
The Role of Citrate Homeostasis in Merkel Cell Carcinoma Pathogenesis

 and
and 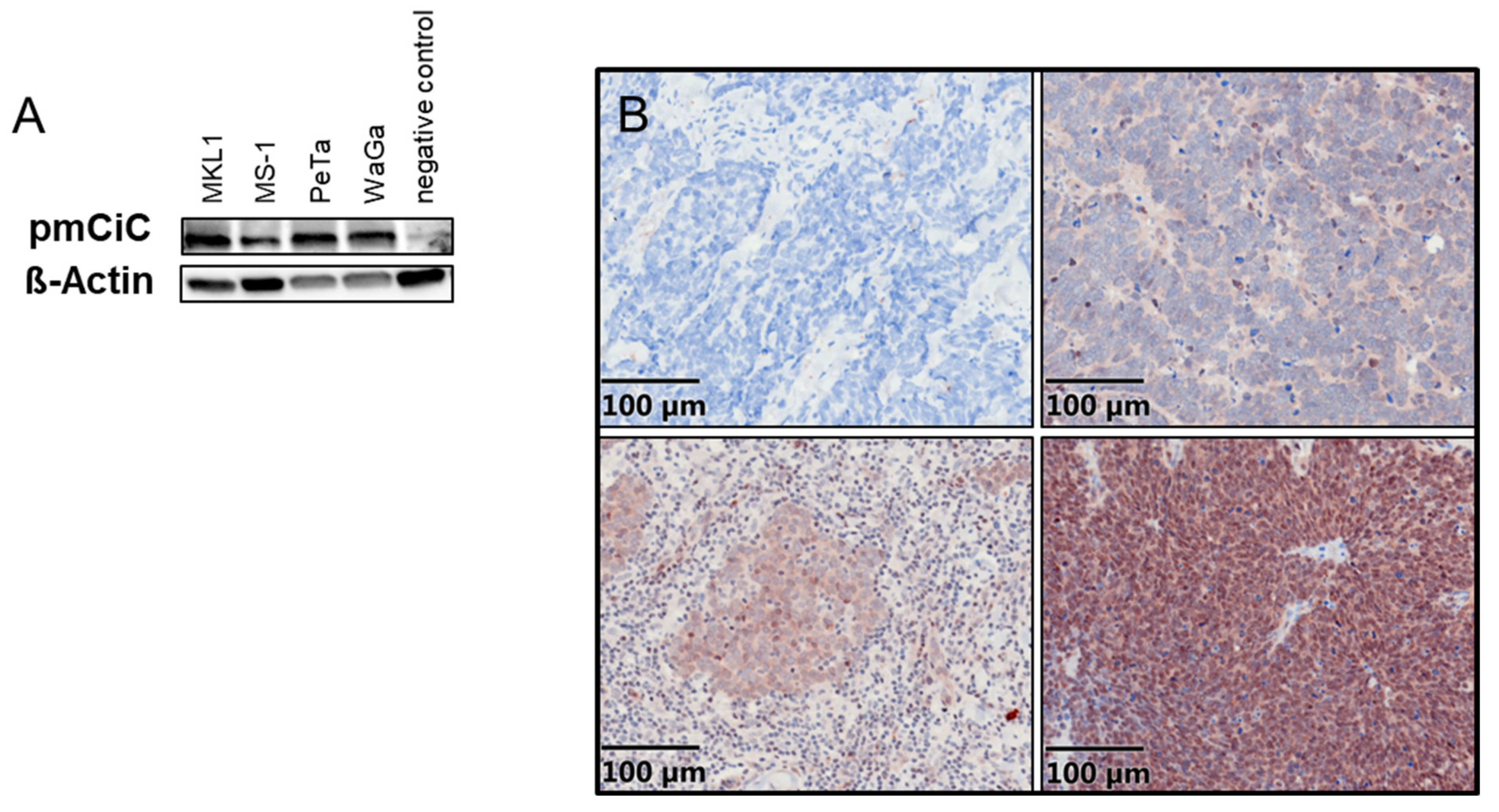{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Expression of PmCiC in Merkel Cell Carcinoma Cells and Merkel Cell Carcinoma Tissues
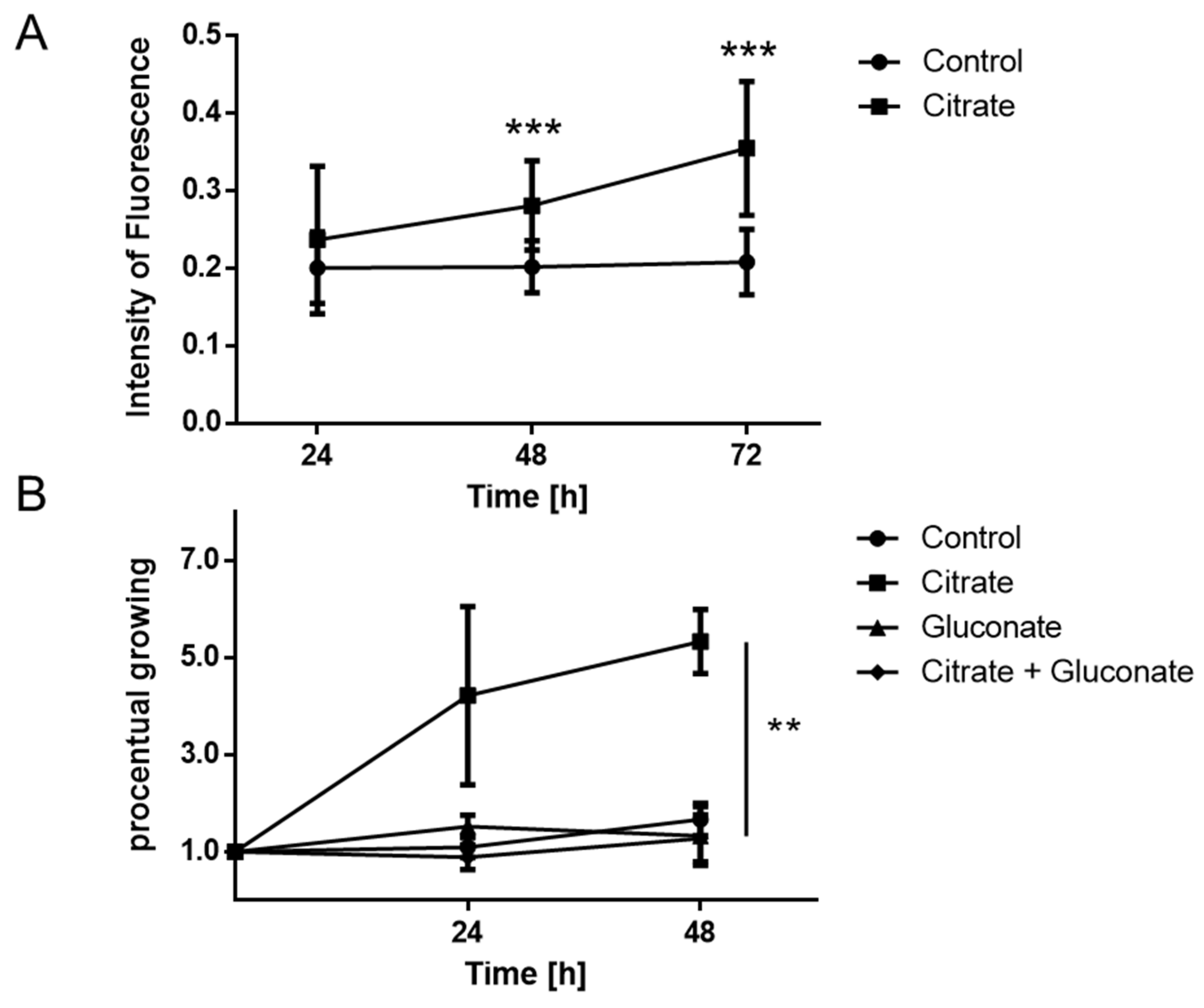
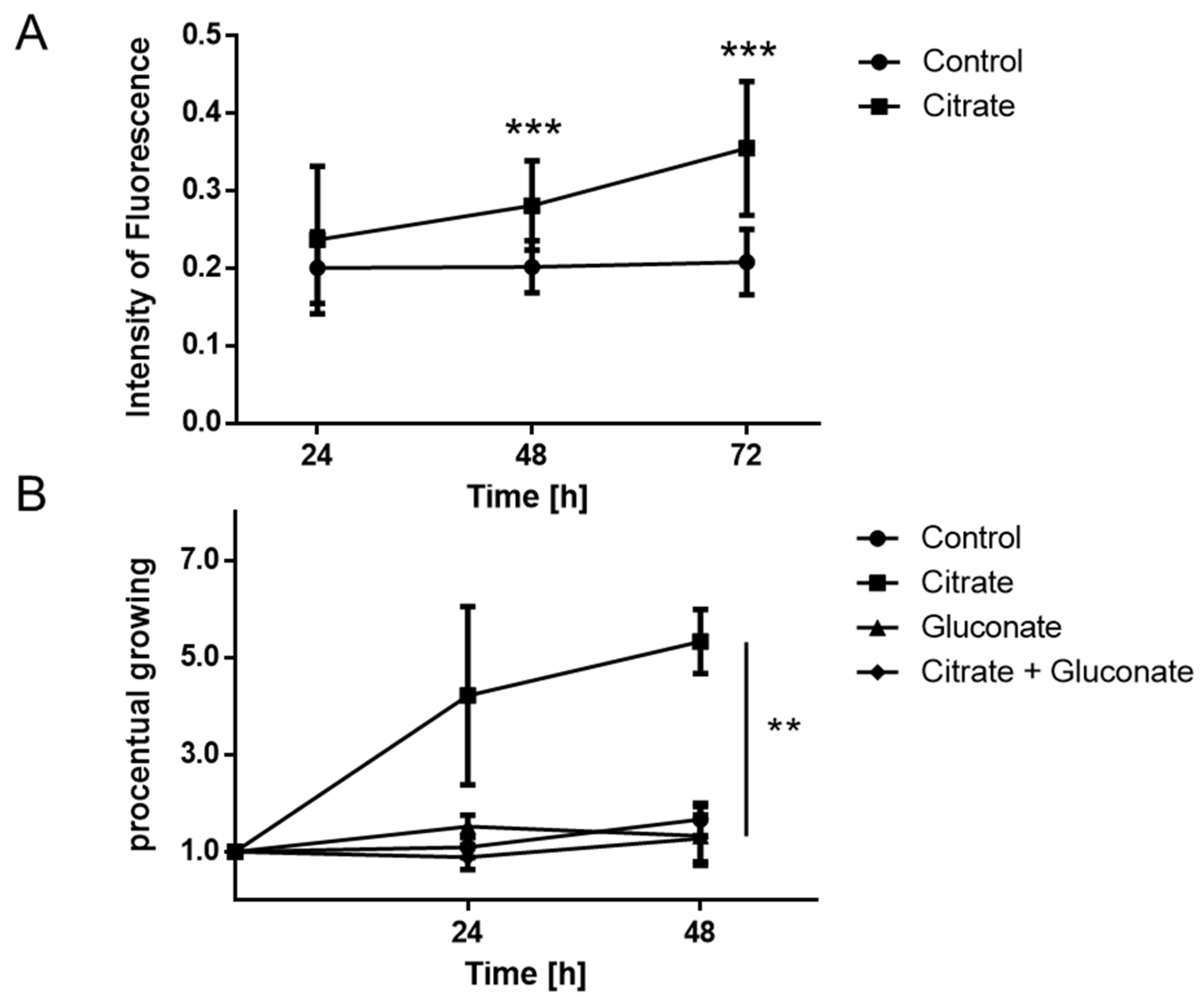
2.2. Proliferation of MCCs Is Increased in the Presence of Extracellular Citrate
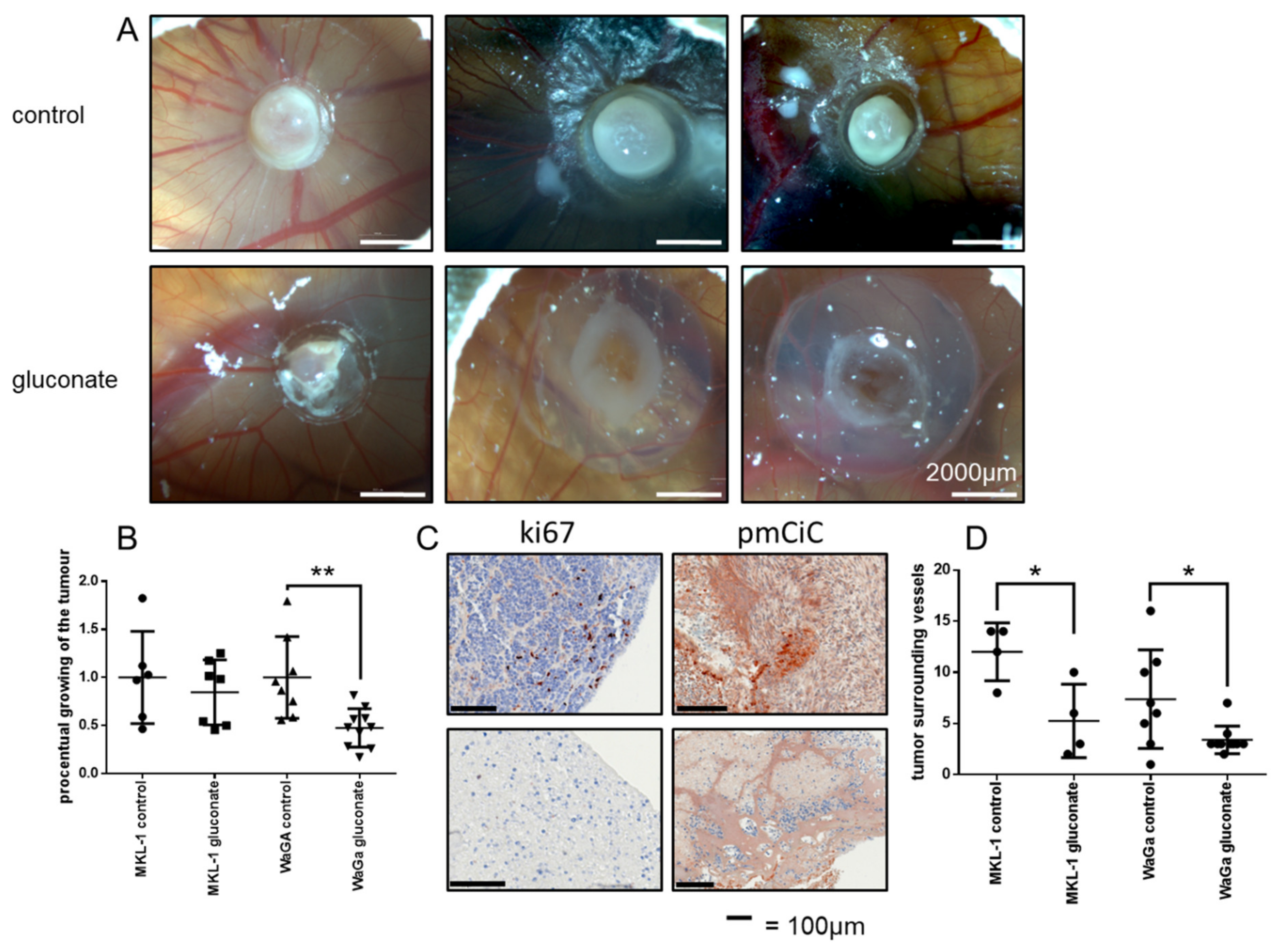
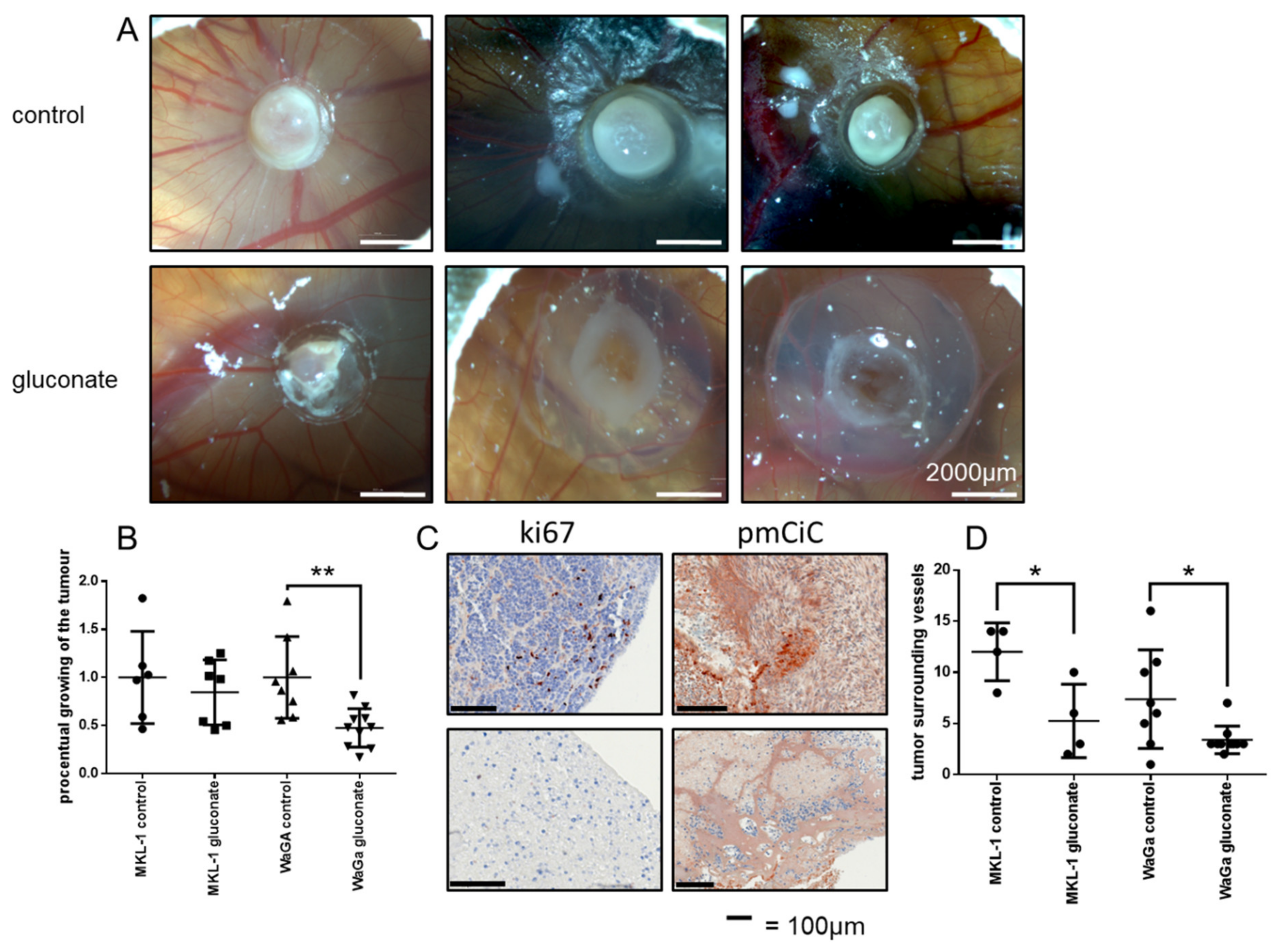
2.3. Gluconate Inhibits Growth of Merkel Cell Carcinomas In Vivo
3. Discussion
4. Materials and Methods
4.1. Immunohistochemistry
4.2. Cell Culture
4.3. Chick Chorioallantoic Membrane Model
4.4. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lin, Z.; McDermott, A.; Shao, L.; Kannan, A.; Morgan, M.; Stack, B.C.; Moreno, M.; Davis, D.A.; A Cornelius, L.; Gao, L. Chronic mTOR activation promotes cell survival in Merkel cell carcinoma. Cancer Lett. 2014, 344, 272–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulitzer, M. Merkel Cell