cancer 2010, 116, 3663–3669. [Google Scholar] [CrossRef] [PubMed]
- Eimer, S.; Dugay, F.; Airiau, K.; Avril, T.; Quillien, V.; Belaud-Rotureau, M.A.; Belloc, F. Cyclopamine Cooperates with EGFR Inhibition to Deplete Stem-like Cancer Cells in Glioblastoma-Derived Spheroid Cultures. Neuro-Oncology 2012, 14, 1441–1451. [Google Scholar] [CrossRef]
- Bar, E.E.; Chaudhry, A.; Lin, A.; Fan, X.; Schreck, K.; Matsui, W.; Piccirillo, S.; Vescovi, A.L.; Dimeco, F.; Olivi, A.; et al. Cyclopamine-Mediated Hedgehog Pathway Inhibition Depletes Stem-Like Cancer Cells in Glioblastoma. Stem Cells 2007, 25, 2524. [Google Scholar] [CrossRef] [PubMed]
- Bureta, C.; Saitoh, Y.; Tokumoto, H.; Sasaki, H.; Maeda, S.; Nagano, S.; Komiya, S.; Taniguchi, N.; Setoguchi, T. Synergistic Effect of Arsenic Trioxide, Vismodegib and Temozolomide on Glioblastoma. Oncol. Rep. 2019, 41, 3404–3412. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Carducci, M.A.; Sepulveda-Sánchez, J.M.; Azaro, A.; Calvo, E.; Seoane, J.; Braña, I.; Sicart, E.; Gueorguieva, I.; Cleverly, A.L.; et al. First-in-Human Dose Study of the Novel Transforming Growth Factor-β Receptor I Kinase Inhibitor LY2157299 Monohydrate in Patients with Advanced Cancer and Glioma. Clin. Cancer Res. 2015, 21, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Kleber, S.; Röhrich, M.; Timke, C.; Han, N.; Tuettenberg, J.; Martin-Villalba, A.; Debus, J.; Peschke, P.; Wirkner, U.; et al. Blockade of TGF-β Signaling by the TGFβR-I Kinase Inhibitor LY2109761 Enhances Radiation Response and Prolongs Survival in Glioblastoma. Cancer Res. 2011, 71, 7155–7167. [Google Scholar] [CrossRef]
- Sareddy, G.R.; Kesanakurti, D.; Kirti, P.B.; Babu, P.P. Nonsteroidal Anti-Inflammatory Drugs Diclofenac and Celecoxib Attenuates Wnt/β-Catenin/Tcf Signaling Pathway in Human Glioblastoma Cells. Neurochem. Res. 2013, 38, 2313–2322. [Google Scholar] [CrossRef] [PubMed]
- Ashizawa, T.; Miyata, H.; Iizuka, A.; Komiyama, M.; Oshita, C.; Kume, A.; Nogami, M.; Yagoto, M.; Ito, I.; Oishi, T.; et al. Effect of the STAT3 Inhibitor STX-0119 on the Proliferation of Cancer Stem-like Cells Derived from Recurrent Glioblastoma. Int. J. Oncol. 2013, 43, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Soroceanu, L.; Singer, E.; Dighe, P.; Sidorov, M.; Limbad, C.; Rodriquez-Brotons, A.; Rix, P.; Woo, R.W.L.; Dickinson, L.; Desprez, P.-Y.; et al. Cannabidiol Inhibits RAD51 and Sensitizes Glioblastoma to Temozolomide in Multiple Orthotopic Tumor Models. Neuro-Oncol. Adv. 2022, 4, vdac019. [Google Scholar] [CrossRef]
- Rath, B.H.; Camphausen, K.; Tofilon, P.J. Glioblastoma Radiosensitization by Pimozide. Transl. Cancer Res. 2016, 5, S1029. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, A.A.; Field, M.; Bushnev, S.; Longo, M.S.; Sugaya, K. The Effects of Histone Deacetylase Inhibitors on Glioblastoma-Derived Stem Cells. J. Mol. Neurosci. 2015, 55, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, O.; Teresa Gentile, M.; Mancini, A.; del Gaudio, N.; di Costanzo, A.; Bajetto, A.; Franco, P.; Altucci, L.; Florio, T.; Patrizia Stoppelli, M.; et al. Histone Deacetylase Inhibitors Impair Vasculogenic Mimicry from Glioblastoma Cells. Cancers 2019, 11, 747. [Google Scholar] [CrossRef] [PubMed]
- Göttlicher, M.; Minucci, S.; Zhu, P.; Krämer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; lo Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic Acid Defines a Novel Class of HDAC Inhibitors Inducing Differentiation of Transformed Cells. EMBO J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, G.D.; Eljamel, S. Impact of Particular Antiepileptic Drugs on the Survival of Patients with Glioblastoma Multiforme. J. Neurosurg. 2013, 118, 859–865. [Google Scholar] [CrossRef]
