Abstract
Pancreatic ductal adenocarcinoma (PDAC) has an extremely poor prognosis and represents a major public health issue, as both its incidence and mortality are expecting to increase steeply over the next years. Effective screening strategies are lacking, and most patients are diagnosed with unresectable disease precluding the only chance of cure. Therapeutic options for advanced disease are limited, and the treatment paradigm is still based on chemotherapy, with a few rare exceptions to targeted therapies. Germline variants in cancer susceptibility genes—particularly those involved in mechanisms of DNA repair—are emerging as promising targets for PDAC treatment and prevention. Hereditary PDAC is part of the spectrum of several syndromic disorders, and germline testing of PDAC patients has relevant implications for broad cancer prevention. Germline aberrations in BRCA1 and BRCA2 genes are predictive biomarkers of response to poly(adenosine diphosphate–ribose) polymerase (PARP) inhibitor olaparib and platinum-based chemotherapy in PDAC, while mutations in mismatch repair genes identify patients suitable for immune checkpoint inhibitors. This review provides a timely and comprehensive overview of germline aberrations in PDAC and their implications for clinical care. It also discusses the need for optimal approaches to better select patients for PARP inhibitor therapy, novel therapeutic opportunities under clinical investigation, and preclinical models for cancer susceptibility and drug discovery.
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a malignant disease with an extremely poor prognosis [1,2]. Both incidence and mortality continue to rise, and PDAC is predicted to soon become the second leading cause of cancer-related death [3,4]. Major efforts in improving surgical outcomes and progress in therapeutic development have only marginally increased the 5-year overall survival (OS) rate of patients with PDAC over the past 5 decades, and is still less than 10% [3]. Much still needs to be improved to impact the burden of this disease. Currently, most patients (up to 80%) are diagnosed with unresectable disease due to non-specific symptoms and a lack of effective screening strategies. Earlier diagnosis may potentially improve outcomes since surgical resection is the only chance of cure [5]. As a consequence, the identification of biomarkers for early detection is an urgent priority. Patients with advanced tumors are treated with chemotherapy with an unselected approach, either in the neoadjuvant or metastatic setting. The most effective combinatorial regimens, based on phase III clinical trials, i.e., FOLFIRINOX (5-fluorouracil, leucovorin, irinotecan, and oxaliplatin) and nab-paclitaxel plus gemcitabine, only marginally improve the OS of patients, which rarely exceeds one year [6,7,8,9,10,11,12,13]. After progression, less than 50% of patients are eligible for further treatments due to the rapid clinical deterioration typical of this disease. Second-line treatments have a very limited impact on clinical and survival outcomes, and clinical trials remain the optimal therapeutic choice in this setting [14].
Multi-omics studies have elucidated the molecular complexity of PDAC, which challenges the development of effective treatments for patients with this tumor [15,16,17,18]. In this context, germline variants in cancer susceptibility genes are emerging as clinically relevant targets for more selective PDAC treatment and prevention. This review summarizes the progress in the field of germline aberrations in PDAC and discusses current challenges, limitations, and implications for clinical care.
2. Germline Variants and PDAC Susceptibility
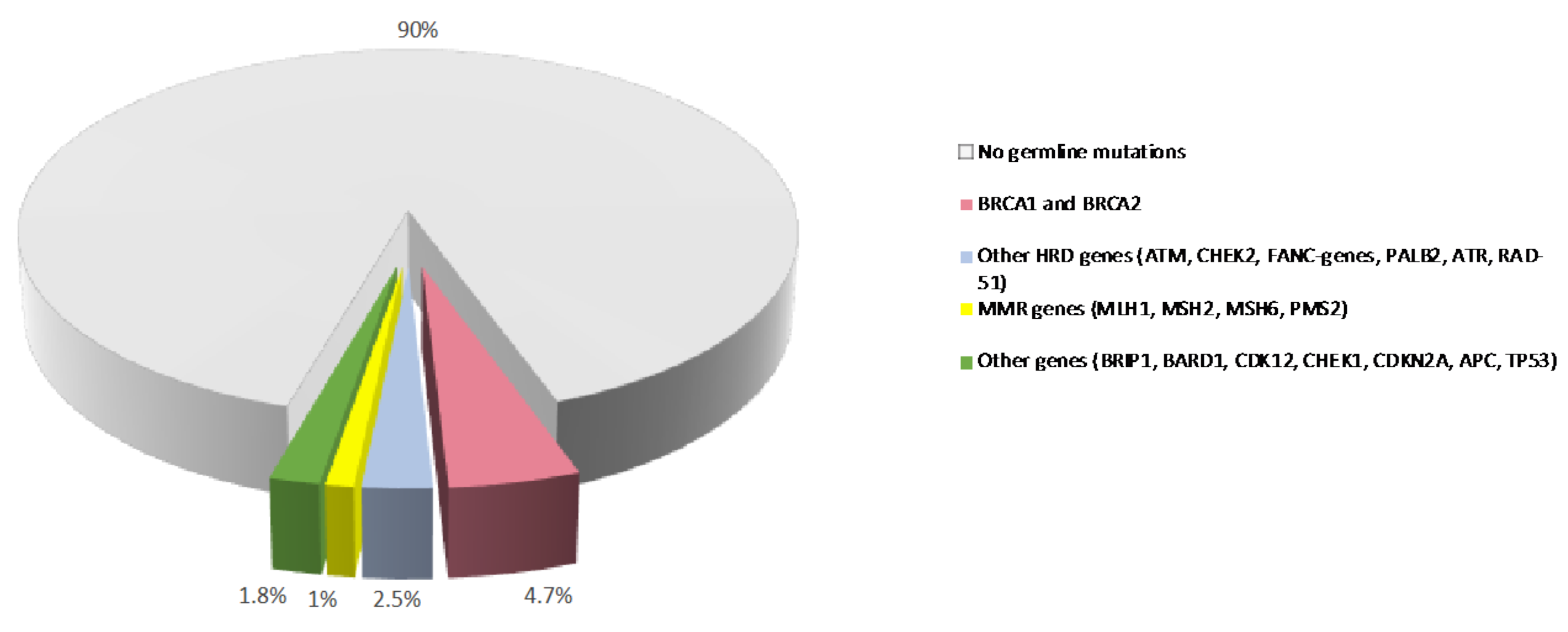
In contrast to somatic mutations, which are acquired during life and arise specifically in tumors, germline variants can be passed from parents to offspring and are associated with hereditary cancer syndromes. Germline pathogenic/likely-pathogenic variants in cancer predisposing genes are prevalent molecular alterations in PDAC (Figure 1) [19,20]. Several studies have shown that 3.8% to 9.7% of patients with PDAC carry a pathogenic germline mutation in genes that predispose them to hereditary cancer syndromes, including familial atypical multiple mole melanoma (CDKN2A), Peutz-Jeghers (STK11), hereditary breast and ovarian cancer (BRCA1, BRCA2, PALB2, ATM), Lynch (MLH1, MSH2, MSH6), and Li-Fraumeni (TP53) syndromes [21,22,23,24,25,26,27,28]. In some large single-center datasets, the prevalence of these alterations is as high as 19.8% [29]. Germline deleterious variants in the hereditary pancreatitis genes PRSS1 and SPINK1 also confer an increased risk of PDAC [30].
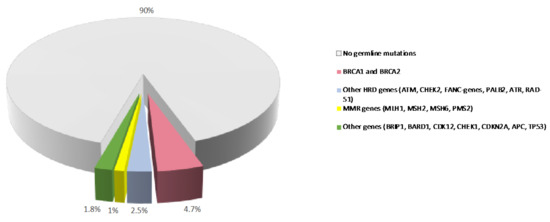
Figure 1.
Prevalence of germline variants in PDAC. Prevalence of germline mutations in PDAC patients from published studies. MMR: mismatch repair. HR: homologous recombination.
Compared with a risk of about 1.5% in the general population, carriers of pathogenic variants in CDKN2A and STK11 have a higher lifetime risk of developing PDAC, which is estimated to be more than 15%. Carriers of pathogenic variants in the breast cancer genes BRCA2, ATM, and PALB2 have a moderate lifetime risk, ranging from 5% to 10%, whereas pathogenic variants in BRCA1 are estimated to confer a lower risk (less than 5%) [31]. More recently, data from a large international consortium of families with hereditary cancer syndromes associated with BRCA germline mutations demonstrated that the relative risk (RR) of PDAC was 2.36 (95% CI, 1.51–3.68) for BRCA1 and 3.34 (95% CI, 2.21–5.06) for BRCA2. The absolute risk of PDAC to age 80 was approximately 2.5% (for both BRCA1 and BRCA2 carriers) [32]. Pathogenic variants in Lynch syndrome genes and TP53 are estimated to confer a moderate pancreatic cancer lifetime risk of about 5–10% [33].
A family history of PDAC also confers an increased risk. Patients with one or more first-degree relatives affected by PDAC are considered familial pancreatic cancer (FPC) cases [34]. In general, in the presence of a first-degree relative family history, the risk of developing PDAC increases with the number of affected relatives (by up to 4, 6, and 32 times for 1, 2, and 3 or more affected relatives, respectively) [35]. Overall, 80–90% of FPC cases are not attributable to a known genetic cause, suggesting the presence of additional genetic factors involved in PDAC susceptibility that have not yet been identified.
The recent broad use of cancer predisposing gene panel testing in clinical practice has allowed for the identification of pathogenic variants in a larger number of candidate cancer susceptibility genes, and also in patients without a family history, the so-called sporadic cases, with mutation rates varying among studies [19,36,37,38]. These findings have supported the current National Comprehensive Cancer Network (NCCN) recommendation of performing extended genetic testing on all patients with a diagnosis of PDAC, regardless of family history or age of onset. Genetic testing should be performed with a comprehensive multi-gene panel, including, at a minimum, the genes ATM, BRCA1, BRCA2, CDKN2A, MLH1, MSH2, MSH6, EPCAM, PALB2, STK11, and TP53 [33].