Carcinoma. Surg. Pathol. Clin. 2017, 10, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.C.; Stang, A.; DeCaprio, J.A.; Cerroni, L.; Lebbé, C.; Veness, M.; Nghiem, P. Merkel cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17077. [Google Scholar] [CrossRef] [PubMed]
- Mauzo, S.H.; Ferrarotto, R.; Bell, D.; Torres-Cabala, C.A.; Tetzlaff, M.T.; Prieto, V.G.; Aung, P. Molecular characteristics and potential therapeutic targets in Merkel cell carcinoma. J. Clin. Pathol. 2016, 69, 382–390. [Google Scholar] [CrossRef]
- Jankowski, M.; Kopinski, P.; Schwartz, R.; Czajkowski, R. Merkel cell carcinoma: Is this a true carcinoma? Exp. Dermatol. 2014, 23, 792–794. [Google Scholar] [CrossRef]
- Fukuhara, M.; Agnarsdóttir, M.; Edqvist, P.-H.; Coter, A.; Ponten, F. SATB2 is expressed in Merkel cell carcinoma. Arch. Dermatol. Res. 2016, 308, 449–454. [Google Scholar] [CrossRef]
- Cassler, N.M.; Merrill, D.; Bichakjian, C.K.; Brownell, I. Merkel Cell Carcinoma Therapeutic Update. Curr. Treat. Options Oncol. 2016, 17, 36. [Google Scholar] [CrossRef] [Green Version]
