
Activation of Oncogenic and Immune-Response Pathways Is Linked to Disease-Specific Survival in Merkel Cell Carcinoma



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients, Clinical Data, and Tissue Samples
2.2. RNA Extraction from FFPE Samples
2.3. 3′ RNA Sequencing
Processing of Sequencing Data
2.4. Identification of DAGs and SAGs
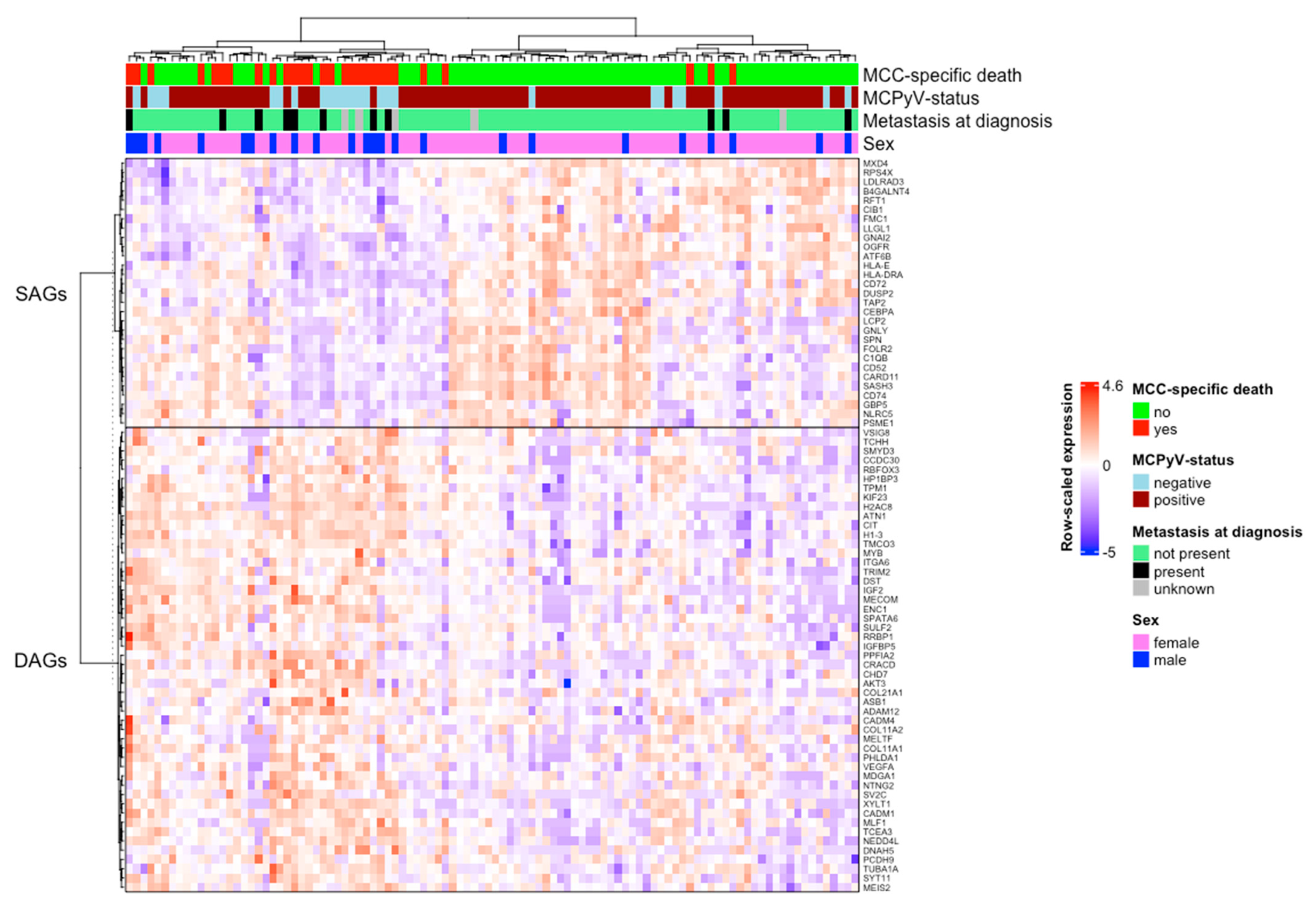
2.5. Clustered Heatmap of Differentially Expressed Genes
2.6. Gene Ontology and Signaling Pathway Analyses of DAGs and SAGs
2.7. Statistical Analysis
2.8. Survival of ≥3 Years as Inclusion Criterion for the Good Prognosis Group
2.9. Differential Expression Test of DAGs Based on MCPyV Status
3. Results
3.1. Overview of Patients
3.2. Identification of DAGs and SAGs
3.3. GO Enrichment and KEGG Signaling Pathway Analysis of DAGs and SAGs
3.4. Survival of ≥3 Years as Inclusion Criterion for the Good Prognosis Group
3.5. Differential Expression Test of DAGs Based on MCPyV Status
3.6. Correlation between Tumor-Infiltrating Lymphocytes and SAGs Related to Antigen Processing and Presentation
3.7. Comparison of FFPE Data to Data from Fresh Frozen Tissues
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kukko, H.; Bohling, T.; Koljonen, V.; Tukiainen, E.; Haglund, C.; Pokhrel, A.; Sankila, R.; Pukkala, E. Merkel Cell Carcinoma—A Population-Based Epidemiological Study in Finland with a Clinical Series of 181 Cases. Eur. J. Cancer 2012, 48, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Chao, P.; Coomarasamy, C.; Mathy, J.A. Epidemiology and Survival of Merkel Cell Carcinoma in New Zealand: A Population-Based Study between 2000 and 2015 with International Comparison. Australas. J. Dermatol. 2019, 60, e284–e291. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, V.; Muralidhar, V.; Margalit, D.N.; Tishler, R.B.; DeCaprio, J.A.; Thakuria, M.; Rabinowits, G.; Schoenfeld, J.D. Merkel Cell Carcinoma: A Population Analysis on Survival. J. Natl. Compr. Cancer Netw. JNCCN 2016, 14, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Sihto, H.; Kukko, H.; Koljonen, V.; Sankila, R.; Bohling, T.; Joensuu, H. Clinical Factors Associated with Merkel Cell Polyomavirus Infection in Merkel Cell Carcinoma. J. Natl. Cancer Inst. 2009, 101, 938–945. [Google Scholar] [CrossRef]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal Integration of a Polyomavirus in Human Merkel Cell Carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [Green Version]
- Sihto, H.; Bohling, T.; Kavola, H.; Koljonen, V.; Salmi, M.; Jalkanen, S.; Joensuu, H. Tumor Infiltrating Immune Cells and Outcome of Merkel Cell Carcinoma: A Population-Based Study. Clin. Cancer Res. 2012, 18, 2872–2881. [Google Scholar] [CrossRef] [Green Version]
- Paulson, K.G.; Iyer, J.G.; Blom, A.; Warton, E.M.; Sokil, M.; Yelistratova, L.; Schuman, L.; Nagase, K.; Bhatia, S.; Asgari, M.M.; et al. Systemic Immune Suppression Predicts Diminished Merkel Cell Carcinoma-Specific Survival Independent of Stage. J. Investig. Dermatol. 2013, 133, 642–646. [Google Scholar] [CrossRef] [Green Version]
- Ricci, C.; Righi, A.; Ambrosi, F.; Gibertoni, D.; Maletta, F.; Uccella, S.; Sessa, F.; Asioli, S.; Pellilli, M.; Maragliano, R.; et al. Prognostic Impact of MCPyV and TIL Subtyping in Merkel Cell Carcinoma: Evidence from a Large European Cohort of 95 Patients. Endocr. Pathol. 2020, 31, 21–32. [Google Scholar] [CrossRef]