The opportunity for extended and universal genetic testing as a standard of care or in the research setting is expanding the probability of identifying clinically actionable germline variants in many genes, but their association with increased risk of PDAC is still uncertain. Indeed, a limited number of proposed candidate susceptibility genes have been consistently associated with an increased risk of PDAC, both in familial and sporadic cases [39,40]. For instance, there is no robust evidence suggesting a significant increased risk of PDAC in mutation carriers of CHEK2 pathogenic variants [40], although these variants are frequently observed in PDAC patients [19]. Overall, the rarity of pathogenic variants makes it very challenging to define reliable population-based risk estimates, and much larger studies are warranted. Targeted sequencing using comprehensive cancer gene panels may represent the best way to accumulate data to improve the genetic risk assessment for known-candidate genes. On the other hand, a broader genomic approach using whole exome sequencing or whole genome sequencing in selected high-risk families may help define “missing heritability” in PDAC.
As for most complex diseases, the role of low-penetrance common single nucleotide polymorphisms (SNPs) has been investigated in PDAC using genome-wide association studies (GWAS). According to the GWAS catalogue (accessed May 2021), a total of 200 associations with pancreatic risk were reported; however, few loci reached GWAS statistical significance at a p-value threshold of 5 × 10−8 and was consistently replicated among many studies (reviewed elsewhere [41]). To date, GWAS-identified loci have been estimated to explain about 4% of the phenotypic variation of PDAC; however, more associated SNPs (up to 1750) are expected to be discovered using larger study populations [29,42]. Since each common variant has a small impact on cancer risk, a polygenic architecture, in which many variants that confer low risk individually act in combination to confer much larger risk in the population, has been suggested as a model of cancer susceptibility. Polygenic risk scores (PRS), developed including GWAS identified loci, and multifactorial risk scores (MRS), developed combining genetic and non-genetic risk factors, were recently shown to improve risk prediction in patients with PDAC [43,44,45]. However, the clinical implementation of these models has yet to be established and deserves further assessment.
3. Implications of Germline Variants Identified in PDAC Patients
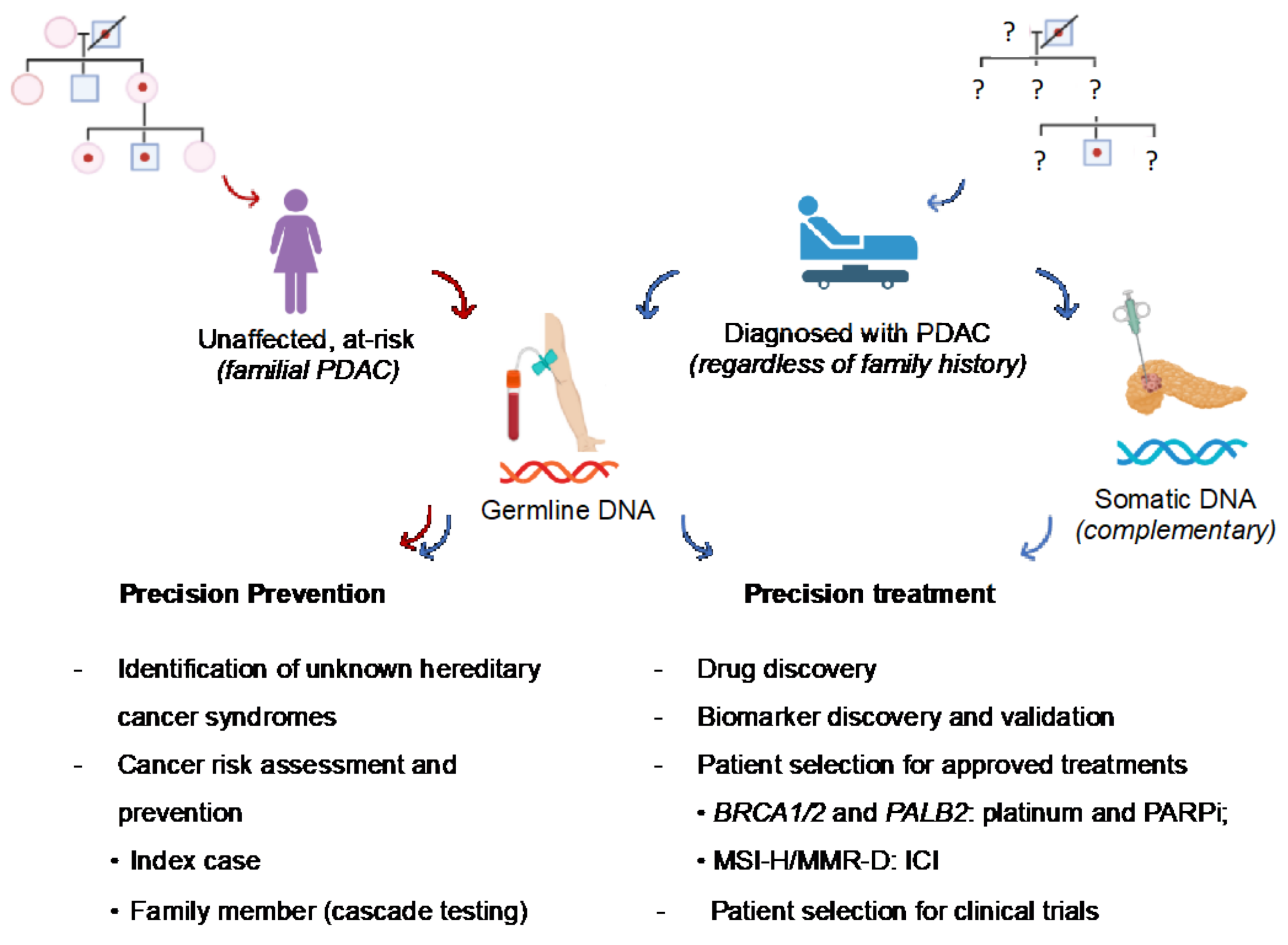
The identification of germline deleterious variants in cancer susceptibility genes in patients with PDAC or healthy subjects with a significant family history of PDAC (at-risk subjects) has relevant implications for cancer prevention and treatment (Figure 2).
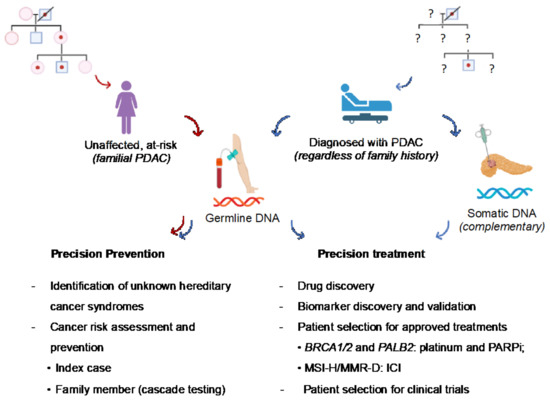
Figure 2.
Overview of the clinical implications of germline variants in PDAC patients and at-risk subjects. Pathogenic germline variants are of key interest in PDAC owing to their therapeutic actionability and implications for cancer prevention (downstream identification of at-risk relatives and possible hereditary cancer syndromes previously unknown in the family). Red arrow: pathway of healthy individuals at risk of PDAC based on family history. Blue arrow: pathway of patients with PDAC who should be tested for germline variants at diagnosis, regardless of family history. Complementary somatic analysis of tumor tissue may help the therapeutic decision (not standard recommendation, only in the research context). MMR-D: mismatch repair-deficiency; MSI-H: microsatellite instability–high; PARPi: poly-ADP (adenosine diphosphate)-ribose polymerase inhibitors; ICI: immune checkpoint inhibitors.
3.1. Preventative Implications
Given that hereditary PDAC is part of the spectrum of several syndromic disorders, germline testing of patients with PDAC has relevant implications for broad cancer prevention. The identification of deleterious variants in well-known cancer susceptibility genes through universal and extended genetic testing, as suggested by recent NCCN guidelines, can potentially become a significant opportunity to reach asymptomatic individuals who are at high risk of certain types of cancers due to genetic predisposition to submit to primary and secondary preventative strategies [46,47]. This can be achieved by applying the strategy of cascade testing, which implies that the direct relatives of mutation carriers are tested in a stepwise manner until all at-risk family members are screened for the specific mutation [48]. As PDAC may share genetic susceptibility with other cancer types (e.g., melanoma, breast, ovarian, colorectal, and prostate cancer), germline testing of healthy relatives through the cascade approach has the potential to indirectly lower overall cancer-related mortality. In this way, PDAC can become a sentinel for the identification of hereditary cancer syndromes previously unknown in the family (Figure 2). Healthy subjects carrying germline variants in BRCA1, BRCA2, PALB2, CDKN2A, ATM, and MMR genes can be referred to as dedicated intensive surveillance programs, which may include risk-reducing measures for several cancer types (breast, ovarian, prostate cancers, colorectal cancer, and melanoma) as standard of care or in research settings to reduce morbidity and mortality due to those syndromes [49].
The management of subjects at risk for PDAC due to familial predispositions is less clear and still debated. First-degree relatives of PDAC patients sharing the same germline deleterious variant are eligible for early detection programs for PDAC. While screening for PDAC is not indicated for the general population due to the relatively low prevalence of this disease, the International Cancer of the Pancreas Screening (CAPS) Consortium [50,51] recommends it to individuals who have a lifetime risk of PDAC > 5% (or a 5-fold increased RR), including those with familial risk only and those diagnosed with predisposing genetic disorders or carrying specific mutations (with or without family history), who may benefit from surgical resection. Familial risk is defined as the presence of (i) at least three affected relatives on the same side of the family, of whom at least one is a first-degree relative; (ii) two affected relatives who are first-degree relatives to each other, of whom at least one is a first-degree relative of the individual to survey; and (iii) at least two affected relatives on the same side of the family, of whom at least one is a first-degree relative [51]. Guidelines of other authoritative international societies, such as those of the American Society of Clinical Oncology [52], NCCN [49], European Society of Medical Oncology [53] and an international panel of experts [54], have also emphasized the importance of PDAC surveillance for high-risk individuals, including those carrying germline pathogenic variants.