- Gaiser, M.R.; Bongiorno, M.; Brownell, I. PD-L1 inhibition with avelumab for metastatic Merkel cell carcinoma. Expert Rev. Clin. Pharmacol. 2018, 11, 345–359. [Google Scholar] [CrossRef] [Green Version]
- Nazemi, M.; Rainero, E. Cross-Talk Between the Tumor Microenvironment, Extracellular Matrix, and Cell Metabolism in Cancer. Front. Oncol. 2020, 10, 239. [Google Scholar] [CrossRef] [Green Version]
- Mycielska, M.E.; Dettmer, K.; Rümmele, P.; Schmidt, K.; Prehn, C.; Milenkovic, V.M.; Jagla, W.; Madej, G.M.; Lantow, M.; Schladt, M.; et al. Extracellular Citrate Affects Critical Elements of Cancer Cell Metabolism and Supports Cancer Development In Vivo. Cancer Res. 2018, 78, 2513–2523. [Google Scholar] [CrossRef] [Green Version]
- Drexler, K.; Schmidt, K.M.; Jordan, K.; Federlin, M.; Milenkovic, V.M.; Liebisch, G.; Artati, A.; Schmidl, C.; Madej, G.; Tokarz, J.; et al. Cancer-associated cells release citrate to support tumour metastatic progression. Life Sci. Alliance 2021, 4, e202000903. [Google Scholar] [CrossRef]
