
Cell-by-Cell: Unlocking Lung Cancer Pathogenesis


Abstract
Simple Summary
Abstract
1. Introduction
2. Epithelial Culprits of LUAD Transformation
2.1. LUAD Cell-of-Origin
2.2. Single-Cell Sequencing Studies Open a Pandora’s Box of Pulmonary Cellular Heterogeneity
3. LUAD Epithelial Plasticity: In Times of Stress, Lineage Commitment Is a Rare Feat
4. Intratumor Heterogeneity at the Single-Cell Level: One Size Does Not Fit All

{kind=link}
{kind=link}
{kind=link}
| LUAD Phenotypes and Features | Disease Stage/Model | Major Discoveries | Ref. |
|---|---|---|---|
| Lineage commitment, plasticity | AAHs, LUADs | AT2 lineage-divergent subset appears in AAHs and up to LUAD development, activated metabolic and stem-cell-like programs. | [40] |
| Advanced stage LUADs | AT2 subset with malignant properties and potential to transition to tumor cells. | [41] | |
| In vivo tracing in Kras; Trp53-driven mouse model of LUAD | Clonal evolution with divergent lineage plasticity programs. | [42] | |
| Metastatic LUADs | Altered ciliated and alveolar differentiation programs. | [45] | |
| Alveolar intermediary/ regenerative states | Late-stage LUADs in response to targeted therapy | New potentially targetable oncogenes. Residual malignant cells are in a primitive regenerative alveolar state. | [43] |
| LUADs | Alveolar and bronchial regenerative state mimicking response to injury. | [44] | |
| Intratumor heterogeneity (ITH) in epithelial, immune, or stromal subsets, and relevance to clinical outcomes and therapy response | Enriched epithelial subsets from LUADs and multiple spatially defined normal tissues | ITH in epithelial lineage plasticity and distinct tumor cells of origin, and ITH in TME that evolved with increasing tumor proximity. | [53,58] |
| Normal lung, LUADs, and metastases | Early pro-tumoral and immunosuppressive TME alterations (stromal and immune) are sustained until later stages. | [45] | |
| Tumor infiltrating myeloid cells in NSCLCs | Comprehensive catalogue of distinct myeloid populations (e.g., neutrophil and DC subsets) linked to survival outcomes. | [66] | |
| NSCLCs including LUADs | ITH is highly linked to tumor-resident neutrophil subpopulations. | [41] | |
| scRNA-seq of low mRNA neutrophil subsets in NSCLCs | Neutrophil signature associated with poor response to ICB. | [67] | |
| scRNA-seq and Cite-Seq in NSCLCs | A prognostically-relevant module for ICB patients based on tumor mutational burden, ectopic antigens, and driver mutations. | [68] | |
| NSCLCs including LUADs | CD8+ T cell pre-exhausted subset associated with better prognosis. | [69] | |
| NSCLCs including LUADs | Increased CD8+ terminally differentiated effector memory or effector cells in tumors. T cell signatures correlated with improved survival. | [70] | |
| LUADs | 52 distinct stromal subtypes, some correlated to patient survival and/or tumor stage. | [71] | |
| LUADs | ITH in tumor histologies, oncogene pathways, and TME patterns signifying distinct prognostic outcomes. | [72] | |
| NSCLCs including LUADs receiving ICB | Responsive tumors associated with precursor exhausted CD8+ T cells with low expression of coinhibitory molecules and high GZMK. | [73] | |
| Late-stage LUAD response to targeted therapy | TME heterogeneity within and across tumors. | [43] | |
| Metastatic LUADs | NK cells sculpt developmental and epithelial plasticity throughout metastatic progression. | [44] | |
