
Focal Adhesion Kinase Provides a Collateral Vulnerability That Can Be Leveraged to Improve mTORC1 Inhibitor Efficacy

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Cell Culture
2.2. Drugs
2.3. In Vivo Studies
2.4. Protein Collection and Western Blots
2.5. RNA Isolation and RNA-seq
2.6. Real-Time Quantitative RT-PCR
2.7. Patient Tumor Samples
2.8. Immunohistochemistry and Scoring
2.9. Cell Growth Assays
2.10. Bioinformatics
2.11. Statistical Methods
3. Results
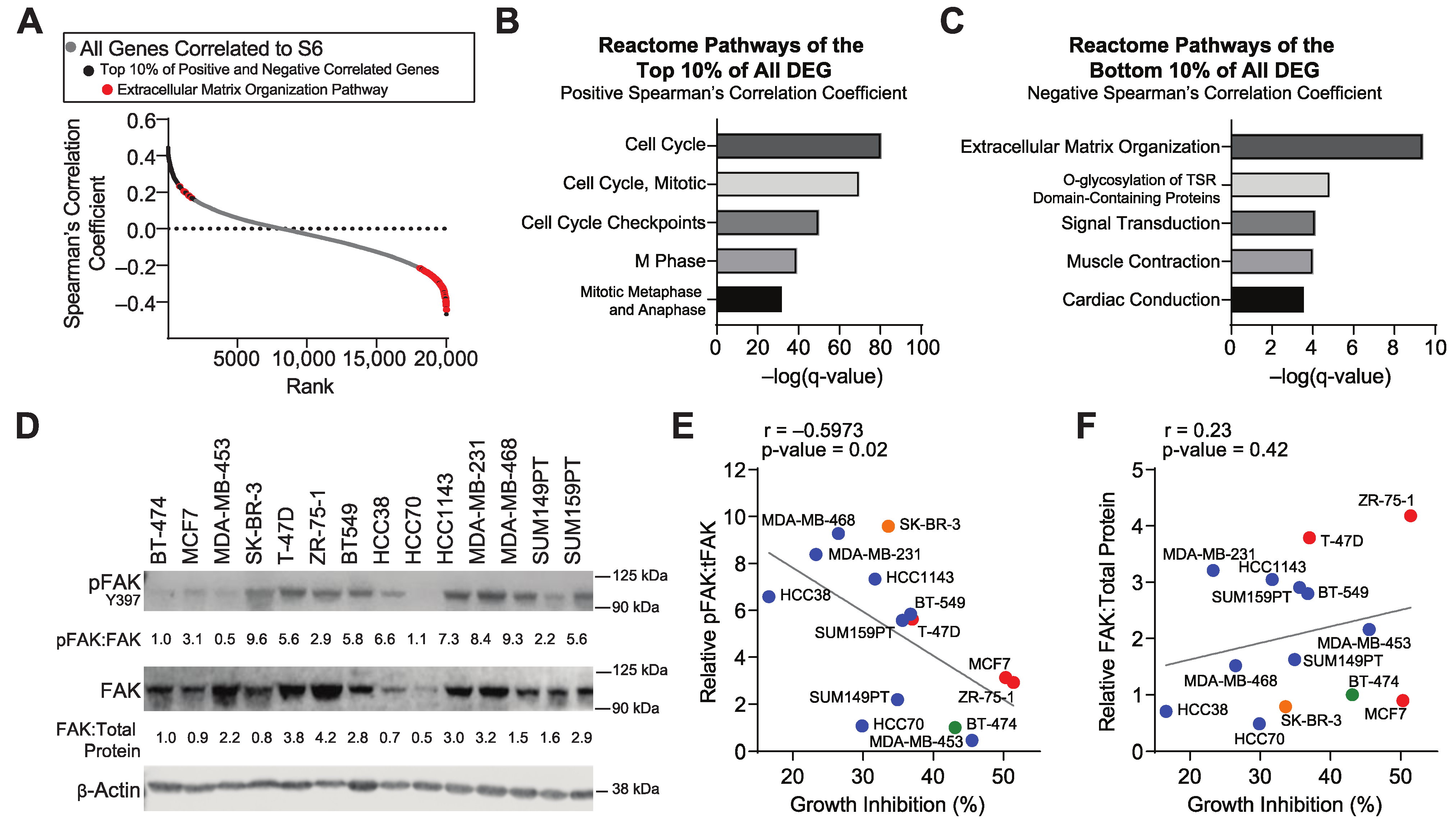
3.1. Inhibition of mTORC1 Signaling in Resistant Tumors Modulates the Expression of Extracellular Matrix Genes
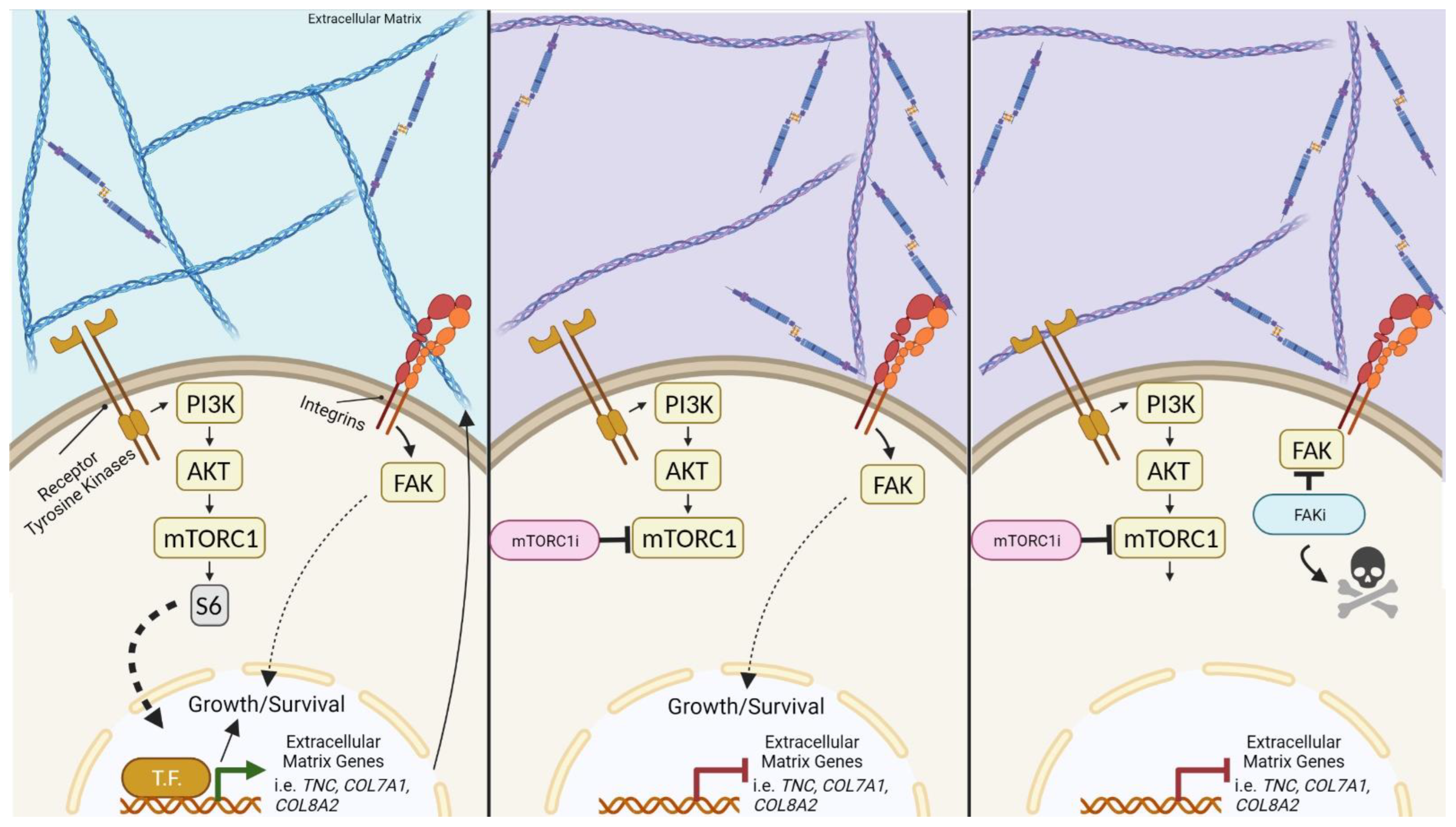
3.2. The Extracellular Matrix and FAK Activation Are Associated with Resistance to mTORC1 Inhibitors
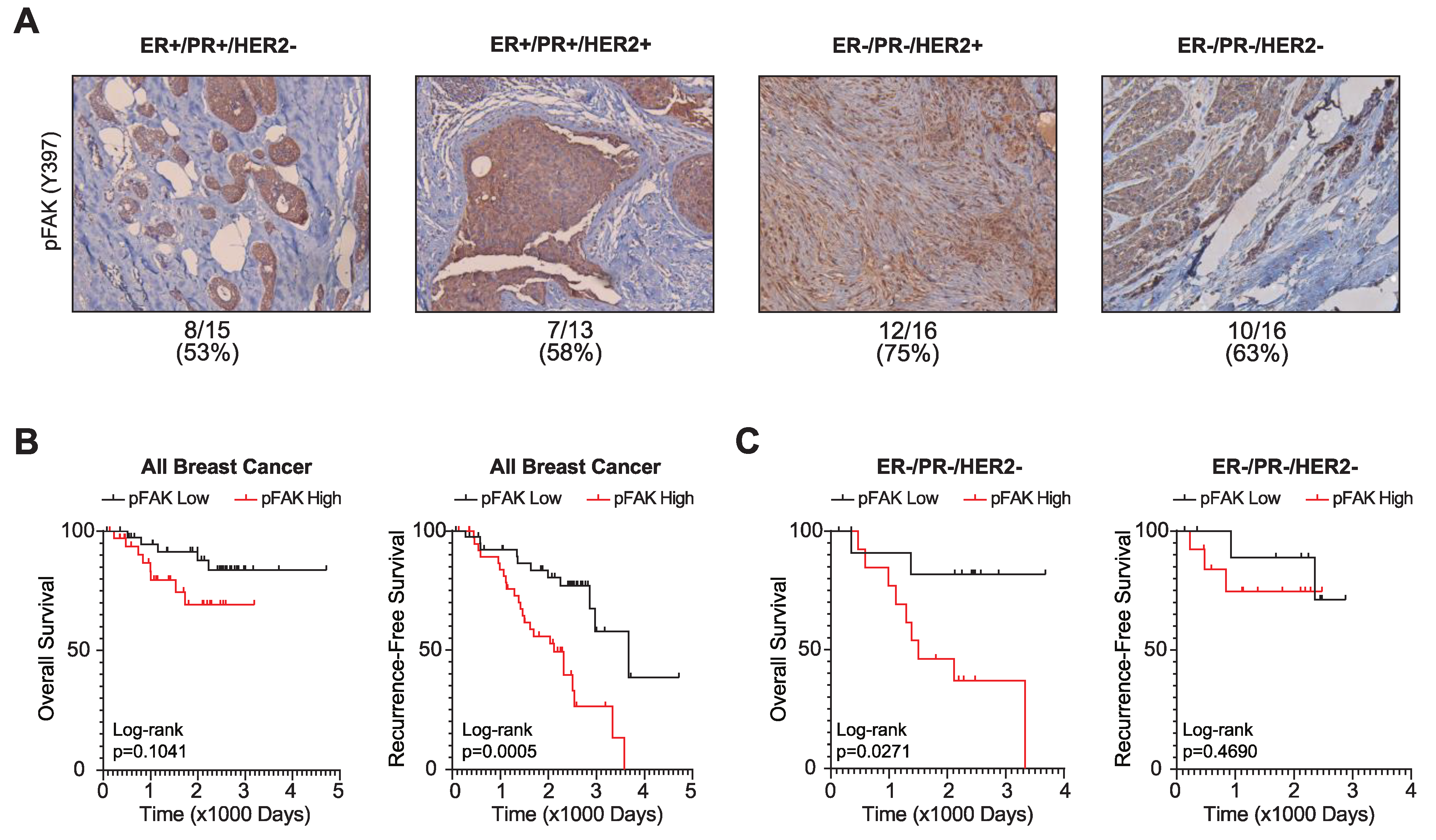
3.3. FAK Activation Is Associated with Poor Breast Cancer Patient Outcomes
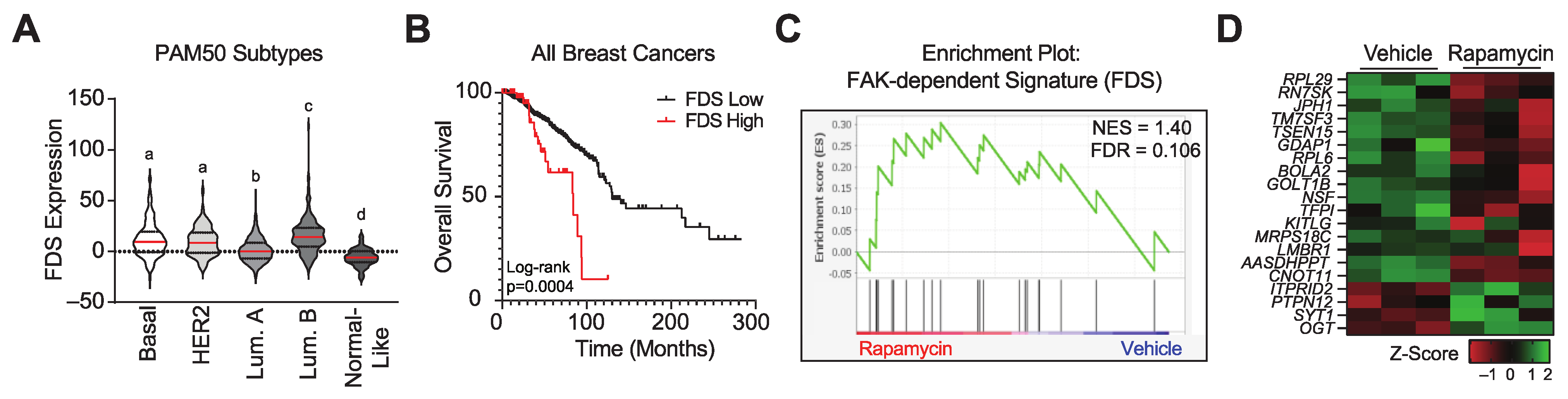
3.4. Pharmacologically Inhibiting mTORC1 Activity Increases the Expression of a FAK Signature Gene Set
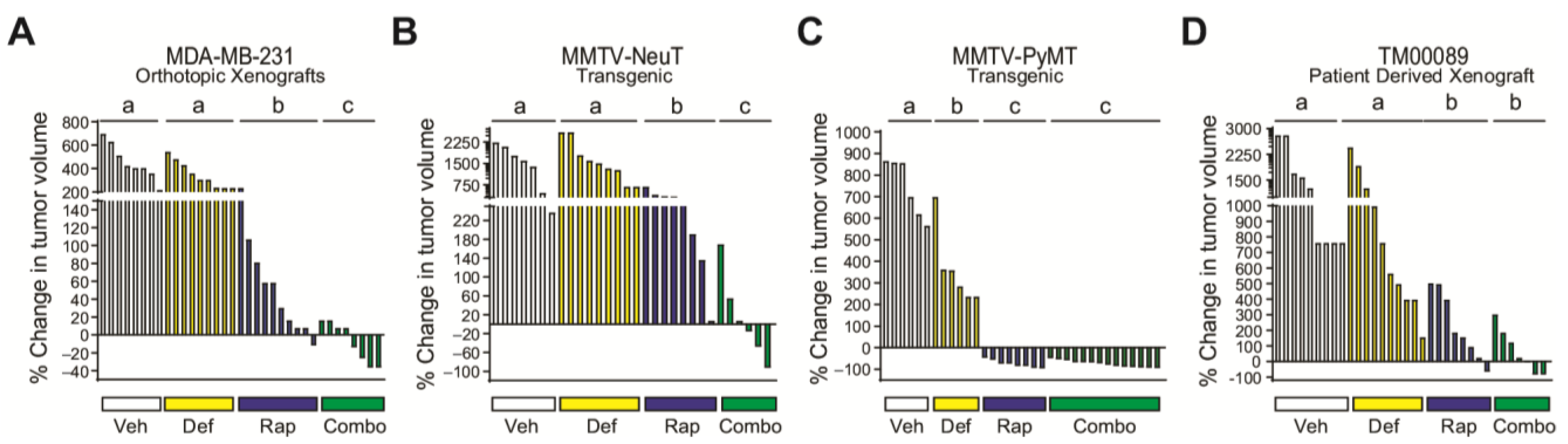
3.5. FAK Blockade Conveys Sensitivity to mTORC1 Inhibition in Resistant/Moderately Resistant Tumors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, J.J.; Loh, K.; Yap, Y.S. PI3K/Akt/mTOR inhibitors in breast cancer. Cancer Biol. Med. 2015, 12, 342–354. [Google Scholar] [PubMed]
- Guerrero-Zotano, A.; Mayer, I.A.; Arteaga, C.L. PI3K/AKT/mTOR: Role in breast cancer progression, drug resistance, and treatment. Cancer Metastasis Rev. 2016, 35, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, I.A.; Arteaga, C.L. The PI3K/AKT Pathway as a Target for Cancer Treatment. Annu. Rev. Med. 2016, 67, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Pernas, S.; Tolaney, S.M. HER2-positive breast cancer: New therapeutic frontiers and overcoming resistance. Ther. Adv. Med. Oncol. 2019, 11, 1758835919833519. [Google Scholar] [CrossRef] [Green Version]
- Paplomata, E.; O’Regan, R. The PI3K/AKT/mTOR pathway in breast cancer: Targets, trials and biomarkers. Ther. Adv. Med. Oncol. 2014, 6, 154–166. [Google Scholar] [CrossRef] [Green Version]
- Markham, A. Alpelisib: First Global Approval. Drugs 2019, 79, 1249–1253. [Google Scholar] [CrossRef]
- Andre, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [Green Version]