- Kerkhof, M.; Dielemans, J.C.M.; van Breemen, M.S.; Zwinkels, H.; Walchenbach, R.; Taphoorn, M.J.; Vecht, C.J. Effect of Valproic Acid on Seizure Control and on Survival in Patients with Glioblastoma Multiforme. Neuro-Oncology 2013, 15, 961–967. [Google Scholar] [CrossRef]
- Asklund, T.; Kvarnbrink, S.; Holmlund, C.; Wibom, C.; Bergenheim, T.; Henriksson, R.; Hedman, H. Synergistic Killing of Glioblastoma Stem-like Cells by Bortezomib and HDAC Inhibitors. Anticancer. Res. 2012, 32, 2407–2413. [Google Scholar]
- Singh, M.M.; Manton, C.A.; Bhat, K.P.; Tsai, W.W.; Aldape, K.; Barton, M.C.; Chandra, J. Inhibition of LSD1 Sensitizes Glioblastoma Cells to Histone Deacetylase Inhibitors. Neuro-Oncology 2011, 13, 894–903. [Google Scholar] [CrossRef]
- Yu, T.; Wang, Y.; Hu, Q.; Wu, W.N.; Wu, Y.; Wei, W.; Han, D.; You, Y.; Lin, N.; Liu, N. The EZH2 Inhibitor GSK343 Suppresses Cancer Stem-like Phenotypes and Reverses Mesenchymal Transition in Glioma Cells. Oncotarget 2017, 8, 98348–98359. [Google Scholar] [CrossRef]
- Jin, X.; Kim, L.J.Y.; Wu, Q.; Wallace, L.C.; Prager, B.C.; Sanvoranart, T.; Gimple, R.C.; Wang, X.; Mack, S.C.; Miller, T.E.; et al. Targeting Glioma Stem Cells through Combined BMI1 and EZH2 Inhibition. Nat. Med. 2017, 23, 1352. [Google Scholar] [CrossRef]
- Wong, H.-K.A.; El Fatimy, R.; Onodera, C.; Wei, Z.; Yi, M.; Mohan, A.; Gowrisankaran, S.; Karmali, P.; Marcusson, E.; Wakimoto, H.; et al. The Cancer Genome Atlas Analysis Predicts MicroRNA for Targeting Cancer Growth and Vascularization in Glioblastoma. Mol. Ther. 2015, 23, 1234. [Google Scholar] [CrossRef] [PubMed]
- Esposito, C.L.; Nuzzo, S.; Kumar, S.A.; Rienzo, A.; Lawrence, C.L.; Pallini, R.; Shaw, L.; Alder, J.E.; Ricci-Vitiani, L.; Catuogno, S.; et al. A Combined MicroRNA-Based Targeted Therapeutic Approach to Eradicate Glioblastoma Stem-like Cells. J. Control. Release 2016, 238, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Ernst, A.; Campos, B.; Meier, J.; Devens, F.; Liesenberg, F.; Wolter, M.; Reifenberger, G.; Herold-Mende, C.; Lichter, P.; Radlwimmer, B. De-Repression of CTGF via the MiR-17-92 Cluster upon Differentiation of Human Glioblastoma Spheroid Cultures. Oncogene 2010, 29, 3411–3422. [Google Scholar] [CrossRef] [PubMed]
- Kalkan, R. Glioblastoma Stem Cells as a New Therapeutic Target for Glioblastoma. Clin. Med. Insights Oncol. 2015, 9, 95. [Google Scholar] [CrossRef]
- Kim, M.M.; Umemura, Y.; Leung, D. Bevacizumab and Glioblastoma: Past, Present, and Future Directions. Cancer J. 2018, 24, 180–186. [Google Scholar] [CrossRef]
- Diaz, R.J.; Ali, S.; Qadir, M.G.; de La Fuente, M.I.; Ivan, M.E.; Komotar, R.J. The Role of Bevacizumab in the Treatment of Glioblastoma. J. Neurooncol. 2017, 133, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Tipping, M.; Eickhoff, J.; Ian Robins, H. Clinical Outcomes in Recurrent Glioblastoma with Bevacizumab Therapy: An Analysis of the Literature. J. Clin. Neurosci. 2017, 44, 101–106. [Google Scholar] [CrossRef]
- Ramezani, S.; Vousooghi, N.; Kapourchali, F.R.; Hadjighasem, M.; Hayat, P.; Amini, N.; Joghataei, M.T. Rolipram Potentiates Bevacizumab-Induced Cell Death in Human Glioblastoma Stem-like Cells. Life Sci. 2017, 173, 11–19. [Google Scholar] [CrossRef]
- Piao, Y.; Liang, J.; Holmes, L.; Zurita, A.J.; Henry, V.; Heymach, J.V.; De Groot, J.F. Glioblastoma Resistance to Anti-VEGF Therapy Is Associated with Myeloid Cell Infiltration, Stem Cell Accumulation, and a Mesenchymal Phenotype. Neuro-Oncology 2012, 14, 1379–1392. [Google Scholar] [CrossRef] [PubMed]