| Early and advanced stage NSCLCs including LUADs | ITH resulting from distinct B cell subtypes (e.g., naïve- or plasma-like B cells) linked to progression and clinical implications in early or late stages. | [74] |
5. The Urgency in Premalignancy
5.1. LUAD’s Earliest Known Preneoplastic Lesions
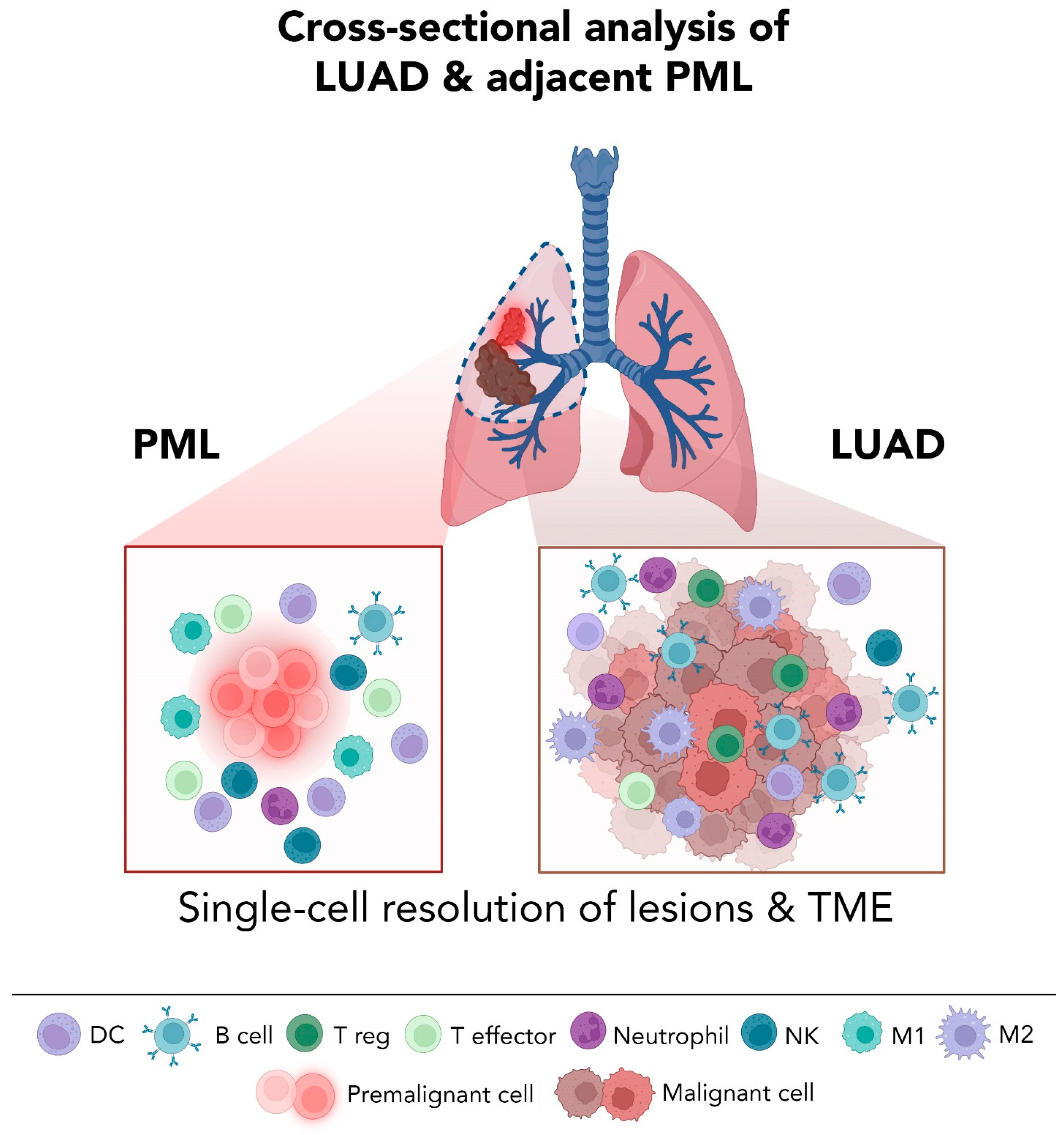
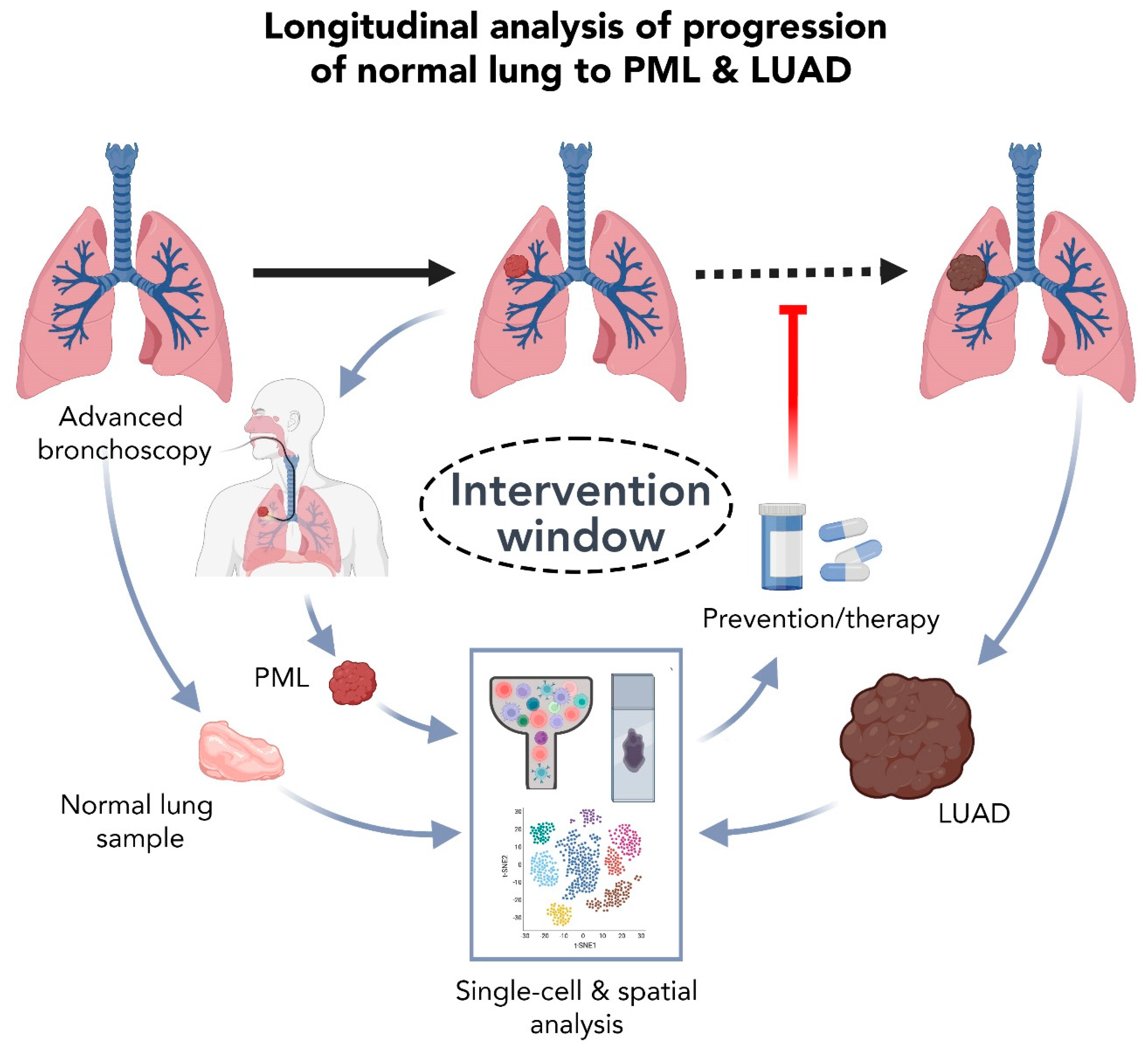
5.2. High Resolution Analysis of Premalignant LUAD Lesions: Promises and Challenges
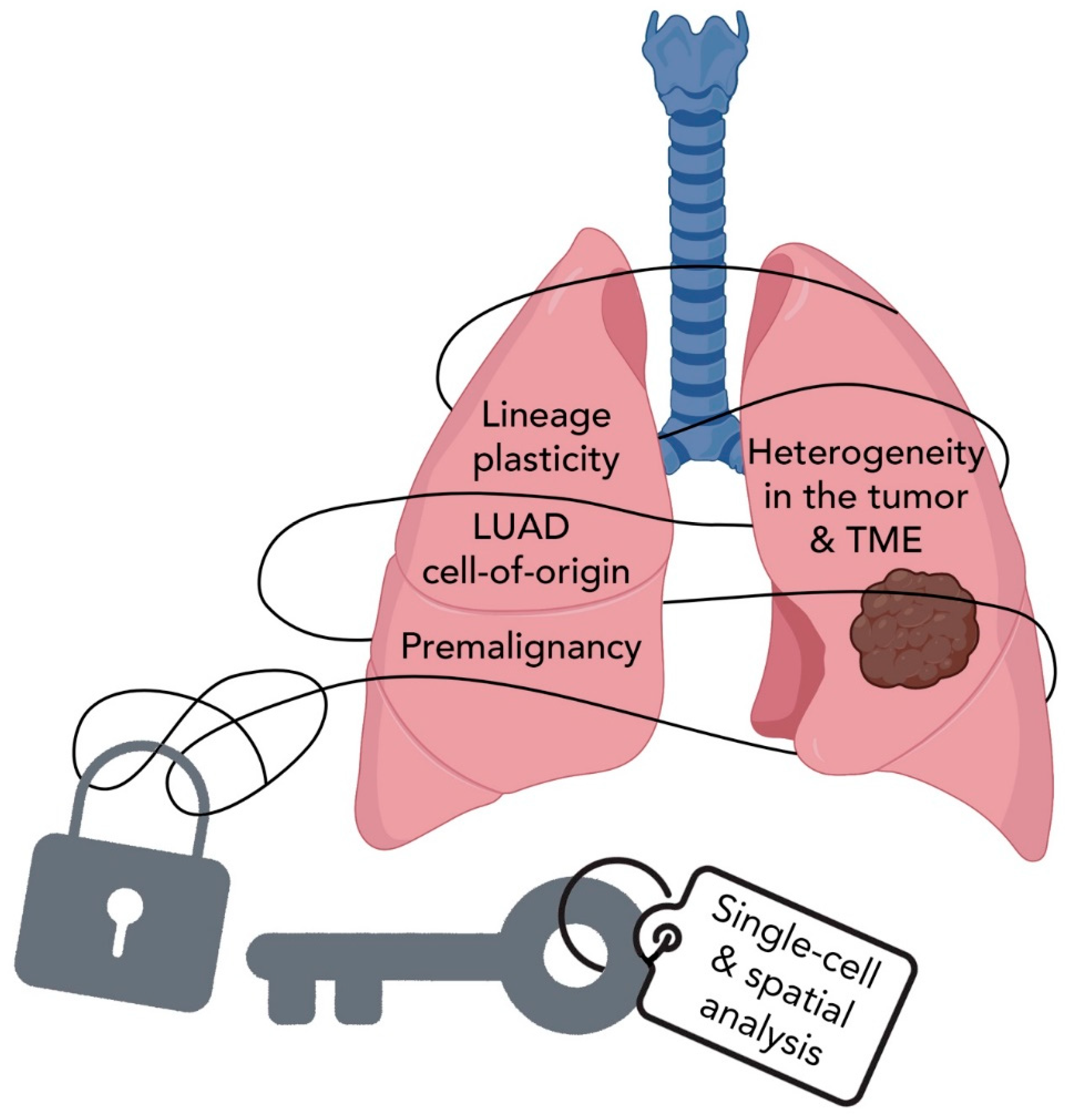
6. Summary and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Goldstraw, P.; Ball, D.; Jett, J.R.; Le Chevalier, T.; Lim, E.; Nicholson, A.G.; Shepherd, F.A. Non-small-cell lung cancer. Lancet 2011, 378, 1727–1740. [Google Scholar] [CrossRef]
- Herbst, R.S.; Heymach, J.V.; Lippman, S.M. Lung cancer. N. Engl. J. Med. 2008, 359, 1367–1380. [Google Scholar] [CrossRef]
- Wistuba, I.I.; Gazdar, A.F. Lung cancer preneoplasia. Annu. Rev. Pathol. 2006, 1, 331–348. [Google Scholar] [CrossRef] [PubMed]
- Aberle, D.R.; Adams, A.M.; Berg, C.D.; Black, W.C.; Clapp, J.D.; Fagerstrom, R.M.; Gareen, I.F.; Gatsonis, C.; Marcus, P.M.; Sicks, J.D. Reduced lung-cancer mortality with low-dose computed tomographic screening. N. Engl. J. Med. 2011, 365, 395–409. [Google Scholar] [CrossRef] [PubMed]
- Forde, P.M.; Chaft, J.E.; Smith, K.N.; Anagnostou, V.; Cottrell, T.R.; Hellmann, M.D.; Zahurak, M.; Yang, S.C.; Jones, D.R.; Broderick, S.; et al. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N. Engl. J. Med. 2018, 378, 1976–1986. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E.; et al. Nivolumab plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef]
- Provencio, M.; Nadal, E.; Insa, A.; García-Campelo, M.R.; Casal-Rubio, J.; Dómine, M.; Majem, M.; Rodríguez-Abreu, D.; Martínez-Martí, A.; De Castro Carpeño, J.; et al. Neoadjuvant chemotherapy and nivolumab in resectable non-small-cell lung cancer (NADIM): An open-label, multicentre, single-arm, phase 2 trial. Lancet Oncol. 2020, 21, 1413–1422. [Google Scholar] [CrossRef]
- Cascone, T.; William, W.N.; Weissferdt, A.; Leung, C.H.; Lin, H.Y.; Pataer, A.; Godoy, M.C.B.; Carter, B.W.; Federico, L.; Reuben, A.; et al. Neoadjuvant nivolumab or nivolumab plus ipilimumab in operable non-small cell lung cancer: The phase 2 randomized NEOSTAR trial. Nat. Med. 2021, 27, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Forde, P.M.; Spicer, J.; Lu, S.; Provencio, M.; Mitsudomi, T.; Awad, M.M.; Felip, E.; Broderick, S.R.; Brahmer, J.R.; Swanson, S.J.; et al. Neoadjuvant Nivolumab plus Chemotherapy in Resectable Lung Cancer. N. Engl. J. Med. 2022, 386, 1973–1985. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.; Leong, T.L. Contemporary Concise Review 2021: Pulmonary nodules from detection to intervention. Respirology 2022. Online ahead of print. [Google Scholar] [CrossRef]
- Chen, W.-W.; Liu, W.; Li, Y.; Wang, J.; Ren, Y.; Wang, G.; Chen, C.; Li, H. Deciphering the Immune–Tumor Interplay During Early-Stage Lung Cancer Development via Single-Cell Technology. Front. Oncol. 2022, 11, 716042. [Google Scholar] [CrossRef]
- Saab, S.; Zalzale, H.; Rahal, Z.; Khalifeh, Y.; Sinjab, A.; Kadara, H. Insights Into Lung Cancer Immune-Based Biology, Prevention, and Treatment. Front. Immunol. 2020, 11, 159. [Google Scholar] [CrossRef] [PubMed]
- Kadara, H.; Scheet, P.; Wistuba, I.I.; Spira, A.E. Early Events in the Molecular Pathogenesis of Lung Cancer. Cancer Prev. Res. 2016, 9, 518–527. [Google Scholar] [CrossRef]