- Rodrik-Outmezguine, V.S.; Chandarlapaty, S.; Pagano, N.C.; Poulikakos, P.I.; Scaltriti, M.; Moskatel, E.; Baselga, J.; Guichard, S.; Rosen, N. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov. 2011, 1, 248–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, S.M.; Goulet, D.R.; Johnson, G.L. Targeting the Breast Cancer Kinome. J. Cell Physiol. 2017, 232, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Pluchino, K.M.; Hall, M.D.; Goldsborough, A.S.; Callaghan, R.; Gottesman, M.M. Collateral sensitivity as a strategy against cancer multidrug resistance. Drug Resist. Updat. 2012, 15, 98–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efferth, T.; Saeed, M.E.M.; Kadioglu, O.; Seo, E.J.; Shirooie, S.; Mbaveng, A.T.; Nabavi, S.M.; Kuete, V. Collateral sensitivity of natural products in drug-resistant cancer cells. Biotechnol. Adv. 2020, 38, 107342. [Google Scholar] [CrossRef] [PubMed]
- Mueller, H.S.; Fowler, C.E.; Dalin, S.; Moiso, E.; Udomlumleart, T.; Garg, S.; Hemann, M.T.; Lees, J.A. Acquired resistance to PRMT5 inhibition induces concomitant collateral sensitivity to paclitaxel. Proc. Natl. Acad. Sci. USA 2021, 118, e2024055118. [Google Scholar] [CrossRef] [PubMed]
- Koh, S.-B.; Ross, K.; Isakoff, S.J.; Melkonjan, N.; He, L.; Matissek, K.J.; Schultz, A.; Mayer, E.L.; Traina, T.A.; Carey, L.A.; et al. RASAL2 Confers Collateral MEK/EGFR Dependency in Chemoresistant Triple-Negative Breast Cancer. Clin. Cancer Res. 2021, 27, 4883–4897. [Google Scholar] [CrossRef]
- Guo, W.; Giancotti, F.G. Integrin signalling during tumour progression. Nat. Rev. Mol. Cell Biol. 2004, 5, 816–826. [Google Scholar] [CrossRef]
- Insua-Rodríguez, J.; Oskarsson, T. The extracellular matrix in breast cancer. Adv. Drug Deliv. Rev. 2016, 97, 41–55. [Google Scholar] [CrossRef]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.Y.; Timpson, P.; Horvath, L.G.; Daly, R.J. FAK signaling in human cancer as a target for therapeutics. Pharmacol. Ther. 2015, 146, 132–149. [Google Scholar] [CrossRef]
- Schwill, M.; Tamaskovic, R.; Gajadhar, A.S.; Kast, F.; White, F.M.; Plückthun, A. Systemic analysis of tyrosine kinase signaling reveals a common adaptive response program in a HER2-positive breast cancer. Sci. Signal. 2019, 12, eaau2875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuhlmiller, T.J.; Miller, S.M.; Zawistowski, J.S.; Nakamura, K.; Beltran, A.S.; Duncan, J.S.; Angus, S.P.; Collins, K.A.; Granger, D.A.; Reuther, R.A.; et al. Inhibition of Lapatinib-Induced Kinome Reprogramming in ERBB2-Positive Breast Cancer by Targeting BET Family Bromodomains. Cell Rep. 2015, 11, 390–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Park, C.C.; Hilsenbeck, S.G.; Ward, R.; Rimawi, M.F.; Wang, Y.C.; Shou, J.; Bissell, M.J.; Osborne, C.K.; Schiff, R. beta1 integrin mediates an alternative survival pathway in breast cancer cells resistant to lapatinib. Breast Cancer Res. 2011, 13, R84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, M.H.; Nam, A.-R.; Park, J.E.; Bang, J.-H.; Bang, Y.-J.; Oh, D.-Y. Resistance Mechanism against Trastuzumab in HER2-Positive Cancer Cells and Its Negation by Src Inhibition. Mol. Cancer Ther. 2017, 16, 1145–1154. [Google Scholar] [CrossRef] [Green Version]