- Butala, A.A.; Jain, V.; Reddy, V.K.; Sebro, R.A.; Song, Y.; Karakousis, G.; Mitchell, T.C.; Lukens, J.N.; Shabason, J.E. Impact of Tumor-Infiltrating Lymphocytes on Overall Survival in Merkel Cell Carcinoma. Oncol. 2021, 26, 63–69. [Google Scholar] [CrossRef]
- Harms, K.L.; Zhao, L.; Johnson, B.; Wang, X.; Carskadon, S.; Palanisamy, N.; Rhodes, D.R.; Mannan, R.; Vo, J.N.; Choi, J.E.; et al. Virus-Positive Merkel Cell Carcinoma Is an Independent Prognostic Group with Distinct Predictive Biomarkers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 2494–2504. [Google Scholar] [CrossRef]
- Paulson, K.G.; Iyer, J.G.; Tegeder, A.R.; Thibodeau, R.; Schelter, J.; Koba, S.; Schrama, D.; Simonson, W.T.; Lemos, B.D.; Byrd, D.R.; et al. Transcriptome-Wide Studies of Merkel Cell Carcinoma and Validation of Intratumoral CD8+ Lymphocyte Invasion as an Independent Predictor of Survival. J. Clin. Oncol. 2011, 29, 1539–1546. [Google Scholar] [CrossRef] [Green Version]
- Nghiem, P.; Bhatia, S.; Lipson, E.J.; Sharfman, W.H.; Kudchadkar, R.R.; Brohl, A.S.; Friedlander, P.A.; Daud, A.; Kluger, H.M.; Reddy, S.A.; et al. Durable Tumor Regression and Overall Survival in Patients with Advanced Merkel Cell Carcinoma Receiving Pembrolizumab as First-Line Therapy. J. Clin. Oncol. 2019, 37, 693–702. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Russell, J.; Hamid, O.; Bhatia, S.; Terheyden, P.; D’Angelo, S.P.; Shih, K.C.; Lebbé, C.; Linette, G.P.; Milella, M.; et al. Avelumab in Patients with Chemotherapy-Refractory Metastatic Merkel Cell Carcinoma: A Multicentre, Single-Group, Open-Label, Phase 2 Trial. Lancet Oncol. 2016, 17, 1374–1385. [Google Scholar] [CrossRef] [Green Version]
- Sahi, H.; Koljonen, V.; Kavola, H.; Haglund, C.; Tukiainen, E.; Sihto, H.; Böhling, T. Bcl-2 Expression Indicates Better Prognosis of Merkel Cell Carcinoma Regardless of the Presence of Merkel Cell Polyomavirus. Virchows Arch. Int. J. Pathol. 2012, 461, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Corley, S.M.; Troy, N.M.; Bosco, A.; Wilkins, M.R. QuantSeq. 3’ Sequencing Combined with Salmon Provides a Fast, Reliable Approach for High Throughput RNA Expression Analysis. Sci. Rep. 2019, 9, 18895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinforma. Oxf. Engl. 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harms, P.W.; Patel, R.M.; Verhaegen, M.E.; Giordano, T.J.; Nash, K.T.; Johnson, C.N.; Daignault, S.; Thomas, D.G.; Gudjonsson, J.E.; Elder, J.T.; et al. Distinct Gene Expression Profiles of Viral- and Nonviral-Associated Merkel Cell Carcinoma Revealed by Transcriptome Analysis. J. Investig. Dermatol. 2013, 133, 936–945. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. The Gene Ontology Consortium. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and Collaborative HTML5 Gene List Enrichment Analysis Tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [Green Version]