Surveillance of these cohorts leads to higher detection rates of PDAC or other pancreatic abnormalities [55]. Although few studies have shown that the surveillance of high-risk subjects could positively impact survival [47,56], it remains uncertain whether PDAC surveillance ultimately reduces PDAC-related mortality. From a clinical standpoint, surveillance should be based on magnetic resonance imaging with cholangiopancreatography, or endoscopic ultrasound, as both have been demonstrated to detect pancreatic disorders at an earlier stage. Regarding the latter, recent reports have shown, unfortunately, that incidental PDAC exists and that 12-month surveillance may sometimes be ineffective [57,58]. In addition, a non-negligible amount of PDAC detected within surveillance is diagnosed at an advanced stage, raising the question of whether this surveillance strategy may be tailored according to an individualized risk-profile [59]. If the screening is negative for pancreatic abnormalities, then it should be repeated annually to possibly improve the early identification of PDAC or pre-malignant lesions. It is not clear at what age pancreatic surveillance should be started. Considering the lifetime risk and anticipation phenomenon that may occur when a germline mutation may be present, 50 years of age (or 10 years earlier than the youngest case of PDAC in the family) is a reasonable cutoff [51,53]. When abnormalities are present, guidelines should be followed for therapeutic management.
In conclusion, the identification of genetic germline variants in PDAC has relevant implications for cancer risk assessment and broad cancer prevention in family members. PDAC surveillance is challenging, and it should not disregard the genetic background of all individuals at risk of PDAC. Ongoing multicenter studies, such as that of the Pancreatic Cancer Early Detection Consortium (PRECEDE), enrolling thousands of individuals at risk of PDAC [60] aims to assess who should be under surveillance and if surveillance ultimately reduces mortality.
3.2. Therapeutic Implications
Emerging data suggest relevant therapeutic implications for PDAC patients with germline pathogenic variants in genes that regulate double-strand break (DSB) repair, such as homologous recombination deficiency (HRD) and MMR (Figure 2). In particular, those in BRCA1, BRCA2, and increasingly PALB2 are the most well characterized and include responses to platinum-based chemotherapy agents and poly-ADP (adenosine diphosphate)-ribose polymerase inhibitor (PARPi) [61,62]. Germline alterations in MMR genes associated with the MSI-high phenotype are predictive of the response to immune checkpoint inhibitors (ICIs) [63,64]. Homologous recombination (HR) is the error-free mechanism of DNA repair that repairs DSB, and its functional defects can be exploited to increase the activity of platinum agents or compounds targeting PARP [65,66]. PARP enzymes are involved in the regulation of multiple cellular processes, including the repair of single-strand DNA breaks through base excision repair [67]. PARPi induces cytotoxic effects by inhibiting PARP enzymes, exploiting a synthetic lethal interaction with defects in HR, most of which are due to BRCA1 or BRCA2 inactivation [68]. The BRCA1 and BRCA2 genes encode critical proteins involved in repairing DSB via HR. The ability to repair double-strand DNA breaks is impaired in cancer cells with deleterious BRCA mutations, resulting in an increased reliance on other DNA damage response (DDR) pathways for survival [69]. As a consequence, they become particularly sensitive to the inhibition of the HR and DDR pathways [69,70]. In patients with germline BRCA mutations, platinum-based chemotherapy and PARP inhibition increase the chance of cancer cell death [71].
A retrospective study of 71 unresectable PDAC patients with BRCA1/2 mutations, showed that those treated with platinum agents achieved significantly longer OS than those treated with non-platinum agents (22 vs. 9 months; p = 0.039) [72]. Although there is no clear evidence for the superiority of first-line platinum agents over non-platinum agents in PDAC patients with germline BRCA1/2 or PALB2 variants, NCCN guidelines recommend FOLFIRINOX or modified FOLFIRINOX or gemcitabine plus cisplatin as first-line chemotherapy for this subgroup of patients [14]. In the phase III POLO study conducted in patients with deleterious or suspected deleterious germline BRCA mutation with metastatic PDAC whose disease had not progressed after at least 16 weeks of first-line platinum-based chemotherapy, subsequent maintenance therapy with PARPi olaparib significantly improved progression-free survival (PFS) versus placebo (7.4 vs. 3.8 months, p = 0.004) with an objective response rate (ORR) of 23% vs. 12%, respectively [12]. These results led to the approval of olaparib in multiple countries as a maintenance therapy after platinum-based first-line treatment in patients with advanced PDAC associated with germline BRCA mutation. Retrospective studies suggest that preoperative platinum-based chemotherapy is most effective in patients with germline BRCA mutation [73,74] highlighting the importance of having BRCA status available at the time of diagnosis, even in patients with early stage disease [75]. In addition, a phase II trial is ongoing to investigate the addition of olaparib following completion of surgery and chemotherapy in patients with resected PDAC and pathogenic mutations in BRCA1, BRCA2, or PALB2 (The APOLLO Trial, ClinicalTrials.gov Identifier: NCT04858334).
Another PARPi, rucaparib, was tested in a phase II study in patients with a pathogenic germline or somatic variant in BRCA1, BRCA2, or PALB2. The results were promising, with a 37% response rate in patients with somatic or germline variants in these genes [76]. PARPi has also been investigated as a monotherapy or in combination with chemotherapy. In phase II trials of patients with advanced PDAC and germline BRCA1 or BRCA2 mutations, olaparib and rucaparib were associated with 21.1% and 21.7% ORR [77,78].
Veliparib was tested in association with gemcitabine and cisplatin in a phase I trial of advanced untreated PDAC patients with BRCA1, BRCA2, and PALB2 mutations, with no significant differences in terms of OS, PFS, or ORR in patients in the investigational arm (treated with veliparib) [79]. A randomized phase II trial of this regimen is currently ongoing (ClinicalTrials.gov Identifier: NCT01585805).
Germline mutations in MMR genes account for around 1% of PDAC cases [80]. MMR increases the fidelity of DNA replication by dealing with the misincorporation of nucleotides [81,82] and relies on highly conserved proteins encoded by the mutS and mutL homologue genes, such as MSH2 and MLH1 [82]. When MMR is defective (dMMR) due to genetic or epigenetic inactivation of MMR genes, the inability to correct DNA replication errors leads to hypermutated genomes with a peculiar mutation pattern affecting dinucleotide repeats, i.e., MSI. dMMR and MSI are observed in sporadic tumors due to double somatic inactivation and also in the context of Lynch syndrome, which is caused by germline mutation of one of the MMR genes (MLH1, MSH2, MSH6, or PMS2) or epigenetic silencing of MSH2 consequent to germline mutation in EPCAM [82]. Although dMMR/MSI is rare in PDAC, it identifies a distinct subgroup of patients with unique clinical, pathological, and genomic features. dMMR PDAC are enriched for several markers of immune activation (including high tumor mutational burden and neoantigen load, chemokine signatures, and cytolytic activity), are less likely to have mutations in usual PDAC driver genes like KRAS and SMAD4, but more likely to have mutations in genes that drive cancers with microsatellite instability like ACV2RA and JAK1 [83].
Anti-PD-1 therapy pembrolizumab showed antitumor activity in dMMR/MSI malignancies regardless of histotype and is now approved for the treatment of patients with unresectable or metastatic dMMR/MSI-high solid tumors, including PDAC, after prior conventional chemotherapies [64]. In a small phase II clinical trial, pembrolizumab achieved an 18% ORR in advanced MSI-high PDAC [63].
There is also basic and translational evidence indicating the synergistic effects of PARPi and ICIs. PARPi-mediated unrepaired DNA breaks modulate the tumor microenvironment by several molecular and cellular mechanisms that might induce a response to ICIs. These include increased genomic instability, activation of immune pathways, and induced PD-L1 expression on cancer cells [84]. Following promising results from breast and ovarian cancer, PARPi are being investigated in combination with ICIs in PDAC patients with HRD, including those with germline mutations (ClinicalTrials.gov Identifier: NCT04666740, NCT04409002).
In conclusion, pathogenic variants in several cancer susceptibility genes are therapeutically actionable with platinum, PARPi, and ICI therapy. Other potential therapeutic targets need to be identified and tested in the clinic to improve the outcomes of patients carrying germline pathogenic variants. There is substantial activity in exploring novel DDR agents and combinations of agents to target these mechanisms in many cancer types. Most notably, the potential ability to generate “synthetic” synthetic lethality, where a drug induces a defect in DDR, can be exploited through the same mechanisms as inherited defects in DDR. Whether non-BRCA mutations in HR genes confer PARPi sensitivity needs to be addressed in the future, although the rarity of the non-BRCA pathogenic variants makes it difficult to evaluate the clinical benefit for those patients.
A selection of interventional clinical trials currently recruiting PDAC patients with germline mutations is reported in Table 1.

Table 1.
Selection of interventional clinical trials for PDAC patients with germline mutations is currently ongoing.