- Piao, Y.; Liang, J.; Holmes, L.; Henry, V.; Sulman, E.; de Groot, J.F. Acquired Resistance to Anti-VEGF Therapy in Glioblastoma Is Associated with a Mesenchymal Transition. Clin. Cancer Res. 2013, 19, 4392–4403. [Google Scholar] [CrossRef]
- Cruz Da Silva, E.; Mercier, M.C.; Etienne-selloum, N.; Dontenwill, M.; Choulier, L. A Systematic Review of Glioblastoma-Targeted Therapies in Phases II, III, IV Clinical Trials. Cancers 2021, 13, 1795. [Google Scholar] [CrossRef]
- Gatto, L.; Franceschi, E.; Tosoni, A.; di Nunno, V.; Maggio, I.; Lodi, R.; Brandes, A.A. IDH Inhibitors and Beyond: The Cornerstone of Targeted Glioma Treatment. Mol. Diagn. Ther. 2021, 25, 457–473. [Google Scholar] [CrossRef] [PubMed]
- Pelaz, S.G.; Jaraíz-Rodríguez, M.; Álvarez-Vázquez, A.; Talaverón, R.; García-Vicente, L.; Flores-Hernández, R.; Gómez de Cedrón, M.; Tabernero, M.; Ramírez de Molina, A.; Lillo, C.; et al. Targeting Metabolic Plasticity in Glioma Stem Cells in Vitro and in Vivo through Specific Inhibition of C-Src by TAT-Cx43 266–283. EBioMedicine 2020, 62, 103134. [Google Scholar] [CrossRef]
- Wang, X.; Yang, K.; Wu, Q.; Kim, L.J.Y.; Morton, A.R.; Gimple, R.C.; Prager, B.C.; Shi, Y.; Zhou, W.; Bhargava, S.; et al. Targeting Pyrimidine Synthesis Accentuates Molecular Therapy Response in Glioblastoma Stem Cells. Sci. Transl. Med. 2019, 11, 4972. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Murphy, B.; Miller, R.; Menon, V.; Banik, N.L.; Giglio, P.; Lindhorst, S.M.; Varma, A.K.; Vandergrift, W.A.; Patel, S.J.; et al. Mechanisms and Clinical Significance of Histone Deacetylase Inhibitors: Epigenetic Glioblastoma Therapy. Anticancer Res. 2015, 35, 615–625. [Google Scholar] [PubMed]
- Hersh, A.M.; Antar, A.; Pennington, Z.; Aygun, N.; Patel, J.; Goldsborough, E.; Porras, J.L.; Elsamadicy, A.A.; Lubelski, D.; Wolinsky, J.-P.; et al. Predictors of Survival and Time to Progression Following Operative Management of Intramedullary Spinal Cord Astrocytomas. J. Neuro-Oncol. 2022, 158, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Mei, L.; Yu, Q.; Xu, C.; Qiu, Y.; Yang, Y.; Shi, K.; Zhang, Q.; Gao, H.; Zhang, Z.; et al. Multifunctional Tandem Peptide Modified Paclitaxel-Loaded Liposomes for the Treatment of Vasculogenic Mimicry and Cancer Stem Cells in Malignant Glioma. ACS Appl. Mater. Interfaces 2015, 7, 16792–16801. [Google Scholar] [CrossRef]
- Bozzato, E.; Bastiancich, C.; Préat, V. Nanomedicine: A Useful Tool against Glioma Stem Cells. Cancers 2021, 13, 9. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Signaling Pathway | Inhibitor | Mechanism of Action | Effects of Inhibition | Clinical Trials | References |
|---|---|---|---|---|---|
| Notch | γ-secretase inhibitors (eg RO4929097, DAPT) | Inhibits proteolytic release of the Notch intracellular domain | Reduces neurosphere growth and GSC markers, prevents tumor growth and improves survival in vivo | NCT01122901, NCT01189240, NCT01269411 | [239] |
| EGFR, PI3K/Akt/mTOR | Metformin | Induces metabolic stress → activates AMPK → inhibits mTOR, activates tumor suppressor FOX03 silenced by signaling pathway | Induces GSC differentiation, inhibits tumor formation and proliferation, improves survival | NCT03243851, NCT02780024, NCT04945148 | [219] |
| Phenformin | Inhibits GSC self-renewal, increases miRNA expression, inhibits tumor growth and improves survival in vivo | [241] | |||