- Kessler, B.E.; Mishall, K.M.; Kellett, M.D.; Clark, E.G.; Pugazhenthi, U.; Pozdeyev, N.; Kim, J.; Tan, A.C.; Schweppe, R.E. Resistance to Src inhibition alters the BRAF-mutant tumor secretome to promote an invasive phenotype and therapeutic escape through a FAK>p130Cas>c-Jun signaling axis. Oncogene 2018, 38, 2565–2579. [Google Scholar] [CrossRef]
- Ichihara, E.; Westover, D.; Meador, C.B.; Yan, Y.; Bauer, J.A.; Lu, P.; Ye, F.; Kulick, A.; de Stanchina, E.; McEwen, R.; et al. SFK/FAK Signaling Attenuates Osimertinib Efficacy in Both Drug-Sensitive and Drug-Resistant Models of EGFR-Mutant Lung Cancer. Cancer Res. 2017, 77, 2990–3000. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.O.; Shin, S.; Karreth, F.A.; Buel, G.R.; Jedrychowski, M.P.; Plas, D.R.; Dedhar, S.; Gygi, S.P.; Roux, P.P.; Dephoure, N.; et al. Focal Adhesion- and IGF1R-Dependent Survival and Migratory Pathways Mediate Tumor Resistance to mTORC1/2 Inhibition. Mol. Cell 2017, 67, 512–527.e514. [Google Scholar] [CrossRef]
- Francois, R.A.; Maeng, K.; Nawab, A.; Kaye, F.J.; Hochwald, S.N.; Zajac-Kaye, M. Targeting Focal Adhesion Kinase and Resistance to mTOR Inhibition in Pancreatic Neuroendocrine Tumors. J. Natl. Cancer Inst. 2015, 107, djv123. [Google Scholar] [CrossRef] [Green Version]
- Shi, P.-J.; Xu, L.-H.; Lin, K.-Y.; Weng, W.-J.; Fang, J.-P. Synergism between the mTOR inhibitor rapamycin and FAK down-regulation in the treatment of acute lymphoblastic leukemia. J. Hematol. Oncol. 2016, 9, 12. [Google Scholar] [CrossRef] [Green Version]
- You, D.; Xin, J.; Volk, A.; Wei, W.; Schmidt, R.; Scurti, G.; Nand, S.; Breuer, E.-K.; Kuo, P.C.; Breslin, P.; et al. FAK Mediates a Compensatory Survival Signal Parallel to PI3K-AKT in PTEN-Null T-ALL Cells. Cell Rep. 2015, 10, 2055–2068. [Google Scholar] [CrossRef] [Green Version]
- Noh, W.-C.; Mondesire, W.H.; Peng, J.; Jian, W.; Zhang, H.; Dong, J.; Mills, G.B.; Hung, M.-C.; Meric-Bernstam, F. Determinants of Rapamycin Sensitivity in Breast Cancer Cells. Clin. Cancer Res. 2004, 10, 1013–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurvitz, S.A.; Kalous, O.; Conklin, D.; Desai, A.J.; Dering, J.; Anderson, L.; O’Brien, N.A.; Kolarova, T.; Finn, R.S.; Linnartz, R.; et al. In vitro activity of the mTOR inhibitor everolimus, in a large panel of breast cancer cell lines and analysis for predictors of response. Breast Cancer Res. Treat. 