4. Challenges in Targeting BRCA Mutations in PDAC
Despite encouraging results, treatment with PARPi in PDAC patients with germline BRCA mutations is challenging due to the primary and secondary therapeutic resistance that inevitably occurs. Inhibition of PARP activity generates DNA lesions, such as collapsed replication forks, which are repaired by HR. Therefore, it is thought that BRCA1/2 deficient or HR-deficient cancer cells are selectively sensitive to PARPi because the collapsed replication forks are not properly repaired in HR-deficient cancer cells. However, the underlying molecular mechanism of how PARPi induces anti-cancer effects has not been fully understood. In addition, the consequence of BRCA1/2 deficiency appears to have a lineage-dependent effect, and it is likely to be seen in the context of other cancer susceptibility gene mutations [85]. A recent study of the lineage dependency of BRCA-mediated phenotypes highlights the possibility that a distinct epigenetic landscape from a different lineage of cancer cells may influence therapeutic responses to PARPi. Likewise, many epigenetic drugs, such as bromodomain inhibitors, HDAC inhibitors, and DNMT inhibitors, sensitize cancer cells to PARPi, likely converting HR-proficient cancer cells to HR-deficient cells, termed ‘induced BRCAness’ [86,87,88,89,90]. Similarly, genetic alterations in epigenetic regulators, such as ARID1A deficiency and ETS fusion, result in increased sensitivity to PARPi in breast and prostate cancer [91,92]. It is also important to note that several epigenetic regulators are frequently mutated in PDAC, and it is therefore highly possible that epigenetic regulators, transcription regulation, and DDR pathways converge in the early stage of PDAC progression and may play a role in PARPi resistance.
Although several mechanisms of resistance to PARPi have been described in breast and ovarian cancer patients, data on PDAC are still limited. The landscape of somatic mutations in BRCA1/2-associated PDAC is essentially indistinguishable from that of sporadic cancers. As it stands, the two types of disease might share the same evolutionary path, including an estimated timeline of two decades from inception to metastatic disease [93,94,95]. Although speculative, a possible implication would be that HR defects are indispensable founding events for tumor maintenance in a subset of germline BRCA1/2 mutation carriers. If these represent possible explanations of primary resistance, the clinical response profile of PDAC patients to PARPi olaparib also suggests the rapid emergence of secondary resistance [12]. Given that patients received platinum prior to PARPi, it would be difficult to gauge the specific contribution of PARPi to secondary resistance in this context. Unfortunately, the confounding factor of prior therapy with platinum is constant in many clinical trials with PARPi in PDAC, as well as in other BRCA1/2-associated cancer types.
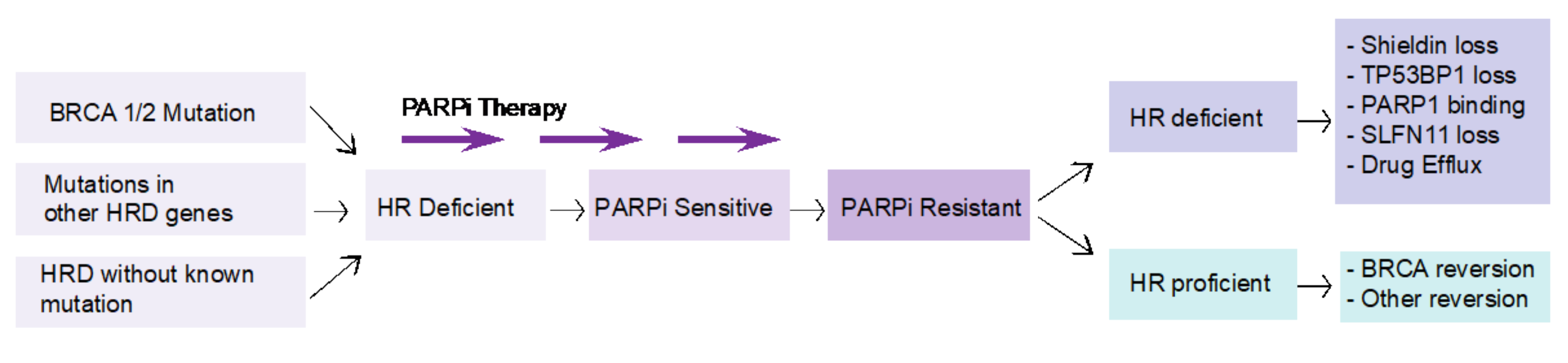
HR deficiency is considered a prerequisite for response to PARP inhibition. Tumors that have acquired resistance to PARP inhibition can be either HR deficient or HR proficient (Figure 3). In HR-proficient tumors, the genetic mutation in the HR gene that results in the HR-deficient phenotype is repaired by a reversion mutation. Secondary mutations either remove the original pathogenic mutation or result in restoration of the open reading frame [96,97,98]. Most of these mutations conferring therapeutic resistance are deletions surrounded by sequence microhomologies, which can be explained by end-joining repair mechanisms [99,100]. Mechanisms of resistance to PARP inhibition where the tumor is still HR deficient are less well understood. Preclinical studies, including forward genetic screens and patient xenograft models, have identified several potential pathways, including loss of activity of the Shieldin complex, TP53BP1 or SLFN11, mutations in PARP1, resulting in loss of PARPi binding, or enhanced drug efflux, resulting in loss of efficacy [101,102,103]. At present, however, robust data corroborating these mechanisms in patients have not been reported. It has been demonstrated that most tumors that develop resistance to PARPi have a hyper-activated ATR/CHK1 pathway. PARP inhibition in BRCA-mutated cancer cells increases reliance on the ATR/CHK1 pathway for genome stability [104]. This provides the rationale for simultaneous inhibition of PARP and ATR/CHK1 pathway. A phase II trial (ClinicalTrials.gov identifier: NCT03462342) is currently investigating olaparib in association with a novel ART inhibitor, AZD6738.
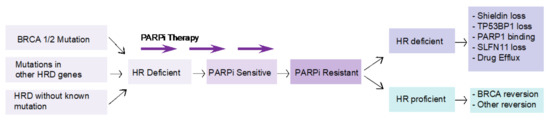
Figure 3.
Mechanisms of resistance to PARPi according to HR status. Homologous recombination (HR) deficiency is considered a prerequisite for response to PARP inhibition. Tumors that have acquired resistance to PARP inhibition can be either HR deficient or HR proficient. In HR-proficient tumors, the genetic mutation in the HR gene that results in the HR-deficient phenotype is repaired by a reversion mutation. Mechanisms of resistance to PARP inhibition where the tumor is still HR deficient include loss of activity of the Shieldin complex, TP53BP1 or SLFN11, mutations in PARP1, or enhanced drug efflux.
In conclusion, treatment with PARPi in PDAC patients with BRCA mutations presents many challenges. Optimal approaches to selecting patients with PDAC for PARPi therapy have yet to be described. Sensitivity to platinum chemotherapy is expected to enrich PARPi sensitivity, although platinum chemotherapy elicits cell death through both HR-dependent and non-HR-dependent mechanisms, thus reducing predictive power. The presence of an inherited mutation in BRCA1/2 or another HR pathway gene may also correlate sub-optimally to PARPi response due to incomplete penetrance. Alternative approaches are based on the detection of the phenotypic consequences of HR deficiency, either at the genomic or functional levels. The genomic scar of HR deficiency is highly characteristic and is likely to represent a strong biomarker of PARPi sensitivity in treatment naïve patients [105]. At present, however, this information can only be acquired through sequencing of the entire, or a large proportion of the cancer genome, which is currently not feasible in routine clinical care for most patients. Surrogate biomarkers for HR deficiency have been reported, such as enumeration of genomic deletions using loss of heterozygosity; however, the sensitivity, specificity, and inter-assay reproducibility of this approach has yet to be determined and is currently the focus of an international harmonization project (https://friendsofcancerresearch.org/hrd, accessed on 8 June 2022). Importantly, genomic scar assays will not distinguish between the two main pathways to PARPi resistance (shown in Figure 3), as tumors that have undergone secondary mutations to reactivate the HR pathway will still carry the genomic scars of HR deficiency. An alternative approach is to deploy a functional assay of HR competence, such as the detection of RAD51 foci in tumor samples [106]. This approach has the potential to select patients for upfront PARPi therapy and to distinguish the key pathways to PARPi resistance, which may be of relevance in informing future lines of therapy.
5. Preclinical and Translational Research in Hereditary PDAC
Preclinical and translational research is essential to improve our understanding of PDAC susceptibility and to facilitate the development of therapeutic strategies for patients with germline mutations. While approximately 10% of PDAC patients may harbor germline mutations, only a few pre-clinical models are commercially available in this field. CAPAN1 is the most commonly used BRCA2-deficient PDAC cell line, harboring BRCA2 c.6774delT truncating mutation [107]. In addition, PL11 and Hs766T harbor genetic alterations in the Fanconi anemia pathway genes FANCC (null mutation) and FANCG (nonsense mutation), respectively [108]. To our knowledge, there are no detailed genomic annotations associated with commercially available PDAC cell lines or other preclinical models, and this hampers the preclinical usage of PDAC cell lines in the context of FPC. Genetically engineered mouse models have been critical for basic PDAC research and the preclinical evaluation of therapeutic strategies. One of the representative PDAC GEMM is the KPC mouse model, which harbors oncogenic KRAS G12D and a gain-of-function p53 R172H or R270H mutation (equivalent to human TP53 R175H or R273H) or p53 null mutation specifically in pancreatic epithelial cells driven by a Pdx1-Cre or Ptf1-Cre transgenic allele [109,110]. Since KRAS and TP53 mutations are the most common genetic alterations in PDAC, putative tumor suppressors or oncogenes have been modeled in the Kras mutant background or Kras/Trp53 double mutant background (reviewed by Guerra and Barbacid [111]). Although many genes are associated with hereditary PDAC, a few genes have been experimentally shown to contribute to PDAC progression in vivo using GEMM. Among cancer predisposing genes, mutations in BRCA2 and ATM play a role in PDAC progression [112,113], whilst the role of other genes remains to be defined. One of the challenges in modeling hereditary PDAC with GEMM is that the generation of conditional knockout alleles for individual cancer predisposing genes is time-consuming. In addition, PDAC mouse models need to be crossed with multiple oncogenic alleles, such as Kras mutations and pancreas-specific Cre alleles. Another critical issue related to hereditary PDAC is that genetic mutations are introduced at the embryonic development stage, which complicates the question of whether cancer predisposing genes play a role in the inception of key driver mutations, such as oncogenic mutations in KRAS in other tumor suppressors. Available GEMMs only allow us to address whether these mutations in cancer predisposing genes can cooperate with oncogenic Kras mutations or other driver mutations for PDAC progression.