- Bob, A.; Nielen, F.; Krediet, J.; Schmitter, J.; Freundt, D.; Terhorst, D.; Röwert-Huber, J.; Kanitakis, J.; Stockfleth, E.; Ulrich, C.; et al. Tumor Vascularization and Clinicopathologic Parameters as Prognostic Factors in Merkel Cell Carcinoma. J. Cancer Res. Clin. Oncol. 2017, 143, 1999–2010. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Figueras, M.-T.; Puig, L.; Musulén, E.; Gilaberte, M.; Lerma, E.; Serrano, S.; Ferrándiz, C.; Ariza, A. Expression Profiles Associated with Aggressive Behavior in Merkel Cell Carcinoma. Mod. Pathol. 2007, 20, 90–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunner, M.; Thurnher, D.; Pammer, J.; Geleff, S.; Heiduschka, G.; Reinisch, C.M.; Petzelbauer, P.; Erovic, B.M. Expression of VEGF-A/C, VEGF-R2, PDGF-Alpha/Beta, c-Kit, EGFR, Her-2/Neu, Mcl-1 and Bmi-1 in Merkel Cell Carcinoma. Mod. Pathol. 2008, 21, 876–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Chen, Z.-H.; Chen, Z.; Tan, D. SULF2 Promotes Tumorigenesis and Inhibits Apoptosis of Cervical Cancer Cells through the ERK/AKT Signaling Pathway. Braz. J. Med. Biol. Res. 2020, 53, e8901. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Han, T.; Zhang, T.; Sun, C. Sulfatase-2 Promotes the Growth and Metastasis of Colorectal Cancer by Activating Akt and Erk1/2 Pathways. Biomed. Pharmacother. 2017, 89, 1370–1377. [Google Scholar] [CrossRef]
- Tu, H.; Li, J.; Lin, L.; Wang, L. COL11A1 Was Involved in Cell Proliferation, Apoptosis and Migration in Non-Small Cell Lung Cancer Cells. J. Investig. Surg. 2021, 34, 664–669. [Google Scholar] [CrossRef]
- Wu, Y.; Tan, X.; Liu, P.; Yang, Y.; Huang, Y.; Liu, X.; Meng, X.; Yu, B.; Wu, M.; Jin, H. ITGA6 and RPSA Synergistically Promote Pancreatic Cancer Invasion and Metastasis via PI3K and MAPK Signaling Pathways. Exp. Cell Res. 2019, 379, 30–47. [Google Scholar] [CrossRef]
- Liu, T.; Wu, X.; Li, Y.; Lu, W.; Zheng, F.; Zhang, C.; Long, Q.; Qiu, H.; Li, Y.; Ge, Q.; et al. RBFOX3 Regulates the Chemosensitivity of Cancer Cells to 5-Fluorouracil via the PI3K/AKT, EMT and Cytochrome-C/Caspase Pathways. Cell. Physiol. Biochem. 2018, 46, 1365–1380. [Google Scholar] [CrossRef]
- Colón-Bolea, P.; Crespo, P. Lysine Methylation in Cancer: SMYD3-MAP3K2 Teaches Us New Lessons in the Ras-ERK Pathway. BioEssays 2014, 36, 1162–1169. [Google Scholar] [CrossRef]
- Wu, C.; Wang, X.; Wu, X.; Chen, X. Ectodermal-neural Cortex 1 Affects the Biological Function of Lung Cancer through the MAPK Pathway. Int. J. Mol. Med. 2021, 47, 79. [Google Scholar] [CrossRef]
- Hafner, C.; Houben, R.; Baeurle, A.; Ritter, C.; Schrama, D.; Landthaler, M.; Becker, J.C. Activation of the PI3K/AKT Pathway in Merkel Cell Carcinoma. PLoS ONE 2012, 7, e31255. [Google Scholar] [CrossRef] [PubMed]
- Dobson, S.J.; Anene, A.; Boyne, J.R.; Mankouri, J.; Macdonald, A.; Whitehouse, A. Merkel Cell Polyomavirus Small Tumour Antigen Activates the P38 MAPK Pathway to Enhance Cellular Motility. Biochem. J. 2020, 477, 2721–2733. [Google Scholar] [CrossRef] [PubMed]
- Rascio, F.; Spadaccino, F.; Rocchetti, M.T.; Castellano, G.; Stallone, G.; Netti, G.S.; Ranieri, E. The Pathogenic Role of PI3K/AKT Pathway in Cancer Onset and Drug Resistance: An Updated Review. Cancers 2021, 13, 3949. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.-J.; Pan, W.-W.; Liu, S.-B.; Shen, Z.-F.; Xu, Y.; Hu, L.-L. ERK/MAPK Signalling Pathway and Tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [Green Version]
- Duan, C.; Allard, J.B. Insulin-Like Growth Factor Binding Protein-5 in Physiology and Disease. Front. Endocrinol. 2020, 11, 100. [Google Scholar] [CrossRef] [Green Version]
- Keehn, C.A.; Saeed, S.; Bickle, K.; Khalil, F.K.; Morgan, M.B. Expression of Insulin-like Growth Factor-I Receptor in Primary Cutaneous Carcinomas. J. Cutan. Pathol. 2004, 31, 368–372. [Google Scholar] [CrossRef]