- Yatabe, Y. EGFR mutations and the terminal respiratory unit. Cancer Metastasis Rev. 2010, 29, 23–36. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Devarakonda, S.; Morgensztern, D.; Govindan, R. Genomic alterations in lung adenocarcinoma. Lancet Oncol. 2015, 16, e342–e351. [Google Scholar] [CrossRef]
- Gould, M.K.; Donington, J.; Lynch, W.R.; Mazzone, P.J.; Midthun, D.E.; Naidich, D.P.; Wiener, R.S. Evaluation of individuals with pulmonary nodules: When is it lung cancer? Diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2013, 143, e93S–e120S. [Google Scholar] [CrossRef]
- Shaw, A.T.; Kirsch, D.G.; Jacks, T. Future of early detection of lung cancer: The role of mouse models. Clin. Cancer Res. 2005, 11, 4999s–5003s. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, Y.; Mashock, M.; Tong, Z.; Mu, X.; Chen, H.; Zhou, X.; Zhang, H.; Zhao, G.; Liu, B.; Li, X. Changing Technologies of RNA Sequencing and Their Applications in Clinical Oncology. Front. Oncol. 2020, 10, 447. [Google Scholar] [CrossRef]
- Meuwissen, R.; Berns, A. Mouse models for human lung cancer. Genes Dev. 2005, 19, 643–664. [Google Scholar] [CrossRef] [PubMed]
- Mainardi, S.; Mijimolle, N.; Francoz, S.; Vicente-Duenas, C.; Sanchez-Garcia, I.; Barbacid, M. Identification of cancer initiating cells in K-Ras driven lung adenocarcinoma. Proc. Natl. Acad. Sci. USA 2014, 111, 255–260. [Google Scholar] [CrossRef]
- Xu, X.; Rock, J.R.; Lu, Y.; Futtner, C.; Schwab, B.; Guinney, J.; Hogan, B.L.; Onaitis, M.W. Evidence for type II cells as cells of origin of K-Ras-induced distal lung adenocarcinoma. Proc. Natl. Acad. Sci. USA 2012, 109, 4910–4915. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Song, H.; Huang, C.; Yao, E.; Gacayan, R.; Xu, S.M.; Chuang, P.T. Alveolar type II cells possess the capability of initiating lung tumor development. PLoS ONE 2012, 7, e53817. [Google Scholar] [CrossRef]
- Sutherland, K.D.; Song, J.Y.; Kwon, M.C.; Proost, N.; Zevenhoven, J.; Berns, A. Multiple cells-of-origin of mutant K-Ras-induced mouse lung adenocarcinoma. Proc. Natl. Acad. Sci. USA 2014, 111, 4952–4957. [Google Scholar] [CrossRef]
- Angelidis, I.; Simon, L.M.; Fernandez, I.E.; Strunz, M.; Mayr, C.H.; Greiffo, F.R.; Tsitsiridis, G.; Ansari, M.; Graf, E.; Strom, T.-M.; et al. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat. Commun. 2019, 10, 963. [Google Scholar] [CrossRef]
- Travaglini, K.J.; Nabhan, A.N.; Penland, L.; Sinha, R.; Gillich, A.; Sit, R.V.; Chang, S.; Conley, S.D.; Mori, Y.; Seita, J.; et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 2020, 587, 619–625. [Google Scholar] [CrossRef]
- Treutlein, B.; Brownfield, D.G.; Wu, A.R.; Neff, N.F.; Mantalas, G.L.; Espinoza, F.H.; Desai, T.J.; Krasnow, M.A.; Quake, S.R. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature 2014, 509, 371–375. [Google Scholar] [CrossRef]
- Sauler, M.; McDonough, J.E.; Adams, T.S.; Kothapalli, N.; Barnthaler, T.; Werder, R.B.; Schupp, J.C.; Nouws, J.; Robertson, M.J.; Coarfa, C.; et al. Characterization of the COPD alveolar niche using single-cell RNA sequencing. Nat. Commun. 2022, 13, 494. [Google Scholar] [CrossRef] [PubMed]
- Plasschaert, L.W.; Žilionis, R.; Choo-Wing, R.; Savova, V.; Knehr, J.; Roma, G.; Klein, A.M.; Jaffe, A.B. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 2018, 560, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Habermann, A.C.; Gutierrez, A.J.; Bui, L.T.; Yahn, S.L.; Winters, N.I.; Calvi, C.L.; Peter, L.; Chung, M.I.; Taylor, C.J.; Jetter, C.; et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1972. [Google Scholar] [CrossRef] [PubMed]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.I.; Ren, Z.; et al. Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1517–1536. [Google Scholar] [CrossRef]