2015, 149, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Dehart, J.P.; Murphy, J.M.; Lim, S.T. Understanding the roles of FAK in cancer: Inhibitors, genetic models, and new insights. J. Histochem. Cytochem. 2015, 63, 114–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yori, J.L.; Lozada, K.L.; Seachrist, D.D.; Mosley, J.D.; Abdul-Karim, F.W.; Booth, C.N.; Flask, C.A.; Keri, R.A. Combined SFK/mTOR Inhibition Prevents Rapamycin-Induced Feedback Activation of AKT and Elicits Efficient Tumor Regression. Cancer Res. 2014, 74, 4762–4771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sizemore, S.T.; Sizemore, G.M.; Booth, C.N.; Thompson, C.L.; Silverman, P.; Bebek, G.; Abdul-Karim, F.W.; Avril, S.; Keri, R.A. Hypomethylation of the MMP7 promoter and increased expression of MMP7 distinguishes the basal-like breast cancer subtype from other triple-negative tumors. Breast Cancer Res. Treat 2014, 146, 25–40. [Google Scholar] [CrossRef] [Green Version]
- Kamburov, A.; Pentchev, K.; Galicka, H.; Wierling, C.; Lehrach, H.; Herwig, R. ConsensusPathDB: Toward a more complete picture of cell biology. Nucleic Acids Res. 2010, 39, D712–D717. [Google Scholar] [CrossRef] [Green Version]
- Kamburov, A.; Wierling, C.; Lehrach, H.; Herwig, R. ConsensusPathDB--a database for integrating human functional interaction networks. Nucleic Acids Res. 2009, 37, D623–D628. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.E.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast Cancer Cell Line Classification and Its Relevance with Breast Tumor Subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vranic, S.; Gatalica, Z.; Wang, Z.-Y. Update on the molecular profile of the MDA-MB-453 cell line as a model for apocrine breast carcinoma studies. Oncol. Lett. 2011, 2, 1131–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iorns, E.; Drews-Elger, K.; Ward, T.M.; Dean, S.; Clarke, J.; Berry, D.; El Ashry, D.; Lippman, M. A New Mouse Model for the Study of Human Breast Cancer Metastasis. PLoS ONE 2012, 7, e47995. [Google Scholar] [CrossRef]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef] [Green Version]
- McLean, G.W.; Carragher, N.O.; Avizienyte, E.; Evans, J.; Brunton, V.G.; Frame, M.C. The role of focal-adhesion kinase in cancer-A new therapeutic opportunity. Nat. Rev. Cancer 2005, 5, 505–515. [Google Scholar] [CrossRef]
- Shen, M.; Jiang, Y.Z.; Wei, Y.; Ell, B.; Sheng, X.; Esposito, M.; Kang, J.; Hang, X.; Zheng, H.; Rowicki, M.; et al. Tinagl1 Suppresses Triple-Negative Breast Cancer Progression and Metastasis by Simultaneously Inhibiting Integrin/FAK and EGFR Signaling. Cancer Cell. 2019, 35, 64–80.e67. [Google Scholar] [CrossRef] [Green Version]