| Sorafenib | Inhibits receptor tyrosine kinases and PI3K/Akt signaling molecules | Reduces stemness markers, induces apoptosis in GSCs; however, use alongside TMZ did not improve efficacy | NCT00544817, NCT00445588, NCT00884416 | [260,261] | |
| Erlotinib | EGFRvIII inhibitor | Reduces GSC proliferation, synergistic effects with cyclopamine | NCT00301418, NCT01110876, NCT00039494, NCT00274833, NCT00387894 | [243,262] | |
| Shh | Cyclopamine | Smo inhibitor | Reduced tumor growth in vitro, depleted GSC population, pretreatment blocked GBM growth in vivo | [93,244,263] | |
| Vismodegib | Smo inhibitor | Anti-tumor agent for solid tumors and medulloblastoma; treatment with arsenic trioxide and TMZ reduced GBM growth in vivo | NCT00980343, NCT03158389 | [264] | |
| Sonidegib (LDE225) | Smo inhibitor | Downregulated Ptch1 and Gli1, delayed GBM growth in vivo, CD133+ cells most sensitive | NCT01576666—included GBM and several advanced solid tumors | [95] | |
| TGF-β | Statins (eg simvastatin, atorvastatin) | Smad3 inhibition | Inhibited GSC growth and prolonged survival in vivo | NCT02029573—examined atorvastatin | [252] |
| Galunisertib (LY2157299) | TGF-β receptor I inhibitor | Disulfiram sensitizes resistant GBM to galunisertib; Phase I study showed clinical benefit in 12/56 patients | NCT01220271 | [253,265] | |
| LY2109761 | Decreases CD44 marker and tumor growth | [254,266] | |||
| Wnt | Non-steroidal anti-inflammatories | Phosphorylates β-catenin → degradation | Diclofenac and celecoxib reduce GBM proliferation in vitro, downregulate β-catenin activity, | NCT00047281, NCT00112502 | [259,267] |
| STAT3 | WP1066 | STAT3 inhibitor | Potentiated effects of radiation, improves survival in vivo | NCT01904123 | [123,249,250] |
| Bazedoxifene | IL-6 receptor inhibitor | Decreases GSC self-renewal capacity, improves survival in vivo | [251] | ||
| Stattic | SH2 domain of STAT3 inhibitor | Sensitizes tumor to radiation and TMZ | [123,130] | ||
| STA-21 | Inhibit STAT3 binding to DNA, prevent neurosphere formation, decrease proliferation | [132] | |||
| S31-201 | |||||
| STX-0119 | STAT3 dimerization inhibitor | Inhibits expression of STAT3 target genes, induces apoptosis, inhibits GSC growth | [268] | ||
| ID-1 | Cannabidiol | Downregulates ID-1 | Reduces GBM invasiveness and self-renewal in vivo, sensitizes GBM to TMZ | NCT01812616, NCT03607643, NCT03529448 | [135,269] |
| Pimozide | Impairs ID-1 deubiquitination → ID-1 degradation | Sensitizes GBM to TMZ and RT, prolongs time to recurrence in vivo | [136,270] | ||
| LY2109761 | TGF-β receptor I inhibitor → reduces ID1 and ID3 | Inhibits GBM growth | [254,266] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hersh, A.M.; Gaitsch, H.; Alomari, S.; Lubelski, D.; Tyler, B.M. Molecular Pathways and Genomic Landscape of Glioblastoma Stem Cells: Opportunities for Targeted Therapy. Cancers 2022, 14, 3743. https://doi.org/10.3390/cancers14153743
Hersh AM, Gaitsch H, Alomari S, Lubelski D, Tyler BM. Molecular Pathways and Genomic Landscape of Glioblastoma Stem Cells: Opportunities for Targeted Therapy. Cancers. 2022; 14(15):3743. https://doi.org/10.3390/cancers14153743
Chicago/Turabian StyleHersh, Andrew M., Hallie Gaitsch, Safwan Alomari, Daniel Lubelski, and Betty M. Tyler. 2022. "Molecular Pathways and Genomic Landscape of Glioblastoma Stem Cells: Opportunities for Targeted Therapy" Cancers 14, no. 15: 3743. https://doi.org/10.3390/cancers14153743
APA StyleHersh, A. M., Gaitsch, H., Alomari, S., Lubelski, D., & Tyler, B. M. (2022). Molecular Pathways and Genomic Landscape of Glioblastoma Stem Cells: Opportunities for Targeted Therapy. Cancers, 14(15), 3743. https://doi.org/10.3390/cancers14153743