- Kervarrec, T.; Gaboriaud, P.; Tallet, A.; Leblond, V.; Arnold, F.; Berthon, P.; Schweinitzer, S.; Larcher, T.; Guyétant, S.; Schrama, D.; et al. VEGF-A Inhibition as a Potential Therapeutic Approach in Merkel Cell Carcinoma. J. Investig. Dermatol. 2019, 139, 736–739. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef] [Green Version]
- Alzahrani, A.S. PI3K/Akt/MTOR Inhibitors in Cancer: At the Bench and Bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef]
- Simpson, A.; Petnga, W.; Macaulay, V.M.; Weyer-Czernilofsky, U.; Bogenrieder, T. Insulin-Like Growth Factor (IGF) Pathway Targeting in Cancer: Role of the IGF Axis and Opportunities for Future Combination Studies. Target. Oncol. 2017, 12, 571–597. [Google Scholar] [CrossRef] [Green Version]
- Fry, E.A.; Inoue, K. C-MYB and DMTF1 in Cancer. Cancer Investig. 2019, 37, 46–65. [Google Scholar] [CrossRef] [PubMed]
- Leick, K.M.; Rodriguez, A.B.; Melssen, M.M.; Benamar, M.; Lindsay, R.S.; Eki, R.; Du, K.-P.; Parlak, M.; Abbas, T.; Engelhard, V.H.; et al. The Barrier Molecules Junction Plakoglobin, Filaggrin, and Dystonin Play Roles in Melanoma Growth and Angiogenesis. Ann. Surg. 2019, 270, 712–722. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-L.; Ji, Y.-M.; Song, R.; Li, X.-N.; Guo, L.-S. KIF23 Promotes Gastric Cancer by Stimulating Cell Proliferation. Dis. Markers 2019, 2019, 9751923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Min, S.; Wang, H.; Shen, Y.; Li, W.; Chen, Y.; Wang, X. MLF1 Protein Is a Potential Therapy Target for Lung Adenocarcinoma. Int. J. Clin. Exp. Pathol. 2018, 11, 3533–3541. [Google Scholar] [PubMed]
- Dunn, L.L.; Sekyere, E.O.; Suryo Rahmanto, Y.; Richardson, D.R. The Function of Melanotransferrin: A Role in Melanoma Cell Proliferation and Tumorigenesis. Carcinogenesis 2006, 27, 2157–2169. [Google Scholar] [CrossRef]
- Liu, Z.; Yan, H.; Yang, Y.; Wei, L.; Xia, S.; Xiu, Y. Down-Regulation of CIT Can Inhibit the Growth of Human Bladder Cancer Cells. Biomed. Pharmacother. 2020, 124, 109830. [Google Scholar] [CrossRef]
- Zha, Y.; Xia, Y.; Ding, J.; Choi, J.-H.; Yang, L.; Dong, Z.; Yan, C.; Huang, S.; Ding, H.-F. MEIS2 Is Essential for Neuroblastoma Cell Survival and Proliferation by Transcriptional Control of M-Phase Progression. Cell Death Dis. 2014, 5, e1417. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Cao, F.; Guo, A.; Chang, W.; Chen, X.; Ma, W.; Gao, X.; Guo, S.; Fu, C.; Zhu, J. Endoplasmic Reticulum Ribosome-Binding Protein 1, RRBP1, Promotes Progression of Colorectal Cancer and Predicts an Unfavourable Prognosis. Br. J. Cancer 2015, 113, 763–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, R.A.C.; Schneider, H.; DeOcesano-Pereira, C.; Lichtenstein, F.; Andrade, F.; Fujita, A.; Trombetta-Lima, M.; Weller, M.; Bowman-Colin, C.; Sogayar, M.C. CHD7 Promotes Glioblastoma Cell Motility and Invasiveness through Transcriptional Modulation of an Invasion Signature. Sci. Rep. 2019, 9, 3952. [Google Scholar] [CrossRef] [Green Version]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An Embryonic Stem Cell-like Gene Expression Signature in Poorly Differentiated Aggressive Human Tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef]