- Tsukui, T.; Sun, K.-H.; Wetter, J.B.; Wilson-Kanamori, J.R.; Hazelwood, L.A.; Henderson, N.C.; Adams, T.S.; Schupp, J.C.; Poli, S.D.; Rosas, I.O.; et al. Collagen-producing lung cell atlas identifies multiple subsets with distinct localization and relevance to fibrosis. Nat. Commun. 2020, 11, 1920. [Google Scholar] [CrossRef]
- Cheung, W.K.C.; Nguyen, D.X. Lineage factors and differentiation states in lung cancer progression. Oncogene 2015, 34, 5771–5780. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Cheung, W.K.; Zhao, M.; Liu, Z.; Stevens, L.E.; Cao, P.D.; Fang, J.E.; Westbrook, T.F.; Nguyen, D.X. Control of alveolar differentiation by the lineage transcription factors GATA6 and HOPX inhibits lung adenocarcinoma metastasis. Cancer Cell 2013, 23, 725–738. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Z.; Zhou, K.; Wang, C.; Jiang, L.; Zhang, L.; Yang, Y.; Luo, W.; Qiao, W.; Wang, G.; et al. Deciphering cell lineage specification of human lung adenocarcinoma with single-cell RNA sequencing. Nat. Commun. 2021, 12, 6500. [Google Scholar] [CrossRef]
- Wu, F.; Fan, J.; He, Y.; Xiong, A.; Yu, J.; Li, Y.; Zhang, Y.; Zhao, W.; Zhou, F.; Li, W.; et al. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat. Commun. 2021, 12, 2540. [Google Scholar] [CrossRef]
- Yang, D.; Jones, M.G.; Naranjo, S.; Rideout, W.M., 3rd; Min, K.H.J.; Ho, R.; Wu, W.; Replogle, J.M.; Page, J.L.; Quinn, J.J.; et al. Lineage tracing reveals the phylodynamics, plasticity, and paths of tumor evolution. Cell 2022, 185, 1905–1923. [Google Scholar] [CrossRef] [PubMed]
- Maynard, A.; McCoach, C.E.; Rotow, J.K.; Harris, L.; Haderk, F.; Kerr, D.L.; Yu, E.A.; Schenk, E.L.; Tan, W.; Zee, A.; et al. Therapy-Induced Evolution of Human Lung Cancer Revealed by Single-Cell RNA Sequencing. Cell 2020, 182, 1232–1251.e1222. [Google Scholar] [CrossRef] [PubMed]
- Laughney, A.M.; Hu, J.; Campbell, N.R.; Bakhoum, S.F.; Setty, M.; Lavallée, V.P.; Xie, Y.; Masilionis, I.; Carr, A.J.; Kottapalli, S.; et al. Regenerative lineages and immune-mediated pruning in lung cancer metastasis. Nat. Med. 2020, 26, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Kim, H.K.; Lee, K.; Hong, Y.; Cho, J.H.; Choi, J.W.; Lee, J.I.; Suh, Y.L.; Ku, B.M.; Eum, H.H.; et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat. Commun. 2020, 11, 2285. [Google Scholar] [CrossRef] [PubMed]
- Morrisey, E.E.; Hogan, B.L. Preparing for the first breath: Genetic and cellular mechanisms in lung development. Dev. Cell 2010, 18, 8–23. [Google Scholar] [CrossRef]
- Maeda, Y.; Davé, V.; Whitsett, J.A. Transcriptional control of lung morphogenesis. Physiol. Rev. 2007, 87, 219–244. [Google Scholar] [CrossRef]
- Penkala, I.J.; Liberti, D.C.; Pankin, J.; Sivakumar, A.; Kremp, M.M.; Jayachandran, S.; Katzen, J.; Leach, J.P.; Windmueller, R.; Stolz, K.; et al. Age-dependent alveolar epithelial plasticity orchestrates lung homeostasis and regeneration. Cell Stem Cell 2021, 28, 1775–1789.e1775. [Google Scholar] [CrossRef]
- Verheyden, J.M.; Sun, X. A transitional stem cell state in the lung. Nat. Cell Biol. 2020, 22, 1025–1026. [Google Scholar] [CrossRef]