In conclusion, various preclinical models of human PDAC are available for basic and translational research, including PDAC cell lines, patient-derived xenografts (PDXs), and patient-derived organoids (PDOs) [114]. Each preclinical model has its own advantages and disadvantages. Thus, the optimal model for each study should be determined based on specific scientific and clinical questions. To evaluate the efficacy of PARPi and other therapies for PDAC patients, a lack of detailed genomic annotations in the currently existing preclinical models is an issue that needs to be addressed. The use of isogenic cell lines or other preclinical models with CRISPR or other genetic engineering approaches is ideal for addressing a mutation-specific drug response in hereditary PDAC. In this way, the possibility of confounding effects that may come from other genetic mutations or other backgrounds can be excluded. In addition, the use of preneoplastic cells (e.g., PanIN-derived organoids) could be useful to address the effect of cancer predisposing gene mutations in the early stage of PDAC progression without generating new GEMM for hereditary PDAC [115].
6. Conclusions
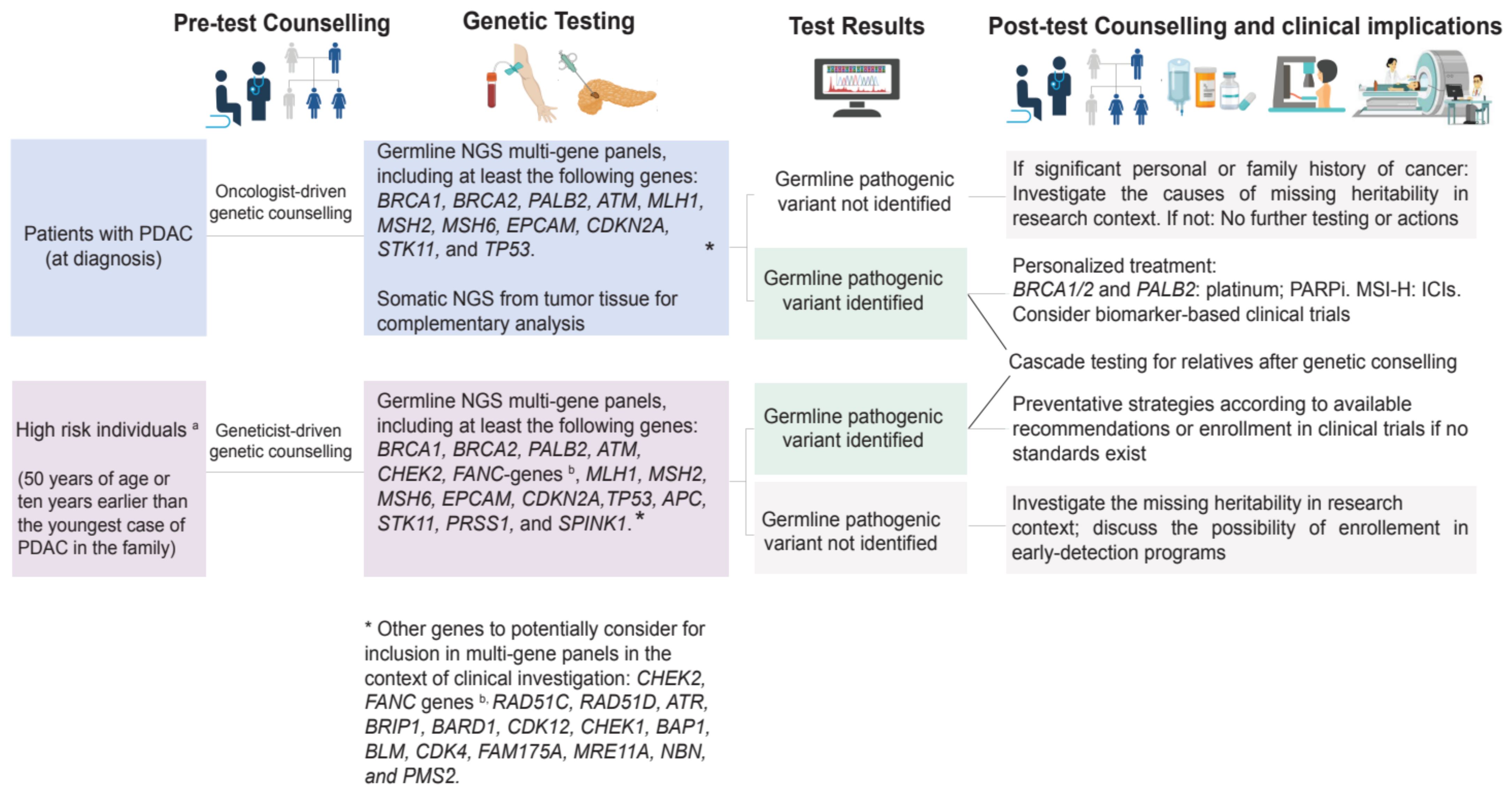
Germline pathogenic variants are prevalent and clinically relevant in PDAC. The American Society of Clinical Oncology and the NCCN guidelines recommend risk assessment and extended germline testing for all individuals with PDAC irrespective of personal or family history of cancer, age, or ethnicity to maximize the opportunity for targeted therapeutic interventions for patients and cancer prevention in the families. The proposed algorithm for germline genetic testing in PDAC and high-risk subjects is shown in Figure 4. Treatment implications include the use of ICI for MMR-d PDAC and PARPi therapy with olaparib as a maintenance strategy in platinum-sensitive BRCA mutation carriers. Whether mutations in other non-BRCA genes can be successfully targeted with PARPi therapies remains to be addressed in both preclinical and clinical settings. Continued research on the actionability of germline variants in PDAC, on the molecular mechanism of how PARPi exerts anti-cancer effects, and on how germline variants contribute to PDAC progression is essential to improve our understanding of PDAC and to facilitate the development of therapeutic and preventative strategies for patients and their families.
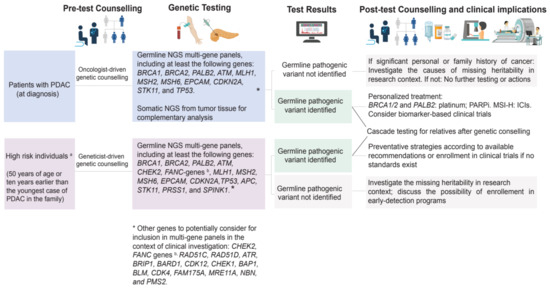
Figure 4.
Suggested algorithm for germline testing for healthy individual at high risk of PDAC and individuals diagnosed with PDAC. MSI-H: microsatellite instability–high; PARPi: poly-ADP (adenosine diphosphate)-ribose polymerase inhibitors; ICIs: immune checkpoint inhibitors.