- Alluri, P.; Newman, L.A. Basal-like and triple-negative breast cancers: Searching for positives among many negatives. Surg. Oncol. Clin. N. Am. 2014, 23, 567–577. [Google Scholar] [CrossRef] [Green Version]
- Muller, W.J.; Arteaga, C.L.; Muthuswamy, S.K.; Siegel, P.M.; Webster, M.A.; Cardiff, R.D.; Meise, K.S.; Li, F.; Halter, S.A.; Coffey, R.J. Synergistic interaction of the Neu proto-oncogene product and transforming growth factor alpha in the mammary epithelium of transgenic mice. Mol. Cell. Biol. 1996, 16, 5726–5736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guy, C.T.; Cardiff, R.D.; Muller, W.J. Induction of mammary tumors by expression of polyomavirus middle T oncogene: A transgenic mouse model for metastatic disease. Mol. Cell. Biol. 1992, 12, 954–961. [Google Scholar] [PubMed] [Green Version]
- Mosley, J.D.; Poirier, J.T.; Seachrist, D.D.; Landis, M.D.; Keri, R.A. Rapamycin inhibits multiple stages of c-Neu/ErbB2 induced tumor progression in a transgenic mouse model of HER2-positive breast cancer. Mol. Cancer Ther. 2007, 6, 2188–2197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steelman, L.S.; Martelli, A.M.; Cocco, L.; Libra, M.; Nicoletti, F.; Abrams, S.L.; McCubrey, J.A. The therapeutic potential of mTOR inhibitors in breast cancer. Br. J. Clin. Pharmacol. 2016, 82, 1189–1212. [Google Scholar] [CrossRef]
- Lebwohl, D.; Anak, O.; Sahmoud, T.; Klimovsky, J.; Elmroth, I.; Haas, T.; Posluszny, J.; Saletan, S.; Berg, W. Development of everolimus, a novel oral mTOR inhibitor, across a spectrum of diseases. Ann. N. Y. Acad. Sci. 2013, 1291, 14–32. [Google Scholar] [CrossRef]
- Chung, C.L.; Lawrence, I.; Hoffman, M.; Elgindi, D.; Nadhan, K.; Potnis, M.; Jin, A.; Sershon, C.; Binnebose, R.; Lorenzini, A.; et al. Topical rapamycin reduces markers of senescence and aging in human skin: An exploratory, prospective, randomized trial. GeroScience 2019, 41, 861–869. [Google Scholar] [CrossRef] [Green Version]
- Shegogue, D.; Trojanowska, M. Mammalian target of rapamycin positively regulates collagen type I production via a phosphatidylinositol 3-kinase-independent pathway. J. Biol. Chem. 2004, 279, 23166–23175. [Google Scholar] [CrossRef] [Green Version]
- Bahrami, B.F.; Ataie-Kachoie, P.; Pourgholami, M.H.; Morris, D.L. p70 Ribosomal protein S6 kinase (Rps6kb1): An update. J. Clin. Pathol. 2014, 67, 1019–1025. [Google Scholar] [CrossRef]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell. Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef] [Green Version]