- Strunz, M.; Simon, L.M.; Ansari, M.; Kathiriya, J.J.; Angelidis, I.; Mayr, C.H.; Tsidiridis, G.; Lange, M.; Mattner, L.F.; Yee, M.; et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat. Commun. 2020, 11, 3559. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Tata, A.; Konkimalla, A.; Katsura, H.; Lee, R.F.; Ou, J.; Banovich, N.E.; Kropski, J.A.; Tata, P.R. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat. Cell Biol. 2020, 22, 934–946. [Google Scholar] [CrossRef]
- Choi, J.; Park, J.E.; Tsagkogeorga, G.; Yanagita, M.; Koo, B.K.; Han, N.; Lee, J.H. Inflammatory Signals Induce AT2 Cell-Derived Damage-Associated Transient Progenitors that Mediate Alveolar Regeneration. Cell Stem Cell 2020, 27, 366–382.e367. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Sinjab, A.; Treekitkarnmongkol, W.; Lu, W.; Serrano, A.G.; Hernandez, S.D.; Rahal, Z.; Cao, X.; Dai, E.; Gomez-Bolanos, L.I.; et al. An atlas of epithelial cell states and plasticity in lung adenocarcinoma. bioRxiv 2022. [Google Scholar] [CrossRef]
- Nicoś, M.; Krawczyk, P. Genetic Clonality as the Hallmark Driving Evolution of Non-Small Cell Lung Cancer. Cancers 2022, 14, 1813. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N. Engl J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef]
- Zhang, J.; Fujimoto, J.; Zhang, J.; Wedge, D.C.; Song, X.; Zhang, J.; Seth, S.; Chow, C.W.; Cao, Y.; Gumbs, C.; et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 2014, 346, 256–259. [Google Scholar] [CrossRef]
- de Bruin, E.C.; McGranahan, N.; Mitter, R.; Salm, M.; Wedge, D.C.; Yates, L.; Jamal-Hanjani, M.; Shafi, S.; Murugaesu, N.; Rowan, A.J.; et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 2014, 346, 251–256. [Google Scholar] [CrossRef]
- Sinjab, A.; Han, G.; Treekitkarnmongkol, W.; Hara, K.; Brennan, P.M.; Dang, M.; Hao, D.; Wang, R.; Dai, E.; Dejima, H.; et al. Resolving the Spatial and Cellular Architecture of Lung Adenocarcinoma by Multiregion Single-Cell Sequencing. Cancer Discov. 2021, 11, 2506. [Google Scholar] [CrossRef]
- Sinjab, A.; Han, G.; Wang, L.; Kadara, H. Field Carcinogenesis in Cancer Evolution: What the Cell Is Going On? Cancer Res. 2020, 80, 4888. [Google Scholar] [CrossRef]
- Dranoff, G. Immunotherapy at large: Balancing tumor immunity and inflammatory pathology. Nat. Med. 2013, 19, 1100–1101. [Google Scholar] [CrossRef]
- Quezada, S.A.; Peggs, K.S.; Simpson, T.R.; Allison, J.P. Shifting the equilibrium in cancer immunoediting: From tumor tolerance to eradication. Immunol Rev. 2011, 241, 104–118. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Schalper, K.A.; Brown, J.; Carvajal-Hausdorf, D.; McLaughlin, J.; Velcheti, V.; Syrigos, K.N.; Herbst, R.S.; Rimm, D.L. Objective measurement and clinical significance of TILs in non-small cell lung cancer. J. Natl. Cancer Inst. 2015, 107, dju435. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Song, C.; Tian, Y.; Yang, X. Single-Cell RNA Sequencing in Lung Cancer: Revealing Phenotype Shaping of Stromal Cells in the Microenvironment. Front. Immunol. 2022, 12, 802080. [Google Scholar] [CrossRef] [PubMed]
- Zilionis, R.; Engblom, C.; Pfirschke, C.; Savova, V.; Zemmour, D.; Saatcioglu, H.D.; Krishnan, I.; Maroni, G.; Meyerovitz, C.V.; Kerwin, C.M.; et al. Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 2019, 50, 1317–1334.e1310. [Google Scholar] [CrossRef]