Author Contributions
Conceptualization, R.C. and A.V.B.; methodology, R.C., V.C., C.-i.H., P.B., S.P., V.S., L.O. and A.V.B.; software, R.C.; resources, R.C., V.C., C.-i.H., P.B. and V.S.; data curation, R.C.; writing—original draft preparation, R.C., V.C., C.-i.H., P.B., S.P., V.S., L.O. and A.V.B.; writing—review and editing, R.C., V.C., C.-i.H., P.B., S.P., V.S., L.O. and A.V.B.; visualization, R.C.; supervision, L.O. and A.V.B.; project administration, R.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
V.S. is supported by Fondazione Umberto Veronesi. The authors would like to thank Fondazione AIRC (Associazione Italiana Ricerca sul Cancro) under IG 2018 ID. 21389 (P.I. Laura Ottini) and Italian Ministry of Education, Universities and Research–Dipartimenti di Eccellenza-L. 232/2016.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Landman, A.; Feetham, L.; Stuckey, D. Working together to reduce the burden of pancreatic cancer. Lancet Oncol. 2020, 21, 334–335. [Google Scholar] [CrossRef]
- Huang, J.; Lok, V.; Ngai, C.H.; Zhang, L.; Yuan, J.; Lao, X.Q.; Ng, K.; Chong, C.; Zheng, Z.J.; Wong, M.C.S. Worldwide Burden of, Risk Factors for, and Trends in Pancreatic Cancer. Gastroenterology 2021, 160, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Wehner, M.R.; Matrisian, L.M.; Nead, K.T. Estimated Projection of US Cancer Incidence and Death to 2040. JAMA Netw. Open 2021, 4, e214708. [Google Scholar] [CrossRef]
- Gaddam, S.; Abboud, Y.; Oh, J.; Samaan, J.S.; Nissen, N.N.; Lu, S.C.; Lo, S.K. Incidence of Pancreatic Cancer by Age and Sex in the US, 2000–2018. JAMA 2021, 326, 2075–2077. [Google Scholar] [CrossRef] [PubMed]
- Gillen, S.; Schuster, T.; Meyer Zum Buschenfelde, C.; Friess, H.; Kleeff, J. Preoperative/neoadjuvant therapy in pancreatic cancer: A systematic review and meta-analysis of response and resection percentages. PLoS Med. 2010, 7, e1000267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nevala-Plagemann, C.; Hidalgo, M.; Garrido-Laguna, I. From state-of-the-art treatments to novel therapies for advanced-stage pancreatic cancer. Nat. Rev. Clin. Oncol. 2020, 17, 108–123. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Chone, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [Green Version]
- Oettle, H.; Riess, H.; Stieler, J.M.; Heil, G.; Schwaner, I.; Seraphin, J.; Gorner, M.; Molle, M.; Greten, T.F.; Lakner, V.; et al. Second-line oxaliplatin, folinic acid, and fluorouracil versus folinic acid and fluorouracil alone for gemcitabine-refractory pancreatic cancer: Outcomes from the CONKO-003 trial. J. Clin. Oncol. 2014, 32, 2423–2429. [Google Scholar] [CrossRef]
- Wang-Gillam, A.; Li, C.P.; Bodoky, G.; Dean, A.; Shan, Y.S.; Jameson, G.; Macarulla, T.; Lee, K.H.; Cunningham, D.; Blanc, J.F.; et al. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): A global, randomised, open-label, phase 3 trial. Lancet 2016, 387, 545–557. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef] [PubMed]
- Tempero, M.A.; Malafa, M.P.; Al-Hawary, M.; Behrman, S.W.; Benson, A.B.; Cardin, D.B.; Chiorean, E.G.; Chung, V.; Czito, B.; Del Chiaro, M.; et al. Pancreatic Adenocarcinoma, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 439–457. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Froeling, F.E.M.; Casolino, R.; Pea, A.; Biankin, A.V.; Chang, D.K. Molecular Subtyping and Precision Medicine for Pancreatic Cancer. J. Clin. Med. 2021, 10, 149. [Google Scholar] [CrossRef]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Casolino, R.; Paiella, S.; Azzolina, D.; Beer, P.A.; Corbo, V.; Lorenzoni, G.; Gregori, D.; Golan, T.; Braconi, C.; Froeling, F.E.M.; et al. Homologous Recombination Deficiency in Pancreatic Cancer: A Systematic Review and Prevalence Meta-Analysis. J. Clin. Oncol. 2021, 39, 2617–2631. [Google Scholar] [CrossRef]
- Gardiner, A.; Kidd, J.; Elias, M.C.; Young, K.; Mabey, B.; Taherian, N.; Cummings, S.; Malafa, M.; Rosenthal, E.; Permuth, J.B. Pancreatic Ductal Carcinoma Risk Associated with Hereditary Cancer-Risk Genes. J. Natl. Cancer Inst. 2022, djac069. [Google Scholar] [CrossRef]
- Jones, S.; Hruban, R.H.; Kamiyama, M.; Borges, M.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Palmisano, E.; Brune, K.; Jaffee, E.M.; et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science 2009, 324, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, N.J.; Jiao, Y.; Yu, J.; Kopelovich, L.; Petersen, G.M.; Bondy, M.L.; Gallinger, S.; Schwartz, A.G.; Syngal, S.; Cote, M.L.; et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2012, 2, 41–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, R.C.; Selander, I.; Connor, A.A.; Selvarajah, S.; Borgida, A.; Briollais, L.; Petersen, G.M.; Lerner-Ellis, J.; Holter, S.; Gallinger, S. Prevalence of germline mutations in cancer predisposition genes in patients with pancreatic cancer. Gastroenterology 2015, 148, 556–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhen, D.B.; Rabe, K.G.; Gallinger, S.; Syngal, S.; Schwartz, A.G.; Goggins, M.G.; Hruban, R.H.; Cote, M.L.; McWilliams, R.R.; Roberts, N.J.; et al. BRCA1, BRCA2, PALB2, and CDKN2A mutations in familial pancreatic cancer: A PACGENE study. Genet. Med. 2015, 17, 569–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macklin, S.K.; Kasi, P.M.; Jackson, J.L.; Hines, S.L. Incidence of Pathogenic Variants in Those with a Family History of Pancreatic Cancer. Front. Oncol. 2018, 8, 330. [Google Scholar] [CrossRef] [PubMed]
- Mocci, E.; Guillen-Ponce, C.; Earl, J.; Marquez, M.; Solera, J.; Salazar-Lopez, M.T.; Calcedo-Arnaiz, C.; Vazquez-Sequeiros, E.; Montans, J.; Munoz-Beltran, M.; et al. PanGen-Fam: Spanish registry of hereditary pancreatic cancer. Eur. J. Cancer 2015, 51, 1911–1917. [Google Scholar] [CrossRef]
- Giardiello, F.M.; Brensinger, J.D.; Tersmette, A.C.; Goodman, S.N.; Petersen, G.M.; Booker, S.V.; Cruz-Correa, M.; Offerhaus, J.A. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 2000, 119, 1447–1453. [Google Scholar] [CrossRef] [Green Version]
- Roberts, N.J.; Norris, A.L.; Petersen, G.M.; Bondy, M.L.; Brand, R.; Gallinger, S.; Kurtz, R.C.; Olson, S.H.; Rustgi, A.K.; Schwartz, A.G.; et al. Whole Genome Sequencing Defines the Genetic Heterogeneity of Familial Pancreatic Cancer. Cancer Discov. 2016, 6, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Childs, E.J.; Mocci, E.; Bracci, P.; Gallinger, S.; Li, D.; Neale, R.E.; Olson, S.H.; Scelo, G.; Bamlet, W.R.; et al. Analysis of Heritability and Genetic Architecture of Pancreatic Cancer: A PanC4 Study. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1238–1245. [Google Scholar] [CrossRef] [Green Version]
- Rebours, V.; Boutron-Ruault, M.C.; Schnee, M.; Ferec, C.; Maire, F.; Hammel, P.; Ruszniewski, P.; Levy, P. Risk of pancreatic adenocarcinoma in patients with hereditary pancreatitis: A national exhaustive series. Am. J. Gastroenterol. 2008, 103, 111–119. [Google Scholar] [CrossRef]
- Zhan, W.; Shelton, C.A.; Greer, P.J.; Brand, R.E.; Whitcomb, D.C. Germline Variants and Risk for Pancreatic Cancer: A Systematic Review and Emerging Concepts. Pancreas 2018, 47, 924–936. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Silvestri, V.; Leslie, G.; Rebbeck, T.R.; Neuhausen, S.L.; Hopper, J.L.; Nielsen, H.R.; Lee, A.; Yang, X.; McGuffog, L.; et al. Cancer Risks Associated with BRCA1 and BRCA2 Pathogenic Variants. J. Clin. Oncol. 2022, 40, JCO2102112. [Google Scholar] [CrossRef] [PubMed]
- Daly, M.B.; Pal, T.; Berry, M.P.; Buys, S.S.; Dickson, P.; Domchek, S.M.; Elkhanany, A.; Friedman, S.; Goggins, M.; Hutton, M.L.; et al. Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 77–102. [Google Scholar] [CrossRef] [PubMed]
- Petersen, G.M. Familial pancreatic cancer. Semin. Oncol. 2016, 43, 548–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, A.P.; Brune, K.A.; Petersen, G.M.; Goggins, M.; Tersmette, A.C.; Offerhaus, G.J.; Griffin, C.; Cameron, J.L.; Yeo, C.J.; Kern, S.; et al. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Res. 2004, 64, 2634–2638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shindo, K.; Yu, J.; Suenaga, M.; Fesharakizadeh, S.; Cho, C.; Macgregor-Das, A.; Siddiqui, A.; Witmer, P.D.; Tamura, K.; Song, T.J.; et al. Deleterious Germline Mutations in Patients with Apparently Sporadic Pancreatic Adenocarcinoma. J. Clin. Oncol. 2017, 35, 3382–3390. [Google Scholar] [CrossRef]
- Hu, C.; LaDuca, H.; Shimelis, H.; Polley, E.C.; Lilyquist, J.; Hart, S.N.; Na, J.; Thomas, A.; Lee, K.Y.; Davis, B.T.; et al. Multigene Hereditary Cancer Panels Reveal High-Risk Pancreatic Cancer Susceptibility Genes. JCO Precis. Oncol. 2018, 2, 1–28. [Google Scholar] [CrossRef]
- Brand, R.; Borazanci, E.; Speare, V.; Dudley, B.; Karloski, E.; Peters, M.L.B.; Stobie, L.; Bahary, N.; Zeh, H.; Zureikat, A.; et al. Prospective study of germline genetic testing in incident cases of pancreatic adenocarcinoma. Cancer 2018, 124, 3520–3527. [Google Scholar] [CrossRef] [Green Version]
- Chaffee, K.G.; Oberg, A.L.; McWilliams, R.R.; Majithia, N.; Allen, B.A.; Kidd, J.; Singh, N.; Hartman, A.R.; Wenstrup, R.J.; Petersen, G.M. Prevalence of germ-line mutations in cancer genes among pancreatic cancer patients with a positive family history. Genet. Med. 2018, 20, 119–127. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Hart, S.N.; Polley, E.C.; Gnanaolivu, R.; Shimelis, H.; Lee, K.Y.; Lilyquist, J.; Na, J.; Moore, R.; Antwi, S.O.; et al. Association Between Inherited Germline Mutations in Cancer Predisposition Genes and Risk of Pancreatic Cancer. JAMA 2018, 319, 2401–2409. [Google Scholar] [CrossRef]
- Gentiluomo, M.; Canzian, F.; Nicolini, A.; Gemignani, F.; Landi, S.; Campa, D. Germline genetic variability in pancreatic cancer risk and prognosis. Semin. Cancer Biol. 2022, 79, 105–131. [Google Scholar] [CrossRef]
- Zhang, Y.D.; Hurson, A.N.; Zhang, H.; Choudhury, P.P.; Easton, D.F.; Milne, R.L.; Simard, J.; Hall, P.; Michailidou, K.; Dennis, J.; et al. Assessment of polygenic architecture and risk prediction based on common variants across fourteen cancers. Nat. Commun. 2020, 11, 3353. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Chen, H.T.; Na, R.; Jiang, D.K.; Lin, X.L.; Yang, F.; Jin, C.; Fu, D.L.; Xu, J.F. Single-nucleotide polymorphisms based genetic risk score in the prediction of pancreatic cancer risk. World J. Gastroenterol. 2020, 26, 3076–3086. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yuan, C.; Babic, A.; Bao, Y.; Clish, C.B.; Pollak, M.N.; Amundadottir, L.T.; Klein, A.P.; Stolzenberg-Solomon, R.Z.; Pandharipande, P.V.; et al. Genetic and Circulating Biomarker Data Improve Risk Prediction for Pancreatic Cancer in the General Population. Cancer Epidemiol. Biomark. Prev. 2020, 29, 999–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galeotti, A.A.; Gentiluomo, M.; Rizzato, C.; Obazee, O.; Neoptolemos, J.P.; Pasquali, C.; Nentwich, M.; Cavestro, G.M.; Pezzilli, R.; Greenhalf, W.; et al. Polygenic and multifactorial scores for pancreatic ductal adenocarcinoma risk prediction. J. Med. Genet. 2021, 58, 369–377. [Google Scholar] [CrossRef]
- Dana-Farber Cancer Institute. What Is Cascade Testing for Hereditary Cancer Syndromes? August 2018. Available online: https://blog.dana-farber.org/insight/2018/08/cascade-testing-hereditary-cancer-syndromes/ (accessed on 19 May 2021).