- Shtivelband, M.I. Everolimus in hormone receptor–positive advanced breast cancer: Targeting receptor-based mechanisms of resistance. Breast 2013, 22, 405–410. [Google Scholar] [CrossRef] [Green Version]
- Schettini, F.; Buono, G.; Trivedi, M.V.; De Placido, S.; Arpino, G.; Giuliano, M. PI3K/mTOR Inhibitors in the Treatment of Luminal Breast Cancer. Why, When and to Whom. Breast Care 2017, 12, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Sun, T.; Shao, Z.; Zhang, Q.; Ouyang, Q.; Tong, Z.; Wang, S.; Luo, Y.; Teng, Y.; Wang, X.; et al. Effectiveness of Adding Everolimus to the First-line Treatment of Advanced Breast Cancer in Premenopausal Women Who Experienced Disease Progression While Receiving Selective Estrogen Receptor Modulators: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2021, 7, e213428. [Google Scholar] [CrossRef] [PubMed]
- Holder, A.M.; Akcakanat, A.; Adkins, F.; Evans, K.; Chen, H.; Wei, C.; Milton, D.R.; Li, Y.; Do, K.-A.; Janku, F.; et al. Epithelial to mesenchymal transition is associated with rapamycin resistance. Oncotarget 2015, 6, 19500–19513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, Z.; Ma, F. Biomarkers of Everolimus Sensitivity in Hormone Receptor-Positive Breast Cancer. J. Breast Cancer 2017, 20, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Omarini, C.; Filieri, M.E.; Bettelli, S.; Manfredini, S.; Kaleci, S.; Caprera, C.; Nasso, C.; Barbolini, M.; Guaitoli, G.; Moscetti, L.; et al. Mutational Profile of Metastatic Breast Cancer Tissue in Patients Treated with Exemestane Plus Everolimus. BioMed Res. Int. 2018, 2018, 3756981. [Google Scholar] [CrossRef]
- Citi, V.; Del Re, M.; Martelli, A.; Calderone, V.; Breschi, M.C.; Danesi, R. Phosphorylation of AKT and ERK1/2 and mutations of PIK3CA and PTEN are predictive of breast cancer cell sensitivity to everolimus in vitro. Cancer Chemother. Pharmacol. 2018, 81, 745–754. [Google Scholar] [CrossRef]
- Hanker, A.B.; Estrada, M.V.; Bianchini, G.; Moore, P.D.; Zhao, J.; Cheng, F.; Koch, J.P.; Gianni, L.; Tyson, D.R.; Sanchez, V.; et al. Extracellular Matrix/Integrin Signaling Promotes Resistance to Combined Inhibition of HER2 and PI3K in HER2(+) Breast Cancer. Cancer Res. 2017, 77, 3280–3292. [Google Scholar] [CrossRef] [Green Version]
- Weigelt, B.; Lo, A.T.; Park, C.C.; Gray, J.W.; Bissell, M.J. HER2 signaling pathway activation and response of breast cancer cells to HER2-targeting agents is dependent strongly on the 3D microenvironment. Breast Cancer Res. Treat. 2009, 122, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Shea, M.P.; O’Leary, K.A.; Wegner, K.A.; Vezina, C.M.; Schuler, L.A. High collagen density augments mTOR-dependent cancer stem cells in ERalpha+ mammary carcinomas, and increases mTOR-independent lung metastases. Cancer Lett. 2018, 433, 1–9. [Google Scholar] [CrossRef]