- Atashzar, M.R.; Baharlou, R.; Karami, J.; Abdollahi, H.; Rezaei, R.; Pourramezan, F.; Zoljalali Moghaddam, S.H. Cancer Stem Cells: A Review from Origin to Therapeutic Implications. J. Cell. Physiol. 2020, 235, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; He, W.; Li, Y.; Xu, N.; Zhu, X.; Lin, Y.; Gou, X. Loss of DUSP2 Predicts a Poor Prognosis in Patients with Bladder Cancer. Hum. Pathol. 2019, 85, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Desuki, A.; Staib, F.; Gockel, I.; Moehler, M.; Lang, H.; Biesterfeld, S.; Maderer, A.; Galle, P.R.; Berger, M.R.; Schimanski, C.C. Loss of LLGL1 Expression Correlates with Diffuse Gastric Cancer and Distant Peritoneal Metastases. Can. J. Gastroenterol. Hepatol. 2019, 2019, 2920493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuphal, S.; Wallner, S.; Schimanski, C.C.; Bataille, F.; Hofer, P.; Strand, S.; Strand, D.; Bosserhoff, A.K. Expression of Hugl-1 Is Strongly Reduced in Malignant Melanoma. Oncogene 2006, 25, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, Z. STAT1 in Cancer: Friend or Foe? Discov. Med. 2017, 24, 19–29. [Google Scholar]
- Nakayama, Y.; Mimura, K.; Tamaki, T.; Shiraishi, K.; Kua, L.-F.; Koh, V.; Ohmori, M.; Kimura, A.; Inoue, S.; Okayama, H.; et al. Phospho-STAT1 Expression as a Potential Biomarker for Anti-PD-1/Anti-PD-L1 Immunotherapy for Breast Cancer. Int. J. Oncol. 2019, 54, 2030–2038. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Ahn, H.J.; Kim, J.K.; Kim, J.; Lee, S.H.; Chae, H.B. Morphine Suppresses Lung Cancer Cell Proliferation Through the Interaction with Opioid Growth Factor Receptor: An In Vitro and Human Lung Tissue Study. Anesth. Analg. 2016, 123, 1429–1436. [Google Scholar] [CrossRef]
- Zagon, I.S.; Donahue, R.; McLaughlin, P.J. Targeting the Opioid Growth Factor: Opioid Growth Factor Receptor Axis for Treatment of Human Ovarian Cancer. Exp. Biol. Med. 2013, 238, 579–587. [Google Scholar] [CrossRef]
- Sigurgeirsson, B.; Emanuelsson, O.; Lundeberg, J. Sequencing Degraded RNA Addressed by 3’ Tag Counting. PLoS ONE 2014, 9, e91851. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
| Characteristic | n = 102 (%) |
|---|---|
| Sex | |
| Male | 24 (24) |
| Female | 78 (76) |
| Age | Range 46–100 |
| ≤50 years | 2 (2.0) |
| 51–69 years | 19 (19) |
| 70–84 years | 52 (51) |
| 85–100 years | 29 (28) |
| Died from MCC | |
| Yes | 28 (27) |
| No | 74 (73) |
| Tumor location | |
| Head and neck | 64 (63) |
| Torso | 10 (10) |
| Limbs | 28 (27) |
| Stage 1 at diagnosis | |
| I | 49 (51) |
| II | 24 (26) |
| III | 8 (8.5) |
| IV | 3 (3.2) |
| Data not available | 8 |
| Metastasis 2 at diagnosis | |
| Present | 11 (11) |
| Not present | 86 (89) |
| Data not available | 5 |
| MCPyV status | |
| Negative | 27 (26) |
| Positive | 75 (74) |
| Variable | HR (95% CI) | p-Value |
|---|---|---|
| MCPyV-negativity | 2.90 (1.28–6.60) | 0.011 |
| Metastasis 1 at diagnosis | 7.78 (3.24–18.70) | <0.001 |
| Male sex | 3.04 (1.37–6.73) | 0.006 |
| Death-Associated Genes | Survival-Associated Genes | ||||||
|---|---|---|---|---|---|---|---|
| Gene | logFC | p-Value | FDR | Gene | logFC | p-Value | FDR |
| TCHH | 2.11 | 8.07 × 106 | 0.010 | GNLY | −2.00 | 2.71 × 105 | 0.017 |
| IGF2 | 1.90 | 2.03 × 105 | 0.014 | CEBPA | −1.82 | 5.43 × 105 | 0.022 |
| DNAH5 | 1.89 | 5.77 × 106 | 0.010 | CARD11 | −1.72 | 1.49 × 104 | 0.032 |
| SV2C | 1.88 | 1.05 × 104 | 0.027 | GBP5 | −1.68 | 3.74 × 105 | 0.018 |