- Vasen, H.; Ibrahim, I.; Ponce, C.G.; Slater, E.P.; Matthai, E.; Carrato, A.; Earl, J.; Robbers, K.; van Mil, A.M.; Potjer, T.; et al. Benefit of Surveillance for Pancreatic Cancer in High-Risk Individuals: Outcome of Long-Term Prospective Follow-Up Studies From Three European Expert Centers. J. Clin. Oncol. 2016, 34, 2010–2019. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, S.; Won, N.Y.; Dotson, W.D.; Wright, S.T.; Roberts, M.C. Barriers and facilitators for cascade testing in genetic conditions: A systematic review. Eur. J. Hum. Genet. 2020, 28, 1631–1644. [Google Scholar] [CrossRef]
- Daly, M.B.; Pilarski, R.; Yurgelun, M.B.; Berry, M.P.; Buys, S.S.; Dickson, P.; Domchek, S.M.; Elkhanany, A.; Friedman, S.; Garber, J.E.; et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 1.2020. J. Natl. Compr. Cancer Netw. 2020, 18, 380–391. [Google Scholar] [CrossRef]
- Canto, M.I.; Harinck, F.; Hruban, R.H.; Offerhaus, G.J.; Poley, J.W.; Kamel, I.; Nio, Y.; Schulick, R.S.; Bassi, C.; Kluijt, I.; et al. International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut 2013, 62, 339–347. [Google Scholar] [CrossRef]
- Goggins, M.; Overbeek, K.A.; Brand, R.; Syngal, S.; Del Chiaro, M.; Bartsch, D.K.; Bassi, C.; Carrato, A.; Farrell, J.; Fishman, E.K.; et al. Management of patients with increased risk for familial pancreatic cancer: Updated recommendations from the International Cancer of the Pancreas Screening (CAPS) Consortium. Gut 2020, 69, 7–17. [Google Scholar] [CrossRef] [Green Version]
- Stoffel, E.M.; McKernin, S.E.; Brand, R.; Canto, M.; Goggins, M.; Moravek, C.; Nagarajan, A.; Petersen, G.M.; Simeone, D.M.; Yurgelun, M.; et al. Evaluating Susceptibility to Pancreatic Cancer: ASCO Provisional Clinical Opinion. J. Clin. Oncol. 2019, 37, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Stjepanovic, N.; Moreira, L.; Carneiro, F.; Balaguer, F.; Cervantes, A.; Balmana, J.; Martinelli, E. Hereditary gastrointestinal cancers: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-updagger. Ann. Oncol. 2019, 30, 1558–1571. [Google Scholar] [CrossRef] [Green Version]
- Pujol, P.; Barberis, M.; Beer, P.; Friedman, E.; Piulats, J.M.; Capoluongo, E.D.; Garcia Foncillas, J.; Ray-Coquard, I.; Penault-Llorca, F.; Foulkes, W.D.; et al. Clinical practice guidelines for BRCA1 and BRCA2 genetic testing. Eur. J. Cancer 2021, 146, 30–47. [Google Scholar] [CrossRef] [PubMed]
- Paiella, S.; Salvia, R.; De Pastena, M.; Pollini, T.; Casetti, L.; Landoni, L.; Esposito, A.; Marchegiani, G.; Malleo, G.; De Marchi, G.; et al. Screening/surveillance programs for pancreatic cancer in familial high-risk individuals: A systematic review and proportion meta-analysis of screening results. Pancreatology 2018, 18, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Canto, M.I.; Almario, J.A.; Schulick, R.D.; Yeo, C.J.; Klein, A.; Blackford, A.; Shin, E.J.; Sanyal, A.; Yenokyan, G.; Lennon, A.M.; et al. Risk of Neoplastic Progression in Individuals at High Risk for Pancreatic Cancer Undergoing Long-Term Surveillance. Gastroenterology 2018, 155, 740–751.E2. [Google Scholar] [CrossRef] [Green Version]
- Overbeek, K.A.; Levink, I.J.M.; Koopmann, B.D.M.; Harinck, F.; Konings, I.; Ausems, M.; Wagner, A.; Fockens, P.; van Eijck, C.H.; Groot Koerkamp, B.; et al. Long-term yield of pancreatic cancer surveillance in high-risk individuals. Gut 2021, 71, 1152–1160.e774. [Google Scholar] [CrossRef]
- Overbeek, K.A.; Goggins, M.G.; Dbouk, M.; Levink, I.J.M.; Koopmann, B.D.M.; Chuidian, M.; Konings, I.; Paiella, S.; Earl, J.; Fockens, P.; et al. Timeline of Development of Pancreatic Cancer and Implications for Successful Early Detection in High-Risk Individuals. Gastroenterology 2022, 162, 772–785. [Google Scholar] [CrossRef]
- Chhoda, A.; Vodusek, Z.; Wattamwar, K.; Mukherjee, E.; Gunderson, C.; Grimshaw, A.; Sharma, A.; Ahuja, N.; Kastrinos, F.; Farrell, J.J. Late-Stage Pancreatic Cancer Detected During High-Risk Individual Surveillance: A Systematic Review and Meta-Analysis. Gastroenterology 2022, 162, 786–798. [Google Scholar] [CrossRef]
- Gonda, T.A.; Everett, J.N.; Wallace, M.; Simeone, D.M.; Consortium, P. Recommendations for a More Organized and Effective Approach to the Early Detection of Pancreatic Cancer from the PRECEDE (Pancreatic Cancer Early Detection) Consortium. Gastroenterology 2021, 161, 1751–1757. [Google Scholar] [CrossRef]
- Principe, D.R. Precision Medicine for BRCA/PALB2-Mutated Pancreatic Cancer and Emerging Strategies to Improve Therapeutic Responses to PARP Inhibition. Cancers 2022, 14, 897. [Google Scholar] [CrossRef]
- Milella, M.; Luchini, C.; Lawlor, R.T.; Johns, A.L.; Casolino, R.; Yoshino, T.; Biankin, A.V. ICGC-ARGO precision medicine: Familial matters in pancreatic cancer. Lancet Oncol. 2022, 23, 25–26. [Google Scholar] [CrossRef]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V.; Kaestner, V.; Mailankody, S. Cancer Drugs Approved Based on Biomarkers and Not Tumor Type-FDA Approval of Pembrolizumab for Mismatch Repair-Deficient Solid Cancers. JAMA Oncol. 2018, 4, 157–158. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Rouleau, M.; Patel, A.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G.G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. [Google Scholar] [CrossRef] [Green Version]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, M.J. Targeting the DNA damage response in cancer. Mol. Cell. 2015, 60, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Ashworth, A. A synthetic lethal therapeutic approach: Poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J. Clin. Oncol. 2008, 26, 3785–3790. [Google Scholar] [CrossRef]
- Mota, A.; Oltra, S.S.; Moreno-Bueno, G. Insight updating of the molecular hallmarks in ovarian carcinoma. EJC Suppl. 2020, 15, 16–26. [Google Scholar] [CrossRef]
- Golan, T.; Kanji, Z.S.; Epelbaum, R.; Devaud, N.; Dagan, E.; Holter, S.; Aderka, D.; Paluch-Shimon, S.; Kaufman, B.; Gershoni-Baruch, R.; et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br. J. Cancer 2014, 111, 1132–1138. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Barenboim, A.; Lahat, G.; Nachmany, I.; Goykhman, Y.; Shacham-Shmueli, E.; Halpern, N.; Brazowski, E.; Geva, R.; Wolf, I.; et al. Increased rate of complete pathologic response after neoadjuvant FOLFIRINOX for BRCA mutation carriers with borderline resectable pancreatic cancer. Ann. Surg. Oncol. 2020, 27, 3963–3970. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Agarwal, P.; Mamtani, R.; Symecko, H.; Spielman, K.; O’Hara, M.; O’Dwyer, P.J.; Schneider, C.; Teitelbaum, U.; Nathanson, K.L.; et al. Retrospective survival analysis of patients with resected pancreatic ductal adenocarcinoma and a germline BRCA or PALB2 mutation. JCO Precis. Oncol. 2019, 3, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Casolino, R.; Braconi, C.; Malleo, G.; Paiella, S.; Bassi, C.; Milella, M.; Dreyer, S.B.; Froeling, F.E.M.; Chang, D.K.; Biankin, A.V.; et al. Reshaping preoperative treatment of pancreatic cancer in the era of precision medicine. Ann. Oncol. 2020, 32, 183–196. [Google Scholar] [CrossRef]
- Reiss, K.A.; Mick, R.; O’Hara, M.H.; Teitelbaum, U.; Karasic, T.B.; Schneider, C.; Cowden, S.; Southwell, T.; Romeo, J.; Izgur, N.; et al. Phase II Study of Maintenance Rucaparib in Patients with Platinum-Sensitive Advanced Pancreatic Cancer and a Pathogenic Germline or Somatic Variant in BRCA1, BRCA2, or PALB2. J. Clin. Oncol. 2021, 39, 2497–2505. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmana, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Shroff, R.T.; Hendifar, A.; McWilliams, R.R.; Geva, R.; Epelbaum, R.; Rolfe, L.; Goble, S.; Lin, K.K.; Biankin, A.V.; Giordano, H.; et al. Rucaparib Monotherapy in Patients with Pancreatic Cancer and a Known Deleterious BRCA Mutation. JCO Precis. Oncol. 2018, 2, 1–15. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Lee, J.W.; Zalupski, M.; Capanu, M.; Park, J.; Golan, T.; Tahover, E.; Lowery, M.A.; Chou, J.F.; Sahai, V.; et al. Randomized, Multicenter, Phase II Trial of Gemcitabine and Cisplatin with or without Veliparib in Patients with Pancreas Adenocarcinoma and a Germline BRCA/PALB2 Mutation. J. Clin. Oncol. 2020, 38, 1378–1388. [Google Scholar] [CrossRef]