- Joyce, M.H.; Lu, C.; James, E.R.; Hegab, R.; Allen, S.C.; Suggs, L.J.; Brock, A. Phenotypic Basis for Matrix Stiffness-Dependent Chemoresistance of Breast Cancer Cells to Doxorubicin. Front. Oncol. 2018, 8, 337. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, H.A.; Wood, J.M.; McSheehy, P.M.; Allegrini, P.R.; Boulay, A.; Brueggen, J.; Littlewood-Evans, A.; Maira, S.M.; Martiny-Baron, G.; Schnell, C.R.; et al. mTOR inhibitor RAD001 (everolimus) has antiangiogenic/vascular properties distinct from a VEGFR tyrosine kinase inhibitor. Clin. Cancer Res. 2009, 15, 1612–1622. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Yi, Q.; Tang, L. The roles of nuclear focal adhesion kinase (FAK) on Cancer: A focused review. J. Exp. Clin. Cancer Res. 2019, 38, 250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedrosa, A.-R.; Bodrug, N.; Gomez-Escudero, J.; Carter, E.P.; Reynolds, L.E.; Georgiou, P.N.; Fernandez, I.; Lees, D.M.; Kostourou, V.; Alexopoulou, A.N.; et al. Tumor Angiogenesis Is Differentially Regulated by Phosphorylation of Endothelial Cell Focal Adhesion Kinase Tyrosines-397 and -861. Cancer Res. 2019, 79, 4371–4386. [Google Scholar] [CrossRef] [Green Version]
- Tavora, B.; Batista, S.; Reynolds, L.E.; Jadeja, S.; Robinson, S.; Kostourou, V.; Hart, I.; Fruttiger, M.; Parsons, M.; Hodivala-Dilke, K.M. Endothelial FAK is required for tumour angiogenesis. EMBO Mol. Med. 2010, 2, 516–528. [Google Scholar] [CrossRef]
- Conciatori, F.; Bazzichetto, C.; Falcone, I.; Pilotto, S.; Bria, E.; Cognetti, F.; Milella, M.; Ciuffreda, L. Role of mTOR Signaling in Tumor Microenvironment: An Overview. Int. J. Mol. Sci. 2018, 19, 2453. [Google Scholar] [CrossRef] [Green Version]
- Gillespie, Z.E.; MacKay, K.; Sander, M.; Trost, B.; Dawicki, W.; Wickramarathna, A.; Gordon, J.; Eramian, M.; Kill, I.R.; Bridger, J.M.; et al. Rapamycin reduces fibroblast proliferation without causing quiescence and induces STAT5A/B-mediated cytokine production. Nucleus 2015, 6, 490–506. [Google Scholar] [CrossRef] [Green Version]
- Heits, N.; Heinze, T.; Bernsmeier, A.; Kerber, J.; Hauser, C.; Becker, T.; Kalthoff, H.; Egberts, J.H.; Braun, F. Influence of mTOR-inhibitors and mycophenolic acid on human cholangiocellular carcinoma and cancer associated fibroblasts. BMC Cancer 2016, 16, 322. [Google Scholar] [CrossRef] [Green Version]