- Salcher, S.; Sturm, G.; Horvath, L.; Untergasser, G.; Fotakis, G.; Panizzolo, E.; Martowicz, A.; Pall, G.; Gamerith, G.; Sykora, M.; et al. High-resolution single-cell atlas reveals diversity and plasticity of tissue-resident neutrophils in non-small cell lung cancer. bioRxiv 2022. [Google Scholar] [CrossRef]
- Leader, A.M.; Grout, J.A.; Maier, B.B.; Nabet, B.Y.; Park, M.D.; Tabachnikova, A.; Chang, C.; Walker, L.; Lansky, A.; Le Berichel, J.; et al. Single-cell analysis of human non-small cell lung cancer lesions refines tumor classification and patient stratification. Cancer Cell 2021, 39, 1594–1609.e1512. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, Y.; Zheng, L.; Zheng, C.; Song, J.; Zhang, Q.; Kang, B.; Liu, Z.; Jin, L.; Xing, R.; et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 2018, 24, 978–985. [Google Scholar] [CrossRef]
- Zheng, L.; Qin, S.; Si, W.; Wang, A.; Xing, B.; Gao, R.; Ren, X.; Wang, L.; Wu, X.; Zhang, J.; et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science 2021, 374, abe6474. [Google Scholar] [CrossRef]
- Lambrechts, D.; Wauters, E.; Boeckx, B.; Aibar, S.; Nittner, D.; Burton, O.; Bassez, A.; Decaluwé, H.; Pircher, A.; Van den Eynde, K.; et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 2018, 24, 1277–1289. [Google Scholar] [CrossRef]
- Bischoff, P.; Trinks, A.; Obermayer, B.; Pett, J.P.; Wiederspahn, J.; Uhlitz, F.; Liang, X.; Lehmann, A.; Jurmeister, P.; Elsner, A.; et al. Single-cell RNA sequencing reveals distinct tumor microenvironmental patterns in lung adenocarcinoma. Oncogene 2021, 40, 6748–6758. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Hu, X.; Feng, K.; Gao, R.; Xue, Z.; Zhang, S.; Zhang, Y.; Corse, E.; Hu, Y.; Han, W.; et al. Temporal single-cell tracing reveals clonal revival and expansion of precursor exhausted T cells during anti-PD-1 therapy in lung cancer. Nat. Cancer 2022, 3, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Tan, Y.; Sun, F.; Hou, L.; Zhang, C.; Ge, T.; Yu, H.; Wu, C.; Zhu, Y.; Duan, L.; et al. Single-cell transcriptome and antigen-immunoglobin analysis reveals the diversity of B cells in non-small cell lung cancer. Genome Biol. 2020, 21, 152. [Google Scholar] [CrossRef]
- Marshall, A.L.; Christiani, D.C. Genetic susceptibility to lung cancer--light at the end of the tunnel? Carcinogenesis 2013, 34, 487–502. [Google Scholar] [CrossRef] [PubMed]
- Wender, R.; Fontham, E.T.; Barrera, E., Jr.; Colditz, G.A.; Church, T.R.; Ettinger, D.S.; Etzioni, R.; Flowers, C.R.; Gazelle, G.S.; Kelsey, D.K.; et al. American Cancer Society lung cancer screening guidelines. CA Cancer J. Clin. 2013, 63, 107–117. [Google Scholar] [CrossRef]
- Sivakumar, S.; San Lucas, F.A.; McDowell, T.L.; Lang, W.; Xu, L.; Fujimoto, J.; Zhang, J.; Futreal, P.A.; Fukuoka, J.; Yatabe, Y.; et al. Genomic landscape of atypical adenomatous hyperplasia reveals divergent modes to lung adenocarcinoma. Cancer Res. 2017, 77, 6119–6130. [Google Scholar] [CrossRef]
- Westra, W.H.; Baas, I.O.; Hruban, R.H.; Askin, F.B.; Wilson, K.; Offerhaus, G.J.; Slebos, R.J. K-ras oncogene activation in atypical alveolar hyperplasias of the human lung. Cancer Res. 1996, 56, 2224–2228. [Google Scholar]
- Wistuba, I.I. Genetics of preneoplasia: Lessons from lung cancer. Curr. Mol. Med. 2007, 7, 3–14. [Google Scholar] [CrossRef]
- Yatabe, Y.; Kosaka, T.; Takahashi, T.; Mitsudomi, T. EGFR mutation is specific for terminal respiratory unit type adenocarcinoma. Am. J. Surg. Pathol. 2005, 29, 633–639. [Google Scholar] [CrossRef]