| COL11A1 | 1.82 | 1.63 × 106 | 0.004 | DUSP2 | −1.61 | 2.63 × 106 | 0.006 |
| PPFIA2 | 1.70 | 6.51 × 106 | 0.010 | LLGL1 | −1.47 | 2.81 × 104 | 0.044 |
| COL21A1 | 1.69 | 3.55 × 105 | 0.018 | CD52 | −1.42 | 1.07 × 104 | 0.027 |
| COL11A2 | 1.68 | 9.38 × 106 | 0.010 | CD72 | −1.31 | 1.22 × 104 | 0.029 |
| CRACD | 1.63 | 7.48 × 105 | 0.023 | TAP2 | −1.29 | 6.97 × 105 | 0.023 |
| RBFOX3 | 1.55 | 1.71 × 105 | 0.013 | FOLR2 | −1.26 | 3.35 × 104 | 0.049 |
| MYB | 1.49 | 1.21 × 107 | 0.001 | LCP2 | −1.25 | 3.59 × 104 | 0.050 |
| MECOM | 1.48 | 2.02 × 105 | 0.014 | NLRC5 | −1.24 | 5.69 × 105 | 0.022 |
| TCEA3 | 1.41 | 9.54 × 105 | 0.025 | SPN | −1.19 | 3.22 × 104 | 0.048 |
| MLF1 | 1.41 | 8.13 × 106 | 0.010 | SASH3 | −1.16 | 1.61 × 104 | 0.033 |
| VSIG8 | 1.40 | 2.33 × 104 | 0.041 | C1QB | −1.10 | 2.19 × 104 | 0.040 |
| MDGA1 | 1.40 | 6.61 × 105 | 0.023 | B4GALNT4 | −1.06 | 3.15 × 104 | 0.047 |
| H2AC8 | 1.39 | 4.70 × 105 | 0.021 | HLA-DRA | −1.05 | 3.05 × 105 | 0.017 |
| TRIM2 | 1.39 | 8.08 × 105 | 0.023 | FMC1 | −1.01 | 2.21 × 104 | 0.040 |
| H1-3 | 1.35 | 2.46 × 104 | 0.042 | RFT1 | −1.01 | 1.38 × 105 | 0.012 |
| MELTF | 1.33 | 7.17 × 105 | 0.023 | CD74 | −1.01 | 1.93 × 104 | 0.036 |
| DST | 1.30 | 7.70 × 107 | 0.004 | LDLRAD3 | −0.92 | 2.79 × 104 | 0.044 |
| PHLDA1 | 1.29 | 7.98 × 105 | 0.023 | CIB1 | −0.85 | 3.51 × 104 | 0.050 |
| ENC1 | 1.29 | 1.25 × 104 | 0.029 | HLA-E | −0.72 | 6.14 × 105 | 0.022 |
| SYT11 | 1.28 | 1.33 × 105 | 0.012 | ATF6B | −0.72 | 2.38 × 104 | 0.041 |
| XYLT1 | 1.25 | 1.18 × 104 | 0.029 | PSME1 | −0.65 | 1.57 × 104 | 0.033 |
| VEGFA | 1.25 | 1.29 × 104 | 0.029 | RPS4X | −0.65 | 5.36 × 105 | 0.022 |
| CADM1 | 1.23 | 3.66 × 105 | 0.018 | MXD4 | −0.63 | 7.98 × 105 | 0.023 |
| NTNG2 | 1.13 | 8.34 × 105 | 0.023 | GNAI2 | −0.56 | 1.55 × 104 | 0.033 |
| NEDD4L | 1.13 | 7.85 × 105 | 0.023 | OGFR | −0.48 | 3.13 × 104 | 0.047 |
| SPATA6 | 1.10 | 2.32 × 104 | 0.041 | ||||
| RRBP1 | 1.09 | 1.43 × 106 | 0.004 | ||||
| KIF23 | 1.02 | 2.66 × 104 | 0.043 | ||||
| SULF2 | 1.01 | 1.41 × 104 | 0.031 | ||||
| CCDC30 | 1.00 | 1.83 × 104 | 0.035 | ||||
| ITGA6 | 0.99 | 3.42 × 104 | 0.049 | ||||
| PCDH9 | 0.98 | 2.96 × 104 | 0.046 | ||||
| SMYD3 | 0.97 | 1.27 × 104 | 0.029 | ||||
| IGFBP5 | 0.96 | 5.95 × 105 | 0.022 | ||||
| ADAM12 | 0.93 | 2.54 × 104 | 0.042 | ||||
| CIT | 0.93 | 8.85 × 105 | 0.024 | ||||
| MEIS2 | 0.91 | 1.42 × 105 | 0.012 | ||||
| ASB1 | 0.84 | 4.80 × 105 | 0.021 | ||||
| ATN1 | 0.83 | 1.75 × 104 | 0.034 | ||||
| TUBA1A | 0.81 | 3.14 × 105 | 0.017 | ||||
| AKT3 | 0.80 | 3.05 × 105 | 0.017 | ||||
| TMCO3 | 0.78 | 9.69 × 105 | 0.025 | ||||
| CADM4 | 0.71 | 1.72 × 104 | 0.034 | ||||
| TPM1 | 0.66 | 3.57 × 104 | 0.050 | ||||
| CHD7 | 0.65 | 1.75 × 104 | 0.034 | ||||
| HP1BP3 | 0.56 | 2.56 × 104 | 0.042 | ||||
| Top 10 GO Biological Process Terms Most Significantly Enriched by Death-Associated Genes | |||
|---|---|---|---|
| GO Term | p-Value | q-Value | Genes |
| chordate embryonic development (GO:0043009) | 3.30 × 107 | <0.001 | [CHD7, IGF2, XYLT1, VEGFA, SULF2] |
| collagen fibril organization (GO:0030199) | 2.82 × 106 | 0.001 | [DST, COL11A1, COL11A2, COL21A1, ITGA6] |
| supramolecular fiber organization (GO:0097435) | 2.52 × 105 | 0.006 | [DST, COL11A1, TPM1, COL11A2, TCHH, COL21A1, ITGA6] |
| in utero embryonic development (GO:0001701) | 3.25 × 105 | 0.006 | [CHD7, IGF2, VEGFA] |