- Duluc, C.; Moatassim-Billah, S.; Chalabi-Dchar, M.; Perraud, A.; Samain, R.; Breibach, F.; Gayral, M.; Cordelier, P.; Delisle, M.B.; Bousquet-Dubouch, M.P.; et al. Pharmacological targeting of the protein synthesis mTOR/4E-BP1 pathway in cancer-associated fibroblasts abrogates pancreatic tumour chemoresistance. EMBO Mol. Med. 2015, 7, 735–753. [Google Scholar] [CrossRef]
- Zaghdoudi, S.; Decaup, E.; Belhabib, I.; Samain, R.; Cassant-Sourdy, S.; Rochotte, J.; Brunel, A.; Schlaepfer, D.; Cros, J.; Neuzillet, C.; et al. FAK activity in cancer-associated fibroblasts is a prognostic marker and a druggable key metastatic player in pancreatic cancer. EMBO Mol. Med. 2020, 12, e12010. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.J.; Hao, M.; Yeo, S.K.; Guan, J.L. FAK signaling in cancer-associated fibroblasts promotes breast cancer cell migration and metastasis by exosomal miRNAs-mediated intercellular communication. Oncogene 2020, 39, 2539–2549. [Google Scholar] [CrossRef] [PubMed]
- Demircioglu, F.; Wang, J.; Candido, J.; Costa, A.S.H.; Casado, P.; de Luxan Delgado, B.; Reynolds, L.E.; Gomez-Escudero, J.; Newport, E.; Rajeeve, V.; et al. Cancer associated fibroblast FAK regulates malignant cell metabolism. Nat. Commun. 2020, 11, 1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.J.; Tsang, J.Y.; Tse, G.M. Tumor Microenvironment in Breast Cancer-Updates on Therapeutic Implications and Pathologic Assessment. Cancers 2021, 13, 4233. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuellar-Vite, L.; Weber-Bonk, K.L.; Abdul-Karim, F.W.; Booth, C.N.; Keri, R.A. Focal Adhesion Kinase Provides a Collateral Vulnerability That Can Be Leveraged to Improve mTORC1 Inhibitor Efficacy. Cancers 2022, 14, 3374. https://doi.org/10.3390/cancers14143374
Cuellar-Vite L, Weber-Bonk KL, Abdul-Karim FW, Booth CN, Keri RA. Focal Adhesion Kinase Provides a Collateral Vulnerability That Can Be Leveraged to Improve mTORC1 Inhibitor Efficacy. Cancers. 2022; 14(14):3374. https://doi.org/10.3390/cancers14143374
Chicago/Turabian StyleCuellar-Vite, Leslie, Kristen L. Weber-Bonk, Fadi W. Abdul-Karim, Christine N. Booth, and Ruth A. Keri. 2022. "Focal Adhesion Kinase Provides a Collateral Vulnerability That Can Be Leveraged to Improve mTORC1 Inhibitor Efficacy" Cancers 14, no. 14: 3374. https://doi.org/10.3390/cancers14143374
APA StyleCuellar-Vite, L., Weber-Bonk, K. L., Abdul-Karim, F. W., Booth, C. N., & Keri, R. A. (2022). Focal Adhesion Kinase Provides a Collateral Vulnerability That Can Be Leveraged to Improve mTORC1 Inhibitor Efficacy. Cancers, 14(14), 3374. https://doi.org/10.3390/cancers14143374