- Luchini, C.; Brosens, L.A.A.; Wood, L.D.; Chatterjee, D.; Shin, J.I.; Sciammarella, C.; Fiadone, G.; Malleo, G.; Salvia, R.; Kryklyva, V.; et al. Comprehensive characterisation of pancreatic ductal adenocarcinoma with microsatellite instability: Histology, molecular pathology and clinical implications. Gut 2021, 70, 148–156. [Google Scholar] [CrossRef]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef]
- Germano, G.; Amirouchene-Angelozzi, N.; Rospo, G.; Bardelli, A. The Clinical Impact of the Genomic Landscape of Mismatch Repair-Deficient Cancers. Cancer Discov. 2018, 8, 1518–1528. [Google Scholar] [CrossRef] [Green Version]
- Grant, R.C.; Denroche, R.; Jang, G.H.; Nowak, K.M.; Zhang, A.; Borgida, A.; Holter, S.; Topham, J.T.; Wilson, J.; Dodd, A.; et al. Clinical and genomic characterisation of mismatch repair deficient pancreatic adenocarcinoma. Gut 2021, 70, 1894–1903. [Google Scholar] [CrossRef] [PubMed]
- Peyraud, F.; Italiano, A. Combined PARP Inhibition and Immune Checkpoint Therapy in Solid Tumors. Cancers 2020, 12, 1502. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, P.; Bandlamudi, C.; Cheng, M.L.; Srinivasan, P.; Chavan, S.S.; Friedman, N.D.; Rosen, E.Y.; Richards, A.L.; Bouvier, N.; Selcuklu, S.D.; et al. Tumour lineage shapes BRCA-mediated phenotypes. Nature 2019, 571, 576–579. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, Y.; Shan, W.; Hu, Z.; Yuan, J.; Pi, J.; Wang, Y.; Fan, L.; Tang, Z.; Li, C.; et al. Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Sci. Transl. Med. 2017, 9, eaal1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, A.L.; Fehling, S.C.; Garcia, P.L.; Gamblin, T.L.; Council, L.N.; van Waardenburg, R.; Yang, E.S.; Bradner, J.E.; Yoon, K.J. The BET inhibitor JQ1 attenuates double-strand break repair and sensitizes models of pancreatic ductal adenocarcinoma to PARP inhibitors. EBioMedicine 2019, 44, 419–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogiwara, H.; Ui, A.; Shiotani, B.; Zou, L.; Yasui, A.; Kohno, T. Curcumin suppresses multiple DNA damage response pathways and has potency as a sensitizer to PARP inhibitor. Carcinogenesis 2013, 34, 2486–2497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muvarak, N.E.; Chowdhury, K.; Xia, L.; Robert, C.; Choi, E.Y.; Cai, Y.; Bellani, M.; Zou, Y.; Singh, Z.N.; Duong, V.H.; et al. Enhancing the Cytotoxic Effects of PARP Inhibitors with DNA Demethylating Agents—A Potential Therapy for Cancer. Cancer Cell 2016, 30, 637–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, R.D.; Gajjar, M.K.; Jensen, K.E.; Hamerlik, P. Enhanced efficacy of combined HDAC and PARP targeting in glioblastoma. Mol. Oncol. 2016, 10, 751–763. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Peng, Y.; Wei, L.; Zhang, W.; Yang, L.; Lan, L.; Kapoor, P.; Ju, Z.; Mo, Q.; Shih, I.M.; et al. ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. Cancer Discov. 2015, 5, 752–767. [Google Scholar] [CrossRef] [Green Version]
- Brenner, J.C.; Ateeq, B.; Li, Y.; Yocum, A.K.; Cao, Q.; Asangani, I.A.; Patel, S.; Wang, X.; Liang, H.; Yu, J.; et al. Mechanistic rationale for inhibition of poly(ADP-ribose) polymerase in ETS gene fusion-positive prostate cancer. Cancer Cell 2011, 19, 664–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, P.J.; Yachida, S.; Mudie, L.J.; Stephens, P.J.; Pleasance, E.D.; Stebbings, L.A.; Morsberger, L.A.; Latimer, C.; McLaren, S.; Lin, M.L.; et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 2010, 467, 1109–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yachida, S.; Iacobuzio-Donahue, C.A. Evolution and dynamics of pancreatic cancer progression. Oncogene 2013, 32, 5253–5260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A.; et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.K.; Harrell, M.I.; Oza, A.M.; Oaknin, A.; Ray-Coquard, I.; Tinker, A.V.; Helman, E.; Radke, M.R.; Say, C.; Vo, L.T.; et al. BRCA Reversion Mutations in Circulating Tumor DNA Predict Primary and Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2019, 9, 210–219. [Google Scholar] [CrossRef] [Green Version]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008, 451, 1111–1115. [Google Scholar] [CrossRef]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef] [Green Version]
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of Single-Strand Annealing and its Role in Genome Maintenance. Trends Genet. 2016, 32, 566–575. [Google Scholar] [CrossRef] [Green Version]
- Swisher, E.M.; Kwan, T.T.; Oza, A.M.; Tinker, A.V.; Ray-Coquard, I.; Oaknin, A.; Coleman, R.L.; Aghajanian, C.; Konecny, G.E.; O’Malley, D.M.; et al. Molecular and clinical determinants of response and resistance to rucaparib for recurrent ovarian cancer treatment in ARIEL2 (Parts 1 and 2). Nat. Commun. 2021, 12, 2487. [Google Scholar] [CrossRef]
- Gupta, R.; Somyajit, K.; Narita, T.; Maskey, E.; Stanlie, A.; Kremer, M.; Typas, D.; Lammers, M.; Mailand, N.; Nussenzweig, A.; et al. DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell 2018, 173, 972–988. [Google Scholar] [CrossRef] [Green Version]
- Mirman, Z.; Lottersberger, F.; Takai, H.; Kibe, T.; Gong, Y.; Takai, K.; Bianchi, A.; Zimmermann, M.; Durocher, D.; de Lange, T. 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polalpha-dependent fill-in. Nature 2018, 560, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Dias, M.P.; Moser, S.C.; Ganesan, S.; Jonkers, J. Understanding and overcoming resistance to PARP inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 773–791. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; George, E.; Ragland, R.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.; Herlyn, M.; Brown, E.; Simpkins, F. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin. Cancer Res. 2017, 23, 3097–3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Castroviejo-Bermejo, M.; Cruz, C.; Llop-Guevara, A.; Gutierrez-Enriquez, S.; Ducy, M.; Ibrahim, Y.H.; Gris-Oliver, A.; Pellegrino, B.; Bruna, A.; Guzman, M.; et al. A RAD51 assay feasible in routine tumor samples calls PARP inhibitor response beyond BRCA mutation. EMBO Mol. Med. 2018, 10, e9172. [Google Scholar] [CrossRef] [PubMed]
- Deer, E.L.; Gonzalez-Hernandez, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010, 39, 425–435. [Google Scholar] [CrossRef] [Green Version]
- van der Heijden, M.S.; Brody, J.R.; Gallmeier, E.; Cunningham, S.C.; Dezentje, D.A.; Shen, D.; Hruban, R.H.; Kern, S.E. Functional defects in the fanconi anemia pathway in pancreatic cancer cells. Am. J. Pathol. 2004, 165, 651–657. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [Green Version]
- Guerra, C.; Barbacid, M. Genetically engineered mouse models of pancreatic adenocarcinoma. Mol. Oncol. 2013, 7, 232–247. [Google Scholar] [CrossRef]
- Skoulidis, F.; Cassidy, L.D.; Pisupati, V.; Jonasson, J.G.; Bjarnason, H.; Eyfjord, J.E.; Karreth, F.A.; Lim, M.; Barber, L.M.; Clatworthy, S.A.; et al. Germline Brca2 heterozygosity promotes Kras(G12D) -driven carcinogenesis in a murine model of familial pancreatic cancer. Cancer Cell 2010, 18, 499–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drosos, Y.; Escobar, D.; Chiang, M.Y.; Roys, K.; Valentine, V.; Valentine, M.B.; Rehg, J.E.; Sahai, V.; Begley, L.A.; Ye, J.; et al. ATM-deficiency increases genomic instability and metastatic potential in a mouse model of pancreatic cancer. Sci. Rep. 2017, 7, 11144. [Google Scholar] [CrossRef]
- Hwang, C.I.; Boj, S.F.; Clevers, H.; Tuveson, D.A. Preclinical models of pancreatic ductal adenocarcinoma. J. Pathol. 2016, 238, 197–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).