| skeletal system development (GO:0001501) | 4.59 × 105 | 0.006 | [CHD7, COL11A2, IGF2, XYLT1, SULF2] |
| extracellular matrix organization (GO:0030198) | 9.89 × 105 | 0.011 | [DST, COL11A1, ADAM12, COL11A2, COL21A1, ITGA6] |
| regulation of vascular associated smooth muscle cell migration (GO:1904752) | 3.98 × 104 | 0.034 | [IGFBP5, TPM1] |
| hemidesmosome assembly (GO:0031581) | 3.98 × 104 | 0.034 | [DST, ITGA6] |
| positive regulation of vascular endothelial cell proliferation (GO:1905564) | 4.69 × 104 | 0.034 | [AKT3, IGF2] |
| heterochromatin organization (GO:0070828) | 5.47 × 104 | 0.034 | [MECOM, HP1BP3] |
| Top 10 GO Molecular Function Terms Most Significantly Enriched by Death-Associated Genes | |||
| vascular endothelial growth factor receptor 2 binding (GO:0043184) | 1.70 × 104 | 0.012 | [CADM4, VEGFA] |
| vascular endothelial growth factor receptor binding (GO:0005172) | 3.98 × 104 | 0.012 | [CADM4, VEGFA] |
| insulin-like growth factor I binding (GO:0031994) | 4.69 × 104 | 0.012 | [IGFBP5, ITGA6] |
| insulin-like growth factor binding (GO:0005520) | 6.30 × 104 | 0.012 | [IGFBP5, ITGA6] |
| histone-lysine N-methyltransferase activity (GO:0018024) | 5.66 × 103 | 0.087 | [MECOM, SMYD3] |
| PDZ domain binding (GO:0030165) | 1.09 × 102 | 0.137 | [CADM1, CIT] |
| neuregulin binding (GO:0038132) | 1.24 × 102 | 0.137 | [ITGA6] |
| myosin light chain binding (GO:0032027) | 1.49 × 102 | 0.140 | [SPATA6] |
| insulin-like growth factor II binding (GO:0031995) | 1.74 × 102 | 0.140 | [IGFBP5] |
| sodium channel inhibitor activity (GO:0019871) | 1.98 × 102 | 0.140 | [NEDD4L] |
| Top 10 GO Biological Process Terms Most Significantly Enriched by Survival-Associated Genes | |||
|---|---|---|---|
| GO Term | p-Value | q-Value | Genes |
| antigen processing and presentation of endogenous peptide antigen (GO:0002483) | 9.87 × 107 | <0.001 | [TAP2, HLA-DRA, HLA-E] |
| positive regulation of immune response (GO:0050778) | 4.03 × 106 | 0.001 | [SASH3, CD74, GBP5, HLA-DRA] |
| positive regulation of innate immune response (GO:0045089) | 1.89 × 105 | 0.003 | [GBP5, NLRC5, HLA-E] |
| T cell receptor signaling pathway (GO:0050852) | 7.63 × 105 | 0.009 | [PSME1, HLA-DRA, LCP2, CARD11] |
| antigen receptor-mediated signaling pathway (GO:0050851) | 1.40 × 104 | 0.012 | [PSME1, HLA-DRA, LCP2, CARD11] |
| antigen processing and presentation of exogenous peptide antigen via MHC class I, TAP-dependent (GO:0002479) | 1.59 × 104 | 0.012 | [TAP2, PSME1, HLA-E] |
| positive regulation of lymphocyte proliferation (GO:0050671) | 1.73 × 104 | 0.012 | [SASH3, CD74, HLA-E] |
| antigen processing and presentation of exogenous peptide antigen via MHC class I (GO:0042590) | 1.94 × 104 | 0.012 | [TAP2, PSME1, HLA-E] |
| positive regulation of alpha-beta T cell activation (GO:0046635) | 3.80 × 104 | 0.020 | [HLA-DRA, HLA-E] |
| antigen processing and presentation of exogenous peptide antigen (GO:0002478) | 4.40 × 104 | 0.021 | [CD74, HLA-DRA, HLA-E] |
| Top 10 GO Molecular Function Terms Most Significantly Enriched by Survival-Associated Genes | |||
| MHC protein binding (GO:0042287) | 9.00 × 106 | 0.001 | [CD74, TAP2, HLA-E] |
| MHC class I protein binding (GO:0042288) | 2.72 × 104 | 0.005 | [TAP2, HLA-E] |
| MHC class II protein complex binding (GO:0023026) | 2.72 × 104 | 0.005 | [CD74, HLA-DRA] |