- Travis, W.D.; Brambilla, E.; Burke, A.P.; Marx, A.; Nicholson, A.G. Introduction to The 2015 World Health Organization Classification of Tumors of the Lung, Pleura, Thymus, and Heart. J. Thorac. Oncol. 2015, 10, 1240–1242. [Google Scholar] [CrossRef]
- Travis, W.D.; Brambilla, E.; Riely, G.J. New pathologic classification of lung cancer: Relevance for clinical practice and clinical trials. J. Clin. Oncol. 2013, 31, 992–1001. [Google Scholar] [CrossRef] [PubMed]
- Ishizumi, T.; McWilliams, A.; MacAulay, C.; Gazdar, A.; Lam, S. Natural history of bronchial preinvasive lesions. Cancer Metastasis Rev. 2010, 29, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Brambilla, E.; Noguchi, M.; Nicholson, A.G.; Geisinger, K.R.; Yatabe, Y.; Beer, D.G.; Powell, C.A.; Riely, G.J.; Van Schil, P.E.; et al. International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J. Thorac. Oncol. 2011, 6, 244–285. [Google Scholar] [CrossRef] [PubMed]
- Kerr, K.M. Pulmonary preinvasive neoplasia. J. Clin. Pathol. 2001, 54, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.M.G.; Jungner, G. Principles and Practice of Screening for Disease; World Health Organization Public Health Papers: Geneva, Switzerland, 1968. [Google Scholar]
- Garfinkel, L.; Silverberg, E. Lung cancer and smoking trends in the United States over the past 25 years. CA Cancer J. Clin. 1991, 41, 137–145. [Google Scholar] [CrossRef]
- Church, T.R.; Black, W.C.; Aberle, D.R.; Berg, C.D.; Clingan, K.L.; Duan, F.; Fagerstrom, R.M.; Gareen, I.F.; Gierada, D.S.; Jones, G.C.; et al. Results of initial low-dose computed tomographic screening for lung cancer. N. Engl. J. Med. 2013, 368, 1980–1991. [Google Scholar] [CrossRef]
- Midthun, D.E. Early detection of lung cancer. F1000Research 2016, 5, Rev-739. [Google Scholar] [CrossRef]
- Rahal, Z.; El Nemr, S.; Sinjab, A.; Chami, H.; Tfayli, A.; Kadara, H. Smoking and Lung Cancer: A Geo-Regional Perspective. Front. Oncol. 2017, 7, 194. [Google Scholar] [CrossRef]
- Tanoue, L.T.; Tanner, N.T.; Gould, M.K.; Silvestri, G.A. Lung cancer screening. Am. J. Respir. Crit. Care Med. 2015, 191, 19–33. [Google Scholar] [CrossRef]
- Vachani, A.; Sequist, L.V.; Spira, A. AJRCCM: 100-Year Anniversary. The Shifting Landscape for Lung Cancer: Past, Present, and Future. Am. J. Respir. Crit. Care Med. 2017, 195, 1150–1160. [Google Scholar] [CrossRef]
- Kim, E.Y.; Cha, Y.J.; Lee, S.H.; Jeong, S.; Choi, Y.J.; Moon, D.H.; Lee, S.; Chang, Y.S. Early lung carcinogenesis and tumor microenvironment observed by single-cell transcriptome analysis. Transl. Oncol. 2022, 15, 101277. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Yang, X.; Shi, Y.; Zhao, M.; Bi, G.; Liang, J.; Chen, Z.; Huang, Y.; Jiang, W.; Lin, Z.; et al. Single-cell transcriptome atlas of lung adenocarcinoma featured with ground glass nodules. Cell Discov. 2020, 6, 69. [Google Scholar] [CrossRef] [PubMed]
- Yanagawa, J.; Tran, L.M.; Fung, E.; Wallace, W.D.; Prosper, A.E.; Fishbein, G.A.; Shea, C.; Hong, R.; Liu, B.; Salehi-Rad, R.; et al. Single-cell characterization of subsolid and solid lesions in the lung adenocarcinoma spectrum. bioRxiv 2020. [Google Scholar] [CrossRef]
- Xing, X.; Yang, F.; Huang, Q.; Guo, H.; Li, J.; Qiu, M.; Bai, F.; Wang, J. Decoding the multicellular ecosystem of lung adenocarcinoma manifested as pulmonary subsolid nodules by single-cell RNA sequencing. Sci. Adv. 2021, 7, eabd9738. [Google Scholar] [CrossRef]