- Mycielska, M.E.; Milenkovic, V.M.; Wetzel, C.H.; Rümmele, P.; Geissler, E. Extracellular Citrate in Health and Disease. Curr. Mol. Med. 2015, 15, 884–891. [Google Scholar] [CrossRef]
- Mycielska, M.E.; Mohr, M.T.J.; Schmidt, K.; Drexler, K.; Rümmele, P.; Haferkamp, S.; Schlitt, H.J.; Gaumann, A.; Adamski, J.; Geissler, E.K. Potential Use of Gluconate in Cancer Therapy. Front. Oncol. 2019, 9, 522. [Google Scholar] [CrossRef]
- Bhat, V.K.; Krump, C.; Bernhart, E.; Becker, J.C.; Sattler, W.; Ghaffari-Tabrizi-Wizsy, N. A short-term in vivo model for Merkel Cell Carcinoma. Exp. Dermatol. 2018, 27, 684–687. [Google Scholar] [CrossRef] [Green Version]
- Röhrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef]
- Icard, P.; Poulain, L.; Lincet, H. Understanding the central role of citrate in the metabolism of cancer cells. Biochim. Biophys. Acta 2012, 1825, 111–116. [Google Scholar] [CrossRef]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.-K.; Jang, H.G.; Jha, A.K.; et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef] [Green Version]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371.e9. [Google Scholar] [CrossRef] [Green Version]