| TAP1 binding (GO:0046978) | 7.23 × 103 | 0.061 | [TAP2] |
| MHC class II protein binding (GO:0042289) | 8.67 × 103 | 0.061 | [CD74] |
| peptide transmembrane transporter activity (GO:1904680) | 1.01 × 102 | 0.061 | [TAP2] |
| MHC class Ib protein binding (GO:0023029) | 1.15 × 102 | 0.061 | [TAP2] |
| CD4 receptor binding (GO:0042609) | 1.15 × 102 | 0.061 | [CD74] |
| natural killer cell lectin-like receptor binding (GO:0046703) | 1.30 × 102 | 0.061 | [HLA-E] |
| guanylate kinase activity (GO:0004385) | 1.30 × 102 | 0.061 | [CARD11] |
| Top 10 KEGG Pathways Most Significantly Enriched by Death-Associated Genes | |||
|---|---|---|---|
| KEGG Pathway Term | p-Value | q-Value | Genes |
| PI3K-Akt signaling pathway | 1.86 × 103 | 0.138 | [MYB, AKT3, IGF2, ITGA6, VEGFA] |
| Protein digestion and absorption | 2.18 × 103 | 0.138 | [COL11A1, COL11A2, COL21A1] |
| Cell adhesion molecules | 6.04 × 103 | 0.164 | [NTNG2, CADM1, ITGA6] |
| MAPK signaling pathway | 6.20 × 103 | 0.164 | [MECOM, AKT3, IGF2, VEGFA] |
| VEGF signaling pathway | 9.57 × 103 | 0.164 | [AKT3, VEGFA] |
| Pathways in cancer | 1.03 × 102 | 0.164 | [MECOM, AKT3, IGF2, ITGA6, VEGFA] |
| Lysine degradation | 1.09 × 102 | 0.164 | [MECOM, SMYD3] |
| Renal cell carcinoma | 1.29 × 102 | 0.164 | [AKT3, VEGFA] |
| Focal adhesion | 1.39 × 102 | 0.164 | [AKT3, ITGA6, VEGFA] |
| Proteoglycans in cancer | 1.46 × 102 | 0.164 | [AKT3, IGF2, VEGFA] |
| Top 10 KEGG Pathways Most Significantly Enriched by Survival-Associated Genes | |||
| Antigen processing and presentation | 8.74 × 108 | <0.001 | [CD74, TAP2, PSME1, HLA-DRA, HLA-E] |
| Human cytomegalovirus infection | 2.97 × 104 | 0.015 | [ATF6B, TAP2, HLA-E, GNAI2] |
| Cell adhesion molecules | 1.26 × 103 | 0.025 | [SPN, HLA-DRA, HLA-E] |
| Phagosome | 1.36 × 103 | 0.025 | [TAP2, HLA-DRA, HLA-E] |
| Allograft rejection | 1.38 × 103 | 0.025 | [HLA-DRA, HLA-E] |
| Graft-versus-host disease | 1.69 × 103 | 0.025 | [HLA-DRA, HLA-E] |
| Type I diabetes mellitus | 1.77 × 103 | 0.025 | [HLA-DRA, HLA-E] |
| Cocaine addiction | 2.29 × 103 | 0.029 | [ATF6B, GNAI2] |
| Autoimmune thyroid disease | 2.67 × 103 | 0.029 | [HLA-DRA, HLA-E] |
| Epstein–Barr virus infection | 3.06 × 103 | 0.029 | [TAP2, HLA-DRA, HLA-E] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sundqvist, B.; Kilpinen, S.; Böhling, T.; Koljonen, V.; Sihto, H. Activation of Oncogenic and Immune-Response Pathways Is Linked to Disease-Specific Survival in Merkel Cell Carcinoma. Cancers 2022, 14, 3591. https://doi.org/10.3390/cancers14153591
Sundqvist B, Kilpinen S, Böhling T, Koljonen V, Sihto H. Activation of Oncogenic and Immune-Response Pathways Is Linked to Disease-Specific Survival in Merkel Cell Carcinoma. Cancers. 2022; 14(15):3591. https://doi.org/10.3390/cancers14153591
Chicago/Turabian StyleSundqvist, Benjamin, Sami Kilpinen, Tom Böhling, Virve Koljonen, and Harri Sihto. 2022. "Activation of Oncogenic and Immune-Response Pathways Is Linked to Disease-Specific Survival in Merkel Cell Carcinoma" Cancers 14, no. 15: 3591. https://doi.org/10.3390/cancers14153591
APA StyleSundqvist, B., Kilpinen, S., Böhling, T., Koljonen, V., & Sihto, H. (2022). Activation of Oncogenic and Immune-Response Pathways Is Linked to Disease-Specific Survival in Merkel Cell Carcinoma. Cancers, 14(15), 3591. https://doi.org/10.3390/cancers14153591