- Lavin, Y.; Kobayashi, S.; Leader, A.; Amir, E.D.; Elefant, N.; Bigenwald, C.; Remark, R.; Sweeney, R.; Becker, C.D.; Levine, J.H.; et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell 2017, 169, 750–765.e717. [Google Scholar] [CrossRef]
- Zugazagoitia, J.; Gupta, S.; Liu, Y.; Fuhrman, K.; Gettinger, S.; Herbst, R.S.; Schalper, K.A.; Rimm, D.L. Biomarkers Associated with Beneficial PD-1 Checkpoint Blockade in Non–Small Cell Lung Cancer (NSCLC) Identified Using High-Plex Digital Spatial Profiling. Clin. Cancer Res. 2020, 26, 4360–4368. [Google Scholar] [CrossRef]
- Nagendran, M.; Riordan, D.P.; Harbury, P.B.; Desai, T.J. Automated cell-type classification in intact tissues by single-cell molecular profiling. eLife 2018, 7, e30510. [Google Scholar] [CrossRef]
- Madissoon, E.; Oliver, A.J.; Kleshchevnikov, V.; Wilbrey-Clark, A.; Polanski, K.; Orsi, A.R.; Mamanova, L.; Bolt, L.; Richoz, N.; Elmentaite, R.; et al. A spatial multi-omics atlas of the human lung reveals a novel immune cell survival niche. bioRxiv 2021. [Google Scholar] [CrossRef]
- Moses, L.; Pachter, L. Museum of spatial transcriptomics. Nat. Methods 2022, 19, 534–546. [Google Scholar] [CrossRef]
- Agrawal, A.; Hogarth, D.K.; Murgu, S. Robotic bronchoscopy for pulmonary lesions: A review of existing technologies and clinical data. J. Thorac. Dis. 2020, 12, 3279–3286. [Google Scholar] [CrossRef]
- Sabath, B.F.; Casal, R.F. Bronchoscopic ablation of peripheral lung tumors. J. Thorac. Dis. 2019, 11, 2628–2638. [Google Scholar] [CrossRef] [PubMed]
- Bae, T.; Tomasini, L.; Mariani, J.; Zhou, B.; Roychowdhury, T.; Franjic, D.; Pletikos, M.; Pattni, R.; Chen, B.J.; Venturini, E.; et al. Different mutational rates and mechanisms in human cells at pregastrulation and neurogenesis. Science 2018, 359, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Behjati, S.; Huch, M.; van Boxtel, R.; Karthaus, W.; Wedge, D.C.; Tamuri, A.U.; Martincorena, I.; Petljak, M.; Alexandrov, L.B.; Gundem, G.; et al. Genome sequencing of normal cells reveals developmental lineages and mutational processes. Nature 2014, 513, 422–425. [Google Scholar] [CrossRef] [PubMed]
- Nanki, K.; Fujii, M.; Shimokawa, M.; Matano, M.; Nishikori, S.; Date, S.; Takano, A.; Toshimitsu, K.; Ohta, Y.; Takahashi, S.; et al. Somatic inflammatory gene mutations in human ulcerative colitis epithelium. Nature 2020, 577, 254–259. [Google Scholar] [CrossRef]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef]
- Lee, B.C.H.; Robinson, P.S.; Coorens, T.H.H.; Yan, H.H.N.; Olafsson, S.; Lee-Six, H.; Sanders, M.A.; Siu, H.C.; Hewinson, J.; Yue, S.S.K.; et al. Mutational landscape of normal epithelial cells in Lynch Syndrome patients. Nat. Commun. 2022, 13, 2710. [Google Scholar] [CrossRef]
- Huang, A.Y.; Lee, E.A. Identification of Somatic Mutations From Bulk and Single-Cell Sequencing Data. Front. Aging 2022, 2, 80. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinjab, A.; Rahal, Z.; Kadara, H. Cell-by-Cell: Unlocking Lung Cancer Pathogenesis. Cancers 2022, 14, 3424. https://doi.org/10.3390/cancers14143424
Sinjab A, Rahal Z, Kadara H. Cell-by-Cell: Unlocking Lung Cancer Pathogenesis. Cancers. 2022; 14(14):3424. https://doi.org/10.3390/cancers14143424
Chicago/Turabian StyleSinjab, Ansam, Zahraa Rahal, and Humam Kadara. 2022. "Cell-by-Cell: Unlocking Lung Cancer Pathogenesis" Cancers 14, no. 14: 3424. https://doi.org/10.3390/cancers14143424
APA StyleSinjab, A., Rahal, Z., & Kadara, H. (2022). Cell-by-Cell: Unlocking Lung Cancer Pathogenesis. Cancers, 14(14), 3424. https://doi.org/10.3390/cancers14143424