- Haferkamp, S.; Drexler, K.; Federlin, M.; Schlitt, H.J.; Berneburg, M.; Adamski, J.; Gaumann, A.; Geissler, E.K.; Ganapathy, V.; Parkinson, E.K.; et al. Extracellular Citrate Fuels Cancer Cell Metabolism and Growth. Front. Cell Dev. Biol. 2020, 8, 602476. [Google Scholar] [CrossRef]
- Kumar, A.; Cordes, T.; Thalacker-Mercer, A.E.; Pajor, A.M.; Murphy, A.N.; Metallo, C.M. NaCT/SLC13A5 facilitates citrate import and metabolism under nutrient-limited conditions. Cell Rep. 2021, 36, 109701. [Google Scholar] [CrossRef]
- Schlüter, C.; Duchrow, M.; Wohlenberg, C.; Becker, M.H.; Key, G.; Flad, H.D.; Gerdes, J. The Cell Proliferation-associated Antigen of Antibody Ki-67: A Very Large, Ubiquitous Nuclear Protein with Numerous Repeated Elements, Representing a New Kind of Cell Cycle-maintaining Proteins. J. CeU Biol. 1993, 123, 513–522. [Google Scholar] [CrossRef]
- Kohl, C.; Aung, T.; Haerteis, S.; Ignatov, A.; Ortmann, O.; Papathemelis, T. The 3D in vivo chorioallantoic membrane model and its role in breast cancer research. J. Cancer Res. Clin. Oncol. 2022, 148, 1033–1043. [Google Scholar] [CrossRef]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. New Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar]
- Unterleuthner, D.; Neuhold, P.; Schwarz, K.; Janker, L.; Neuditschko, B.; Nivarthi, H.; Crncec, I.; Kramer, N.; Unger, C.; Hengstschläger, M.; et al. Cancer-associated fibroblast-derived WNT2 increases tumor angiogenesis in colon cancer. Angiogenesis 2020, 23, 159–177. [Google Scholar] [CrossRef] [Green Version]
- Pion, E.; Asam, C.; Feder, A.-L.; Felthaus, O.; Heidekrueger, P.I.; Prantl, L.; Haerteis, S.; Aung, T. Laser speckle contrast analysis (LASCA) technology for the semiquantitative measurement of angiogenesis in in-ovo-tumor-model. Microvasc. Res. 2021, 133, 104072. [Google Scholar] [CrossRef]
- Kohl, C.; Aung, T.; Haerteis, S.; Papathemelis, T. Assessment of breast cancer primary tumor material in a 3D in vivo model. Clin. Hemorheol. Microcirc. 2021, 79, 157–166. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drexler, K.; Schwertner, B.; Haerteis, S.; Aung, T.; Berneburg, M.; Geissler, E.K.; Mycielska, M.E.; Haferkamp, S. The Role of Citrate Homeostasis in Merkel Cell Carcinoma Pathogenesis. Cancers 2022, 14, 3425. https://doi.org/10.3390/cancers14143425
Drexler K, Schwertner B, Haerteis S, Aung T, Berneburg M, Geissler EK, Mycielska ME, Haferkamp S. The Role of Citrate Homeostasis in Merkel Cell Carcinoma Pathogenesis. Cancers. 2022; 14(14):3425. https://doi.org/10.3390/cancers14143425
Chicago/Turabian StyleDrexler, Konstantin, Barbara Schwertner, Silke Haerteis, Thiha Aung, Mark Berneburg, Edward K. Geissler, Maria E. Mycielska, and Sebastian Haferkamp. 2022. "The Role of Citrate Homeostasis in Merkel Cell Carcinoma Pathogenesis" Cancers 14, no. 14: 3425. https://doi.org/10.3390/cancers14143425
APA StyleDrexler, K., Schwertner, B., Haerteis, S., Aung, T., Berneburg, M., Geissler, E. K., Mycielska, M. E., & Haferkamp, S. (2022). The Role of Citrate Homeostasis in Merkel Cell Carcinoma Pathogenesis. Cancers, 14(14), 3425. https://doi.org/10.3390/cancers14143425